 Marina I. Arleevskaya
Marina I. Arleevskaya Olga A. Kravtsova
Olga A. Kravtsova Julie Lemerle
Julie Lemerle Yves Renaudineau
Yves Renaudineau Anatoly P. Tsibulkin1
Anatoly P. Tsibulkin1

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 17 August 2016
Sec. Microbial Immunology
Volume 7 - 2016 | https://doi.org/10.3389/fmicb.2016.01296
This article is part of the Research TopicMicrobial and environmental factors in autoimmune and inflammatory diseasesView all 18 articles
The pathogenesis of rheumatoid arthritis (RA), similar to development of a majority of inflammatory and autoimmune disorders, is largely due to an inappropriate or inadequate immune response to environmental challenges. Among these challenges, infectious agents are the undisputed leaders. Since the 1870s, an impressive list of microorganisms suspected of provoking RA has formed, and the list is still growing. Although a definite causative link between a specific infectious agent and the disease has not been established, several arguments support such a possibility. First, in the absence of a defined pathogen, the spectrum of triggering agents may include polymicrobial communities or the cumulative effect of several bacterial/viral factors. Second, the range of infectious episodes (i.e., clinical manifestations caused by pathogens) may vary in the process of RA development from preclinical to late-stage disease. Third, infectious agents might not trigger RA in all cases, but trigger it in a certain subset of the cases, or the disease onset may arise from an unfortunate combination of infections along with, for example, psychological stress and/or chronic joint tissue microtrauma. Fourth, genetic differences may have a role in the disease onset. In this review, two aspects of the problem of “microorganisms and RA” are debated. First, is there an acquired immune deficiency and, in turn, susceptibility to infections in RA patients due to the too frequent and too lengthy infections, which at last break the tolerance of self antigens? Or, second, is there a congenital deficiency in tolerance and inflammation control, which may occur even with ordinary infection frequency and duration?
Rheumatoid arthritis (RA) pathogenesis, in the most general sense, is largely due to an inadequate immune response in genetically predisposed individuals to environmental challenges, with bacteria and viruses (latent carrier or clinical signs of infection) being the undisputed leaders among these environmental challenges (Brooks et al., 2010; Le Dantec et al., 2015b).
The microbial concept of RA triggering agents has been discussed since the 1870s (Benedek, 2006). But, in spite of this long history, a direct role of microorganisms in the disease is controversial, while the suspected pathogen list is still growing (Table 1). The fact is that it is impossible to make a causal link between a specific pathogen and the disease. In light of these disappointing results in understanding RA, there are calls for even larger studies with the use of more advanced technologies (Silman and Pearson, 2002; Carty et al., 2004).

TABLE 1. Pathogens suspected of triggering rheumatoid arthritis (RA).
First, in the absence of a defined pathogen, the spectrum of microorganisms involved in triggering RA may include polymicrobial communities or the cumulative effect of several bacterial/viral factors. The simultaneous effects of various pathogens on the immune system in an RA patient may have different direct effects. It has been demonstrated that specific infections (gastrointestinal and urogenital), hypothetically associated with the changes in the gut microbiome, could diminish or increase the risk of RA (Sandberg et al., 2015).
Second, the spectrum of bacteria and viruses, as well as the reaction of the immune system to such pathogens, with or without clinical manifestations of infection, may vary from preclinical to late-stage RA: such that any of numerous pathogens may trigger RA at a preclinical stage, but subsequently lose their influence in the advanced stage (Arleevskaya et al., 2014). Moreover, the delicate balance between pathogens and the anti-infection immune response of an RA patient may be disturbed by several factors including the limited mobility of the patients as well as the drug therapy selected.
Third, infections might not trigger RA in all cases, but only in a subset of the cases. For example, co-occurence with psychological stress and/or chronic joint tissue microtrauma might be needed. Indeed, in Swedish and Finish cohorts only 16–20% of the patients reported infection in the early phase of RA (Leirisalo-Repo, 2005); whereas in a Russian cohort this index was as high as 75% (Arleevskaya et al., 2014). The age at disease onset also seems to be important since younger patients attributed their RA to a previous infection more often than did older patients. (Soderlin et al., 2011).
Fourth, the differences in trigger factors might reflect variations in the pathogenetic mechanisms. Indeed, RA is recognized as being a multigene disorder with a huge number of genetic polymorphisms contributing to the disease pathogenesis. The differences in trigger factors may also be due to geographic, life style, drug or chemical exposures, and ethnic differences (Konsta et al., 2015; Le Dantec et al., 2015a; Lemerle et al., 2016). The diagnostic category of RA includes multiple subtypes of the disease and the different phenotypes of RA might be due to the different genotypes. In fact, the set of clinical signs, which has been known since 1782 (Jonsson and Helgason, 1996), and which we call “RA” may be the outcome of a variety of pathogenic pathways, or in other words, it is a syndrome, i.e., a group of symptoms that arise as a consequence of a number of different diseases (Weyand et al., 1998).
Accordingly, two aspects of the problem of “pathogens and RA” versus “RA and infections” are still debated. Is there an acquired immune deficiency in RA patients caused by too frequent and prolonged infections, which break tolerance of self-antigens? Or, is there a congenital deficiency of the tolerance and inflammation control, which may occur even with ordinary infection frequency and duration?
The results of various studies testing the acquired or the genetically determined predisposition to infections in RA are rather contradictory. These differences can be explained by several reasons. First, the conflicting results may be explained in part by features of the patient groups studied, including the particular therapeutic approach used and the specific types of infections tracked by the authors (Vandenbroucke et al., 1987; Widdifield et al., 2013; Sandberg et al., 2015).
Second, evolution of the immune system reaction to pathogens during RA development is usually not taken into account. Our 10-year follow-up showed that both early stage RA patients and their relatives suffer from more frequent and prolonged minor infections than those individuals without autoimmune diseases in their family history (Arleevskaya et al., 2014). A gradual decrease in the frequency and duration of the infectious episodes was observed in RA patients at a later stage, when they were taken under observation at an early stage and observed for longer than 3 years. It was also observed from this cohort that the frequency and duration of the infectious episodes increased even more in the year before the RA onset, and that all the relatives who developed RA during the observation (i.e., included in the study at the pre-clinical stage) had a pronounced infectious syndrome (Arleevskaya et al., 2014). It is noteworthy that Germano et al. (2014) reported an association between the infection risk and disease activity, while these authors also supported the hypothesis that the infectious syndrome decrease with RA duration. With this in mind, we speculate that there is probably both a congenital and or an acquired deficiency of the anti-infection defense leading to the frequent and prolonged minor infections in early RA patients and their relatives. Attempts to eradicate the infections eventually lead to a certain amount of success, but at the cost of RA onset due to an inappropriate activation/inhibition of various key parts of the immune system. It should be noted, that in late stage RA, microbial colonization (including the increased frequency of heavy Escherichia coli and Staphylococcus aureus colonization) persists (Arleevskaya et al., 2014). So, in late stage RA, despite the reduction of clinical signs of frequent and prolonged infectious episodes, there are still laboratory signs of dysbiosis. Thus, a delicate balance of microflora and the immune host defense might be disrupted at any time, for example, when there is a change in the therapy. This hypothesis is indirectly confirmed by the data, indicating that a history of previous infections appeared to be among the risk factors for the infectious complications during infliximab and disease-modifying anti-rheumatic drug therapy (Widdifield et al., 2013). We interpret these data in such a way that, in RA patients with a deficiency in their anti-infection defenses, which has been manifested earlier in any infections and compensated for to some extent later, the risk of renewal of infections still remains high.
There are two possible approaches to the problem of “infections and RA”. One of them, being of particular importance for practicing rheumatologists, aims to study the susceptibility to infections as a prognostic factor for the infectious complications in RA therapy. The goal is to study all patients without exception, including those with a variety of reasons for the development of infection complications, even in the absence of RA (such as leucopenia, low mobility, diabetes, or other comorbiditiesl; Doran et al., 2002; Soderlin et al., 2011). The results of these studies indicate an increased susceptibility to infections in RA patients. The second approach is to study the role of infections in the provocation and persistence of RA activity. In this case, it seems logical (albeit with obvious damage to the completeness of coverage of the problem) not to extend the study on the patients with the concomitant additional risk factors for the infection development.
There is a definite list of possible mechanisms by which the pathogen/host interactions could trigger pathological autoimmune responses: molecular mimicry, epitope spreading, polyclonal lymphocyte activation, bystander activation, and viral persistence (Bach, 2005; Kivity et al., 2009; Vojdani, 2014; Floreani et al., 2015).
Molecular mimicry occurs when foreign antigens bear sufficient structural similarity to self-antigens (i.e., similar epitopes). As a result, an immune response to pathogens could lead to cross reactivity with self-antigens. The similarity of pathogen and self-antigen structures is a widespread phenomenon. By exact peptide matching analysis, it was demonstrated that all human proteins harbor a bacterial penta- or hexa-peptide motif (Trost et al., 2010). Moreover, the study of pathogenic and non-pathogenic proteomes from Vibrio cholera and Mycobacterium tuberculosis up to Lactobacillus acidophilus and Bifidobacterium adolescentis showed that the bacterial pathogenicity does not affect the cross-reactivity with human proteins. So, molecular mimicry would seem to be too commonplace to shift on it the blame for the development of autoimmune diseases. In the inflammatory foci a variety of bacterial proteins undergo several different types of post-translational modifications, and most of these proteins have motifs similar to those found in human polypeptides.
Epitope spreading is a phenomenon in which the immune T or B cell response extends beyond the original epitope. In general, epitope spreading is quite a beneficial phenomenon, contributing to the ability of the immune system to attack multiple pathogens (Powell and Black, 2001). Epitope spreading develops in the process of an ongoing immune reaction during an acute or persistent infection and following tissue destruction. Epitope spreading optimizes protection against newly encountered pathogens and assists in clearing inflammatory sites of damaged endogenous proteins of the body’s own tissues. Its success is due to the ability to quickly target new epitopes and the ability to adequately destroy pathogens and clear inflammatory sites. It appears evident that, due to RA development, abnormalities occur in the mechanisms that would otherwise constrain epitope spreading only to the diversity of foreign antigens and prevent development of an immune response to self-epitopes. A complete review of these abnormalities and self-epitopes is beyond the scope of the present discussion. We’ll briefly examine only some aspects. Epitope spreading can result from a change in a protein structure. One well-known example, protein citrullination, which is a conversion of arginine to citrulline by peptidyl arginine deaminase (PAD), could be due to development of an immune reaction against the original protein or its citrullinated form, but may also arise against other citrullinated proteins.
Currently interest in the well-established link between periodontitis and RA (Helminen-Pakkala, 1968) is going through a renaissance. This reignited interest is due to clarification of the role of citrullinated peptides as epitopes with pathogenic significance in RA, and identification of the ability of Porphyromonas gingivalis, the main pathogen in chronic periodontitis, to citrullinate proteins by using its own PAD (Maresz et al., 2013). Thus, this situation represents a clear example of an autoimmune disease triggered by infection via molecular mimicry and epitope spreading mechanisms. When activated, B lymphocytes initially producing antibodies (Ab) against the citrullinated P. gingivalis enolase, start producing Ab against citrullinated human enolase, and then against other citrullinated endogenous proteins (reviewed in Lundberg et al., 2010). Quirke et al. (2015) compared the incidence in RA patients of increased levels of serum Ab to arginine containing proteins (enolase, vimentin, fibrinogen), and to the citrullinated modifications according to the occurrence of lung bacterial infections. As compared to RA control patients, lung infections were associated with an elevated level of Ab to citrullinated proteins.
Microbial molecules can directly induce proliferation and differentiation of T- and B-lymphocytes regardless of their antigen specificity. In the model of murine Trypanosoma cruzi infection, it was shown that the number of high rate immunoglobulin secreting cells of both IgM and IgG classes increased up to 100 fold each and a variety of effector T cell activities were revealed, with both Ab and cellular immune reactions being predominantly non-specific (Arala-Chaves et al., 1992). Similar observations have been recorded in various viral, bacterial and fungal infections. The authors provided the results of their experiments, showing that as much as 95% of Ab produced in response to an infectious stimulus fail to bind to the antigens used for the immunization. The polyclonal activation normally produced excessive amounts of usually low-affinity Abs, which are directed against foreign and self-antigens. With regards to expression of the anti-immunoglobulin G Abs (also known as rheumatoid factor, RF) in human RF-positive Tg murine models, results differed according to the timing of the infection. On the one hand and following acute influenza viral infection, an abortive activation of RF-positive B cells and no increase in RF production characterized infected mice as compared to the uninfected ones (Woods et al., 2007); while, on the other hand, the same group also reported that chronic bacterial infection in the RF-positive Tg mice was associated with increased RF production, which resulted from polyclonal lymphocyte activation (Soulas et al., 2005).
In RA, peripheral blood and synovial lymphocyte polyclonal activation is well documented, although its extent is limited since hypergammaglobulinemia does not occur in these patients (Becker et al., 1990; Bucht et al., 1992; Yamamoto et al., 1992; Outschoorn et al., 1993; Brown et al., 1995). When testing the serum levels of Ab free light chains (as a biomarker of B cell activation) in a large cohort of the general population, Deng et al. (2015) reported elevated levels of the polyclonal free light chains 3–5 years before clinical onset of RA, an elevated level that remains during the follow-up of the patients. The authors pointed out that the elevation was moderate, and failed to demonstrate any correlation with mortality in the RA group.
A number of the results demonstrate that the situation with the polyclonal lymphocyte activation in RA is complicated. The data of recent studies, which tested the isotype distribution, antigen specifity and affinity of the serum and synovial fluid RFs and anti-CCP Abs as well as the expression of the inherently autoreactive idiotope 9G4 in preclinical, early and advanced stages of RA, failed to clarify whether such a process is crucial in RA development (Moyes et al., 1996; Verpoort et al., 2006, Ioan-Facsinay et al., 2008; Cambridge et al., 2014, Falkenburg et al., 2015). When analyzing these data, it looks like the anti-citrullinated Abs are produced due to the ongoing immune response to citrullinated proteins, while RFs appear later and persist due to both the polyclonal and antigen-driven activation of B lymphocytes (Renaudineau et al., 2005). Cambridge et al. (2014), speculated that “in RA, autoreactive B cell specificities escape deletion, receptor editing or anergy early in their development, ultimately giving rise to a population of B cells which can also survive entry into the mature B cell compartment in the periphery.”
Another important aspect is the imperfection of the polyclonal activation of peripheral blood B-lymphocytes from RA patients. The cell response to bacterial peptidoglycan, pokeweed mitogen, or phorbol myristate acetate stimulation was significantly reduced compared to controls (Abrahamsen et al., 1978; Pardo et al., 1984). Synovial fluid lymphocytes also proliferated poorly when stimulated polyclonally (Abrahamsen et al., 1978; Petersen, 1988). Finally, RA lymphocytes stimulated by the polyclonal B cell activator Epstein-Barr virus (EBV), produced less IgM than controls after 1 week and showed increasing IgM production between 14 and 21 days, whereas, in normal lymphocytes, IgM production decreased during this period (Irving et al., 1985).
Autoreactive cells may be expanded and activated by non-specific means or by a combination of non-specific effects with self antigens being released and presented in an inflammatory environment created by infection (Kim B. et al., 2006). Most studies examining non-cognate T cell responses have focused on CD8 T cells, primarily in viral infection models with the stimuli being the products of infected cells, or inflammatory molecules generated by chronically virus-infected tissues or the factors produced by the cells, activated during the viral molecules interaction with toll-like receptors (Miller et al., 1997; Horwitz et al., 1998; Toubi and Shoenfeld, 2004; O’Donnell and McSorley, 2014). However, bacteria, in particular the intra-macrophage bacteria, also may be responsible for the bystander activation (Das et al., 2000; McSorley, 2014; O’Donnell and McSorley, 2014). One of the main factors provoking bystander activation is related to proinflammatory cytokines. At least in experiments studying this phenomenon, the proinflammatory cytokine cocktails or any of these cytokines are typically used for the cell stimulation (Unutmaz et al., 1994; Brennan et al., 2008). Cytokine stimulation of naive and resting memory T cells results in the proliferation of the lymphocytes and display of the effector functions of the memory T cells as measured by lymphokine synthesis and help stimulate immunoglobulin production by B cells (Unutmaz et al., 1994). At the same time, T cells activated by cytokines in the absence of T cell receptor stimulation also activate monocytes and monocyte cytokine production (Sebbag et al., 1997). Evidently these processes occur in RA pathogenesis (Sebbag et al., 1997; Brennan et al., 2008).
Recent results from genome wide association studies (GWAS) support a contributory role of genetic and epigenetic factors in the development of RA (Konsta et al., 2015). The most well-studied genetic example is related to the RA associated DRB1∗01 and ∗04 alleles (shared epitopes), which are effective, when expressed, in binding citrullinated peptides, and presenting them to T cell receptors which is the optimal scenario for development of an immune response against citrullinated peptides, as well as for activation of the proinflammatory Th1 cytokine production (Tian et al., 1998; Rosloniec et al., 2002b; Auger and Roudier, 2005; Gourraud et al., 2006; Ohnishi et al., 2006). Moreover in RA patients, an increased expression of the HLA-DR molecules has been reported leading to a significant low-affinity peptide presentation and activation of autoreactive peripheral T cells (Kerlan-Candon et al., 2001; Rosloniec et al., 2002a; Hill et al., 2003; Auger and Roudier, 2005; Gourraud et al., 2006, 2007; Ohnishi et al., 2006).
With regards to associations between infections and HLA-DR peculiarities in RA, HLA-DRB1∗0404 was associated with a low frequency of occurence of T cells specific for EBV gp110, a replicative phase glycoprotein which is critical for the EBV infection control (Toussirot et al., 1999). While opposite to this, HLA-DRB1∗07, an allele associated with reduced risk of developing RA, was associated with the highest frequencies of the peripheral blood T-lymphocytes specific for gp110. Therefore, it’s not surprising that EBV DNA and EBV-encoded RNA I transcripts are present significantly more often in synovial tissue of patients positive for the shared epitope (especially HLA-DRB1∗0404–positive; Saal et al., 1999). When considering HLA-DRB1 alleles predisposing to the development of recurrent herpes lymphocytic meningitis and CMV reactivation, results were more contrasted with the characterization of both RA-associated non share epitopes HLA-DRB1∗09 and DRB1∗15, and RA-associated share epitopes HLA-DRB1∗01/∗04 (Du et al., 2007; Kekik et al., 2009; Kallio-Laine et al., 2010; Acar et al., 2014).
Other gene polymorphisms associated with RA development are mainly related to imperfect control of lymphocyte activity (PTPN22, CTLA-4, BTLA, and others) (Plenge, 2009; Oki et al., 2011). Due to these SNPs the immune response becomes difficult to manage (Figure 1). The impaired clearance of inflammatory sites caused by an imbalance of the pro-oxidant and anti-oxidant factors and inadequate activity of several enzymes involved in remodeling of the extracellular matrix may also be affected by mutations in the genes of the corresponding factors (Mattey et al., 1999, 2000; Nemec et al., 2006; Ling et al., 2007).
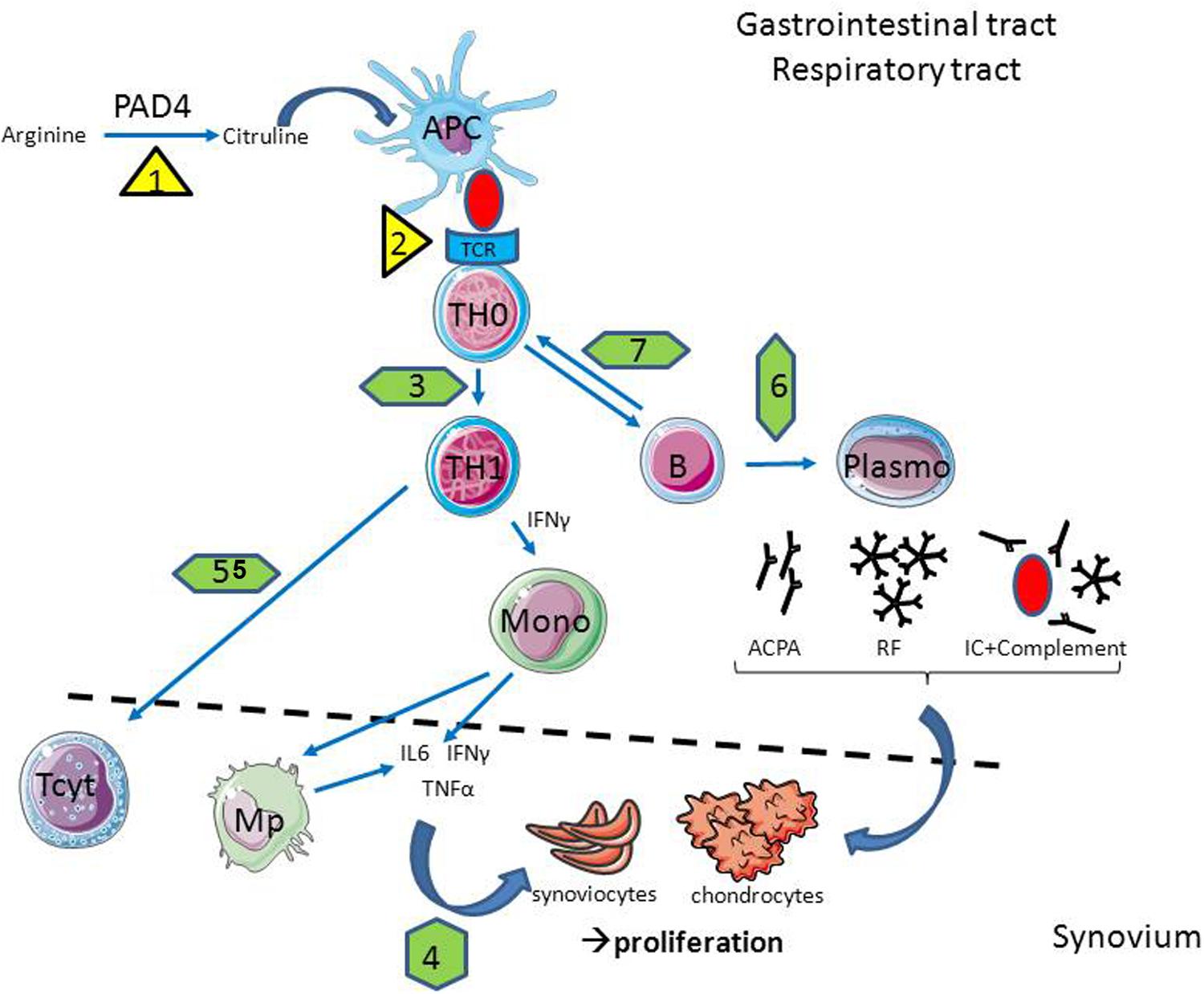
FIGURE 1. In case of infection, the cellular and molecular players of rheumatoid arthritis (RA) are affected. Infections are able to promote citrullinisation (1) and antigen presentation (2) through HLA-DR overexpression in antigen presenting cells (APC); to interfere with T helper (Th) polarization (3) and cytokine production (4); to reduce lymphocytotoxic activity (5) as well as antibody production (6) and antigen presentation by B cells (7). Abbreviations: Mono, Monocyte; Mp – macrophage; TCR, T cell receptor; PAD4, peptidyl arginine deaminase 4; ACPA, anti-citrullinated protein antibodies; IL, interleukine; IFNγ, interferon gamma; TNFα, tumor necrosis factor alpha; IC, immune complex; RF, rheumatoid factor.
For all the diversity of RA-associated gene SNPs, an enrichment of these RA-associated genes was found in particular in two pathways: the NF-κB and the JAK-STAT signaling cascades (Diogo et al., 2014; Messemaker et al., 2015).
The NF-κB pathway (Figure 2) is considered to be a prototypical proinflammatory signaling pathway controlling both RA pathogenesis and viral infection, due to the expression of proinflammatory genes of chemokines, cytokines, receptors, apoptotic regulators, intracellular signaling molecules, and transcription factors (Hinz et al., 2002; Lawrence, 2009). Indeed, stimulation of the NF-κB signaling pathway and inhibition of TNF-related apoptosis (impaired in RA even without this additional exposure) potentiate RA development. Such a pathway could be amplified in the case of infections with CMV and EBV, since the viruses need live, functional and activated “lymphocytes” which could be achieved by controlling the same NF-κB signaling pathway and, in turn, by blocking TNF-related apoptosis (Goodkin et al., 2003).
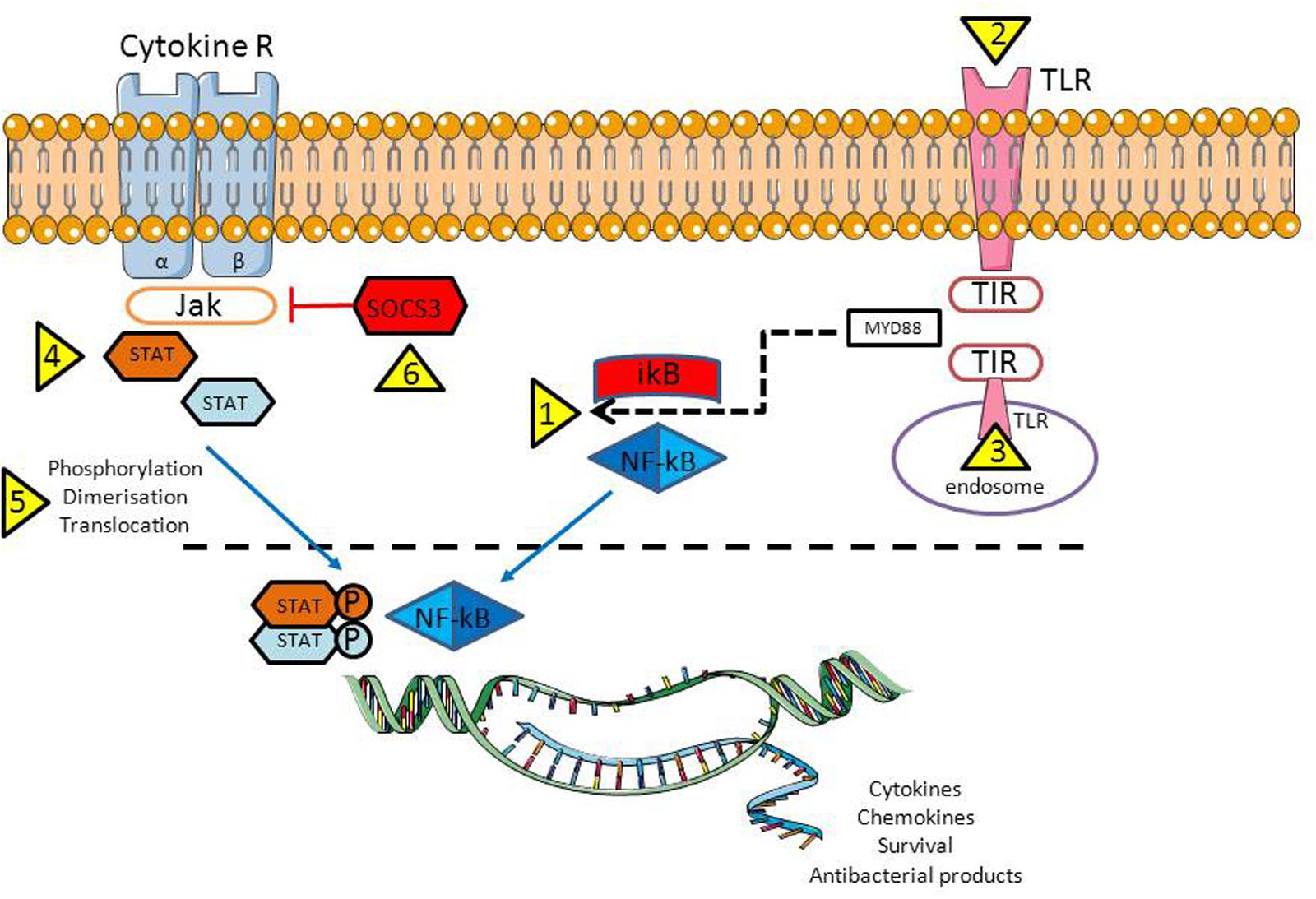
FIGURE 2. Activation of the NF-κB and JAK-STAT signaling pathways are crucial for proper antiviral response. However, in case of infection, bacteria/viruses have a number of opportunities to inhibit both pathways: trigger NF-κB translocation from cytoplasm into the nuclei (1) through plasma membrane (2, bacteria) or endosomal (3, viruses) TLR activation; by activing various STAT family members that being important for the virus replication (4), by controlling STAT activation/translocation to the nucleus (5); and last but not least by reducing the activity of the suppressor of cytokine signaling-3 (SOCS3), a host negative regulator of the JAK/STAT pathway (6). Abbreviations: TLR, Toll like receptor; TIR, Toll/IL-1R; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor kappa B; IkB – inhibitor of the NF-κB kinase; P – phosphate residue (activated form); Cytokine R, a cytokine receptor with β1 and β2 subunits; JAK, Janus tyrosine kinase familly; STAT, signal transducer and activator of transcription family; SOCS3, suppressor of cytokine signaling 3.
STATs are a family of proteins which are latent transcription factors activated by extracellular signaling ligands such as cytokines, growth factors and hormones (Abroun et al., 2015). The principal RA players: IL6, TNF-α as well as IFNs, exert their biological functions through the JAK/STAT signaling pathway (Figure 2), proteins which are being overexpressed in the various cells in RA (Rottapel, 2001; Shouda et al., 2001). STAT proteins become activated in the cytoplasm by Janus kinases (JAK), a family of tyrosine kinases. These signaling pathways have diverse biological functions, which include participating in inflammation and cell differentiation, proliferation, development and apoptosis. In particular, TNF-α production and NF-κB pathway activation can occur through JAK/STAT signaling (Ahmad et al., 2015). At the same time NF-κB positively regulates STAT5a expression and signaling pathways, and promotes persistent activation (Prosch et al., 2003).
Worldwide rates of either HSV-1 or HSV-2 in adults are found to be 60–95% (Chayavichitsilp et al., 2009), and due to such a high level of exposure, some authors have suggested that these viruses should be considered to be part of the normal microbiotic flora (Grinde, 2013). However, diagnosis of an HSV infection based on the presence of viral DNA in blood cells does not provide comprehensive information since HSVs can take up life-time residency in nerve cells during the latency phase and are transported to the mucosa during reactivation phases (Grinde, 2013). The latency versus reactivation strategy of the Herpesviridae is defined by the balance between viral proliferation and the ability of the immune system to clear the virus (Grinde, 2013). During the latent state, the viral genome is packaged by histones and copied by the host cell’s DNA polymerases, along with the chromosomes, primarily when the cell engages in mitosis (Knipe and Cliffe, 2008; Grinde, 2013). This contrasts with lytic replication in which the viral DNA polymerase is engaged, reflecting a viral takeover of the cell. Reactivation may be provoked by various factors, leading to activation of demethylation and histone modification processes, processes that appear to be excessive in RA (Bottini and Firestein, 2013; Wada et al., 2014, de Andres et al., 2015). As a whole, the virus’s tactics to avoid immune detection and establish latency works in a significant population – up to 80% in human adults for HSV-1 and about 40% for HSV2 (Akhtar and Shukla, 2009).
According to our data the ratio of people with clinical signs of HSV exacerbation (criteria reviewed in Grinde, 2013) is significantly increased in the early stage of RA as well as among the first degree relatives of these patients (Arleevskaya et al., 2014). However, when considering HSV1/2 DNA detection in blood, joint tissues and synovial fluid cells, an infrequent presence of HSV1/2 infection has been reported in the patients with early diseases (Zhang et al., 1993; Stahl et al., 2000). In late stage RA, the prevalence of the serum IgG anti-HSV antibodies and the viral DNA presence in the blood and synovial fluid cells were similar to that seen in controls (Us et al., 2011). The data on the serum IgM anti-HSV antibody prevalence in RA are contradictory and could not help resolve the discordance observed between the biology and clinical observations (Kurbanov and Mamedov, 2009; Us et al., 2011).
Since spreading of the virus from cell to cell depends on specific receptors, the presence of these receptors plays a critical role in viral infection and, in turn, in disease exacerbation (Akhtar and Shukla, 2009). Among them, the herpes virus entry mediator (HVEM), a member of the tumor necrosis factor receptor superfamily, serves as one of the entry receptors of HSV (Gavrieli et al., 2006; Cai and Freeman, 2009). In RA, HVEM is overexpressed and contributes to the proliferation and activation of synovial fibroblasts (Ishida et al., 2008). This factor is overexpressed on most cell types found in RA synovial tissues (Kang et al., 2007; Shang et al., 2012). Serum levels of soluble HVEM are increased in RA as well (Jung et al., 2003). So, HVEM overexpression in RA is suspected of contributing to HSV dissemination and to the RA progression directly or indirectly via reception of the viruses.
The epidermal growth factor receptor (EGFR) is another receptor by which HSV enters a cell. EGFR is found to be overexpressed on a variety of synovial tissue cells, and the receptor gene overexpression in bone marrow-derived mononuclear cells is due to an RA-associated SNP (Nakano et al., 2005; Nakamura et al., 2006; Lo et al., 2012; Yuan et al., 2013; Xu et al., 2015).
The capacity of the immune system to effectively control Herpesviridae is determined by the adequacy of the virus (i) to directly interact with the immune system through the pattern recognition receptors (PPR), (ii) to modulate cytokine and chemokine production, and (iii) to control invariant natural killers (iNK) T cells, CD8+ cytotoxic T cells (IFN-γ, TNF-α, cytolytic molecules including perforin and granzymes), and the ability of the immune system to establish and maintain a pool of HSV-specific memory/effector CD8(+) T-lymphocytes (Figure 3).
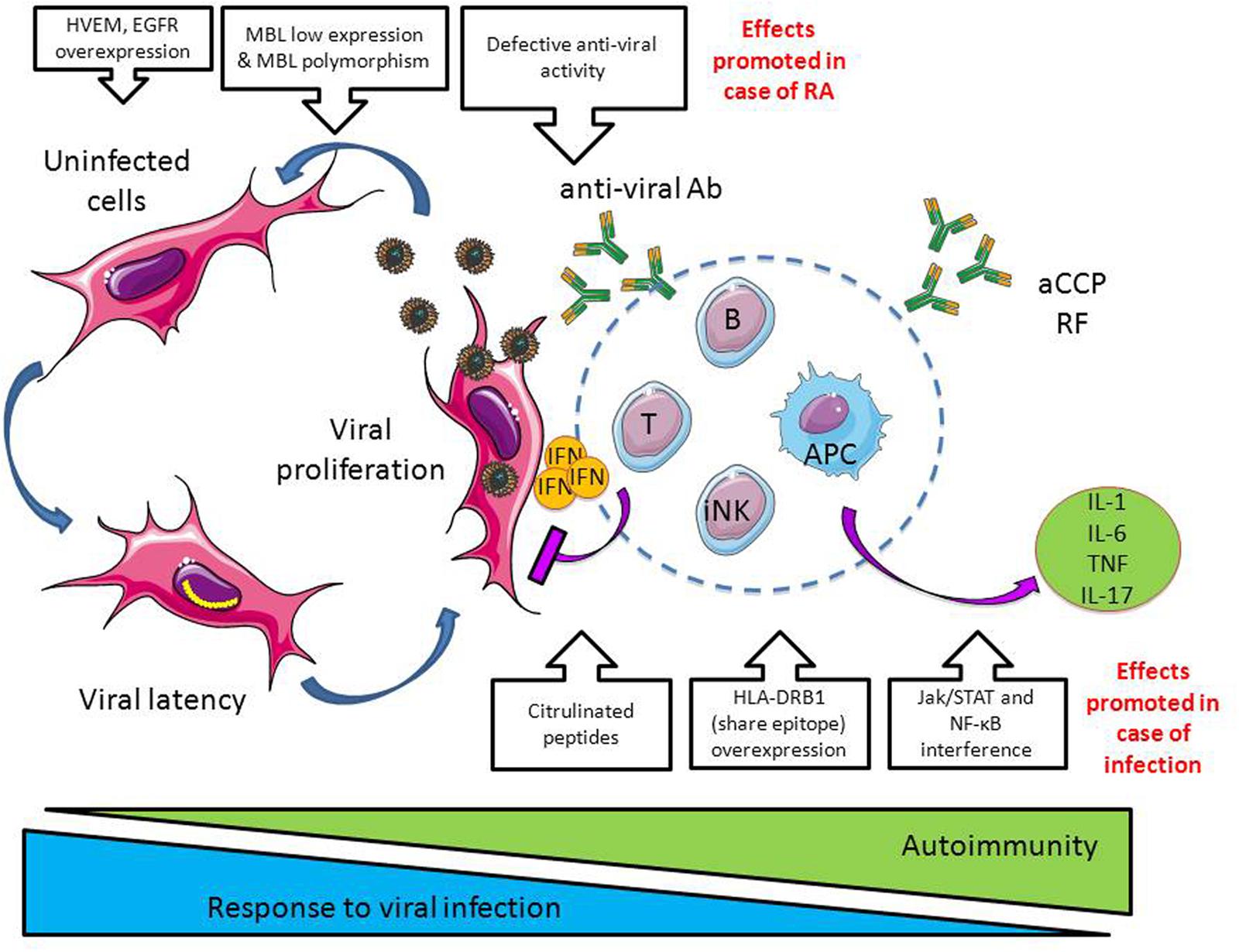
FIGURE 3. Dangerous liaisons between infections and RA. In case of viral infections such as observed with Herpesviridae, the immune system limits viral infection by producing proinflammatory cytokines (e.g., interferon, IFN), anti-viral antibodies and through the activation of cytotoxic cells such as CD8+ T cells and innate natural killer cells (iNK). In case of RA, a defective antiviral activity is observed together with factors promoting Herpesviridae propagation (e.g., herpes virus entry mediator, HVEM, and epidermal growth factor receptor, EGFR, overexpression; downregulation of the opsonic factor MBL, mannose binding lectin). At the opposite, infections promote autoimmunity by inducing protein citrulinisation, by promoting HLA-DR overexpression, and by interfering with the pathways involved in the production of cytokines and chemokines, Jak/STAT and NF-κB (see Figure 2).
The viruses are recognized by PPR, among which the mannose receptors are considered to be the most important (Summerfield et al., 1995; Seppanen et al., 2009). Some preliminary evidence suggests that mannose receptors may be overexpressed on immune cells of myeloid lineage present in blood and synovial tissue from RA patients (Put et al., 2013).
One member of the secreted soluble PPR family, the mannose-binding lectin (MBL), is a complement component and an opsonic factor binding to HSV. Serum MBL levels vary according to the history of the disease. In early RA, low MBL levels were revealed, which are associated with a higher risk of developing early erosive RA and higher levels of IgM RF and CRP (Jacobsen et al., 2001; Saevarsdottir et al., 2001), and later, the MBL levels are increased (Saevarsdottir et al., 2007). Given the likely increase of MBL expression during RA evolution from early to late stage, the data by Olsen et al are of interest, showing that in the early stage of RA, the MBL gene is down regulated more than threefold compared with late stage RA (Olsen et al., 2004). The importance of MBL in RA physiopathology is reinforced by the observation of a nucleotide polymorphism (SNP) at MBL gene associated RA susceptibility (Ip et al., 2000; Jacobsen et al., 2001), and with HSV infection reccurences, possibly due to impaired recognition of the viruses (Seppanen et al., 2009).
The initial stages of Herpesviridae infection are predominantly influenced by the activity of iNK cells to produce type I interferons (IFN), which limit the spread of viruses. The other inhibitors of the infection are macrophages (producing IFNs, TNF-α, and IL-6), and lymphocytes that are instrumental in active immune surveillance by producing IFN-γ (Khanna et al., 2003; Khanna et al., 2004; Egan et al., 2013). Activated CD4+ and CD8+ T cells play a pivotal role in clearing the primary infection (Nash, 2000; Egan et al., 2013). After resolution of the primary infection, a small proportion of the primed specific T cell population generates a stable long-term memory cell pool, which activates during reactivation of a latent infection (Crough et al., 2005). Both nonspecific and specific CD8+ T cells infiltrate and persist within virus infected cells, the specific CD8+ T-lymphocytes being the prevailing ones, expressing a late effector memory phenotype and being activated by stimulation from the infected cells (Borysiewicz et al., 1988; Posavad et al., 2000; van Lint et al., 2005; Verjans et al., 2007; Zhu et al., 2007). It appears that the principal role of B cells in the immune response to Herpesviridae infection is not to produce neutralizing antibodies but instead to present antigens and secrete cytokines (Deshpande et al., 2000; Youinou et al., 2005, 2009; Gazeau et al., 2015).
While the IFN-γ levels were increased in the synovial fluid of early and late stage RA patients, the serum levels of the cytokines were close to that seen in controls (Bucht et al., 1996). Some authors also pointed out that, although IFN-γ was revealed in the synovial fluid and membrane, IFN-γ T cell producers were absent from peripheral blood and locally present in the joint tissues in small quantities (Ridderstad et al., 1991; De Keyser et al., 1995). T-helper 1 (Th1) cells rather than the antiviral CD8+ cytotoxic T-lymphocyte subset represent the main cellular source of IFN-γ in RA (Schuerwegh et al., 1999). Moreover, experiments using killed influenza virus (Berg et al., 2000), EBV lytic/latent peptide epitopes (Klatt et al., 2005) also support a defective capacity of the immune system to produce IFN-γ in response to viral stimulation. As a consequence, it has been proposed that a low serum level of IFN-γ may be an important factor for recurrence and reactivation of the virus (Minami et al., 2002; Davis et al., 2013; Deng et al., 2015; Motamedifar et al., 2015). A link between cytokine production and HLA alleles associated with an increased risk of RA development, HLA DRB1∗0401 and DQ8, has been also established (Taneja, 2015).
At the molecular level, Herpesviridae produce proteins that are effective to control various STAT family members and the translocation of NF-κB from the cytoplasm into the nuclei, which is necessary for (re)activation of the virus (Tsavachidou et al., 2001; Goodkin et al., 2003; Kuchipudi, 2015). As an example, STAT1 regulates the expression of the HSV-1 latency-associated transcript by interacting with its promoter (Kriesel et al., 2004). However, the STAT family proteins also regulate gene expression of a number of antiviral factors. This was elegantly demonstrated in a STAT3-deficient mouse model infected with HSV-1 that produced less IFN-γ and virus-specific CD8+ T cells (Yu et al., 2013). Another consequence of an infection by HSV-1 is its capacity to produce the suppressor of cytokine signaling-3 (SOCS3), a host negative regulator of the JAK/STAT pathway, which, in turn, inhibits the IFN-γ capacity to induce phosphorylation of JAK kinases (Yokota et al., 2004). As a whole, Herpesviridae have the opportunity to inhibit proinflammatory cytokine production (Melroe et al., 2004; Cox et al., 2015) through the control of the T cell response (Toussirot et al., 1999; Toussirot et al., 2000; Klatt et al., 2005). Such effect contributes to the reactivation of the Herpesviridae infection.
Another gene that is remarkably down regulated in early RA is the CC chemokine receptor 1 (CCR1), which regulates leukocyte migration to infection sites. Inhibition of this factor in herpes-infected mice caused a decreased and shortened recruitment of natural killer cells and led to an impaired antiviral response with a significantly higher viral level, inspite of the markedly enhanced levels of pro-inflammatory cytokines (Sorensen and Paludan, 2004). It is noteworthy that Olsen et al. (2004) concluded that, in total, the early RA signature showed some overlap with that seen in the normal immune response to viral antigens.
The iNK subset is a minor population of the innate-like T-lymphocytes. They serve as an early source of cytokines (Opasawatchai and Matangkasombut, 2015). When stimulated by the altered glycolipids of the infected host cells, iNKT rapidly begin to release a variety of cytokines and chemokines, including IFNs and TNF-α, and exert direct cytolysis (Chang et al., 2007; Matsuda et al., 2008; Tessmer et al., 2009; Horst et al., 2012). An adequate response of these cells is required for control of the viral load and protection from the massive tissue damage that can occure in cases of severe viral infection (Grubor-Bauk et al., 2003; Grubor-Bauk et al., 2008). The iNK cells have specific value in keeping the virus in latency.
Several studies have unambiguously reported qualitative and quantitative abnormalities in the iNKT population in early stage RA. Mansour et al. (2015) showed that, in early stage RA, circulating iNKTs were reduced and their frequency was inversely correlated with the disease activity score. The proliferative iNKT response was also defective. Functional iNKT alterations were due to a skewed iNKT-TCR repertoire with a selective reduction of high-affinity clones. Furthermore, the high-affinity iNKTs exhibited an altered functional Th profile with Th1 (in treatment-naive) or Th2-like (in treated patients) phenotype, compared to Th0-like Th profiles exhibited by high-affinity iNKTs in controls. Cell cytotoxicity of iNK in early stage RA is also reduced (Taylor et al., 1993). It is noteworthy that in late stage RA the data, obtained by various authors were controversial: the number of the iNK and their functions were found to be increased, decreased, or didn’t differ from that seen in controls (McChesney and Bankhurst, 1986; Taylor et al., 1993; Aggarwal et al., 2014).
The population of the virus specific CD8+ T-lymphocytes and their cytotoxic activity focused on infected cells or the separate Herpesviridae peptide epitopes is decreased both in the early and late stage RA in contrast to an abundant and hyperactivated whole population of CD8+ T cells of various specificities (Moss et al., 1983; Gaston et al., 1986; Toussirot et al., 2000; Shimojima et al., 2008; Carvalheiro et al., 2015).
Obviously, the ideal immune system response to bacterial/viral aggression should be based on the principles of a reasonable adequacy and a perfect balance between all parts of the immune system. However, for some patients such a delicate equilibrium could be broken, leading to the development of RA. Several factors are suspected for this including:
1. A susceptibility to bacterial and viral infections greater than in the general population;
2. An imbalance of the immune system greater than in the general population;
3. An incapacity to control the inflammatory reactions, and this becomes one of the major factors provoking RA development;
4. Last but not least, disharmony in the relationship of microorganisms and the immune system of an individual predisposed to developing RA, which may be due to both genetic and epigenetic problems.
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are thankful to M. P. Shulaeva (Microbiology Department of Kazan State Medical Academy) and R. Aminov (Technical University of Denmark, Denmark) for consultation on several issues of classification of microorganisms and viruses. Authors express thanks to Dr. W. H. Brooks (University of South Florida, FL, USA) for comments and editorial assistance. We are also grateful to Simone Forest and Geneviève Michel for their help in typing the paper.
Abrahamsen, T. G., Froland, S. S., and Natvig, J. B. (1978). In vitro mitogen stimulation of synovial fluid lymphocytes from rheumatoid arthritis and juvenile rheumatoid arthritis patients: dissociation between the response to antigens and polyclonal mitogens. Scand. J. Immunol. 7, 81–90. doi: 10.1111/j.1365-3083.1978.tb00429.x
Abroun, S., Saki, N., Ahmadvand, M., Asghari, F., Salari, F., and Rahim, F. (2015). STATs: an old story. Yet mesmerizing. Cell J. 17, 395–411.
Acar, K., Aki, S. Z., Ozkurt, Z. N., Bozdayi, G., Rota, S., and Turkoz Sucak, G. (2014). Factors associated with cytomegalovirus reactivation following allogeneic hematopoietic stem cell transplantation: human leukocyte antigens might be among the risk factors. Turk. J. Haematol. 31, 276–285. doi: 10.4274/tjh.2013.0244
Aggarwal, A., Sharma, A., and Bhatnagar, A. (2014). Role of cytolytic impairment of natural killer and natural killer T-cell populations in rheumatoid arthritis. Clin. Rheumatol. 33, 1067–1078. doi: 10.1007/s10067-014-2641-z
Ahmad, S. F., Ansari, M. A., Zoheir, K. M., Bakheet, S. A., Korashy, H. M., Nadeem, A., et al. (2015). Regulation of TNF-alpha and NF-kappaB activation through the JAK/STAT signaling pathway downstream of histamine 4 receptor in a rat model of LPS-induced joint inflammation. Immunobiology 220, 889–898. doi: 10.1016/j.imbio.2015.01.008
Akhtar, J., and Shukla, D. (2009). Viral entry mechanisms: cellular and viral mediators of herpes simplex virus entry. FEBS J. 276, 7228–7236. doi: 10.1111/j.1742-4658.2009.07402.x
Arala-Chaves, M., Lima, M. R., Coutinho, A., Pena-Rossi, C., and Minoprio, P. (1992). V-region-related and -unrelated immunosuppression accompanying infections. Mem. Inst. Oswaldo Cruz 87(Suppl. 5), 35–41. doi: 10.1590/S0074-02761992000900005
Arleevskaya, M. I., Gabdoulkhakova, A. G., Filina, Y. V., Miftakhova, R. R., Bredberg, A., and Tsybulkin, A. P. (2014). A transient peak of infections during onset of rheumatoid arthritis: a 10-year prospective cohort study. BMJ Open 4:e005254. doi: 10.1136/bmjopen-2014-005254
Auger, I., and Roudier, J. (2005). Interaction between HSP73 and HLA-DRB1∗0401: implications for the development of rheumatoid arthritis. Immunol. Res. 31, 261–266. doi: 10.1385/IR:31:3:261
Bach, J. F. (2005). Infections and autoimmune diseases. J. Autoimmun. 25(Suppl.), 74–80. doi: 10.1016/j.jaut.2005.09.024
Becker, H., Weber, C., Renelt, M., and Federlin, K. (1990). [Polyclonal activated circulating B-lymphocytes in patients with rheumatoid arthritis]. Z. Rheumatol. 49, 34–38.
Benedek, T. G. (2006). The history of bacteriologic concepts of rheumatic fever and rheumatoid arthritis. Semin. Arthritis Rheum. 36, 109–123. doi: 10.1016/j.semarthrit.2006.05.001
Berg, L., Ronnelid, J., Sanjeevi, C. B., Lampa, J., and Klareskog, L. (2000). Interferon-gamma production in response to in vitro stimulation with collagen type II in rheumatoid arthritis is associated with HLA-DRB1(∗)0401 and HLA-DQ8. Arthritis Res. 2, 75–84. doi: 10.1186/ar71
Borysiewicz, L. K., Graham, S., Hickling, J. K., Mason, P. D., and Sissons, J. G. (1988). Human cytomegalovirus-specific cytotoxic T cells: their precursor frequency and stage specificity. Eur. J. Immunol. 18, 269–275. doi: 10.1002/eji.1830180214
Bottini, N., and Firestein, G. S. (2013). Epigenetics in rheumatoid arthritis: a primer for rheumatologists. Curr. Rheumatol. Rep. 15:372. doi: 10.1007/s11926-013-0372-9
Brennan, F. M., Smith, N. M., Owen, S., Li, C., Amjadi, P., Green, P., et al. (2008). Resting CD4+ effector memory T cells are precursors of bystander-activated effectors: a surrogate model of rheumatoid arthritis synovial T-cell function. Arthritis Res. Ther. 10:R36. doi: 10.1186/ar2390
Brooks, W. H., Le Dantec, C., Pers, J. O., Youinou, P., and Renaudineau, Y. (2010). Epigenetics and autoimmunity. J. Autoimmun. 34, J207–J219. doi: 10.1016/j.jaut.2009.12.006
Brown, C. M., Fitzgerald, K. J., Moyes, S. P., Mageed, R. A., Williams, D. G., and Maini, R. N. (1995). Sequence analysis of immunoglobulin heavy-chain variable region genes from the synovium of a rheumatoid arthritis patient shows little evidence of mutation but diverse CDR3. Immunology 84, 367–374.
Bucht, A., Larsson, P., Weisbrot, L., Thorne, C., Pisa, P., Smedegard, G., et al. (1996). Expression of interferon-gamma (IFN-gamma), IL-10, IL-12 and transforming growth factor-beta (TGF-beta) mRNA in synovial fluid cells from patients in the early and late phases of rheumatoid arthritis (RA). Clin. Exp. Immunol. 103, 357–367. doi: 10.1111/j.1365-2249.1996.tb08288.x
Bucht, A., Soderstrom, K., Hultman, T., Uhlen, M., Nilsson, E., Kiessling, R., et al. (1992). T cell receptor diversity and activation markers in the V delta 1 subset of rheumatoid synovial fluid and peripheral blood T lymphocytes. Eur. J. Immunol. 22, 567–574. doi: 10.1002/eji.1830220240
Cai, G., and Freeman, G. J. (2009). The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol. Rev. 229, 244–258. doi: 10.1111/j.1600-065X.2009.00783.x
Cambridge, G., Moura, R. A., Santos, T., Khawaja, A. A., Polido-Pereira, J., Canhao, H., et al. (2014). Expression of the inherently autoreactive idiotope 9G4 on autoantibodies to citrullinated peptides and on rheumatoid factors in patients with early and established rheumatoid arthritis. PLoS ONE 9:e107513. doi: 10.1371/journal.pone.0107513
Carter, J. D., Espinoza, L. R., Inman, R. D., Sneed, K. B., Ricca, L. R., Vasey, F. B., et al. (2010). Combination antibiotics as a treatment for chronic Chlamydia-induced reactive arthritis: a double-blind, placebo-controlled, prospective trial. Arthritis Rheum. 62, 1298–1307. doi: 10.1002/art.27394
Carty, S. M., Snowden, N., and Silman, A. J. (2004). Should infection still be considered as the most likely triggering factor for rheumatoid arthritis? Ann. Rheum. Dis. 63(Suppl. 2), ii46–ii49. doi: 10.1136/ard.2004.028241
Carvalheiro, H., Duarte, C., Silva-Cardoso, S., da Silva, J. A., and Souto-Carneiro, M. M. (2015). CD8+ T cell profiles in patients with rheumatoid arthritis and their relationship to disease activity. Arthritis Rheumatol. 67, 363–371. doi: 10.1002/art.38941
Caselli, M., Pazzi, P., LaCorte, R., Aleotti, A., Trevisani, L., and Stabellini, G. (1989). Campylobacter-like organisms, nonsteroidal anti-inflammatory drugs and gastric lesions in patients with rheumatoid arthritis. Digestion 44, 101–104. doi: 10.1159/000199898
Chang, Y. J., Huang, J. R., Tsai, Y. C., Hung, J. T., Wu, D., Fujio, M., et al. (2007). Potent immune-modulating and anticancer effects of NKT cell stimulatory glycolipids. Proc. Natl. Acad. Sci. U.S.A. 104, 10299–10304. doi: 10.1073/pnas.0703824104
Chayavichitsilp, P., Buckwalter, J. V., Krakowski, A. C., and Friedlander, S. F. (2009). Herpes simplex. Pediatr. Rev. 30, 119–129; quiz 130. doi: 10.1542/pir.30-4-119
Cox, J. E., McClure, L. V., Goga, A., and Sullivan, C. S. (2015). Pan-viral-microRNA screening identifies interferon inhibition as a common function of diverse viruses. Proc. Natl. Acad. Sci. U.S.A. 112, 1856–1861. doi: 10.1073/pnas.1417891112
Crough, T., Burrows, J. M., Fazou, C., Walker, S., Davenport, M. P., and Khanna, R. (2005). Contemporaneous fluctuations in T cell responses to persistent herpes virus infections. Eur. J. Immunol. 35, 139–149. doi: 10.1002/eji.200425548
Das, G., Vohra, H., Saha, B., and Mishra, G. C. (2000). Th1-specific bystander costimulation imparts resistance against Mycobacterium tuberculosis infection. Scand. J. Immunol. 52, 515–518. doi: 10.1046/j.1365-3083.2000.00808.x
Davis, J. M., Knutson, K. L., Strausbauch, M. A., Green, A. B., Crowson, C. S., Therneau, T. M., et al. (2013). Immune response profiling in early rheumatoid arthritis: discovery of a novel interaction of treatment response with viral immunity. Arthritis Res. Ther. 15:R199. doi: 10.1186/ar4389
de Andres, M. C., Perez-Pampin, E., Calaza, M., Santaclara, F. J., Ortea, I., Gomez-Reino, J. J., et al. (2015). Assessment of global DNA methylation in peripheral blood cell subpopulations of early rheumatoid arthritis before and after methotrexate. Arthritis Res. Ther. 17:233. doi: 10.1186/s13075-015-0748-5
De Keyser, F., Elewaut, D., Vermeersch, J., De Wever, N., Cuvelier, C., and Veys, E. M. (1995). The role of T cells in rheumatoid arthritis. Clin. Rheumatol. 14(Suppl. 2), 5–9. doi: 10.1007/BF02215850
Deng, X., Crowson, C. S., Rajkumar, S. V., Dispenzieri, A., Larson, D. R., Therneau, T. M., et al. (2015). Elevation of serum immunoglobulin free light chains during the preclinical period of rheumatoid arthritis. J. Rheumatol. 42, 181–187. doi: 10.3899/jrheum.140543
Deshpande, S. P., Kumaraguru, U., and Rouse, B. T. (2000). Dual role of B cells in mediating innate and acquired immunity to herpes simplex virus infections. Cell Immunol. 202, 79–87. doi: 10.1006/cimm.2000.1666
Diogo, D., Okada, Y., and Plenge, R. M. (2014). Genome-wide association studies to advance our understanding of critical cell types and pathways in rheumatoid arthritis: recent findings and challenges. Curr. Opin. Rheumatol. 26, 85–92. doi: 10.1097/BOR.0000000000000012
Doran, M. F., Crowson, C. S., Pond, G. R., O’Fallon, W. M., and Gabriel, S. E. (2002). Predictors of infection in rheumatoid arthritis. Arthritis Rheumatol. 46, 2294–2300. doi: 10.1002/art.10529
Du, J., Liu, J., Gu, J., and Zhu, P. (2007). HLA-DRB1∗09 is associated with increased incidence of cytomegalovirus infection and disease after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 13, 1417–1421. doi: 10.1016/j.bbmt.2007.09.003
Egan, K. P., Wu, S., Wigdahl, B., and Jennings, S. R. (2013). Immunological control of herpes simplex virus infections. J. Neurovirol. 19, 328–345. doi: 10.1007/s13365-013-0189-3
Falkenburg, W. J., van Schaardenburg, D., Ooijevaar-de Heer, P., Wolbink, G., and Rispens, T. (2015). IgG subclass specificity discriminates restricted IgM rheumatoid factor responses from more mature ACPA-associated or isotype-switched IgA responses. Arthritis Rheumatol. 67, 3124–3134. doi: 10.1002/art.39299
Floreani, A., Leung, P. S., and Gershwin, M. E. (2015). Environmental basis of autoimmunity. Clin. Rev. Allergy Immunol. 50, 287–300.
Gaston, J. S., Rickinson, A. B., Yao, Q. Y., and Epstein, M. A. (1986). The abnormal cytotoxic T cell response to Epstein-Barr virus in rheumatoid arthritis is correlated with disease activity and occurs in other arthropathies. Ann. Rheum. Dis. 45, 932–936. doi: 10.1136/ard.45.11.932
Gavrieli, M., Sedy, J., Nelson, C. A., and Murphy, K. M. (2006). BTLA and HVEM cross talk regulates inhibition and costimulation. Adv. Immunol. 92, 157–185. doi: 10.1016/S0065-2776(06)92004-5
Gazeau, P., Devauchelle-Pensec, V., Pochard, P., Pers, J. O., Saraux, A., Renaudineau, Y., et al. (2015). Abatacept efficacy in rheumatoid arthritis is dependent upon baseline blood B-cell levels. Rheumatology (Oxford) 55, 1138–1140. doi: 10.1093/rheumatology/kev417
Germano, V., Cattaruzza, M. S., Osborn, J., Tarantino, A., Di Rosa, R., Salemi, S., et al. (2014). Infection risk in rheumatoid arthritis and spondyloarthropathy patients under treatment with DMARDs, corticosteroids and TNF-alpha antagonists. J. Transl. Med. 12:77. doi: 10.1186/1479-5876-12-77
Goodkin, M. L., Ting, A. T., and Blaho, J. A. (2003). NF-kappaB is required for apoptosis prevention during herpes simplex virus type 1 infection. J. Virol. 77, 7261–7280. doi: 10.1128/JVI.77.13.7261-7280.2003
Gourraud, P. A., Boyer, J. F., Barnetche, T., Abbal, M., Cambon-Thomsen, A., Cantagrel, A., et al. (2006). A new classification of HLA-DRB1 alleles differentiates predisposing and protective alleles for rheumatoid arthritis structural severity. Arthritis Rheumatol. 54, 593–599. doi: 10.1002/art.21630
Gourraud, P. A., Dieudé, P., Boyer, J. F., Nogueira, L., Cambon-Thomsen, A., and Mazières, B. (2007). A new classification of HLA-DRB1 alleles differentiates predisposing and protective alleles for autoantibody production in rheumatoid arthritis. Arthritis Res. Ther. 9:R27. doi: 10.1186/ar2131
Grinde, B. (2013). Herpesviruses: latency and reactivation - viral strategies and host response. J. Oral Microbiol. 5, 1–9. doi: 10.3402/jom.v5i0.22766
Grubor-Bauk, B., Arthur, J. L., and Mayrhofer, G. (2008). Importance of NKT cells in resistance to herpes simplex virus, fate of virus-infected neurons, and level of latency in mice. J. Virol. 82, 11073–11083. doi: 10.1128/JVI.00205-08
Grubor-Bauk, B., Simmons, A., Mayrhofer, G., and Speck, P. G. (2003). Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NKT cells expressing the semivariant V alpha 14-J alpha 281 TCR. J. Immunol. 170, 1430–1434. doi: 10.4049/jimmunol.170.3.1430
Hart, H., and Marmion, B. P. (1977). Rubella virus and rheumatoid arthritis. Ann. Rheum. Dis. 36, 3–12. doi: 10.1136/ard.36.1.3
Helminen-Pakkala, E. (1968). [Periodontal conditions and articular rheumatism are connected. Comparison, I., of the conditions]. Suom. Hammaslaak. Toim. 64, 217–218.
Hermann, E., Mayet, W. J., Klein, O., Lohse, A. W., Trautwein, C., Michiels, I., et al. (1991). Candida arthritis: cellular immune responses of synovial fluid and peripheral blood lymphocytes to Candida albicans. Ann. Rheum. Dis. 50, 697–701. doi: 10.1136/ard.50.10.697
Hill, J. A., Ichim, T. E., Kusznieruk, K. P., Li, M., Huang, X., Yan, X., et al. (2003). Immune modulation by silencing IL-12 production in dendritic cells using small interfering RNA. J. Immunol. 171, 691–696. doi: 10.4049/jimmunol.171.2.691
Hinz, M., Lemke, P., Anagnostopoulos, I., Hacker, C., Krappmann, D., Mathas, S., et al. (2002). Nuclear factor kappaB-dependent gene expression profiling of Hodgkin’s disease tumor cells, pathogenetic significance, and link to constitutive signal transducer and activator of transcription 5a activity. J. Exp. Med. 196, 605–617. doi: 10.1084/jem.20020062
Horowitz, S., Evinson, B., Borer, A., and Horowitz, J. (2000). Mycoplasma fermentans in rheumatoid arthritis and other inflammatory arthritides. J. Rheumatol. 27, 2747–2753.
Horst, D., Geerdink, R. J., Gram, A. M., Stoppelenburg, A. J., and Ressing, M. E. (2012). Hiding lipid presentation: viral interference with CD1d-restricted invariant natural killer T (iNKT) cell activation. Viruses 4, 2379–2399. doi: 10.3390/v4102379
Horwitz, M. S., Bradley, L. M., Harbertson, J., Krahl, T., Lee, J., and Sarvetnick, N. (1998). Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat. Med. 4, 781–785. doi: 10.1038/nm0798-781
Imai, D., Holden, K., Velazquez, E. M., Feng, S., Hodzic, E., and Barthold, S. W. (2013). Influence of arthritis-related protein (BBF01) on infectivity of Borrelia burgdorferi B31. BMC Microbiol. 13:100. doi: 10.1186/1471-2180-13-100
Ioan-Facsinay, A., Willemze, A., Robinson, D. B., Peschken, C. A., Markland, J., van der Woude, D., et al. (2008). Marked differences in fine specificity and isotype usage of the anti-citrullinated protein antibody in health and disease. Arthritis Rheum. 58, 3000–3008. doi: 10.1002/art.23763
Ip, W. K., Lau, Y. L., Chan, S. Y., Mok, C. C., Chan, D., Tong, K. K., et al. (2000). Mannose-binding lectin and rheumatoid arthritis in southern Chinese. Arthritis Rheum. 43, 1679–1687. doi: 10.1002/1529-0131(200008)43:8<1679::AID-ANR3>3.3.CO;2-4
Irving, W. L., Walker, P. R., and Lydyard, P. M. (1985). Abnormal responses of rheumatoid arthritis lymphocytes to Epstein-Barr virus infection in vitro: evidence for multiple defects. Ann. Rheum. Dis. 44, 462–468. doi: 10.1136/ard.44.7.462
Ishida, S., Yamane, S., Ochi, T., Nakano, S., Mori, T., Juji, T., et al. (2008). LIGHT induces cell proliferation and inflammatory responses of rheumatoid arthritis synovial fibroblasts via lymphotoxin beta receptor. J. Rheumatol. 35, 960–968.
Jacobsen, S., Madsen, H. O., Klarlund, M., Jensen, T., Skjodt, H., Jensen, K. E., et al. (2001). The influence of mannose binding lectin polymorphisms on disease outcome in early polyarthritis. TIRA Group. J. Rheumatol. 28, 935–942.
Jonsson, H., and Helgason, J. (1996). Rheumatoid arthritis in an Icelandic textbook from 1782. Scand. J. Rheumatol. 25, 134–137. doi: 10.3109/03009749609080002
Jung, H. W., La, S. J., Kim, J. Y., Heo, S. K., Kim, J. Y., Wang, S., et al. (2003). High levels of soluble herpes virus entry mediator in sera of patients with allergic and autoimmune diseases. Exp. Mol. Med. 35, 501–508. doi: 10.1038/emm.2003.65
Kallio-Laine, K., Seppanen, M., Aittoniemi, J., Kautiainen, H., Seppala, I., Valtonen, V., et al. (2010). HLA-DRB1∗01 allele and low plasma immunoglobulin G1 concentration may predispose to herpes-associated recurrent lymphocytic meningitis. Hum. Immunol. 71, 179–181. doi: 10.1016/j.humimm.2009.10.014
Kang, Y. M., Kim, S. Y., Kang, J. H., Han, S. W., Nam, E. J., Kyung, H. S., et al. (2007). LIGHT up-regulated on B lymphocytes and monocytes in rheumatoid arthritis mediates cellular adhesion and metalloproteinase production by synoviocytes. Arthritis Rheum. 56, 1106–1117. doi: 10.1002/art.22493
Kekik, C., Besisik, S. K., Seyhun, Y., Oguz, F. S., Sargin, D., and Carin, M. N. (2009). Relationship between HLA tissue type, CMV infection, and acute graft-vs-host disease after allogeneic hematopoietic stem cell transplantation: single-center experience. Transplant Proc. 41, 3859–3862. doi: 10.1016/j.transproceed.2009.04.017
Kerlan-Candon, S., Louis-Plence, P., Wiedemann, A., Combe, B., Clot, J., Eliaou, J. F., et al. (2001). Specific overexpression of rheumatoid arthritis-associated HLA-DR alleles and presentation of low-affinity peptides. Arthritis Rheum. 44, 1281–1292. doi: 10.1002/1529-0131(200106)44:6<1281::AID-ART220>3.0.CO;2-2
Khanna, K. M., Bonneau, R. H., Kinchington, P. R., and Hendricks, R. L. (2003). Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity 18, 593–603. doi: 10.1016/S1074-7613(03)00112-2
Khanna, K. M., Lepisto, A. J., Decman, V., and Hendricks, R. L. (2004). Immune control of herpes simplex virus during latency. Curr. Opin. Immunol. 16, 463–469. doi: 10.1016/j.coi.2004.05.003
Kim, B., Kaistha, S. D., and Rouse, B. T. (2006). Viruses and autoimmunity. Autoimmunity 39, 71–77. doi: 10.1080/08916930500484708
Kim, H. R., Kim, E. Y., Cerny, J., and Moudgil, K. D. (2006). Antibody responses to mycobacterial and self heat shock protein 65 in autoimmune arthritis: epitope specificity and implication in pathogenesis. J. Immunol. 177, 6634–6641. doi: 10.4049/jimmunol.177.10.6634
Kivity, S., Agmon-Levin, N., Blank, M., and Shoenfeld, Y. (2009). Infections and autoimmunity–friends or foes? Trends Immunol. 30, 409–414. doi: 10.1016/j.it.2009.05.005
Klatt, T., Ouyang, Q., Flad, T., Koetter, I., Buhring, H. J., Kalbacher, H., et al. (2005). Expansion of peripheral CD8+ CD28- T cells in response to Epstein-Barr virus in patients with rheumatoid arthritis. J. Rheumatol. 32, 239–251.
Knipe, D. M., and Cliffe, A. (2008). Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 6, 211–221. doi: 10.1038/nrmicro1794
Konsta, O. D., Le Dantec, C., Brooks, W. H., and Renaudineau, Y. (2015). Genetics and epigenetics of autoimmune diseases. eLS 1–9. doi: 10.1002/9780470015902.a0023593
Kriesel, J. D., Jones, B. B., Dahms, K. M., and Spruance, S. L. (2004). STAT1 binds to the herpes simplex virus type 1 latency-associated transcript promoter. J. Neurovirol. 10, 12–20. doi: 10.1080/13550280490261680
Kuchipudi, S. V. (2015). The complex role of STAT3 in viral infections. J. Immunol. Res. 2015:272359. doi: 10.1155/2015/272359
Kurbanov, S. A., and Mamedov, M. K. (2009). [Serological markers of viral infections in patients with rheumatoid arthritis]. Georgian Med. News 166, 65–67.
Lawrence, T. (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1:a001651. doi: 10.1101/cshperspect.a001651
Le Dantec, C., Gazeau, P., Mukherjee, S., Brooks, W. H., and Renaudineau, Y. (2015a). “How the environment influences epigenetics, DNA methylation, and autoimmune diseases,” in Epigenetics and Dermatology, eds Q. Lu, C. C. Chang, and B. C. Richardson (Amsterdam: Elsevier), 467–485.
Le Dantec, C., Vallet, S., Brooks, W. H., and Renaudineau, Y. (2015b). Human endogenous retrovirus group E and its involvement in diseases. Viruses 7, 1238–1257. doi: 10.3390/v7031238
Leirisalo-Repo, M. (2005). Early arthritis and infection. Curr. Opin. Rheumatol. 17, 433–439. doi: 10.1097/01.bor.0000166388.47604.8b
Lemerle, J., Arleevskaya, M. I., Brooks, W. H., and Renaudineau, Y. (2016). Effects of environmental factors and omega-3 fatty acids on rheumatoid arthritis. Ann. Joint 1, 1–4. doi: 10.21037/aoj.2016.06.02
Li, S., Yu, Y., Yue, Y., Zhang, Z., and Su, K. (2013). Microbial Infection and Rheumatoid Arthritis. J. Clin. Cell Immunol. 4:174.
Ling, S., Li, Z., Borschukova, O., Xiao, L., Pumpens, P., and Holoshitz, J. (2007). The rheumatoid arthritis shared epitope increases cellular susceptibility to oxidative stress by antagonizing an adenosine-mediated anti-oxidative pathway. Arthritis Res. Ther. 9:R5. doi: 10.1186/ar2111
Lo, S. F., Wan, L., Lin, H. C., Huang, C. M., Chen, S. Y., Liu, S. C., et al. (2012). Association of rheumatoid arthritis risk with EGFR genetic polymorphisms in Taiwan’s Han Chinese population. Rheumatol. Int. 32, 2301–2306. doi: 10.1007/s00296-011-1961-4
Lundberg, K., Wegner, N., Yucel-Lindberg, T., and Venables, P. J. (2010). Periodontitis in RA-the citrullinated enolase connection. Nat. Rev. Rheumatol. 6, 727–730. doi: 10.1038/nrrheum.2010.139
Mansour, S., Tocheva, A. S., Sanderson, J. P., Goulston, L. M., Platten, H., Serhal, L., et al. (2015). Structural and functional changes of the invariant NKT clonal repertoire in early rheumatoid arthritis. J. Immunol. 195, 5582–5591. doi: 10.4049/jimmunol.1501092
Maresz, K. J., Hellvard, A., Sroka, A., Adamowicz, K., Bielecka, E., Koziel, J., et al. (2013). Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLoS Pathog. 9:e1003627. doi: 10.1371/journal.ppat.1003627
Martinez-Martinez, R. E., Abud-Mendoza, C., Patino-Marin, N., Rizo-Rodriguez, J. C., Little, J. W., and Loyola-Rodríguez, J. P. (2009). Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J. Clin. Periodontol. 36, 1004–1010. doi: 10.1111/j.1600-051X.2009.01496.x
Maslova, I. I., Khorobrykh, N. E., Ushakova, M. A., Ovechko, N. N., and Murav’ev Iu, V. (2004). [Antibodies to Yersinia enterocolitica and Proteus mirabilis in blood sera of rheumatoid arthritis patients]. Zh. Mikrobiol. Epidemiol. Immunobiol. 4, 71–72.
Matsuda, J. L., Mallevaey, T., Scott-Browne, J., and Gapin, L. (2008). CD1d-restricted iNKT cells, the ‘Swiss-Army knife’ of the immune system. Curr. Opin. Immunol. 20, 358–368. doi: 10.1016/j.coi.2008.03.018
Mattey, D. L., Hassell, A. B., Dawes, P. T., Jones, P. W., Yengi, L., Alldersea, J., et al. (2000). Influence of polymorphism in the manganese superoxide dismutase locus on disease outcome in rheumatoid arthritis: evidence for interaction with glutathione S-transferase genes. Arthritis Rheum. 43, 859–864. doi: 10.1002/1529-0131(200004)43:4<859::AID-ANR17>3.0.CO;2-Y
Mattey, D. L., Hassell, A. B., Plant, M., Dawes, P. T., Ollier, W. R., Jones, P. W., et al. (1999). Association of polymorphism in glutathione S-transferase loci with susceptibility and outcome in rheumatoid arthritis: comparison with the shared epitope. Ann. Rheum. Dis. 58, 164–168. doi: 10.1136/ard.58.3.164
McChesney, M. B., and Bankhurst, A. D. (1986). Cytotoxic mechanisms in vitro against Epstein-Barr virus infected lymphoblastoid cell lines in rheumatoid arthritis. Ann. Rheum. Dis. 45, 546–552. doi: 10.1136/ard.45.7.546
McSorley, S. J. (2014). The role of non-cognate T Cell stimulation during intracellular bacterial infection. Front. Immunol. 5:319. doi: 10.3389/fimmu.2014.00319
Melroe, G. T., DeLuca, N. A., and Knipe, D. M. (2004). Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 78, 8411–8420. doi: 10.1128/JVI.78.16.8411-8420.2004
Messemaker, T. C., Huizinga, T. W., and Kurreeman, F. (2015). Immunogenetics of rheumatoid arthritis: understanding functional implications. J. Autoimmun. 64, 74–81. doi: 10.1016/j.jaut.2015.07.007
Miller, S. D., Vanderlugt, C. L., Begolka, W. S., Pao, W., Yauch, R. L., Neville, K. L., et al. (1997). Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 3, 1133–1136. doi: 10.1038/nm1097-1133
Minami, M., Kita, M., Yan, X. Q., Yamamoto, T., Iida, T., Sekikawa, K., et al. (2002). Role of IFN-gamma and tumor necrosis factor-alpha in herpes simplex virus type 1 infection. J. Interferon Cytokine Res. 22, 671–676. doi: 10.1089/10799900260100150
Moss, D. J., Klestov, A., Burrows, S., and Kane, R. G. (1983). A comparison of Epstein-Barr virus-specific T-cell immunity in rheumatoid arthritis and osteoarthritis patients. Aust. J. Exp. Biol. Med. Sci. 61(Pt 5), 509–516. doi: 10.1038/icb.1983.48
Motamedifar, M., Sarvari, J., Ebrahimpour, A., and Emami, A. (2015). Symptomatic reactivation of HSV infection correlates with decreased serum levels of TNF-alpha. Iran J. Immunol. 12, 27–34.
Moyes, S. P., Brown, C. M., Scott, B. B., Maini, R. N., and Mageed, R. A. (1996). Analysis of V kappa genes in rheumatoid arthritis (RA) synovial B lymphocytes provides evidence for both polyclonal activation and antigen-driven selection. Clin. Exp. Immunol 105, 89–98. doi: 10.1046/j.1365-2249.1996.d01-735.x
Nakamura, N., Shimaoka, Y., Tougan, T., Onda, H., Okuzaki, D., Zhao, H., et al. (2006). Isolation and expression profiling of genes upregulated in bone marrow-derived mononuclear cells of rheumatoid arthritis patients. DNA Res. 13, 169–183. doi: 10.1093/dnares/dsl006
Nakano, K., Asano, R., Tsumoto, K., Kwon, H., Goins, W. F., Kumagai, I., et al. (2005). Herpes simplex virus targeting to the EGF receptor by a gD-specific soluble bridging molecule. Mol. Ther. 11, 617–626. doi: 10.1016/j.ymthe.2004.12.012
Nash, A. A. (2000). T cells and the regulation of herpes simplex virus latency and reactivation. J. Exp. Med. 191, 1455–1458. doi: 10.1084/jem.191.9.1455
Nemec, P., Goldbergova, M., Swobodnik, T., Polaskova, D., Soucek, M., and Vasku, A. (2006). [Polymorphism of gene promotor region for MMP-2 in rheumatoid arthritis]. Vnitr Lek 52, 348–354.
Newkirk, M. M., Goldbach-Mansky, R., Senior, B. W., Klippel, J., Schumacher, H. R. Jr., and El-Gabalawy, H. S. (2005). Elevated levels of IgM and IgA antibodies to Proteus mirabilis and IgM antibodies to Escherichia coli are associated with early rheumatoid factor (RF)-positive rheumatoid arthritis. Rheumatology (Oxford) 44, 1433–1441. doi: 10.1093/rheumatology/kei036
O’Donnell, H., and McSorley, S. J. (2014). Salmonella as a model for non-cognate Th1 cell stimulation. Front. Immunol. 5:621. doi: 10.3389/fimmu.2014.00621
Ogrendik, M. (2009). Rheumatoid arthritis is linked to oral bacteria: etiological association. Mod. Rheumatol. 19, 453–456. doi: 10.1007/s10165-009-0194-9
Ohnishi, Y., Tsutsumi, A., Matsumoto, I., Goto, D., Ito, S., Kuwana, M., et al. (2006). Altered peptide ligands control type II collagen-reactive T cells from rheumatoid arthritis patients. Mod. Rheumatol. 16, 226–228. doi: 10.3109/s10165-006-0495-1
Oki, M., Watanabe, N., Owada, T., Oya, Y., Ikeda, K., Saito, Y., et al. (2011). A functional polymorphism in B and T lymphocyte attenuator is associated with susceptibility to rheumatoid arthritis. Clin. Dev. Immunol. 2011:305656. doi: 10.1155/2011/305656
Olsen, N., Sokka, T., Seehorn, C. L., Kraft, B., Maas, K., Moore, J., et al. (2004). A gene expression signature for recent onset rheumatoid arthritis in peripheral blood mononuclear cells. Ann. Rheum. Dis. 63, 1387–1392. doi: 10.1136/ard.2003.017194
Opasawatchai, A., and Matangkasombut, P. (2015). iNKT cells and their potential lipid ligands during viral infection. Front. Immunol. 6:378. doi: 10.3389/fimmu.2015.00378
Outschoorn, I., Rowley, M. J., Cook, A. D., and Mackay, I. R. (1993). Subclasses of immunoglobulins and autoantibodies in autoimmune diseases. Clin. Immunol. Immunopathol. 66, 59–66. doi: 10.1006/clin.1993.1008
Pardo, I., Carafa, C., Dziarski, R., and Levinson, A. I. (1984). Analysis of in vitro polyclonal B cell differentiation responses to bacterial peptidoglycan and pokeweed mitogen in rheumatoid arthritis. Clin. Exp. Immunol. 56, 253–262.
Petersen, J. (1988). B lymphocyte function in patients with rheumatoid arthritis: impact of regulatory T lymphocytes and macrophages–modulation by antirheumatic drugs. Dan. Med. Bull. 35, 140–157.
Phillips, P. E. (1986). Infectious agents in the pathogenesis of rheumatoid arthritis. Semin. Arthritis Rheum. 16, 1–10. doi: 10.1016/0049-0172(86)90038-7
Plenge, R. M. (2009). Recent progress in rheumatoid arthritis genetics: one step towards improved patient care. Curr. Opin. Rheumatol. 21, 262–271. doi: 10.1097/BOR.0b013e32832a2e2d
Posavad, C. M., Huang, M. L., Barcy, S., Koelle, D. M., and Corey, L. (2000). Long term persistence of herpes simplex virus-specific CD8+ CTL in persons with frequently recurring genital herpes. J. Immunol. 165, 1146–1152. doi: 10.4049/jimmunol.165.2.1146
Powell, A. M., and Black, M. M. (2001). Epitope spreading: protection from pathogens, but propagation of autoimmunity? Clin. Exp. Dermatol. 26, 427–433. doi: 10.1046/j.1365-2230.2001.00852.x
Prosch, S., Priemer, C., Hoflich, C., Liebenthaf, C., Babel, N., Kruger, D. H., et al. (2003). Proteasome inhibitors: a novel tool to suppress human cytomegalovirus replication and virus-induced immune modulation. Antivir. Ther. 8, 555–567.
Put, S., Schoonooghe, S., Devoogdt, N., Schurgers, E., Avau, A., Mitera, T., et al. (2013). SPECT imaging of joint inflammation with Nanobodies targeting the macrophage mannose receptor in a mouse model for rheumatoid arthritis. J. Nucl. Med. 54, 807–814. doi: 10.2967/jnumed.112.111781
Quirke, A. M., Perry, E., Cartwright, A., Kelly, C., De Soyza, A., Eggleton, P., et al. (2015). Bronchiectasis is a model for chronic bacterial infection inducing autoimmunity in rheumatoid arthritis. Arthritis Rheumatol. 67, 2335–2342. doi: 10.1002/art.39226
Renaudineau, Y., Jamin, C., Saraux, A., and Youinou, P. (2005). Rheumatoid factor on a daily basis. Autoimmunity 38, 11–16. doi: 10.1080/08916930400022574
Ridderstad, A., Abedi-Valugerdi, M., and Moller, E. (1991). Cytokines in rheumatoid arthritis. Ann. Med. 23, 219–223. doi: 10.3109/07853899109148051
Rosloniec, E. F., Latham, K., and Guedez, Y. B. (2002a). Paradoxical roles of IFN-gamma in models of Th1-mediated autoimmunity. Arthritis Res. 4, 333–336. doi: 10.1186/ar432
Rosloniec, E. F., Whittington, K. B., Zaller, D. M., and Kang, A. H. (2002b). HLA-DR1 (DRB1∗0101) and DR4 (DRB1∗0401) use the same anchor residues for binding an immunodominant peptide derived from human type II collagen. J. Immunol. 168, 253–259. doi: 10.4049/jimmunol.168.1.253
Rottapel, R. (2001). Putting the brakes on arthritis: can suppressors of cytokine signaling (SOCS) suppress rheumatoid arthritis? J. Clin. Invest. 108, 1745–1747. doi: 10.1172/JCI200114661
Saal, J. G., Krimmel, M., Steidle, M., Gerneth, F., Wagner, S., Fritz, P., et al. (1999). Synovial Epstein-Barr virus infection increases the risk of rheumatoid arthritis in individuals with the shared HLA-DR4 epitope. Arthritis Rheum. 42, 1485–1496. doi: 10.1002/1529-0131(199907)42:7<1485::AID-ANR24>3.0.CO;2-7
Saevarsdottir, S., Steinsson, K., Grondal, G., and Valdimarsson, H. (2007). Patients with rheumatoid arthritis have higher levels of mannan-binding lectin than their first-degree relatives and unrelated controls. J. Rheumatol. 34, 1692–1695.
Saevarsdottir, S., Vikingsdottir, T., Vikingsson, A., Manfredsdottir, V., Geirsson, A. J., and Valdimarsson, H. (2001). Low mannose binding lectin predicts poor prognosis in patients with early rheumatoid arthritis. A prospective study. J. Rheumatol. 28, 728–734.
Sandberg, M. E., Bengtsson, C., Klareskog, L., Alfredsson, L., and Saevarsdottir, S. (2015). Recent infections are associated with decreased risk of rheumatoid arthritis: a population-based case-control study. Ann. Rheum. Dis. 74, 904–907. doi: 10.1136/annrheumdis-2014-206493
Schuerwegh, A. J., De Clerck, L. S., De Schutter, L., Bridts, C. H., Verbruggen, A., and Stevens, W. J. (1999). Flow cytometric detection of type 1 (IL-2, IFN-gamma) and type 2 (IL-4, IL-5) cytokines in T-helper and T-suppressor/cytotoxic cells in rheumatoid arthritis, allergic asthma and atopic dermatitis. Cytokine 11, 783–788. doi: 10.1006/cyto.1998.0483
Sebbag, M., Parry, S. L., Brennan, F. M., and Feldmann, M. (1997). Cytokine stimulation of T lymphocytes regulates their capacity to induce monocyte production of tumor necrosis factor-alpha, but not interleukin-10: possible relevance to pathophysiology of rheumatoid arthritis. Eur. J. Immunol. 27, 624–632. doi: 10.1002/eji.1830270308
Seppanen, M., Lokki, M. L., Lappalainen, M., Hiltunen-Back, E., Rovio, A. T., Kares, S., et al. (2009). Mannose-binding lectin 2 gene polymorphism in recurrent herpes simplex virus 2 infection. Hum. Immunol. 70, 218–221. doi: 10.1016/j.humimm.2009.01.022
Shadidi, K. R., Thompson, K. M., Henriksen, J. E., Natvig, J. B., and Aarvak, T. (2002). Association of antigen specificity and migratory capacity of memory T cells in rheumatoid arthritis. Scand. J. Immunol. 55, 274–283. doi: 10.1046/j.0300-9475.2002.01036.x
Shang, Y., Guo, G., Cui, Q., Li, J., Ruan, Z., and Chen, Y. (2012). The expression and anatomical distribution of BTLA and its ligand HVEM in rheumatoid synovium. Inflammation 35, 1102–1112. doi: 10.1007/s10753-011-9417-2
Shimojima, Y., Matsuda, M., Ishii, W., Gono, T., and Ikeda, S. (2008). Analysis of peripheral blood lymphocytes using flow cytometry in polymyalgia rheumatica, RS3PE and early rheumatoid arthritis. Clin. Exp. Rheumatol. 26, 1079–1082.
Shouda, T., Yoshida, T., Hanada, T., Wakioka, T., Oishi, M., Miyoshi, K., et al. (2001). Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J. Clin. Invest. 108, 1781–1788. doi: 10.1172/JCI13568
Silman, A. J., and Pearson, J. E. (2002). Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 4(Suppl. 3), S265–S272. doi: 10.1186/ar578
Soderlin, M. K., Bergsten, U., Svensson, B., and BARFOT Study Group (2011). Patient-reported events preceding the onset of rheumatoid arthritis: possible clues to aetiology. Musculoskeletal Care 9, 25–31. doi: 10.1002/msc.193
Sorensen, L. N., and Paludan, S. R. (2004). Blocking CC chemokine receptor (CCR) 1 and CCR5 during herpes simplex virus type 2 infection in vivo impairs host defence and perturbs the cytokine response. Scand. J. Immunol. 59, 321–333. doi: 10.1111/j.0300-9475.2004.01399.x
Soulas, P., Woods, A., Jaulhac, B., Knapp, A. M., Pasquali, J. L., Martin, T., et al. (2005). Autoantigen, innate immunity, and T cells cooperate to break B cell tolerance during bacterial infection. J. Clin. Invest. 115, 2257–2267. doi: 10.1172/JCI24646
Stahl, H. D., Hubner, B., Seidl, B., Liebert, U. G., van der Heijden, I. M., Wilbrink, B., et al. (2000). Detection of multiple viral DNA species in synovial tissue and fluid of patients with early arthritis. Ann. Rheum. Dis. 59, 342–346. doi: 10.1136/ard.59.5.342
Summerfield, J. A., Ryder, S., Sumiya, M., Thursz, M., Gorchein, A., Monteil, M. A., et al. (1995). Mannose binding protein gene mutations associated with unusual and severe infections in adults. Lancet 345, 886–889. doi: 10.1016/S0140-6736(95)90009-8
Sutliff, W. D., Shepard, R., and Dunham, W. B. (1953). Acute Leptospira pomona arthritis and myocarditis. Ann. Intern. Med. 39, 134–140. doi: 10.7326/0003-4819-39-1-134
Taneja, V. (2015). Cytokines pre-determined by genetic factors are involved in pathogenesis of Rheumatoid arthritis. Cytokine 75, 216–221. doi: 10.1016/j.cyto.2014.11.028
Taylor, H. G., Nixon, N. B., and Dawes, P. T. (1993). Natural killer cell cytotoxicity and alpha-interferon in early rheumatoid arthritis. Scand. J. Rheumatol. 22, 280–283. doi: 10.3109/03009749309095139
Tessmer, M. S., Fatima, A., Paget, C., Trottein, F., and Brossay, L. (2009). NKT cell immune responses to viral infection. Expert Opin. Ther. Targets 13, 153–162. doi: 10.1517/14712590802653601
Tian, X., Jiang, M., and Qiu, C. (1998). HLA-DRB1 genes in 5 rheumatic disease multi-case families. Chin. Med. J. (Engl.) 111, 208–210.
Toubi, E., and Shoenfeld, Y. (2004). Toll-like receptors and their role in the development of autoimmune diseases. Autoimmunity 37, 183–188. doi: 10.1080/08916930410001704944
Toussirot, E., Auger, I., Roudier, C., Luka, J., Wendling, D., Tiberghien, P., et al. (1999). HLA-DR polymorphism influences T-cell precursor frequencies to Epstein-Barr virus (EBV) gp110: implications for the association of HLA-DR antigens with rheumatoid arthritis. Tissue Antigens 54, 146–152. doi: 10.1034/j.1399-0039.1999.540205.x
Toussirot, E., Wendling, D., Tiberghien, P., Luka, J., and Roudier, J. (2000). Decreased T cell precursor frequencies to Epstein-Barr virus glycoprotein Gp110 in peripheral blood correlate with disease activity and severity in patients with rheumatoid arthritis. Ann. Rheum. Dis. 59, 533–538. doi: 10.1136/ard.59.7.533
Trost, B., Lucchese, G., Stufano, A., Bickis, M., Kusalik, A., and Kanduc, D. (2010). No human protein is exempt from bacterial motifs, not even one. Self Nonself 1, 328–334. doi: 10.4161/self.1.4.13315
Tsavachidou, D., Podrzucki, W., Seykora, J., and Berger, S. L. (2001). Gene array analysis reveals changes in peripheral nervous system gene expression following stimuli that result in reactivation of latent herpes simplex virus type 1: induction of transcription factor Bcl-3. J. Virol. 75, 9909–9917. doi: 10.1128/JVI.75.20.9909-9917.2001
Unutmaz, D., Pileri, P., and Abrignani, S. (1994). Antigen-independent activation of naive and memory resting T cells by a cytokine combination. J. Exp. Med. 180, 1159–1164. doi: 10.1084/jem.180.3.1159
Us, T., Cetin, E., Kasifoglu, N., Kasifoglu, T., and Akgun, Y. (2011). [Investigation of Epstein-Barr virus and herpes simplex virus markers by serological and molecular methods in patients with rheumatoid arthritis and systemic lupus erythematosus]. Mikrobiyol. Bul. 45, 677–683.
van Lint, A. L., Kleinert, L., Clarke, S. R., Stock, A., Heath, W. R., and Carbone, F. R. (2005). Latent infection with herpes simplex virus is associated with ongoing CD8+ T-cell stimulation by parenchymal cells within sensory ganglia. J. Virol. 79, 14843–14851. doi: 10.1128/JVI.79.23.14843-14851.2005
Vandenbroucke, J. P., Kaaks, R., Valkenburg, H. A., Boersma, J. W., Cats, A., Festen, J. J., et al. (1987). Frequency of infections among rheumatoid arthritis patients, before and after disease onset. Arthritis Rheum. 30, 810–813. doi: 10.1002/art.1780300711
Verjans, G. M., Hintzen, R. Q., van Dun, J. M., Poot, A., Milikan, J. C., Laman, J. D., et al. (2007). Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc. Natl. Acad. Sci. U.S.A. 104, 3496–3501. doi: 10.1073/pnas.0610847104
Verpoort, K. N., Jol-van der Zijde, C. M., Papendrecht-van der Voort, E. A., Ioan-Facsinay, A., Drijfhout, J. W., and van Tol, M. J. (2006). Isotype distribution of anti-cyclic citrullinated peptide antibodies in undifferentiated arthritis and rheumatoid arthritis reflects an ongoing immune response. Arthritis Rheum. 54, 3799–3808. doi: 10.1002/art.22279
Vojdani, A. (2014). A potential link between environmental triggers and autoimmunity. Autoimmune Dis. 2014:437231. doi: 10.1155/2014/437231
Wada, T. T., Araki, Y., Sato, K., Aizaki, Y., Yokota, K., Kim, Y. T., et al. (2014). Aberrant histone acetylation contributes to elevated interleukin-6 production in rheumatoid arthritis synovial fibroblasts. Biochem. Biophys. Res. Commun. 444, 682–686. doi: 10.1016/j.bbrc.2014.01.195
Weyand, C. M., Klimiuk, P. A., and Goronzy, J. J. (1998). Heterogeneity of rheumatoid arthritis: from phenotypes to genotypes. Springer Semin. Immunopathol. 20, 5–22. doi: 10.1007/BF00831996
Widdifield, J., Bernatsky, S., Paterson, J. M., Gunraj, N., Thorne, J. C., Pope, J., et al. (2013). Serious infections in a population-based cohort of 86,039 seniors with rheumatoid arthritis. Arthritis Care Res. (Hoboken) 65, 353–361. doi: 10.1002/acr.21812
Wilson, C., Tiwana, H., and Ebringer, A. (2000). Molecular mimicry between HLA-DR alleles associated with rheumatoid arthritis and Proteus mirabilis as the Aetiological basis for autoimmunity. Microbes Infect. 2, 1489–1496. doi: 10.1016/S1286-4579(00)01303-4
Woods, A., Monneaux, F., Soulas-Sprauel, P., Muller, S., Martin, T., Korganow, A. S., et al. (2007). Influenza virus-induced type I interferon leads to polyclonal B-cell activation but does not break down B-cell tolerance. J. Virol. 81, 12525–12534. doi: 10.1128/JVI.00839-07
Wu, H. J., Ivanov, I. I., Darce, J., Hattori, K., Shima, T., Umesaki, Y., et al. (2010). Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32, 815–827. doi: 10.1016/j.immuni.2010.06.001
Xu, Y., Huang, Y., Cai, D., Liu, J., and Cao, X. (2015). Analysis of differences in the molecular mechanism of rheumatoid arthritis and osteoarthritis based on integration of gene expression profiles. Immunol. Lett. 168, 246–253. doi: 10.1016/j.imlet.2015.09.011
Yamamoto, K., Sakoda, H., Nakajima, T., Kato, T., Okubo, M., Dohi, M., et al. (1992). Accumulation of multiple T cell clonotypes in the synovial lesions of patients with rheumatoid arthritis revealed by a novel clonality analysis. Int. Immunol. 4, 1219–1223. doi: 10.1093/intimm/4.11.1219
Yokota, S., Yokosawa, N., Okabayashi, T., Suzutani, T., Miura, S., Jimbow, K., et al. (2004). Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 78, 6282–6286. doi: 10.1128/JVI.78.12.6282-6286.2004
Youinou, P., Jamin, C., Pers, J. O., Berthou, C., Saraux, A., and Renaudineau, Y. (2005). B lymphocytes are required for development and treatment of autoimmune diseases. Ann. N. Y. Acad. Sci. 1050, 19–33. doi: 10.1196/annals.1313.003
Youinou, P., Taher, T. E., Pers, J. O., Mageed, R. A., and Renaudineau, Y. (2009). B lymphocyte cytokines and rheumatic autoimmune disease. Arthritis Rheum. 60, 1873–1880. doi: 10.1002/art.24665
Yu, C. R., Dambuza, I. M., Lee, Y. J., Frank, G. M., and Egwuagu, C. E. (2013). STAT3 regulates proliferation and survival of CD8+ T cells: enhances effector responses to HSV-1 infection, and inhibits IL-10+ regulatory CD8+ T cells in autoimmune uveitis. Mediators Inflamm. 2013:359674. doi: 10.1155/2013/359674
Yuan, F. L., Li, X., Lu, W. G., Sun, J. M., Jiang, D. L., and Xu, R. S. (2013). Epidermal growth factor receptor (EGFR) as a therapeutic target in rheumatoid arthritis. Clin. Rheumatol. 32, 289–292. doi: 10.1007/s10067-012-2119-9
Zahiri Yeganeh, S., Ataee, R. A., Alishiri, G. H., and Movahedi, M. (2015). Bacteriological and molecular assessment of staphylococcal enterotoxin e in the blood of patients with rheumatoid arthritis. Jundishapur J. Microbiol. 8:e16621. doi: 10.5812/jjm.16621
Zhang, L., Nikkari, S., Skurnik, M., Ziegler, T., Luukkainen, R., Mottonen, T., et al. (1993). Detection of herpesviruses by polymerase chain reaction in lymphocytes from patients with rheumatoid arthritis. Arthritis Rheum. 36, 1080–1086. doi: 10.1002/art.1780360808
Keywords: rheumatoid arthritis, immune system provocation, Infection, microorganisms, viruses
Citation: Arleevskaya MI, Kravtsova OA, Lemerle J, Renaudineau Y and Tsibulkin AP (2016) How Rheumatoid Arthritis Can Result from Provocation of the Immune System by Microorganisms and Viruses. Front. Microbiol. 7:1296. doi: 10.3389/fmicb.2016.01296
Received: 01 December 2015; Accepted: 05 August 2016;
Published: 17 August 2016.
Edited by:
Hao Shen, Perelman School of Medicine, USACopyright © 2016 Arleevskaya, Kravtsova, Lemerle, Renaudineau and Tsibulkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina I. Arleevskaya, bWFybGVldkBtYWlsLnJ1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.