
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 10 January 2017
Sec. Infectious Agents and Disease
Volume 7 - 2016 | https://doi.org/10.3389/fmicb.2016.02156
 Michelle L. Mendonca1,2
Michelle L. Mendonca1,2 Jake C. Szamosi2
Jake C. Szamosi2 Anne-Marie Lacroix1,2
Anne-Marie Lacroix1,2 Michelle E. Fontes1,2
Michelle E. Fontes1,2 Dawn M. Bowdish3,4
Dawn M. Bowdish3,4 Michael G. Surette1,2,4,5*
Michael G. Surette1,2,4,5*The Streptococcus Invasion Locus (Sil) was first described in Streptococcus pyogenes and Streptococcus pneumoniae, where it has been implicated in virulence. The two-component peptide signaling system consists of the SilA response regulator and SilB histidine kinase along with the SilCR signaling peptide and SilD/E export/processing proteins. The presence of an associated bacteriocin region suggests this system may play a role in competitive interactions with other microbes. Comparative analysis of 42 Streptococcus Anginosus/Milleri Group (SAG) genomes reveals this to be a hot spot for genomic variability. A cluster of bacteriocin/immunity genes is found adjacent to the sil system in most SAG isolates (typically 6–10 per strain). In addition, there were two distinct SilCR peptides identified in this group, denoted here as SilCRSAG-A and SilCRSAG-B, with corresponding alleles in silB. Our analysis of the 42 sil loci showed that SilCRSAG-A is only found in Streptococcus intermedius while all three species can carry SilCRSAG-B. In S. intermedius B196, a putative SilA operator is located upstream of bacteriocin gene clusters, implicating the sil system in regulation of microbe–microbe interactions at mucosal surfaces where the group resides. We demonstrate that S. intermedius B196 responds to its cognate SilCRSAG-A, and, less effectively, to SilCRSAG-B released by other Anginosus group members, to produce putative bacteriocins and inhibit the growth of a sensitive strain of S. constellatus.
The Streptococcus Anginosus/Milleri Group (SAG) is a group of three distinct yet closely related species: S. anginosus, S. constellatus, and S. intermedius. The species S. anginosus is divided into S. anginosus subsp. anginosus and S. anginosus subsp. whileyi while the species S. constellatus is divided into S. constellatus subsp. constellatus, S. constellatus subsp. pharyngis and S. constellatus subsp. viborgensis (Jensen et al., 2013). In humans, the SAG lead a dual lifestyle as both commensals and pathogens. These strains can be found asymptomatically colonizing the oral cavity as well as the gastrointestinal and urogenital tracts (Poole and Wilson, 1979; Whiley et al., 1992; Jacobs et al., 1995). However, they are also important pyogenic pathogens involved in empyema and soft tissue abscesses (Ruoff, 1988; Whiley et al., 1992; Coman et al., 1995; Laupland et al., 2006; Ripley et al., 2006; Siegman-Igra et al., 2012; Asam and Spellerberg, 2014) as well as infections of the lower airways (Shinzato and Saito, 1995; Parkins et al., 2008; Sibley et al., 2008). The SAG can be difficult to culture and identify (Ruoff, 1988; Sibley et al., 2010) and are underappreciated pathogens. They have been associated with more invasive pyogenic infections than Group A and Group B Streptococcus combined (Laupland et al., 2006; Siegman-Igra et al., 2012).
Host colonization involves competition with resident microorganisms. One mechanism by which streptococci inhibit closely related bacteria is through short peptides called bacteriocins (Dawid et al., 2007). Variable bacteriocin and putative bacteriocin genes are found adjacent to the sil locus in GAS, and these are predicted to be under the regulatory control of SilA (Belotserkovsky et al., 2009). The Streptococcus Invasion locus (sil) system was first identified in Group A Streptococci (GAS), where a transposon insertion in the sil system attenuated virulence in a murine model (Hidalgo-Grass et al., 2002). The core signaling system has been characterized and contains the cell–cell signaling peptide SilCR, a peptide processing/export system (SilD/E), and two component sensing system (SilA/B). Upon sensing pheromone peptide SilCR, the histidine kinase SilB phosphorylates the response regulator SilA, upregulating genes involved in SilCR production. These include the SilCR peptide itself and ABC transporters, SilD and SilE. SilC is encoded on the antisense strand of silCR and its expression represses SilA-inducible genes in GAS (Hidalgo-Grass et al., 2002; Eran et al., 2007). SilA/B transcriptional regulation is uncharacterized, but induction of the sil system is dependent on their presence and expression (Eran et al., 2007). The system also includes a putative CAAX protease, thought to be involved in immunity against or maturation of bacteriocins.
The sil system has been implicated in virulence in GAS (Hidalgo-Grass et al., 2002; Salim et al., 2008). Disruption of silC leads to attenuation in a murine model (Hidalgo-Grass et al., 2002). As SilC represses SilCR induction, the lack of the pheromone peptide appears to favor pathogenesis. However, interspecies communication occurring between GAS and Group G streptococci (GGS) via SilCR peptide positively regulates SilCR and bacteriocins (Belotserkovsky et al., 2009). Thus, regulation of this system in streptococci is complex and can impact pathogenesis in multiple ways.
The sil system has not been characterized in SAG. Here, we describe the sil system in 42 SAG genomes, outlining species-specific variation and identification of novel putative bacteriocins. We note this locus as a hotspot for genetic variability with most strains featuring a large cluster of putative bacteriocin/immunity genes. We demonstrate that the inhibitory activity of SilCR-dependent putative bacteriocins can give S. intermedius a competitive advantage.
Forty-four assembled SAG genomes were used for our analysis (Supplementary Table S1). Seven of these were previously published complete, closed genomes (C1050, C1051, C818, C232, C270, C238, B196; Olson et al., 2013), and 26 were unpublished draft genomes (see below). The strains of these 26 new genomes have been described in Kaiser et al. (2014). The SAG sil regions have been deposited in GenBank (accession numbers KY315440-KY315481) (Supplementary Table S1). Eleven draft genomes were also downloaded from NCBI (Benson et al., 2015) (ADME01000005.1, AP013072.1, AJKN01000015.1, AICP01000048.1, AFXO01000004.1, NC 018073.1, AFUP01000001.1, AICQ01000033.1, AFXN01000007.1, AREF01000001.1, AECT01000012.1).
The in-house draft genomes were all sequenced using NexteraXT libraries and Illumina MiSeq using either 150 or 250 bp paired-end reads. The sequences were assembled using our in-house genome assembly pipeline [Surette Lab Assembly Pipeline (SLAP)]. SLAP preprocesses the reads by trimming adapters with Cutadapt (Martin, 2011) and performing quality trimming and error correction with sga preprocess and sga correct (Simpson and Durbin, 2012). It then estimates an optimal kmer size using an in-house adaptation of the program Kmergenie (Chikhi and Medvedev, 2014), and calls four separate assemblers [MaSuRCA (Zimin et al., 2013), IDBA (Peng et al., 2011), SPAdes (Bankevich et al., 2012), and Velvet (Zerbino and Birney, 2008)] to produce four candidate assemblies. The assemblies are then scaffolded using SSPACE (Boetzer et al., 2011) and quality metrics are provided to the user via FastQC (Andrews, 2010) for read quality and QUAST (Gurevich et al., 2013) for assembly quality. For our purposes, the best assembly was chosen for each strain based on N50 value and total assembly length. All in-house code is available from the authors upon request.
We assigned our samples to strain clusters following Jensen et al. (2013). We found each of the seven housekeeping genes used in the paper in all of our strains. The SAG accession numbers for map (KY274883 – KY274924), pfl (KY274925 – KY274966), ppaC (KY274967 – KY275008), pyk (KY275009 – KY275050) rpoB (KY275051 – KY275092), sodA (KY275093 – KY275133), and tuf (KY275134 – KY275175) are as indicated. All genes were present in all samples with the exception of sodA, which was absent from C1051. These genes were aligned with the trimmed sequences used by Jensen et al. (2013), trimmed to the same length, and a phylogenetic tree was constructed using MEGA 7.0.20 using default parameters with the following exceptions: we used a minimum-evolution model with 500 bootstrap replicates and pairwise deletion for missing data.
The putative sil system was identified in the published genome of S. intermedius B196 (Accession number: NC_022246.1) based on its structural similarity to the GAS and GGS sil locus. We used megablast (Altschul et al., 1997) to find each gene from this putative sil locus in the 44 additional SAG genomes. We found the genes together on a single contig in 42 genomes (aside from S. intermedius B196) and split across multiple contigs in two genomes (M1 and C424, which we excluded from our analysis). A list of the 42 strains used for our analysis is provided in Supplementary Table S1. The 42 putative sil regions were annotated from the BLAST hits using in-house Python and Biopython scripts (van Rossum and Drake, 2001; Cock et al., 2009) with manual adjustments, and the annotations were visualized and compared using Geneious (Kearse et al., 2012).
A phylogenetic tree of the sil locus was generated using MrBayes (Huelsenbeck and Ronquist, 2001; Altekar et al., 2004). Each gene (silA through silD) was aligned individually using MUSCLE (Edgar, 2004) and the alignments were concatenated (with gaps in place of the nucleotides for missing genes), providing a single alignment of all genes. The genes were each assigned their own partition in the MrBayes model, with a 4-by-4 General Time Reversible evolutionary model (Tavare, 1986) and invariant/gamma rates distribution (Yang, 1994) for each partition. Since many sequences were missing individual genes, the presence or absence of each gene was encoded as binary “standard” data in a separate partition (Lewis, 2001). The trees were run until the average standard deviation of the split was less than 0.01 and the consensus tree was visualized using the Interactive Tree of Life (iTOL) (Letunic and Bork, 2016).
The putative bacteriocin region adjacent to the sil region was annotated using the online version of antiSMASH (Blin et al., 2013, 2014) accessed November of 2013. Only the results of the antiSMASH-internal GLIMMER (Delcher et al., 1999) annotation were used. In order to determine how variable the accessory regions are, the sequences from the GLIMMER hits were translated and preliminary clustering was undertaken using OrthoMCL (Fischer et al., 2011). This produced 16 putative orthologous groups, here referred to as ORF1 through ORF16. To identify any pseudogenes or related ORFs not discovered by GLIMMER, we manually divided each cluster into smaller groups based on the MUSCLE alignment of the cluster (if multiple groups were apparent) and generated a consensus sequence for each group. We searched naively for each of these consensus sequences in all of the bacteriocin regions (tolerant to 20% mismatch at the amino acid level).
We generated a tree for each of the 16 ORF clusters produced by OrthoMCL using FastTree (Price et al., 2009). The location of each putative accessory gene in each strain was marked and patterns of synteny were manually examined.
We searched for each ORF in the non-redundant protein sequences (nr) database with Blastx (Altschul et al., 1997) to identify putative functions. In addition, each ORF was searched against the BACTIBASE database (Hammami et al., 2007, 2010) of bacteriocins to identify hits with existing bacteriocins.
We cultured S. intermedius B196 and S. constellatus M505 on Todd Hewitt agar with 0.5% yeast extract (THY) and incubated at 37°C in a 5% CO2 incubator for 3 days. We inoculated THY broth with colonies and incubated either at 5% CO2 or anaerobically (5% CO2, 5% H2, 90% N2) at 37°C overnight. We used broth cultures to conduct further experiments. THY supplemented with 75 μg/ml spectinomycin (THY-spec) was used to grow our knockout strain (see below). We used Escherichia coli Top10 chemically competent cells (Life Technologies) during cloning. E. coli carrying desired knockout constructs grew on Luria-Bertani agar with 100 μg/mL spectinomycin (LB-spec).
Binding of response regulator SilA to direct repeats in GAS and GGS affects expression of sil locus components as well as putative bacteriocins (Hidalgo-Grass et al., 2002). The SilA binding site in GAS (ACCATTCATG-11bp-ACCTTTTAAG) (Belotserkovsky et al., 2009) was used as a query to identify putative sites in S. intermedius B196 using the motif search in Geneious (Kearse et al., 2012). The ClustalW 2.1 alignment of these sites (Goujon et al., 2010) was used to generate an S. intermedius B196 consensus sequence, visualized using WebLogo (Crooks et al., 2004).
We constructed a deletion mutant of silCR in S. intermedius B196. First, we cloned the spectinomycin resistance marker from pDL278 (Dunny et al., 1991) along with its promoter into pUC19 with primers specF and specR as shown in Table 1. We then cloned the upstream and downstream regions of silCR in S. intermedius on either side of a spectinomycin resistance marker in pUC19 using primers SilCRupF, SilCRupR, SilCRdownF, and SilCRdownR (Table 1). We amplified the cassette (silCR upstream: specR: silCR downstream) using PCR with primers SilCRupF and SilCRdownR and purified it. Our laboratory has found that S. intermedius B196 is naturally competent and can be transformed using the competence stimulating peptide, ComC (DSRIRMGFDFSKLFGK) (data not shown). An overnight THY broth culture was diluted 1000 fold in 500 μL THY and incubated for 2 h at 37°C in 5% CO2 before adding competence peptide (10 ng) and purified cassette DNA (500 ng). The reaction was incubated in normal growth conditions for an hour before plating the transformation reaction on THY- spec. We incubated the plates for 2 days anaerobically at 37°C and screened colonies using PCR and sequencing to verify the deletion.

TABLE 1. Primers used for silCR knockout construction and Real time PCR.
To assess bacteriocin activity, we adapted top-agar overlay experiments (Kormin et al., 2001; Maricic and Dawid, 2014). S. intermedius B196 (the bacteriocin producer) and mutant strain were grown in an overnight anaerobic broth culture as previously described. A volume of 4 μL of the overnight culture was spotted on THY agar and incubated in the anaerobe chamber at 37°C for 2 days. In some cases, synthetic SilCR peptide was spotted along with culture. SilCR peptide was synthesized by RS Synthesis, LLC (Louisville). The amount of peptide added was dependent on the experiment. S. constellatus M505, which lacks the bacteriocin cluster and caax gene, was used as a bacteriocin-sensitive strain. This was grown anaerobically in THY for 24 h. Top agar (THY) was prepared using 1.5% agar as this higher percentage improved the visualization of the zone of clearing. A 100 μL of overnight broth culture of S. constellatus M505 was added to 5 mL molten Top agar and inverted three times before pouring onto the agar plates with S. intermedius spots. Plates were incubated for 1–2 days at 5% CO2 at 37°C.
To assess SilCR dependent expression of putative bacteriocins, we designed primers for specific genes in the sil locus and reference genes (molecular chaperone dnaK and recombinase recA) (Table 1). Biological replicates of S. intermedius B196 and mutant were grown and spotted on THY agar as described, with and without synthetic SilCR peptide. After 16 h of growth in the anaerobic chamber at 37°C, bacterial spots were resuspended in RNAlater Solution (Ambion). Bacterial RNA was purified using enzymatic lysis, TRIzol (Invitrogen) and the RNeasy mini kit (Qiagen) as described in Fei et al., 2016. To eliminate DNA contamination, samples were treated with the RNase free DNase set (Qiagen) and subsequently purified using the RNeasy mini columns (Qiagen). RNA was normalized using readings from the Nanodrop prior to cDNA preparation using the SuperScript® III first-strand cDNA synthesis system for RT-PCR (Invitrogen) as per manufacturer’s instructions. SsoFastTM Evagreen® Supermix (Bio-Rad) was used for real time PCR as per manufacturer’s instructions. Real-time PCR was conducted on the CFX96 TouchTM Real-time PCR detection system (Bio-Rad). Thermal cycling conditions included an initial 95°C for 30 s and 39 cycles of 95°C for 5 s and 55°C for 5 s. The gene expression level of sample groups was normalized to wild type S. intermedius B196 without SilCR peptide and calculated using the ΔΔCT method in CFX manager (Bio-Rad).
We have previously sequenced and annotated the genome for the clinically isolated strain S. intermedius B196 (Olson et al., 2013). We identified a region with structural similarity to the sil region previously identified in GAS and GGS. This region contains five of the six genes included in the GAS/GGS sil locus (silA, silB, silCR, silD, and silE), with the same relative positioning and orientation (Figure 1A). The locus is bounded by a putative D-Ala-D-Ala carboxypeptidase (SIR_RS15545) at one end and sodA at the other. Adjacent to the carboxipeptidase is a conserved hypothetical protein (SIR_RS15550) that has a thioredoxin-like domain. This is followed by 13 small ORFs containing putative bacteriocins, followed by a gene with homology to CAAX proteases. Downstream of the CAAX protease are the sil genes including silA, silB, silCR, silD, and silE. We did not find evidence for a counterpart to silC in S. intermedius B196 based on nucleotide similarity to GAS silC. While there is an open reading frame (ORF) on the antisense strand of SilCR, there is no evidence that it is transcribed or plays any role in sil regulation. More research is required to conclude if this is silC.
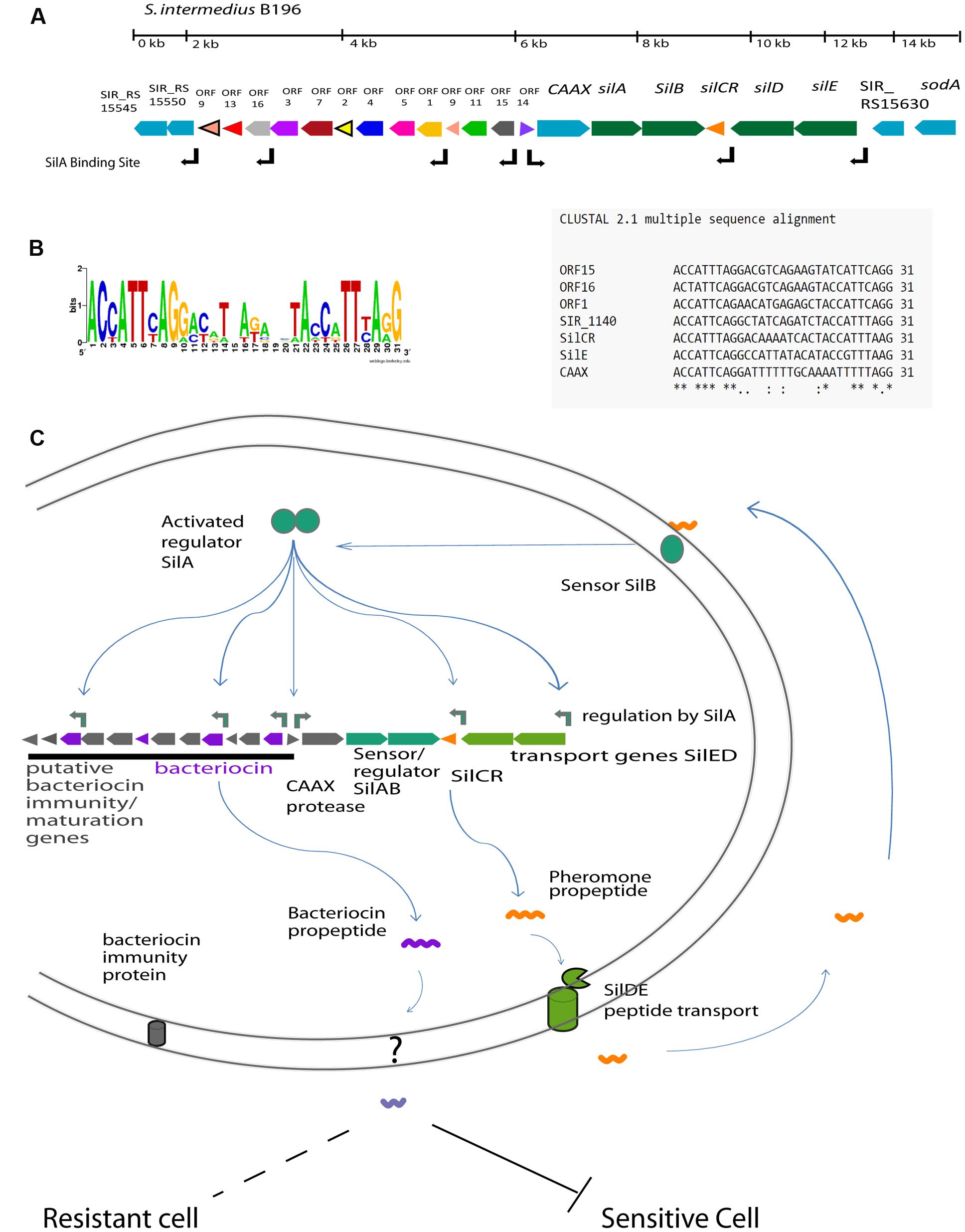
FIGURE 1. The predicted Sil system in Streptococcus intermedius B196. (A) The organization of the sil locus showing the signal peptide gene (SilCR) and signaling transduction genes (silAB and silED) in green. The associated locus genes including putative CAAX protease is shown in blue and bacteriocin associated gene clusters are indicated in color. The positions of predicted SilA binding sites are as indicated. (B) The predicted SilA binding site and consensus sequence. (C) Schematic illustration of the sil regulatory network in the Streptococcus Anginosus Group based on the sil system in Group A Streptococcus (Eran et al., 2007; Belotserkovsky et al., 2009).
Comparison between the S. intermedius B196 and GAS sil systems revealed sequence divergence despite the overall similar organization of the sil loci. Although, the putative sil region in S. intermedius B196 shares its structure with that found in Streptococcus pyogenes AF493605.1 (Hidalgo-Grass et al., 2002), these genes have low rates of sequence identity. S. intermedius B196 SilA is only 49.4% identical (85.7% similar) to GAS SilA in amino acid sequence (60.0% nucleotide identity). B196 SilB is 34.6% identical and 73.9% similar to GAS SilB (53.7% nucleotide identity). B196 SilCR is 28.0% identical and 68.0% similar to GAS SilCR (47.8% nucleotide identity). B196 SilD is 56.2% identical and 90.3% similar to GAS SilD (63.6% nucleotide identity). Finally, B196 SilE is 74.5% identical and 88.6% similar to GAS SilE (68.5% nucleotide identity).
In other streptococci, the sil system and putative bacteriocins are transcriptionally regulated by response regulator SilA and the SilCR pheromone peptide (Hidalgo-Grass et al., 2002). The SilA binding site in GAS (ACCATTCATG-11bp-ACCTTTTAAG) was used to find putative SilA binding sites in the S. intermedius B196 sil locus, as highlighted in Figure 1A. Direct repeats were found upstream of silCR, silE, caax, SIR_RS15550 and in the accessory region upstream of ORF1, ORF15, and ORF16. The predicted operator was conserved in all of these genes except CAAX protease, which had inconsistencies in the second repeat (Figure 1B).
A schematic for the hypothetical regulation of the sil system in SAG is shown in Figure 1C. The predicted regulatory network functions as follows. SilCR is produced by SAG species and exported. Sensing of extracellular SilCR peptide induces SilB to autophosphorylate and phosphorylate response regulator SilA. SilA, in turn, induces expression of the seven genes indicated in Figure 1B. Induction of these genes is predicted to amplify the response via SilCR dependent autoregulation and induce production of bacteriocins which can inhibit closely related bacteria.
We investigated the role of the sil system in inter-species competition using S. intermedius B196 as a model. A putative SilA binding site was found upstream of putative bacteriocins ORF1, ORF15, and ORF16 (Figure 1). To investigate whether SilCR regulates bacteriocin expression in S. intermedius B196, we constructed a silCR deletion mutant. We assayed bacteriocin activity by spotting S. intermedius B196 and its mutant on THY agar and after growth, applying a top agar overlay of a sensitive strain, S. constellatus M505. Strain M505 was chosen because it lacks the sil accessory region (see below) and we therefore predicted it would lack immunity genes and be sensitive to SilCR-dependent bacteriocins produced by B196. When M505 was overlaid in top agar over B196, a clear zone of growth inhibition was observed (Figure 2A). This activity is lost in the B196 ΔsilCR mutant (S. intermedius B196 ΔsilCR; Figure 2A). Exogenous addition of the synthetic SilCR peptide from B196 (SilCRSAG-A) restored the wild type phenotype, demonstrating that SilCR induces production of an inhibitor of strain M505.
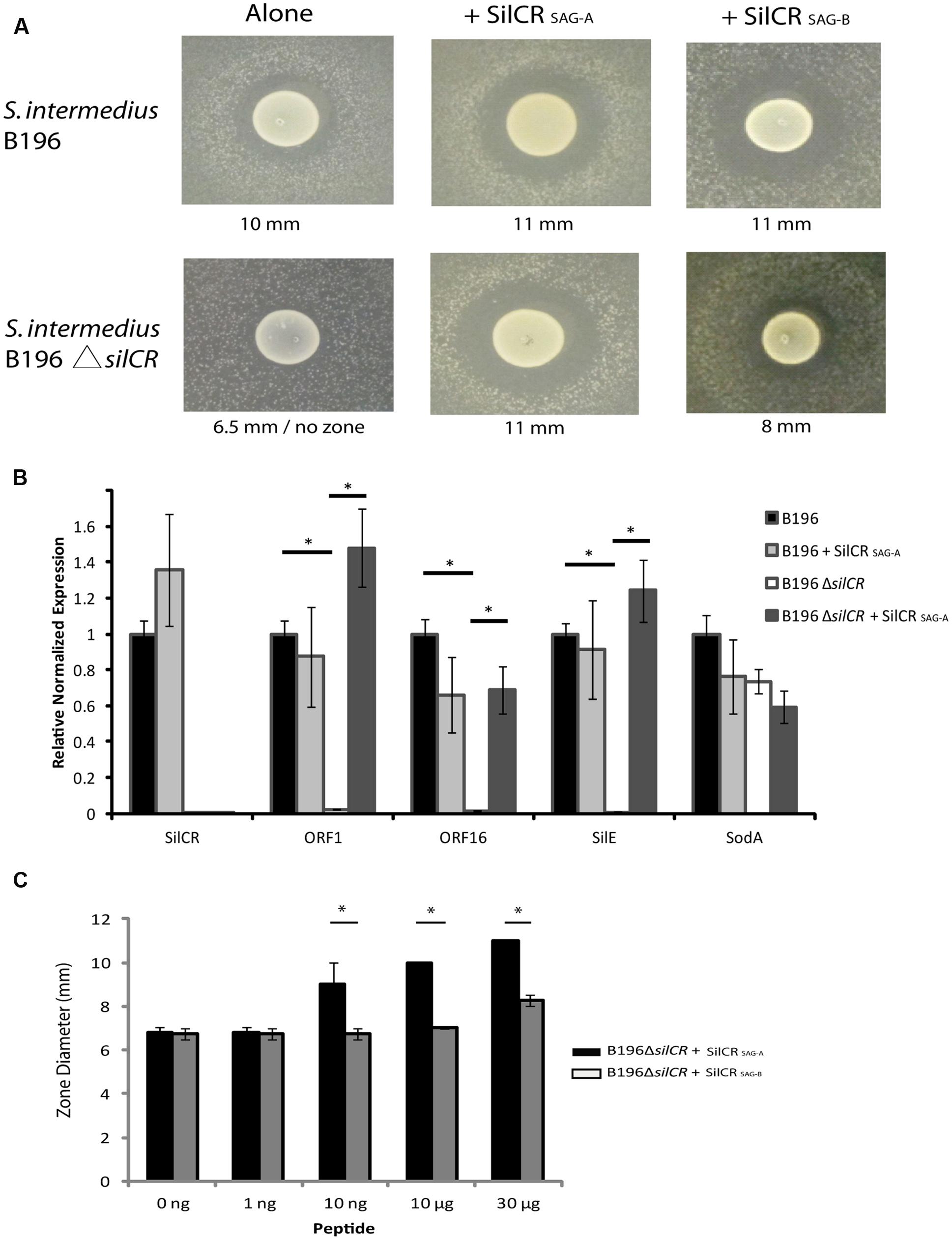
FIGURE 2. Bacteriocin-mediated competition in S. intermedius is controlled by the SilCR peptide. The gene for the pheromone peptide SilCR was deleted in S. intermedius B196 (S. intermedius B196 ΔsilCR). (A) Competition in the wild type and mutant was ascertained using an overlay of sensitive strain S. constellatus M505. The two identified SAG SilCR peptides were exogenously added to wild type and mutant and the diameter of the inhibition zone measured as shown. (B) Relative reverse transcription PCR on B196 and B196ΔsilCR for experiment shown in (A) without the M505 overlay (∗p < 0.005). (C) A concentration dependent induction of bacteriocin activity by SilCR peptides A and B in S. intermedius B196. Note the B196 colony diameter is ∼6.5 mm. SilCRSAG-A induced zones are significantly larger than those with SilCRSAG-B (∗p < 0.005).
In order to identify whether deletion of silCR affected gene expression of the putative bacteriocins ORF1 and ORF16, we conducted relative real-time PCR on the wild type and mutant strains (Figure 2B). We used spotted B196 and B196 ΔsilCR with and without exogenously added SilCRSAG-A for our analysis. Deletion of silCR downregulated expression of gene silE and putative bacteriocins ORF1 and ORF16 in the samples tested. Addition of SilCR SAG-A to the knockout induced expression of ORF1, ORF16 and silE by 61-, 43-, and 138-fold respectively; demonstrating that SilCR regulates expression of putative bacteriocins ORF1 and ORF16 and these are putatively involved in inhibition of S. constellatus M505.
We investigated the distribution of the sil system in SAG. Forty four SAG strains were used in our analysis: a combination of in-house sequenced genomes and those available online. Only genomes containing the sil genes on a single contig were analyzed (42 of the 44 genomes; as listed in Supplementary Table S1). We used the multilocus sequence analysis (MLSA) described by Jensen et al. (2013) to assign our strains into seven designated clusters for SAG. Our MLSA tree reproduced the clusters found by Jensen et al. (2013) with our new strains neatly nested within six of the seven clusters. Strain cluster assignments are included in Figure 3 and the MLSA phylogeny is shown in Supplementary Figure S3.
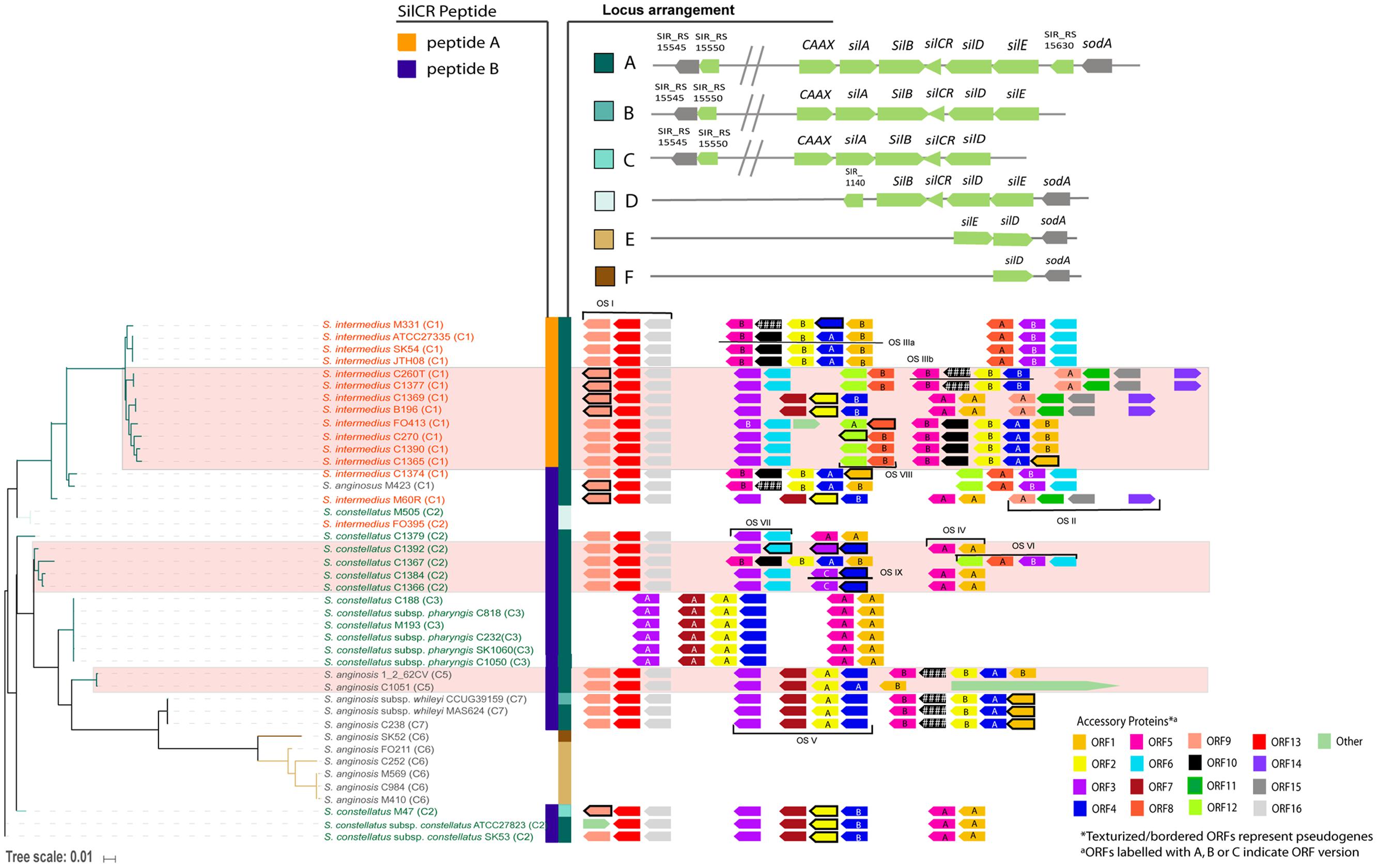
FIGURE 3. sil locus heterogeneity in the Streptococcus Anginosus Group (SAG). A tree was constructed based on sil genes in 42 strains. Six locus arrangements were found (shown in locus arrangement legend with the locus annotation). Hashes (//) in the annotation indicate a putative bacteriocin accessory region. Two SilCR peptides were identified, shown in the peptide legend. SAG species text is color coded (S. intermedius in orange, S. anginosus in gray, and S. constellatus in green). The accessory region ORFs are shown with the tree. Some ORFs have several versions, indicated by the letter inside the ORF. Genes that have acquired mutations leading to pseudogenes in some strains and are indicated in the figure.
Initial investigation of the sil sequences divided SAG strains into two groups. In 24 of the 42 genomes, both silB and silCR appeared truncated; silB at the 5′ end and silCR at the 3′ end. Further examination, however, found that this 5′ end of silB was well-conserved in 22 of the 24 strains. In addition, results from a BLAST search of this region showed high identity with histidine kinases, which is consistent with the putative function of SilB. Indeed, the gene had been annotated as such in at least one of the published genomes in our data set (protein accession YP 008494978 from genome C232), implying that we had found a variation of silB. These 22 strains also had a well-conserved region following their truncated silCR. A stop codon located 150 bp from the start of silCR was used to define the variation of putative silCR. Thus, we identified two distinct SilCR peptides in SAG, SilCRSAG-A and SilCRSAG-B (previously truncated version), with predicted mature amino acid sequences GWLEDLLKHFSGYNSLTKGDSNNTLG and GWLEDLFSPYLKKYKLGKLGQPDLG, respectively. Notably, each SilCR peptide allele associated exclusively with a corresponding variant of histidine kinase SilB. This suggests that the response by a strain could be dependent on the kinase variant. The conserved 5′ end of SilCR propeptide is hypothetically cleaved at the double glycine residues to produce the mature SilCR peptide.
To investigate whether S. intermedius B196 could respond to the alternative peptide, SilCRSAG-B was added to the knockout (Figure 2A). Despite high peptide concentrations, SilCRSAG-B induced a smaller inhibitory zone in M505 than did SilCRSAG-A (8 mm vs. 11 mm). To further test the peptide dependent bacteriocin induction of strain B196, several concentrations of each peptide were used. We found that higher concentrations of SilCRSAG-B were required to produce inhibitory zones comparable to those produced by SilCRSAG-A (Figure 2C). Thus, while strain B196 can sense and respond to both pheromone SilCR peptides produced by SAG strains, its inhibitor production is peptide- and concentration- dependent requiring high concentration of the non-cognate signaling peptide to elicit a response.
To investigate the phylogeny of the sil locus in the 42 SAG genomes, the gene presence, sequence, and orientation were used to construct a tree (Figure 3). The tree of sil genes corresponded well with SAG species. Six different organizations of the sil locus were noted in SAG, as depicted in the locus arrangement legend in Figure 3. The type of SilCR peptide the strain produced (SilCRSAG-A vs. SilCRSAG-B) is also indicated in the figure. Strains encoding SilCRSAG-A have low diversity overall and are monophyletically nested within S. intermedius, while all three species can encode SilCRSAG-B. In 32 of the 42 sil loci, we identified the canonical arrangement of the sil locus (locus arrangement A in Figure 3). This arrangement is found in strains carrying SilCRSAG-A and SilCRSAG-B and is the arrangement seen in S. intermedius B196 (Figure 1A). The additional sil locus arrangement groups are characterized by the loss of genes and when SilCR is present, only included SilCRSAG-B. Locus group B is defined by an intact sil region but is altered next to silE with sodA absent and includes a single S. anginosus strain. The other arrangements of the sil region (C-D) are represented by deletions leading to loss of core sil genes. Group C consists of a single S. constellatus strain and is defined by missing silE and sodA. Group D is represented by one S. constellatus strain and one S. intermedius strain and is characterized by missing a number of sil locus components including the putative bacteriocin region, CAAX protease and silA response regulator. Group E is composed of only S. anginosus strains, and is characterized by the lack of all sil genes and accessory proteins except silD, silE, and sodA, with an inversion in silD and silE relative to sodA. Group F includes a single S. anginosus strain and is identical to Group E but missing silE. Overall, the S. anginosus strains had the most variation in composition of the sil locus with all locus groups represented except Groups C and D.
We identified a few strains where one or more sil genes had undergone inactivating mutations. A number of these pseudogenes appear to be the result of a few single mutation events and are consistent with the sil tree (Figure 3). In strains SK54, ATCC27335, and JTH08 (sister taxa in our sil gene phylogeny), silCR has a single-nucleotide deletion at base 12. In strains C1366 and C1384 (also sister taxa), silCR has a different single-nucleotide deletion at base 14. The sil system is otherwise intact in these strains. In strains C188, C232, and C818 (three of six sisters in an unresolved node in our phylogeny), the secretion protein silD has a single-nucleotide deletion at base 1314. These strains also have otherwise-intact sil regions. In addition to these, a small number of individual strains do not share sil gene inactivation events with other strains in our data set. Strain M410 has a partial sil region (containing silE, silD, and sodA only) and a single nucleotide deletion at base 972 in silE. Strain CCUG39159 has an intact sil region and an SNP that introduces a stop codon at base 1342. Strain M47 appears to have undergone a larger genome rearrangement event, with a 70 Kb region intervening between partial silD and silE genes. Strains M423, 1_2_62CV, and C270 have different frameshift mutation events in non-sil genes (SIR_RS15550 for the first, CAAX for the latter two). Finally, strain SK1060 appears to be degenerate in several places; the SIR_RS15550 homolog contains a single nucleotide insertion at base 95, while CAAX, silA, silB, and silD contain deletions at bases 558, 18, 136, and 1107 respectively.
The Streptococcus sil system controls expression of competitive bacteriocins in GAS (Eran et al., 2007; Belotserkovsky et al., 2009; Armstrong et al., 2016). In S. intermedius B196, we observe that bacteriocin activity is dependent on this signaling system. We discovered a highly variable accessory region in all strains carrying sil locus groups A-C (34 of the 42 genomes). We investigated the hypothetical ORFs in this region for putative bacteriocin and immunity genes. Sixteen distinct orthologous groups were identified using OrthoMCL. A detailed similarity comparison within each orthologous group is included in the Supplementary Similarity Files. The amino acid identity heatmap for each ORF is shown in the Supplementary Figures. In general, while some ORFs are highly conserved within SAG, others are not. For example, ORF13 is conserved with 97% or higher amino acid identity across 28 strains while ORF2 is clearly divided into two groups with one group having 44% amino acid identity to the other. Using this classification scheme, it was apparent that some strains had two copies of predicted ORFs (e.g., ORF4 in S. anginosus 1_2_62CV; Figure 3). It is important to note that the two copies are not identical and, given their patterns of synteny, it is not likely that they are functionally equivalent.
We assigned putative functions to the 16 ORFs identified based on similarity to hits in the non-redundant protein sequence database using BlastX (NCBI). These are summarized in Table 2. In total, six putative bacteriocins were identified (ORF1, ORF2, ORF6, ORF8, ORF15, and ORF16). Of the six bacteriocins, two were highly similar to existing bacteriocins (ORF2 is similar to bovicin 255 and ORF15 is similar to ThmA bacteriocin). The non-bacteriocin ORFs may have chaperone or immunity functions. ORFs 4 and 7 had high similarity with a bacteriocin secretion protein and a bacteriocin immunity gene, respectively. ORF3 has a putative role in replication. ORF14 shares some nucleotide identity with a tRNA (cytidine/uridine-2′-O-)- methyltransferase. It is unknown whether ORF14 serves a similar function in SAG. Six of the putative ORFs did not have predicted functions (ORFs 5, 9, 10, 11, 12, and 13). Characterization of these genes is required to gain insight into their role. However, some speculation based on the patterns of co-occurrence with other genes is possible. Several of the strains have acquired mutations in some of these accessory genes that has led to pseudogenes. These include two predicted bacteriocin genes (ORF1 and ORF2), several genes with no assigned function (ORF3, ORF9, ORF10, and ORF12) and one gene assigned to bacteriocin secretion function (ORF4). No pseudogenes were detected in any of the predicted immunity genes.
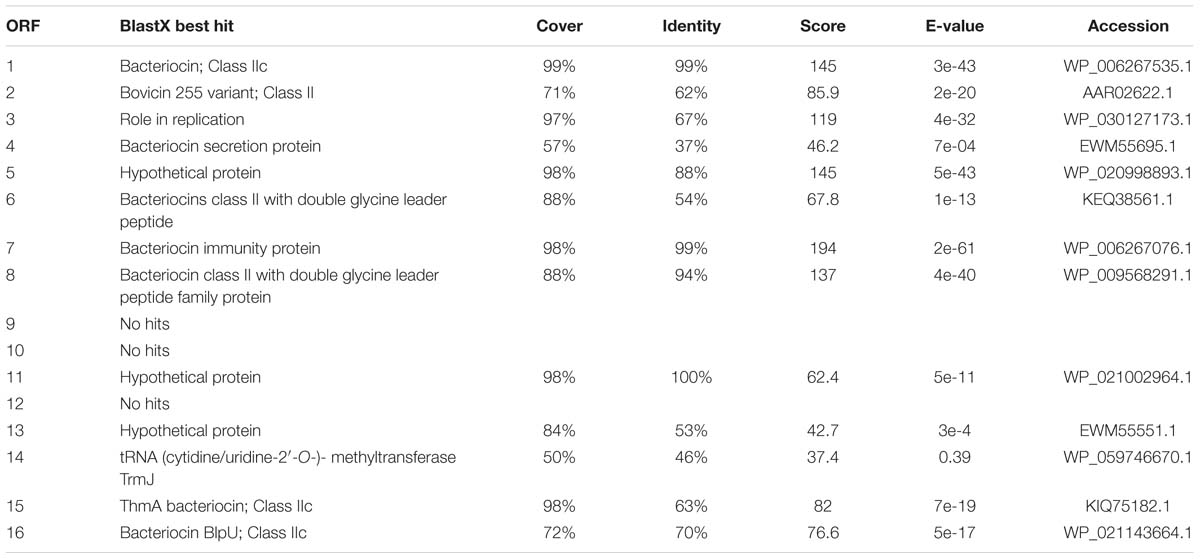
TABLE 2. Bioinformatic analysis of identified putative bacteriocin accessory region ORFs using a sequence search against the non-redundant protein sequence database using BlastX (NCBI).
There were some clear patterns of synteny in the accessory regions of the sil loci (Figure 3). These became more apparent after taking the subclusters of each ORF into consideration. We describe the syntenic groupings of ORFs as “ORF sets” (OS) because the operon structures have not been established for these strains. In combination with our putative functional assignments, we attempted to characterize these ORF sets (Supplementary Figure S1) in order to group bacteriocins with co-occurring genes that may be functionally associated with them. All ORF sets except OS IX contain at least one putative bacteriocin (Supplementary Figure S1). Three ORFs occurred only once each across all strains and are labeled “other” (Figure 3). Putative bacteriocin ORF1A is exclusively found adjacent to ORF5A, while ORF1B is separated from ORF5B by bacteriocin ORF2 and its associated genes. The two versions of putative bacteriocin ORF2 were found in two distinct clusters, OS III and OS V. ORF2 is always associated with a version of the putative secretion protein, ORF4. It is also always adjacent to either the putative immunity gene ORF7 or the unknown ORF10, suggesting an immunity function for the latter. Putative bacteriocin ORF6 is always found adjacent to ORF3, implying ORF3 may confer immunity against ORF6. A version of ORF3 is also found associated with ORF2 in OS V. It may be that ORF3, rather than ORF7, confers immunity to ORF2A. Putative bacteriocin ORF8 was found adjacent to ORF12 in most strains (although ORF12 has been mutated to a pseudogene in some of these strains). ORF12 was not present in strains lacking ORF8 and may therefore be only required in the presence of ORF8. Uncharacterized ORF9 is associated with two putative bacteriocins (ORF15 and 16) in two separate ORF sets (OS I and OS II). In each case, the genes are also associated with a hypothetical protein of unknown function (ORFs 11 and 13, respectively). None of the strains carried all six putative bacteriocins, implying that each strain may be susceptible to at least one bacteriocin. A number of S. intermedius strains had five predicted bacteriocins (Figure 3) implying that this species has high competitive potential. S. constellatus C1367 and S. anginosus C423 also had five predicted bacteriocins.
The arrangements of ORF sets within the accessory regions correspond imperfectly with the sil tree, indicating a large amount of recombination or horizontal gene transfer is occurring within this region. OS I is found in a majority of strains. The six strains lacking OS I form a monophyletic group with low genetic distance in the sil region, indicating a recent loss event. Areas of apparent recombination are highlighted with pink boxes in Figure 3. OS I occurs at one end of the bacteriocin accessory region in the strains where it is present. OS II is found on five genomes, in two positions relative to other ORF sets. OS IV typically appears at the downstream end of accessory regions where it is found (except in strains C1369 and B196), while OS III may be at the downstream end or may be upstream of OS VI. OS V occurs either upstream of OS II or OS III. In either case it is directly downstream of OS I.
In GAS and GGS, the Streptococcus sil system senses and responds to pheromone peptide SilCR with induction of endogenous SilCR production and expression of bacteriocins. In some streptococci, expression of silCR is inhibited by expression of silC, which is encoded on the antisense strand at the 3′ end of silCR. In those organisms, it has been shown that silC expression can repress SilCR-activated promoters (Hidalgo-Grass et al., 2002; Eran et al., 2007). No conserved silC ORF across all silCR+ strains of SAG was found and more research is required to identify if there is a silC variant in SAG.
The prevalence of the sil locus within GAS is low, with only 4 out of the 19 fully sequenced genomes of GAS carrying it. Remnants of the locus have been left behind in strains that do not (Kizy and Neely, 2009; Michael-Gayego et al., 2013; Jimenez and Federle, 2014). The prevalence is higher in GGS, with all sequenced strains carrying a functional SilCR gene (Belotserkovsky et al., 2009; Michael-Gayego et al., 2013). Our analysis of SAG showed that the majority of the strains sequenced carry the locus. Locus arrangement Groups A and B in Figure 3 shows that all components of the locus are present in most SAG strains, although individual genes do appear to be degenerate in some strains. The other groups included few strains and, like GAS, showed loss of some sil genes. Further study is needed to determine whether the lack of a fully functional sil system affects a strain’s competitive fitness or ability to colonize or invade.
In SAG, considerably higher genetic heterogeneity was seen in the bacteriocin accessory region than in the sil gene region. S. anginosus displayed the most variation in the presence of sil genes, implying that the locus may not be under stabilizing selection and may not be required for competition in this species. Conversely, most S. intermedius and S. constellatus strains were included in locus group A, suggesting that the sil locus may be more competitively necessary in these species, at least in the clinical context from which these strains were isolated.
The bacteriocins identified in the SAG sil locus have not been characterized or directly associated with intra- or interspecies competition; however, the identification of six putative bacteriocins within the sil locus in SAG is a new finding. Bacteriocins can mediate competition with closely related bacteria and can provide a competitive advantage during in vivo colonization experiments (Dawid et al., 2007). ORF2 is a homolog of bovicin 255, which has been shown to inhibit growth of select streptococci (Whitford et al., 2001). ORF15 is homologous to thermophilin 13 bacteriocin (ThmA), which is produced by S. thermophilus and can inhibit a broad range of Gram-positive bacteria including spore formers (Marciset et al., 1997). In addition to these genes, four more putative bacteriocins were identified and remain to be characterized. The species selectivity of these putative bacteriocins in SAG is currently being investigated.
Interspecies SilCR induction of SilA-responsive genes has been described previously between GAS and GGS (Belotserkovsky et al., 2009). Inter- and intra- species competition in streptococci can occur in a polymicrobial environment such as the oral cavity. The hypothetical mature SilCRSAG-A and SilCRSAG-B peptides are 26 and 25 amino acids respectively, while the mature GAS peptide is 17. Both SAG immature peptides have the same six amino acids at their 5′ end but vary at their 3′ end. The SAG SilCR peptides are not very similar to the GAS and GGS SilCR peptides in either sequence or length and it remains to be determined if these species’ sil systems are cross-reactive.
Our data demonstrates that S. intermedius B196 can sense and respond to SilCRSAG-B, which can be carried by all three SAG species; however, a much higher concentration of the foreign peptide SilCRSAG-B was required to rescue the phenotype in B196ΔsilCR than the B196-native version (SilCRSAG-A). This implies a divergent co-evolution of the silCR and silB genes in the SAG-A clade, with selection for self-detection.
SilCR expression has been shown to attenuate virulence in a mouse model of necrotizing fasciitis (Hidalgo-Grass et al., 2002, 2004). However, SilA has also been shown to promote expression of virulence-associated genes including streptolysin S, iron transporter SiaA and serine protease ScpC (Salim et al., 2008). It is unknown whether the sil system in SAG affects its pathogenicity. This system is unusual in that it can contribute to both infection in the host and bacterial competition depending on the environment. Given the host-specificity of SAG, and the commensal and pathogenic roles it can play, further analysis of this system could deepen our understanding of SAG competition and its associated clinical diseases.
All authors listed have made substantial, direct and intellectual contribution to the work and approved it for publication.
This work was supported by a grant for the Canadian Institutes of Health Research (CIHR) to MS. MS and DB were supported by a Canada Research Chair in Interdisciplinary Microbiome Research and Canada Research Chair in Aging & Immunity, respectively.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank members of the Bowdish and Surette labs for helpful discussion.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.02156/full#supplementary-material
Altekar, G., Dwarkadas, S., Huelsenbeck, J. P., and Ronquist, F. (2004). Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics 20, 407–415. doi: 10.1093/bioinformatics/btg427
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Armstrong, B. D., Herfst, C. A., Tonial, N. C., Wakabayashi, A. T., Zeppa, J. J., and McCormick, J. K. (2016). Identification of a two-component Class IIb bacteriocin in Streptococcus pyogenes by recombinase-based in vivo expression technology. Sci. Rep. 6:636233. doi: 10.1038/srep36233
Asam, D., and Spellerberg, B. (2014). Molecular pathogenicity of Streptococcus anginosus. Mol. Oral. Microbiol. 29, 145–155. doi: 10.1111/omi.12056
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Belotserkovsky, I., Baruch, M., Peer, A., Dov, E., Ravins, M., Mishalian, I., et al. (2009). Functional analysis of the quorum-sensing streptococcal invasion locus (sil). PLoS Pathog. 5:e1000651. doi: 10.1371/journal.ppat.1000651
Benson, D. A., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Sayers, E. W. (2015). GenBank. Nucleic Acids Res. 43, D30–D35. doi: 10.1093/nar/gku1216
Blin, K., Kazempour, D., Wohlleben, W., and Weber, T. (2014). Improved lanthipeptide detection and prediction for antiSMASH. PLoS ONE 9:e89420. doi: 10.1371/journal.pone.0089420
Blin, K., Medema, M. H., Kazempour, D., Fischbach, M. A., Breitling, R., Takano, E., et al. (2013). antiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 41, W204–W212. doi: 10.1093/nar/gkt449
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Chikhi, R., and Medvedev, P. (2014). Informed and automated k-mer size selection for genome assembly. Bioinformatics 30, 31–37. doi: 10.1093/bioinformatics/btt310
Cock, P. J. A., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
Coman, G., Pânzaru, C., Diculencu, D., Gotia, D., Cârlan, M., Dahorea, C., et al. (1995). Pyogenic infections with different locations caused by Streptococcus anginosus alone or in association with anaerobic bacteria. Rev. Med. Chir. Soc. Med. Nat. Iasi 99, 215–219.
Crooks, G. E., Hon, G., Chandonia, J.-M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Dawid, S., Roche, A. M., and Weiser, J. N. (2007). The blp bacteriocins of Streptococcus pneumoniae mediate intraspecies competition both in vitro and in vivo. Infect. Immun. 75, 443–451. doi: 10.1128/IAI.01775-05
Delcher, A., Harmon, D., Kasif, S., White, O., and Salzberg, S. L. (1999). Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27, 4636–4641. doi: 10.1093/nar/27.23.4636
Dunny, G. M., Lee, L. N., and LeBlanc, D. J. (1991). Improved electroporation and cloning vector system for gram-positive bacteria. Appl. Environ. Microbiol. 57, 1194–1201.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eran, Y., Getter, Y., Baruch, M., Belotserkovsky, I., Padalon, G., Mishalian, I., et al. (2007). Transcriptional regulation of the sil locus by the SilCR signalling peptide and its implications on group A streptococcus virulence. Mol. Microbiol. 63, 1209–1222. doi: 10.1111/j.1365-2958.2007.05581.x
Fei, F., Mendonca, M. L., McCarry, B. E., Bowdish, D. M. E., and Surette, M. G. (2016). Metabolic and transcriptomic profiling of Streptococcus intermedius during aerobic and anaerobic growth. Metabolomics 12:46. doi: 10.1007/s11306-016-0966-0
Fischer, S., Brunk, B. P., Chen, F., Gao, X., Harb, O. S., Iodice, J. B., et al. (2011). Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinformatics Chapter 6, 1–19. doi: 10.1002/0471250953.bi0612s35
Goujon, M., McWilliam, H., Li, W., Valentin, F., Squizzato, S., Paern, J., et al. (2010). A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699. doi: 10.1093/nar/gkq313
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hammami, R., Zouhir, A., Ben Hamida, J., Fliss, I., Jack, R., Tagg, J., et al. (2007). BACTIBASE: a new web-accessible database for bacteriocin characterization. BMC Microbiol. 7:89. doi: 10.1186/1471-2180-7-89
Hammami, R., Zouhir, A., Le Lay, C., Ben Hamida, J., Fliss, I., Riley, M., et al. (2010). BACTIBASE second release: a database and tool platform for bacteriocin characterization. BMC Microbiol. 10:22. doi: 10.1186/1471-2180-10-22
Hidalgo-Grass, C., Dan-Goor, M., Maly, A., Eran, Y., Kwinn, L. A., Nizet, V., et al. (2004). Effect of a bacterial pheromone peptide on host chemokine degradation in group A streptococcal necrotising soft-tissue infections. Lancet 363, 696–703. doi: 10.1016/S0140-6736(04)15643-2
Hidalgo-Grass, C., Ravins, M., Dan-Goor, M., Jaffe, J., Moses, A. E., and Hanski, E. (2002). A locus of group A Streptococcus involved in invasive disease and DNA transfer. Mol. Microbiol. 46, 87–99. doi: 10.1046/j.1365-2958.2002.03127.x
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Jacobs, J. A., Pietersen, H. G., Stobberingh, E. E., and Soeters, P. B. (1995). Streptococcus anginosus, Streptococcus constellatus and Streptococcus intermedius. Clinical relevance, hemolytic and serologic characteristics. Am. J. Clin. Pathol. 104, 547–553. doi: 10.1093/ajcp/104.5.547
Jensen, A., Hoshino, T., and Kilian, M. (2013). Taxonomy of the Anginosus group of the genus Streptococcus and description of Streptococcus anginosus subsp. whileyi subsp. nov. and Streptococcus constellatus subsp. viborgensis subsp. nov. Int. J. Syst. Evol. Microbiol. 63, 2506–2519. doi: 10.1099/ijs.0.043232-0
Jimenez, J. C., and Federle, M. J. (2014). Quorum sensing in group A Streptococcus. Front. Cell Infect. Microbiol. 4:127. doi: 10.3389/fcimb.2014.00127
Kaiser, J. C., Verschoor, C. P., Surette, M. G., and Bowdish, D. M. (2014). Host cytokine responses distinguish invasive from airway isolates of the Streptococcus milleri/anginosis group. BMC Infect. Dis. 14:498. doi: 10.1186/1471-2334-14-498
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kizy, A. E., and Neely, M. N. (2009). First Streptococcus pyogenes signature-tagged mutagenesis screen identifies novel virulence determinants. Infect. Immun. 77, 1854–1865. doi: 10.1128/IAI.01306-08
Kormin, S., Rusul, G., Radu, S., and Ling, F. H. (2001). Bacteriocin-producing lactic Acid bacteria isolated from traditional fermented food. Malays. J. Med. Sci. MJMS 8, 63–68.
Laupland, K. B., Ross, T., Church, D. L., and Gregson, D. B. (2006). Population-based surveillance of invasive pyogenic streptococcal infection in a large Canadian region. Clin. Microbiol. Infect. 12, 224–230. doi: 10.1111/j.1469-0691.2005.01345.x
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Lewis, P. O. (2001). A likelihood approach to estimating phylogeny from discrete morphological character data. Syst. Biol. 50, 913–925. doi: 10.1080/106351501753462876
Marciset, O., Jeronimus-Stratingh, M. C., Mollet, B., and Poolman, B. (1997). Thermophilin 13, a nontypical antilisterial poration complex bacteriocin, that functions without a receptor. J. Biol. Chem. 272, 14277–14284. doi: 10.1074/jbc.272.22.14277
Maricic, N., and Dawid, S. (2014). Using the overlay assay to qualitatively measure bacterial production of and sensitivity to pneumococcal bacteriocins. J. Vis. Exp. 91:e51876. doi: 10.3791/51876
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17:10. doi: 10.14806/ej.17.1.200
Michael-Gayego, A., Dan-Goor, M., Jaffe, J., Hidalgo-Grass, C., and Moses, A. E. (2013). Characterization of sil in invasive group A and G Streptococci: antibodies against bacterial pheromone peptide SilCR result in severe infection. Infect. Immun. 81, 4121–4127. doi: 10.1128/IAI.00359-13
Olson, A. B., Kent, H., Sibley, C. D., Grinwis, M. E., Mabon, P., Ouellette, C., et al. (2013). Phylogenetic relationship and virulence inference of Streptococcus anginosus Group: curated annotation and whole-genome comparative analysis support distinct species designation. BMC Genomics 14:895. doi: 10.1186/1471-2164-14-895
Parkins, M. D., Sibley, C. D., Surette, M. G., and Rabin, H. R. (2008). The Streptococcus milleri group–an unrecognized cause of disease in cystic fibrosis: a case series and literature review. Pediatr. Pulmonol. 43, 490–497. doi: 10.1002/ppul.20809
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2011). Meta-IDBA: a de Novo assembler for metagenomic data. Bioinformatics 27, i94–i101. doi: 10.1093/bioinformatics/btr216
Poole, P. M., and Wilson, G. (1979). Occurrence and cultural features of Streptococcus milleri in various body sites. J. Clin. Pathol. 32, 764–768. doi: 10.1136/jcp.32.8.764
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Ripley, R. T., Cothren, C. C., Moore, E. E., Long, J., Johnson, J. L., and Haenel, J. B. (2006). Streptococcus milleri infections of the pleural space: operative management predominates. Am. J. Surg. 192, 817–821. doi: 10.1016/j.amjsurg.2006.08.050
Ruoff, K. L. (1988). Streptococcus anginosus ("Streptococcus milleri"): the unrecognized pathogen. Clin. Microbiol. Rev. 1, 102–108. doi: 10.1128/CMR.1.1.102
Salim, K. Y., de Azavedo, J. C., Bast, D. J., and Cvitkovitch, D. G. (2008). Regulation of sagA, siaA and scpC by SilCR, a putative signaling peptide of Streptococcus pyogenes. FEMS Microbiol. Lett. 289, 119–125. doi: 10.1111/j.1574-6968.2008.01375.x
Shinzato, T., and Saito, A. (1995). The Streptococcus milleri group as a cause of pulmonary infections. Clin. Infect. Dis. 21(Suppl. 3), S238–S243. doi: 10.1093/clind/21.Supplement_3.S238
Sibley, C. D., Grinwis, M. E., Field, T. R., Parkins, M. D., Norgaard, J. C., Gregson, D. B., et al. (2010). McKay agar enables routine quantification of the “Streptococcus milleri” group in cystic fibrosis patients. J. Med. Microbiol. 59, 534–540. doi: 10.1099/jmm.0.016592-0
Sibley, C. D., Parkins, M. D., Rabin, H. R., Duan, K., Norgaard, J. C., and Surette, M. G. (2008). A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc. Natl. Acad. Sci. U.S.A. 105, 15070–15075. doi: 10.1073/pnas.0804326105
Siegman-Igra, Y., Azmon, Y., and Schwartz, D. (2012). Milleri group streptococcus–a stepchild in the viridans family. Eur. J. Clin. Microbiol. Infect. Dis. 31, 2453–2459. doi: 10.1007/s10096-012-1589-7
Simpson, J. T., and Durbin, R. (2012). Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 22, 549–556. doi: 10.1101/gr.126953.111
Tavare, S. (1986). Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 17, 57–86.
van Rossum, G., and Drake, F. L. (2001). Python Reference Manual. Virginia: PythonLabs. Available at: http://www.python.org
Whiley, R. A., Beighton, D., Winstanley, T. G., Fraser, H. Y., and Hardie, J. M. (1992). Streptococcus intermedius, Streptococcus constellatus, and Streptococcus anginosus (the Streptococcus milleri group): association with different body sites and clinical infections. J. Clin. Microbiol. 30, 243–244.
Whitford, M. F., McPherson, M. A., Forster, R. J., and Teather, R. M. (2001). Identification of bacteriocin-like inhibitors from rumen Streptococcus spp. and isolation and characterization of bovicin 255. Appl. Environ. Microbiol. 67, 569–574. doi: 10.1128/AEM.67.2.569-574.2001
Yang, Z. (1994). Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39, 306–314. doi: 10.1007/BF00160154
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Keywords: Streptococcus Milleri Group, Streptococcus Anginosus Group, sil system, quorum sensing, bacteriocins, cell–cell signaling
Citation: Mendonca ML, Szamosi JC, Lacroix A-M, Fontes ME, Bowdish DM and Surette MG (2017) The sil Locus in Streptococcus Anginosus Group: Interspecies Competition and a Hotspot of Genetic Diversity. Front. Microbiol. 7:2156. doi: 10.3389/fmicb.2016.02156
Received: 18 September 2016; Accepted: 21 December 2016;
Published: 10 January 2017.
Edited by:
Fumito Maruyama, Kyoto University, JapanReviewed by:
Shinya Watanabe, Jichi Medical University, JapanCopyright © 2017 Mendonca, Szamosi, Lacroix, Fontes, Bowdish and Surette. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael G. Surette, c3VyZXR0ZUBtY21hc3Rlci5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.