 Callum J. Cooper
Callum J. Cooper Mohammadali Khan Mirzaei
Mohammadali Khan Mirzaei Anders S. Nilsson
Anders S. Nilsson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 03 August 2016
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 7 - 2016 | https://doi.org/10.3389/fmicb.2016.01209
This article is part of the Research Topic Phage therapy: past, present and future View all 38 articles
The global rise of multi-drug resistant bacteria has resulted in the notion that an “antibiotic apocalypse” is fast approaching. This has led to a number of well publicized calls for global funding initiatives to develop new antibacterial agents. The long clinical history of phage therapy in Eastern Europe, combined with more recent in vitro and in vivo success, demonstrates the potential for whole phage or phage based antibacterial agents. To date, no whole phage or phage derived products are approved for human therapeutic use in the EU or USA. There are at least three reasons for this: (i) phages possess different biological, physical, and pharmacological properties compared to conventional antibiotics. Phages need to replicate in order to achieve a viable antibacterial effect, resulting in complex pharmacodynamics/pharmacokinetics. (ii) The specificity of individual phages requires multiple phages to treat single species infections, often as part of complex cocktails. (iii) The current approval process for antibacterial agents has evolved with the development of chemically based drugs at its core, and is not suitable for phages. Due to similarities with conventional antibiotics, phage derived products such as endolysins are suitable for approval under current processes as biological therapeutic proteins. These criteria render the approval of phages for clinical use theoretically possible but not economically viable. In this review, pitfalls of the current approval process will be discussed for whole phage and phage derived products, in addition to the utilization of alternative approval pathways including adaptive licensing and “Right to try” legislation.
The discovery of penicillin in 1928 heralded a dynamic shift in modern medicine with antibiotics quickly becoming one of the linchpins of modern medicine (Zaffiri et al., 2012). However, in the 1960s, the “golden era” of the identification of novel antibiotics ended with modern development focusing on the modification of existing drugs (Nathan and Cars, 2014) with only four multinational pharma companies maintaining antibiotic divisions (Fair and Tor, 2014). This lack of interest is not only due to the difficulties in discovering new antibiotic classes but also decreasing financial returns within drug development (Scannell et al., 2012). It is particularly true for anti-infective agents, where a median 10 day drug-treatment costs ~US$ 85 for non-HIV anti-microbial drugs compared to ~US$ 848 for anti-neoplastic drugs (Falagas et al., 2006). When compared to the total number of drugs granted regulatory approval, anti-infective drugs represent due to a poor return on overall investment which stifles their development (Figure 1; Piddock, 2012).
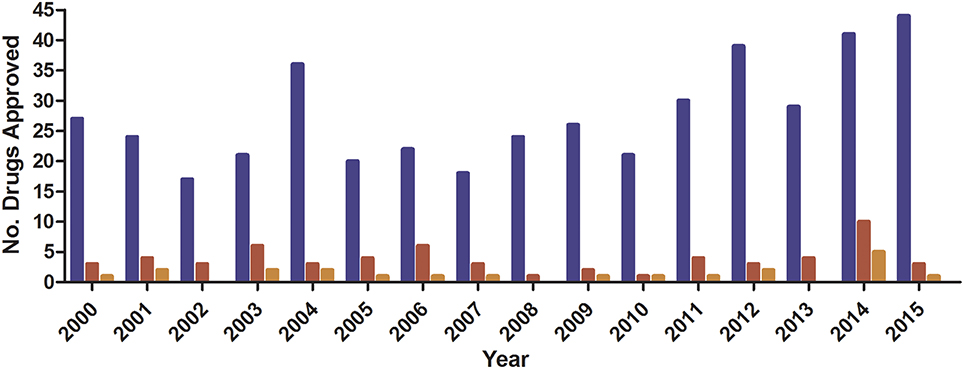
Figure 1. FDA Novel Drug Approval 2011–2015. Data obtained from (http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm430302.htm).  , Total;
, Total;  , anti-infective;
, anti-infective;  , anti-bacterial.
, anti-bacterial.
Despite public concern about increasing levels of antibiotic resistance, antibiotic consumption continues to increase, particularly in the BRIC (Brazil, Russia, India, China) nations (Van Boeckel et al., 2014). The ability to obtain antibiotics without prescription, their subsequent misuse by patients (Li, 2014), and the continued use of antibiotics as a growth promoter in agriculture (Cully, 2014), has contributed to an increase in the number and scale of multi-drug resistant infections (Molton et al., 2013). Increased consumption and fervid media reporting has generated huge interest in the development of new antibacterial agents and has led to the formation of a number of working groups, such as the US Food and Drug Administration's (FDA) Antibacterial Drug Development Task Force (http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm317207.htm; accessed 13th June 2016) or the Biotechs from Europe innovating in Anti-Microbial resistance (BEAM) Alliance (http://beam-alliance.eu/; accessed 13th June 2016). These groups have called for financial incentives and patent extensions to be applied to antibacterial drug development in order to stimulate research (Sonderholm, 2009; Laxminarayan and Powers, 2011; Wise, 2015).
Interest in phage therapy (i.e., the clinical use of bacteriophage based therapeutic products in humans), has been traditionally confined to academic groups and a few clinical centers in Eastern Europe, most notably the ELIAVA Institute in Tbilisi, Republic of Georgia. However, the specificity of phages and the enormous variation in human—bacteria—phage combinations will lead to an immense number of obligatory clinical trials if they are to be considered as a viable alternative to antibiotics. This constitutes one of the primary obstacles for the industrial development of phage based therapeutic products, in addition to concerns over intellectual property protection. Nevertheless, commercial interest has been piqued in the form of small BioPharma companies (Table 1), but significant interest from multinational pharmaceuticals is still lacking. These small BioPharma companies have enabled a number of commercial phage products to be approved for use in reducing food contaminants (Endersen et al., 2014), but widespread use of phage therapeutics in humans remains elusive in the West (Kingwell, 2015).

Table 1. Summary of current phage based products in development for the treatment of human disease.
The societal need for new antibacterial agents, and the knowledge that phage therapy may work in practice, requires the engagement of commercial entities to further develop phage based products rather than proceeding as a purely academic enterprise. However, what appears to limit the development of phage products for human use is primarily associated with development costs and regulations. Through the application of new or refined regulations the development of phage based pharmaceutical products may become faster and more commercially attractive for companies.
In this article, the current requirements for the development and approval of new antibacterial drugs are described with emphasis placed on the challenges faced by phages and phage based products. Potential alternative or additional approval pathways within existing and proposed legislation and how phage therapy could benefit from these pathways are also discussed.
Since the early part of the 20th Century, bacteriophages have been used to treat a range of different bacterial infections (Kutter et al., 2010). However, since the introduction and success of antibiotics in the mid-20th century, interest in phages as antimicrobial agents within Western Europe and the US has waned. Increasing problems with antibiotic resistant bacterial infections has led to alternative strategies being sought. This has in turn revitalized research into bacteriophages and their derived products as antibacterial agents (Oliveira et al., 2015). Phage therapy possesses advantages and disadvantages when compared to conventional antibiotics. These advantages include the ability of phages to self-replicate in the presence of a suitable bacterial host. They act with minimal disruption to the local microbiota and are relatively easy to isolate from environmental sources, while the limited host range of lytic phages may detract from their overall clinical usefulness. Although the advantages and disadvantages of phage therapy have been briefly highlighted here, they are discussed extensively elsewhere (Loc-Carrillo and Abedon, 2011; Nilsson, 2014; Kutter et al., 2015).
Virulent phages have been isolated from a variety of environments and proven in vitro to be efficient against a large number of bacterial species (Mattila et al., 2015; Salem et al., 2015; Sauder et al., 2016). In vivo testing has shown that phages can be used to treat various types of infections in animal models (Hawkins et al., 2010; Dufour et al., 2015; Ghorbani-Nezami et al., 2015; Holguín et al., 2015; Galtier et al., 2016) and also in humans (Kutter et al., 2010; Abedon et al., 2011; Chanishvili, 2012; Rose et al., 2014; Abedon, 2015). Results from these more recent in vitro and in vivo trials have led to a deeper understanding of the unique nature of phage therapy but have also highlighted the need for further research into their pharmacokinetics and pharmacodynamics (PD/PK).
Often compared to conventional antibiotics in the lay press, the capability to kill bacteria is the only similarity that whole phages and antibiotics share. Therefore, whole phage therapy is often complicated by additional factors and as such possesses unique pharmacokinetics and pharmacodynamics that remain poorly understood. Amongst these unique characteristics is the poor diffusion of phages as the result of their immense size when compared to antibiotics (~106 times larger). This means that whole phages cannot be administered in high concentrations (>1010 PFU). In order to provide an equivalent amount of “drug” compared to a 10 day course of penicillin (assuming equal minimum inhibitory concentrations and non-replicating phages) over 100 Kg of phages would be required (Bancroft and Freifelder, 1970; Nilsson, 2014). This lack of diffusion and restricted dosage concentration can be offset by the ability of phages to replicate upon finding their target organism.
As with all antimicrobial agents, the ever present shadow of resistance is particularly relevant to whole phage therapies where bacterial exposure to phages provides a co-selective pressure to develop and evade resistance. In the case of conventional antibiotics, targets are often essential metabolic functions, while phages and phage derived products (e.g., endolysins) primarily target surface structures, among the most rapidly changing features in bacteria. The epoch spanning co-evolutionary arms race between phages and bacteria have also resulted in the development of a number of distinct and constantly evolving anti-phage systems, most famously CRISPR-Cas systems (Horvath and Barrangou, 2010), to protect bacteria from infection by phages. In addition to CRISPR-Cas systems, other bacterial resistance mechanisms exist including phage exclusion and restriction modification systems, and have been discussed extensively elsewhere (Hyman and Abedon, 2010). While such systems present a threat to the overall efficacy of a whole phage therapeutic, they are not universally distributed in bacterial species (Grissa et al., 2007; Burstein et al., 2016) and phages also develop counter measures to these resistance mechanisms (Maxwell, 2016).
Although humans are routinely exposed to phages on a daily basis, concern persists over their immunogenicity and overall safety, presenting an additional stumbling block for the adoption of phage therapy. High doses of phage proteins can elicit unwanted side effects from stimulation of the immune system (Gorski et al., 2012; Dabrowska et al., 2014). Due to their classification as biological therapeutics (Rose et al., 2014), both whole phage therapies and therapies based on phage derived products will need to be manufactured under current good manufacturing practices (cGMP) and also adhere to current pharmacopeia requirements that are based on the type of application. This will require not only large scale manufacturing in inert suspension media, something being addressed by small biopharma, but also production of ultrapure preparations conforming to strict endotoxin requirements (<0.5 EU/mL for subcutaneous injections).
The regulatory foundation for clinical studies and clinical trials in humans is to ethically establish the potential toxicity, efficacy and side effects of new drugs and to prioritize the health of the participants over the generation of results. It is equally important that sufficient data support the claim of potential benefits and that these benefits outweigh anticipated risks. Clinical studies and trials should be carried out in a scientifically correct and transparent manner, be designed to result in trustworthy data and assess the pharmacological properties of the new drug in a stepwise process adapted to available information.
In the current paper, a number of points will be discussed that specifically impact upon individual licensing pathways. In addition, there are a number of pre-clinical testing issues which need to be addressed prior to use in patients regardless of the approval process. These include the need to develop standard testing protocols such as those found in antibiotic (e.g., BSAC or EUCAST; Brown et al., 2016) or microbicide biocide testing (e.g., ASTM E2197) to ensure consistency in results. There is an ongoing shift from classical qualitative assays such as the spot test (host range is assessed by plaque formation) to more quantitative methods such as the efficacy of plating (number of plaques on a target strain compared to number of plaques on the routine host; Khan Mirzaei and Nilsson, 2015), protocols still vary between laboratories. The creation of international standards would ensure the reliability and reproducibility of data.
Standard “efficacy” criteria are utilized by companies seeking to claim activity for chemical microbicides against a particular pathogen (e.g., a defined strain of S. aureus as an analog for MRSA). These activities are often under defined environmental and test conditions utilizing a reference strain as the target (e.g., ASTM E2197)1. Currently no such standardized criteria exist for whole phages. Although whole phages are unlikely to achieve such large reductions in short time periods (usually ≥5log10 reduction in <5 min), suitable criteria could be established. These criteria could be based upon a defined lower kill level, the persistence of antibacterial activity over prolonged periods of time, or other virulence characteristics (Borysowski et al., 2014). Such criteria would enable multiple libraries based on lytic activity to be assembled for custom made therapies.
Clinical trials in the United States must be carried out in accordance with laws in the United States Code, title 21, chapter 9; the Federal Food, Drug and Cosmetic Act and in particular part A of subchapter V: Drugs and Devices (http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/FDCActChapterVDrugsandDevices/default.htm#Part_A; accessed 13th June 2016) under the jurisdiction of the FDA and have influenced the regulations of many other countries due to their comprehensive nature. Within the EU, clinical trials are currently performed in accordance to the Clinical Trials Directive (EU-CTD). This should be superseded in 2016 by the simplified and updated Clinical Trials Regulation (EU-CTR; http://ec.europa.eu/health/human-use/clinical-trials/directive/index_en.htm; accessed 13th June 2016), allowing a single application in one member state to apply to all EU member states which would participate in the trial.
Clinical trials are often broken into four distinct phases (Figure 2) following successful pre-clinical studies. These phases increase in complexity and size as a product moves closer to approval. Should approval be granted, all products are then subjected to rigorous routine review as they are used. These stages have been reviewed in detail elsewhere (Pocock, 1983) and are summarized in Table 2.
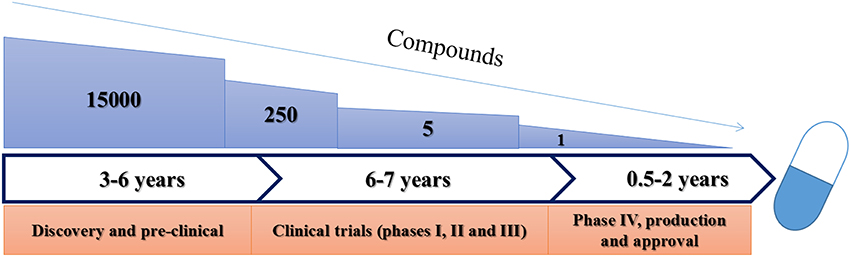
Figure 2. Schematic representation of current FDA approval procedures for anti-infective drugs.
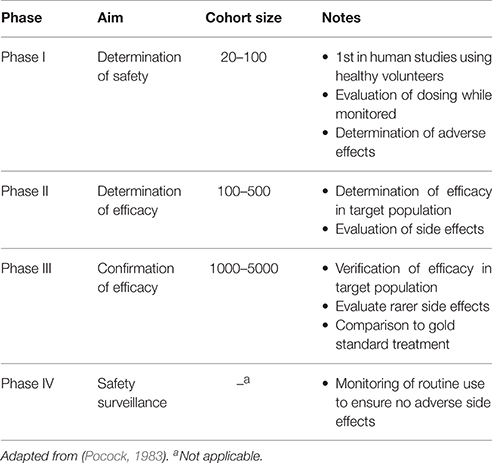
Table 2. Summary of clinical trials testing requirements.
It is estimated that ~US$2.6 billion is required to successfully go from concept to an approved drug (Mullard, 2014). Although some of the initial screening has been taken up by academia (mainly pre-clinical and development work), a significant investment of time and resources is still required from pharmaceutical companies. When compared to drugs developed to treat chronic conditions (e.g., statins), the development of antibacterial agents is economically unviable, due to their comparatively short usage time.
In response to the pressing need to develop new, safe and effective antibacterial agents, additional legislation has been introduced which attempts to modify the approval process, including limited population approval and the incentivization of antibacterial drug development (Brown, 2013; Bax and Green, 2015).
Both whole phages and their derived products will be subjected to the same rigorous clinical trials process as antibiotics. They are also classified as “Therapeutic biological products” and thus subject to the Food, Drug, and Cosmetic Act (http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/FDCActChapterVDrugsandDevices/default.htm#Part_A; accessed 13th June 2016) and also the Public Health Service Act (http://www.fda.gov/RegulatoryInformation/Legislation/ucm148717.htm; accessed 13th June 2016) and under EU Directive 2001/83/EC (Rose et al., 2014; Pelfrene et al., 2016) and would require additional controls over the manufacturing process. Should any changes be made to the manufacturing process, extensive comparability testing would be required to confirm the consistency of the product (Chirino and Mire-Sluis, 2004). Regulators, such as the EMA, are aware of the additional issues faced by phage therapeutics and believe that dialogue with developers will contribute toward a solution (Kingwell, 2015).
For ease of reference, a number of assumptions have been made in the current article as follows:
• A single strictly virulent phage or phage derived product (e.g., endolysin) is selected from pre-clinical studies which produces a suitable level of bacterial reduction for the intended application.
• The phage or phage derived product has been subjected to appropriate pre-clinical in vivo testing to determine the toxicity, immunogenicity, and dosing of the treatment.
• All products should be produced according to cGMP, as determined by local regulators, as well as being soluble in, and compatible with, commonly used physiological solutions (e.g., saline) or other physiologically inactive media.
• All components of a phage or phage product cocktail must have been shown to be acceptable as stated above for single phages. In addition, the dynamics between the individual components of the cocktail should also be assessed prior to use.
Initial clinical testing (Phase I) will not vary greatly between antibiotics, whole phages or their derived products and as such should be relatively simple to perform with the appropriate approvals. The routine exposure of humans to phages provides the immune system with a low level of circulating phage-specific antibodies (Kucharewica-Krukowska and Slopek, 1987) and subsequent exposure as part of a therapy may compound this. Indeed a number of in vitro and in vivo studies have shown that phages stimulate the innate and adaptive immune systems in a phage specific manner (Dabrowska et al., 2014; Majewska et al., 2015) and potentially in a protein specific manner (Dabrowska et al., 2014). However, a number of studies performed with cocktails of whole phages have suggested that phages are harmless when ingested (Table 3; Bruttin and Brussow, 2005; Sarker et al., 2012) or applied topically (Rhoads et al., 2009). This lack of response may be in part due to the degradation of phages as they transit the digestive system, reducing the number that come into contact with immunostimulatory cells (Abedon, 2015), but also due to varying degrees of sensitivity between different cell types.
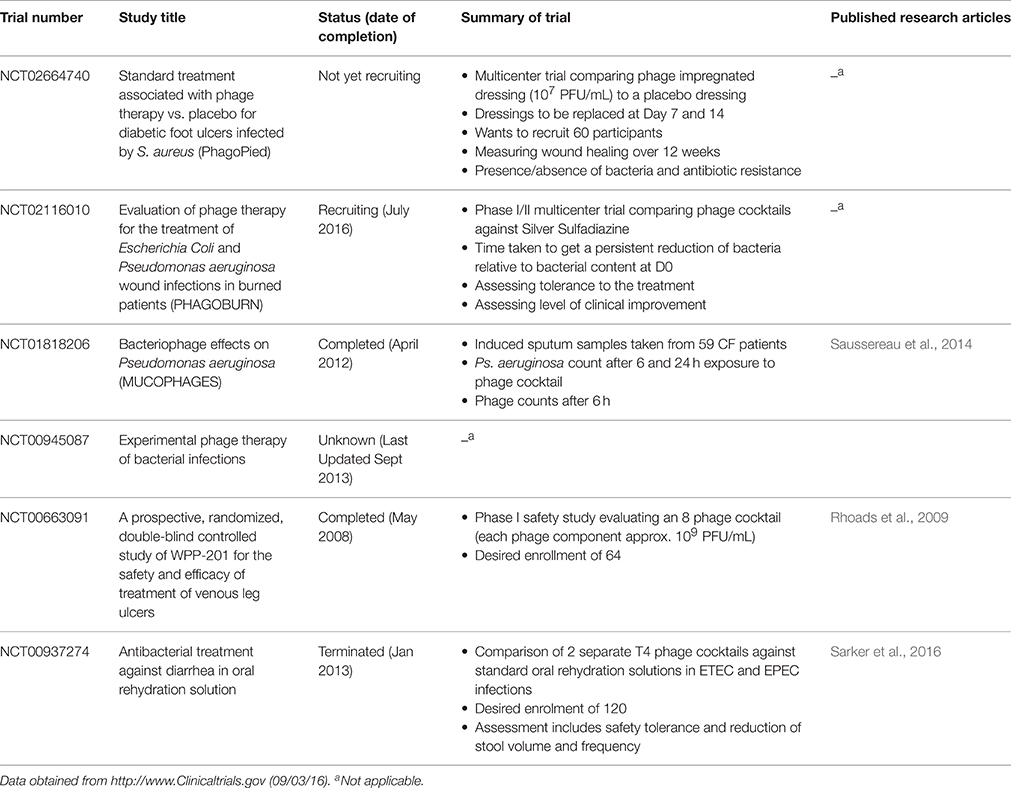
Table 3. Current clinical trials for phage based therapy in humans.
Despite this initial suggestion of safety under Phase I conditions, potential safety issues will remain during Phases II–IV as “rarer” side effects are sought. At these stages, drugs under evaluation are subjected to trials of efficacy in a population who suffer from the particular disease under investigation. Due to the nature of the lytic phage lifecycle, it is not inconceivable that the active replication of phages at a site of infection could produce side effects, such as toxic shock, as bacterial debris is released. Such issues could potentially be anticipated and avoided by the selection of phages which exhibit different properties such as lower virulence or through the combination of phages with conventional antibiotics. Although this may suggest that whole replicating phage therapies could be consigned to topical application further research is required, particularly if the issue is addressed through the incorporation of anti-endotoxin adjuvants (de Tejada et al., 2015; Valera et al., 2015).
During Phase II and III studies additional complications arise when trying to recruit a statistically relevant homogenous population to study (Rose et al., 2014). In the case of diseases caused by a single bacterial species (e.g., cholera), this may be due to a low incidence in the general population or, and more likely, the disease can be caused by multiple organisms (e.g., diabetic foot ulcers). It is therefore likely that clinical trials on phages will be based on long term or multi-site studies in order to obtain representative population sizes and could be facilitated by the introduction of the EU-CTR. The introduction of the EU-CTR could also enable trials to be coordinated from specialist phage therapy centers allowing for the distribution of specialist knowledge and products.
Although the combination of multiple phages into a cocktail compensates for a limited host range (Bruttin and Brussow, 2005) a number of compromises are made. The increased complexity of multi-phage cocktails will dilute the concentration of the individual phage components due to their size, and also introduce competition of phages for binding sites, both of which could compromise the treatment (Nilsson, 2014).
Pre-made phage cocktails can be designed to target either against uncharacterized (multiple phages targeting multiple bacterial species), or typed bacterial infections (multiple phages targeting a single bacterial species). Additionally, patient specific cocktails can be produced in which phages are selected from a pre-existing library against the patient's specific strain (Pirnay et al., 2011). While the manufacture of pre-made cocktails would be tightly controlled and mass produced under cGMP in order to satisfy regulatory requirements (Parracho et al., 2012; Rose et al., 2014) it could decrease the production cost per dose. Pre-made cocktails would also require supplementary approval as cocktail components are modified to compensate for the development of bacterial resistance. The clinical usefulness of pre-made cocktails would be limited due to the shifting nature of epidemic strains; however, the rate at which resistance develops under therapeutic conditions is currently unknown. In theory at least, a pre-defined cocktail should be able to successfully navigate the current regulatory process assuming appropriate non-inferiority (drugs under investigation possess similar levels of activity compared to standard treatment) trial designs. However, it remains unclear if additional approval would be required as components of the cocktail change.
Although patient specific cocktails may provide better overall results (due to the tailored nature of the treatment) these would present additional challenges in order to gain regulatory approval. Classical trials of patient specific cocktails would have to be designed to target specified bacterial strains within the same species, further reducing the available population which could be recruited and require multi centered trials to be undertaken. In theory at least individual approvals would be required due to their unique composition. In order to compensate for the variety of potentially infectious strains, patient specific cocktails would require libraries of pre-approved phages to be developed. This would allow cocktails to be assembled on a case by case basis, currently an unprecedented move, although the stockpiling of vaccines and some antitoxins could be considered to be a suitable analog (Bodas et al., 2012; Martin et al., 2012).
As previously mentioned, the overall cost for the complete (pre-clinical to Phase III) development of a novel drug is astronomical (estimated to be US$2.6 billion; Mullard, 2014) and represents a significant obstacle for the broad evaluation of phage therapy in human populations. This cost would be on a per cocktail basis (assuming cocktails were pre-defined and manufactured) and probably equate to those encountered by an antibiotic or phage derived protein. When new strains arise, for which the pre-defined cocktail is not approved, treatment could still be carried out under compassionate usage or “off license.” However, in the case of patient specific cocktails, approvals may have to be obtained on a phage by phage basis, prior to combination into cocktails and could potentially, increase overall costs by orders of magnitude.
The biology and unique, but poorly understood, PD/PK of whole phage based therapies, may in actuality reduce their viability as antibiotic replacements for the treatment of bacterial infections in humans. In the case of PD, their large size and poor diffusion through non-aqueous mediums would present challenges if used as a systemic treatment. This would require potentially huge doses of phage to be administered in order to achieve a therapeutic effect. In terms of pharmacokinetics, large phage doses would be cleared efficiently from the body by the immune system and could prevent the establishment of a productive phage infection. The administration of large doses of phages would increase the probability of phages being able to reach the site of infection prior to being removed by the immune system. In addition to this, the overall immunostimulatory capacity of phage could be reduced through complex formulation by masking the phages (Kim et al., 2008) or through modification of phages to alter immunostimulatory proteins (Dabrowska et al., 2014).
Although many of the issues raised here have been presented as phase specific, they in fact transcend the individual trial phases and represent incompatibilities within the current approvals process itself. Indeed, for patient specific cocktails, it is highly improbable that current regulations would allow for the approval of a library rather than requiring the approval of each individual phage, drastically increasing the overall cost.
Conversely, phage derived products (e.g., endolysins) may address some of these limitations and have attracted some attention from commercial entities (Table 2). Despite being defined as “therapeutic biological products” their activity kinetics, and probably approval pathways, would be more akin to antibiotics than whole phage cocktails. The ability to produce them as recombinant proteins in a non-target vector means that overall manufacturing and purification processes could be adapted from currently existing methods (such as those used to produce insulin). This would allow for classification at a substantially higher level of detail than is possible with whole phage cocktails. The activity of endolysins has also been shown to transcend single bacterial strains in Gram positive pathogens such as S. aureus (Fischetti, 2016). This would not only enable trials to be performed on larger populations but could also increase their attractiveness to larger scale pharma companies, as they could be used to target multiple conditions caused by a particular bacterial species. However, they would be of limited effectiveness against polymicrobial infections. Many of the derived endolysins currently described in the literature target Gram positive pathogens (Fischetti, 2010; Nakonieczna et al., 2015). Further research is needed to evaluate their efficacy against Gram negative pathogens due to differences in cell wall composition as well as more research on Gram negative specific endolysins (Dong et al., 2015; Oliveira et al., 2016). Additionally, other phage derived proteins such as holins and tailspikes may provide suitable alternatives to endolysins for Gram negative pathogens (Saier and Reddy, 2015).
The process for gaining regulatory approval for novel antibiotics is a long and time consuming process, which is further complicated when whole phages are applied to these proceedings. Although the process itself is optimized for certain types of drugs, regulators believe that the current trials legislation is “adequate” for use with bacteriophage based therapies (Verbeken et al., 2012; Pelfrene et al., 2016). However, many researchers engaged in the field actively disagree with this, as the current approvals procedures are too rigid and too costly in terms of time and money. They have more recently suggested that current pathways need to be modified or novel pathways need to be developed for use with phages (Verbeken et al., 2014; Kutter et al., 2015; Young and Gill, 2015).
Multiple initiatives have been taken by both the FDA and Europe Medicines Agency (EMA) to simplify and shorten the approvals process for drugs while maintaining standards. They are not designed for antibacterial drugs or, more specifically, phage based therapeutics. These frameworks include an EMA pilot project on adaptive licensing initiated in 2014 (AL or adaptive pathways) and encompasses six undisclosed products. The pilot study, which is due to report later in 2016, seeks to investigate how current regulations can be optimized for the approval of new drugs in cases where there is a high medical need. The pilot also seeks to determine which criteria should apply for drugs that can be approved in a graduated simplified process.
Introduced to the US Senate in January 2015, the Promise for Antibiotics and Therapeutics for Health (PATH) Act (S.185; https://www.congress.gov/bill/114th-congress/senate-bill/185/text; accessed 13th June 2016) is an amendment to section 506 of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 356). This amendment proposes the creation of a “limited population pathway for antibacterial drugs” that will allow for the approval of antibacterial drugs within a highly defined population without the need for clinical trials through the development of a Benefit-Risk profile that reflects the “severity, rarity, or prevalence” of the infection. Although the decision making process could inevitably be informed by both traditional (e.g., survival), alternative (e.g., bacterial clearance), and small clinical data sets, the process may also take into consideration other supplementary information such as non-clinical susceptibility and pharmacokinetic data. However, the supplementary set of considerations will be decided on a drug by drug basis. Once approved, product labeling will reflect the limited population that the drug can be used on and subjected to post approval monitoring. Subsequent approval for use within a wider population can be sought, but it is not clear if this would require full clinical trials or if off license use would be permitted.
AL pathways are iterative processes in which treatment outcomes are used to inform the ongoing trial, through the involvement of all stakeholders, as well as input from independent scientific advice. The iterative process can, as in the EMA pilot project, be based on different conditions including: (i) the introduction of more steps, starting with a small highly defined patient population which is expanded as more information becomes available (ii) conditional approval for a product that is granted based on existing data, or (iii) adopting a centralized compassionate use of a new drug.
When compared to classical trials, AL pathways may provide additional flexibility that would enable whole phage therapeutics and their derived products to be approved for clinical use. These opportunities would come in the form of initial limited population testing and potentially through the use of non-traditional surrogate endpoints.
Regardless of the pathway employed, phage clinical trials will inevitably consist of a series of compromises due to the complex interplay between infection type, causative agent and therapeutic strategy. Trial outcomes will also shift depending on additional priorities such as clinical need, scope of the trial, anticipated efficacy of treatment, and overall cost. In addition to this, trials could be based around a multitude of different formulations, each possessing advantages and disadvantages (Figure 3).
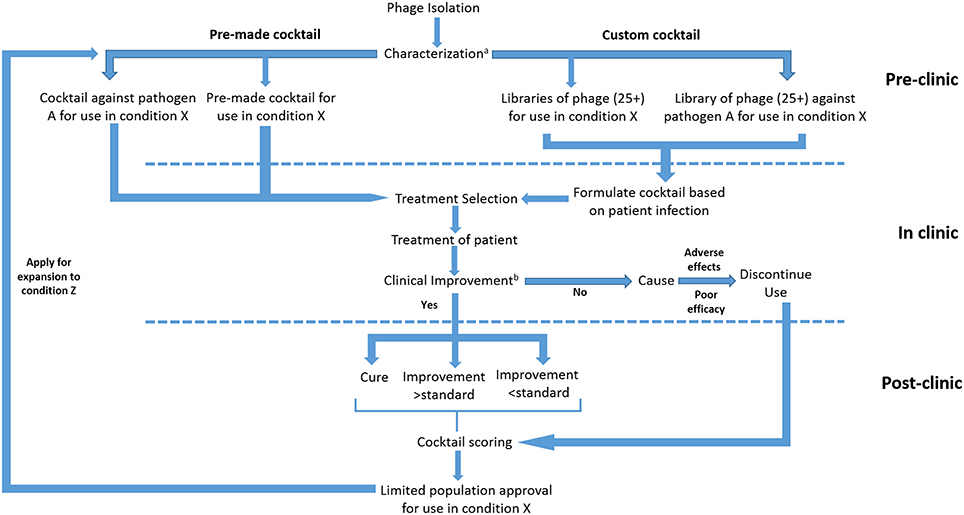
Figure 3. Proposed submission of whole phage based products under an adaptive licensing framework. aCharacterization to be based on genotypic analysis and lytic activity. bImprovement to be characterized on the basis of classical and surrogate endpoints.
As previously mentioned, phage therapy may involve treatment with single phages, phage derived products e.g., endolysins, or cocktails. Host range considerations would limit the available population that could be treated with single phage preparations, whereas the formation of phage cocktails would be able to increase the number of patients that could be treated. The number of potential recruits for trials could also be increased for disease states which cause seasonal outbreaks of a single clonal type in a confined area (e.g., hospital acquired Clostridium difficilie infections; Furuya-Kanamori et al., 2015). Trial participants could be easily recruited for clinical trials of a single phage, particularly if the target organism is a commonly occurring pathogen (e.g., MRSA). The recruitment process would be also be facilitated by a centralized approvals process (e.g., EU-CTR) which would enable multi-centered trials to be performed with a single application. However, many infecting bacterial strains are temporally or spatially restricted, limiting the availability of participants.
Phage cocktails, on the other hand, can increase the number of strains susceptible to infection by one or more of the components which make it possible to target common bacterial infections caused by different strains in different patients and thus facilitate the recruitment of participants. Cocktails can be pre-made and targeted against common pathogenic strains or custom made, either upon the emergence of a particular strain or to fit the requirements of individual patients (Pirnay et al., 2011).
Highly defined pre-made cocktails should be able to fit into existing AL frameworks as it would be relatively simple to define a limited population, although this would not be able to compensate for differences between causative strains. Should the causative strain be specified (e.g., Pseudomonas aeruginosa PAO1) this would further limit the population which could be recruited, therefore a multi-centered approach would be needed and could be expedited by the modified EU-CTR. To compensate for shifting global trends, clinically relevant bacterial collections should be assembled and distributed which would enable easier isolation of suitable phages. It should also be noted that pre-defined cocktails are less flexible to the rise and fall of new bacterial strains and would ultimately be susceptible to the development of bacterial resistance. The overall efficacy would decrease over time in which a new cocktail would need to be developed and approved for use.
Custom made cocktails are one way to address the development of bacterial resistance against phages. In the case of bacteria which harbor phage resistance systems (e.g., CRISPR), phages encoding specific anti-defense mechanisms e.g., anti-CRISPR systems (Bondy-Denomy et al., 2013) could be given higher priority even if their infection characteristics are not as good as other phages. Several phages targeting different surface receptors could be applied simultaneously or serially, resulting in a synergistic effect and could reduce the potential for resistance developing (Schmerer et al., 2014) although other criteria could be used for the selection of cocktail components (Chan and Abedon, 2012). The inducement of synergy between phages would also be a good strategy for long term treatment of deep infections. In such infections, phages would have difficulty reaching their targets and would be cleared by the immune system reducing their overall number. By inducing synergy a smaller overall number of phages would be required to reach the site of infection as each phage component would be able to establish a productive infection. However, as with all phage trials there are arguments that the overall number of participants in custom cocktail trials would be limited to just one (Eichler et al., 2015) as infecting strains, and therefore cocktail composition, would vary on a patient to patient basis. As such it may be advantageous to inform AL trials based on a pre-characterized library of phages against a defined pathogen in a defined condition (e.g., Ps. aeruginosa burn infections), although this would require a significant redesign of approvals processes.
A long-term possibility for the implementation of whole phage therapeutics would be to create phage libraries containing the most efficient phages against the most severe pathogens, e.g., multiresistant Gram-negative bacteria. This could be done by initially pooling existing phage banks (most of them maintained by research institutions) into common libraries, followed by the continuous isolation and addition of new phages. Copies of the library would be stored in national phage depositories or major hospitals. This would shorten the time for finding highly efficient matching phages and assembling cocktails as well as facilitate an approval process. Individual phages would initially be characterized in vitro (as with most isolation and characterization papers) using structural, genomic (i.e., absence of lysogenic properties), host range, and efficacy analyses (Malki et al., 2015; Sauder et al., 2016). However, and more importantly, the pre-characterization could also establish safety and efficacy including suitable in vivo testing in animal models before phages are added to the library. It is important to ensure that even phages which do not meet the required in vitro efficacy criteria (e.g., insufficient lytic activity) are not discarded as they may perform better in vivo (e.g., less immunostimulatory), or possess greater efficacy against future epidemic strains.
The assembly of pre-approved phage libraries could potentially prove advantageous as it would allow for the creation of multiple cocktails to target an individual infection type within a single library thereby increasing the population size that could be recruited. Only the cocktails themselves would need to be tested for safety and efficacy since individual phages in the library would already be fully tested. This approach would also allow for the creation of additional libraries for phages which appear to be less active or possess a restricted host range. Indeed the possibility of creating multiple or tiered libraries (as is the case with multiple lines of antibiotics) would allow additional flexibility as resistance to individual phages develops. However, as the complexity of cocktails increases to treat polymicrobial infections, or as multiple tiered libraries are assembled, the overall cost and time required to complete trials would increase.
It should be noted that the concept of pre-approved libraries would require a radical shift in the thought processes of regulatory agencies and would require the development of new assessment criteria. These criteria could include the absence of non-toxicity regardless of the combination of phages used, in combination with a “minimal” activity level of each phage. Such a shift in thought process would also ultimately lead to the development and approval of libraries of phage derived antibacterial proteins. By approving both phages and their derived proteins as libraries rather than on an individual basis, the overall number of trials would be reduced due to increased flexibility of the drug and potentially an increased treatment success rate.
Following the successful formation of a pre-characterized library or specified cocktail, patients would be recruited on the basis of confirmed infection type (i.e., a specified bacterial agent in a specified disease state). As in the EMA pilot studies, the trial would progress iteratively, starting with small groups of participants and resolving uncertainties before expanding the trial into new populations. Adverse effects influenced by rare human genetic traits may be problematic for this type of AL approach, but further research is required. As a consequence, the initial (human) testing of phages or cocktails from the libraries should be conducted in non-life threatening topical infections (e.g., diabetic foot infections). Any target infection should also have at least one additional treatment available as a safety precaution. Should the application of the cocktail result in Serious Adverse Events/Serious Adverse reactions, Suspected Unexpected Serious Adverse Reactions or no measurable clinical benefit, the decision to remove a patient from the treatment should be made quickly and alternative treatment applied as soon as possible.
The necessary expansion from small groups of trial participants to larger cohorts during adaptive pathway trials would be contingent on the outcomes that can be assessed given the actual cohort size. Classical clinical trials often use well-defined and clinically relevant endpoints like patient survival time, resolution of infection, decrease in lesion size or perceived symptoms, but surrogate endpoints based on biomarkers for indirect assessment can also be applied (Fleming and Powers, 2012).
In the initial stages, classical endpoints should be included in adaptive pathway phage therapy trials, but pharmacological assessment will be as important. Surrogate endpoints will probably be of greater importance for phage therapy trials. Increase in phage titers, reduction of bacteria load or infection parameters (e.g., CRP) and absence of additional pro-inflammatory responses may indicate that the treatment has a positive effect and that the study can be widened. Therefore, phage trials under AL would be designed as non-inferiority trials, in which the intervention is compared to the conventional therapy (e.g., antibiotics) to establish a similar level of overall effect (D'Agostino et al., 2003) rather than having to demonstrate superiority.
If applied in a pre-formed library format for patient specific cocktails, individual phage components could potentially be scored to further inform and develop the library. Such a scoring mechanism would not only require the treatment outcome to be established, but the actual role of the individual phage in that outcome and would require the ability to differentiate between the components of the cocktail.
AL pathways offer many possibilities for the approval of whole phages. However, each of these different avenues require compromises which will subsequently impact the efficacy of the treatment and on the overall cost and time required to complete trials (Table 4). In the case of cocktails the time and cost to reach the clinic will increase significantly if it is necessary to approve each individual component of the cocktail separately. However, if radical action is taken and libraries of phages against specified pathogens are approved, this could potentially counter the increased cost and time.
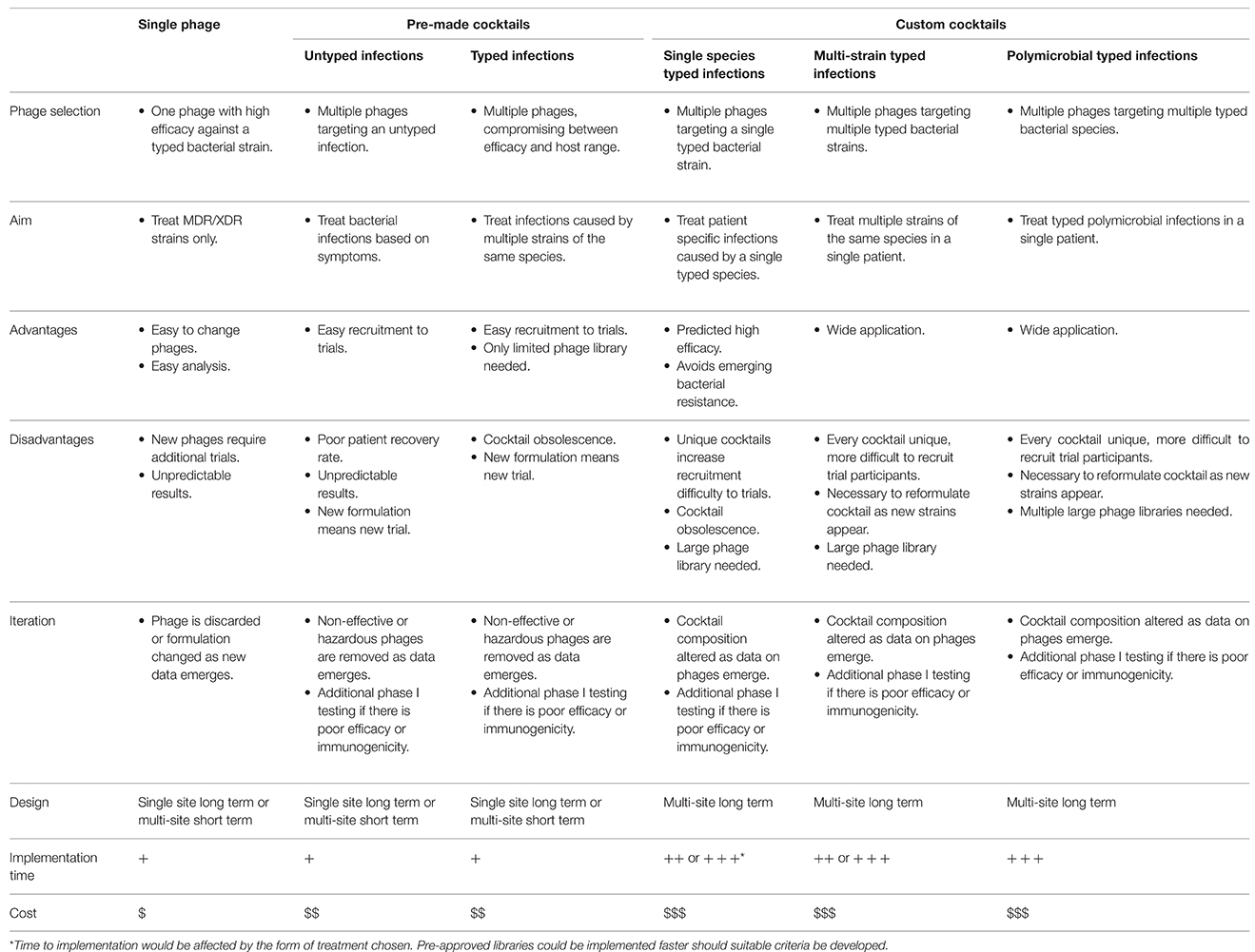
Table 4. The implementation of adaptive licensing pathways for single phage and pre-made or custom phage cocktails.
Although it is likely that phage derived proteins will not suffer adversely when trying to gain regulatory approval, AL pathways could still prove beneficial. The use of a limited population approach would enable data to be obtained while informing future clinical studies of different disease states, particularly if developers are interested in systemic application. In addition to this, classical and surrogate endpoints could be utilized that could be derived from antibiotic trials (Cornely et al., 2012; Verduri et al., 2015).
The Code of Federal Regulations (CFR) Title 21, Chapter I, Subchapter D, Part 312, Subpart I (http://www.ecfr.gov/cgi-bin/text-idx?SID=43f054659224216924a6379ef9602c2b&mc=true&tpl=/ecfrbrowse/Title21/21tab_02.tpl; accessed 13th June 2016) and European Regulation 726/2004/EC (http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:136:0001:0033:en:PDF; accessed 13th June 2016) govern the expanded access of non-approved medications to patients that have been submitted for approval by regulatory agencies. Although this is often under compassionate use guidelines where no alternative treatment exists (Whitfield et al., 2010) there are increasing calls to amend the laws in the US to make this process easier through additional legislation such as the H.R 3012 (Right to Try Act; https://www.congress.gov/bill/114th-congress/house-bill/3012; accessed 13th June 2016). Introduced to Congress during July 2015, H.R. 3012 would allow Phase I experimental drugs, biological products, or devices to be used in terminally ill patients.
Although such regulations would allow for the use of phages on individual patients in the US under Part 312.310, expansion to an intermediate patient population size (Part 312.315) would be possible, but potentially difficult. While there have been a limited number of cases in which phage cocktails have been approved for compassionate use this process is not routine (Rhoads et al., 2009; Khawaldeh et al., 2011). However, should resistance rates continue to increase, and the number of available drugs decrease further, phage therapy may be the only remaining therapeutic option.
In addition to cGMP and other specific requirements, informed consent from patients is required for compassionate use. While this should be relatively simple, the lack of public awareness surrounding phages may be detrimental to recruitment. This lack of public awareness could be circumvented by the formation of trials in EU member states possessing specialized phage centers (such as the Institute of Immunology and Experimental therapy in Wroclaw Poland) as part of an EU-CTR application in combination with compassionate usage in other parts of Europe which has been suggested by researchers in the field (Kutter et al., 2015).
The clinical use of phages in specialized centers in Eastern Europe has generated immense amounts of data, little of which is published in Western scientific literature (Miedzybrodzki et al., 2012). As some of these centers now lie within the EU, data generated post membership should conform to Western standards and could potentially form the basis of a meta-analysis or systematic review of clinical phage therapy when combined with more recent trial data that has been generated.
In order to utilize such data that has been generated prior to EU membership, or for countries whose regulatory frameworks may differ to EU and FDA standards, criteria would need to be established in order to assess the overall quality of the work performed. This could be done on the basis of the achievement of an appropriate clinical outcome (i.e., duration of hospitalization or resolution of infection) or an assessment of the methodology and trial design prior to incorporation into meta analyses.
Despite the pressing need to develop new antibacterial agents, the approval rate of anti-bacterial drugs remains low when compared to other forms of drug due in part to both economic and scientific issues. Phage derived products, such as endolysins, are likely to be suitable for classical clinical trials procedures due to their similarities with conventional antibiotics. However, in the case of whole phage therapies, currently available mechanisms are not suitable, requiring large patient cohorts and extensive resources. In this article the limitations of current clinical approval pathways, as well as possible alternative pathways for the approval of phage therapy, have been discussed and summarized (Table 5).
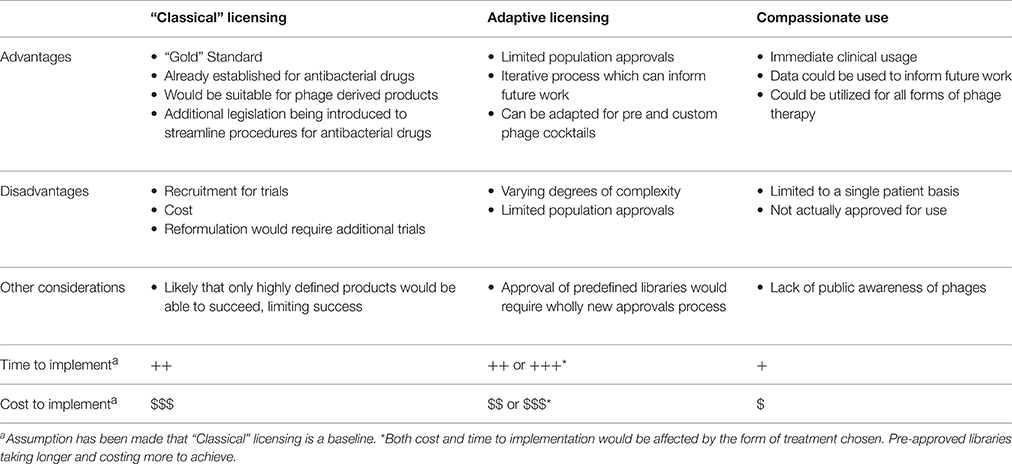
Table 5. Summary of current and possible alternative pathways as applied to whole phage and phage derived therapeutics.
While the current article is by no means exhaustive on every potential pathway that could be employed for the approval of phage based therapies for human use, it hopefully sparks discussion and debate on the nature of clinical trials and the need for more flexible regulations when dealing with phages and their derived products. For phages that are genetically similar (>95%) this could include additional accelerated or automatic approval pathways. This would be particularly useful for those phages whose major components (e.g., capsids) are identical but whose host range and efficacy is influenced by small changes in tail fiber composition (Ando et al., 2015; Goren et al., 2015). However, genetic engineering of whole phages (Ando et al., 2015), or the creation of wholly artificial phages from sequence data (Smith et al., 2003), would require approvals pathways to be revisited in the future as these technologies approach clinical readiness. In the case of customizable cocktails taken from pre-licensed libraries, suitable regulatory criteria need to be developed. This would in essence separate phage approvals from the normal biological therapeutics approvals and would require a substantial shift in the collective mindset of regulatory agencies.
Perhaps the most radical possibility would be to establish a centralized phage bank under governmental control from which phages could be isolated, collated, tested, and distributed on a case by case basis to be used in compassionate use treatments or AL trials. Data and treatment outcomes could then be collated by the phage bank to provide greater insight into phage therapy as a whole. This would not only remove the commercial element to development, but would provide direct to clinic access for therapies and also enable a greater degree of control to be exerted over treatment potentially reducing phage resistance rates. Such a system could be developed within existing public health organizations (e.g., Public Health England, UK or Folkhälsomyndigheten, Sweden) as these organizations are responsible for collating data on antibiotic resistance trends and provide reference laboratory facilities, but would require a substantial initial investment to establish and thus may be unpalatable in the current economic climate.
While no phage specific approvals pathway currently exists, such a pathway could be developed with suitable engagement between regulators and researchers. This pathway could be based on existing guidelines, where products which have been successfully completed phase I clinical studies are applied on a case by case basis under compassionate use guidelines. However, in order to gain widespread adoption it may be better to base phage approvals on AL principles, whereby approval is granted for small specified populations which can then be expanded upon as post-approval data is gathered.
Regardless of the pathway implemented, the overall cost of drug development and the poor return on investment of antibacterial agents will remain one of the defining development issues. Due to the abundance of phages in the environment, patents may be circumvented relatively easily as new phages are isolated and would therefore reduce the potential level of interest from traditional pharmaceutical companies.
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was partly funded by the Olle Engkvist byggmästare foundation (ASN). The authors would also like to thank Harsha Siani for proof reading the manuscript.
1. ^ASTM E2197-11, Standard Quantitative Disk Carrier Test Method for Determining the Bactericidal, Virucidal, Fungicidal, Mycobactericidal and Sporicidal Activities of Liquid Chemical Germicides, ASTM International, West Conshohocken, PA, (2011), www.astm.org.
Abedon, S. T. (2015). Phage therapy of pulmonary infections. Bacteriophage 5:e1020260. doi: 10.1080/21597081.2015.1020260
Abedon, S. T., Kuhl, S. J., Blasdel, B. G., and Kutter, E. M. (2011). Phage treatment of human infections. Bacteriophage 1, 66–85. doi: 10.4161/bact.1.2.15845
Ando, H., Lemire, S., Pires, D. P., and Lu, T. K. (2015). Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst. 1, 187–196. doi: 10.1101/020891
Bancroft, F. C., and Freifelder, D. (1970). Molecular weights of coliphages and coliphage DNA. I. Measurement of the molecular weight of bacteriophage T7 by high-speed equilibrium centrifugation. J. Mol. Biol. 54, 537–546. doi: 10.1016/0022-2836(70)90124-5
Bax, R., and Green, S. (2015). Antibiotics: the changing regulatory and pharmaceutical industry paradigm. J. Antimicrob. Chemother. 70, 1281–1284. doi: 10.1093/jac/dku572
Bodas, M., Yuval, L., Zadok, R., Hess, Z., Haran, B., Kaplan, M., et al. (2012). Shelf-life extension program (SLEP) as a significant contributor to strategic national stockpile maintenance: the Israeli experience with ciprofloxacin. Biosecur. Bioterror 10, 182–187. doi: 10.1089/bsp.2011.0107
Bondy-Denomy, J., Pawluk, A., Maxwell, K. L., and Davidson, A. R. (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432. doi: 10.1038/nature11723
Borysowski, J., Gorski, A., and Miedzybrodzki, R. (2014). Phage Therapy: Current Research and Applications. Norfolk, UK: Caister Academic Press.
Brown, D. F., Wooton, M., and Howe, R. A. (2016). Antimicrobial susceptibility testing breakpoints and methods from BSAC to EUCAST. J. Antimicrob. Chemother. 71, 3–5. doi: 10.1093/jac/dkv287
Brown, E. D. (2013). Is the GAIN Act a turning point in new antibiotic discovery? Can. J. Microbiol. 59, 153–156. doi: 10.1139/cjm-2013-0089
Bruttin, A., and Brussow, H. (2005). Human volunteers receiving Escherichia coli phage T4 orally: a safety test of phage therapy. Antimicrob. Agents Chemother. 49, 2874–2878. doi: 10.1128/AAC.49.7.2874-2878.2005
Burstein, D., Sun, C. L., Brown, C. T., Sharon, I., Anantharaman, K., Probst, A. J., et al. (2016). Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat. Commun. 7:10613. doi: 10.1038/ncomms10613
Chan, B. K., and Abedon, S. T. (2012). Phage therapy pharmacology: phage cocktails. Adv. Appl. Microbiol. 78, 1–23. doi: 10.1016/B978-0-12-394805-2.00001-4
Chanishvili, N. (2012). A Literature Review of the Practical Application of Bacteriophage Research. New York, NY: Nova Biomedical Books.
Chirino, A. J., and Mire-Sluis, A. (2004). Characterizing biological products and assessing comparability following manufacturing changes. Nat. Biotechnol. 22, 1383–1391. doi: 10.1038/nbt1030
Cornely, O. A., Crook, D. W., Esposito, R., Poirier, A., Somero, M. S., Weiss, K., et al. (2012). Fidaxomicin versus vancomycin for infection with Clostridium difficile in Europe, Canada, and the USA: a double-blind, non-inferiority, randomised controlled trial. Lancet Infect. Dis. 12, 281–289. doi: 10.1016/S1473-3099(11)70374-7
Dabrowska, K., Miernikiewicz, P., Piotrowicz, A., Hodyra, K., Owczarek, B., Lecion, D., et al. (2014). Immunogenicity studies of proteins forming the T4 phage head surface. J. Virol. 88, 12551–12557. doi: 10.1128/JVI.02043-14
D'Agostino, R. B., Massaro, J. M., and Sullivan, L. M. (2003). Non-inferiority trials: design concepts and issues–the encounters of academic consultants in statistics. Stat. Med. 22, 169–186. doi: 10.1002/sim.1425
de Tejada, G. M., Heinbockel, L., Ferrer-Espada, R., Heine, H., Alexander, C., Bárcena-Varela, S., et al. (2015). Lipoproteins/peptides are sepsis-inducing toxins from bacteria that can be neutralized by synthetic anti-endotoxin peptides. Sci. Rep. 5:14292. doi: 10.1038/srep14292
Dong, H., Zhu, C., Chen, J., Ye, X., and Huang, Y. P. (2015). Antibacterial Activity of Stenotrophomonas maltophilia Endolysin P28 against both Gram-positive and Gram-negative Bacteria. Front. Microbiol. 6:1299. doi: 10.3389/fmicb.2015.01299
Dufour, N., Debarbieux, L., Fromentin, M., and Ricard, J. D. (2015). Treatment of highly virulent extraintestinal pathogenic Escherichia coli pneumonia with bacteriophages. Crit. Care Med. 43, e190–e198. doi: 10.1097/CCM.0000000000000968
Eichler, H. G., Baird, L. G., Barker, R., Bloechl-Daum, B., Børlum-Kristensen, F., Brown, J., et al. (2015). From adaptive licensing to adaptive pathways: delivering a flexible life-span approach to bring new drugs to patients. Clin. Pharmacol. Ther. 97, 234–246. doi: 10.1002/cpt.59
Endersen, L., O'Mahony, J., Hill, C., Ross, R. P., McAuliffe, O., and Coffey, A. (2014). Phage therapy in the food industry. Annu. Rev. Food Sci. Technol. 5, 327–349. doi: 10.1146/annurev-food-030713-092415
Fair, R. J., and Tor, Y. (2014). Antibiotics and bacterial resistance in the 21st century. Perspect. Medicin. Chem. 6, 25–64. doi: 10.4137/PMC.S14459
Falagas, M. E., Fragoulis, K. N., and Karydis, I. (2006). A comparative study on the cost of new antibiotics and drugs of other therapeutic categories. PLoS ONE 1:e11. doi: 10.1371/journal.pone.0000011
Fischetti, V. A. (2010). Bacteriophage endolysins: a novel anti-infective to control Gram-positive pathogens. Int. J. Med. Microbiol. 300, 357–362. doi: 10.1016/j.ijmm.2010.04.002
Fischetti, V. A. (2016). Lysin therapy for Staphylococcus aureus and other bacterial pathogens. Curr. Top Microbiol. Immunol. doi: 10.1007/82_2015_5005. [Epub ahead of print].
Fleming, T. R., and Powers, J. H. (2012). Biomarkers and surrogate endpoints in clinical trials. Stat. Med. 31, 2973–2984. doi: 10.1002/sim.5403
Furuya-Kanamori, L., McKenzie, S. J., Yakob, L., Clark, J., Paterson, D. L., Riley, T. V., et al. (2015). Clostridium difficile infection seasonality: patterns across hemispheres and continents–A systematic review. PLoS ONE 10:e0120730. doi: 10.1371/journal.pone.0120730
Galtier, M., De Sordi, L., Maura, D., Arachchi, H., Volant, S., Dillies, M. A., et al. (2016). Bacteriophages to reduce gut carriage of antibiotic resistant uropathogens with low impact on microbiota composition. Environ. Microbiol. 18, 2237–2245. doi: 10.1111/1462-2920.13284
Ghorbani-Nezami, S., Leblanc, L., Yost, D. G., and Amy, P. S. (2015). Phage therapy is effective in protecting honeybee larvae from American Foulbrood disease. J. Insect. Sci. 15, 84. doi: 10.1093/jisesa/iev051
Goren, M. G., Yosef, I., and Qimron, U. (2015). Programming bacteriophages by swapping their specificity determinants. Trends Microbiol. 23, 744–746. doi: 10.1016/j.tim.2015.10.006
Gorski, A., Miedzybrodzki, R., Borysowski, J., Dabrowska, K., Wierzbicki, P., Ohams, M., et al. (2012). Phage as a modulator of immune responses: practical implications for phage therapy. Adv. Virus Res. 83, 41–71. doi: 10.1016/B978-0-12-394438-2.00002-5
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 8:172. doi: 10.1186/1471-2105-8-172
Hawkins, C., Harper, D., Burch, D., Anggård, E., and Soothill, J. (2010). Topical treatment of Pseudomonas aeruginosa otitis of dogs with a bacteriophage mixture: a before/after clinical trial. Vet. Microbiol. 146, 309–313. doi: 10.1016/j.vetmic.2010.05.014
Holguín, A. V., Rangel, G., Clavijo, V., Prada, C., Mantilla, M., Gomez, M. C., et al. (2015). Phage ΦPan70, a putative temperate phage, controls Pseudomonas aeruginosa in planktonic, biofilm and burn mouse model assays. Viruses 7, 4602–4623. doi: 10.3390/v7082835
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. doi: 10.1126/science.1179555
Hyman, P., and Abedon, S. T. (2010). Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 70, 217–248. doi: 10.1016/S0065-2164(10)70007-1
Khan Mirzaei, M., and Nilsson, A. S. (2015). Isolation of phages for phage therapy: a comparison of spot tests and efficiency of plating analyses for determination of host range and efficacy. PLoS ONE 10:e0118557. doi: 10.1371/journal.pone.0118557
Khawaldeh, A., Morales, S., Dillon, B., Alavidze, Z., Ginn, A. N., Thomas, L., et al. (2011). Bacteriophage therapy for refractory Pseudomonas aeruginosa urinary tract infection. J. Med. Microbiol. 60, 1697–1700. doi: 10.1099/jmm.0.029744-0
Kim, K. P., Cha, J. D., Jang, E. H., Klumpp, J., Hagens, S., and Hardt, W. D. (2008). PEGylation of bacteriophages increases blood circulation time and reduces T-helper type 1 immune response. Microb. Biotechnol. 1, 247–257. doi: 10.1111/j.1751-7915.2008.00028.x
Kingwell, K. (2015). Bacteriophage therapies re-enter clinical trials. Nat. Rev. Drug Discov. 14, 515–516. doi: 10.1038/nrd4695
Kucharewica-Krukowska, A., and Slopek, S. (1987). Immunogenic effect of bacteriophage in patients subjected to phage therapy. Arch Immunol. Ther. Exp. (Warsz.) 35, 553–561.
Kutter, E. M., De Vos, D., Gvasalia, G., Alvidze, Z., Gogokhia, L., Kuhl, S., et al. (2010). Phage therapy in clinical practice: treatment of human infections. Curr. Pharm. Biotechnol. 11, 69–86. doi: 10.2174/138920110790725401
Kutter, E. M., Kuhl, S. J., and Abedon, S. T. (2015). Re-establishing a place for phage therapy in western medicine. Future Microbiol. 10, 685–688. doi: 10.2217/fmb.15.28
Laxminarayan, R., and Powers, J. H. (2011). Antibacterial R&D incentives. Nat. Rev. Drug Discov. 10, 727–728. doi: 10.1038/nrd3560
Li, Y. (2014). China's misuse of antibiotics should be curbed. BMJ 348:g1083. doi: 10.1136/bmj.g1083
Loc-Carrillo, C., and Abedon, S. T. (2011). Pros and cons of phage therapy. Bacteriophage 1, 111–114. doi: 10.4161/bact.1.2.14590
Majewska, J., Beta, W., Lecion, D., Hodyra-Stefaniak, K., Kłopot, A., Kaźmierczak, Z., et al. (2015). Oral application of T4 phage induces weak antibody production in the gut and in the blood. Viruses 7, 4783–4799. doi: 10.3390/v7082845
Malki, K., Kula, A., Bruder, K., Sible, E., Hatzopoulos, T., Steidel, S., et al. (2015). Bacteriophages isolated from Lake Michigan demonstrate broad host-range across several bacterial phyla. Virol. J. 12, 164. doi: 10.1186/s12985-015-0395-0
Martin, S., Costa, A., and Perea, W. (2012). Stockpiling oral cholera vaccine. Bull. World Health Org. 90, 714–714. doi: 10.2471/BLT.12.112433
Mattila, S., Ruotsalainen, P., and Jalasvuori, M. (2015). On-demand isolation of bacteriophages against drug-resistant bacteria for personalized phage therapy. Front. Microbiol. 6:1271. doi: 10.3389/fmicb.2015.01271
Maxwell, K. L. (2016). Phages fight back: inactivation of the CRISPR-cas bacterial immune system by Anti-CRISPR proteins. PLoS Pathog. 12:e1005282. doi: 10.1371/journal.ppat.1005282
Miedzybrodzki, R., Boroysowski, J., Weber-Dabrowska, B., Fortuna, W., Letkiewicz, S., Szufnarowski, K., et al. (2012). Clinical aspects of phage therapy. Adv. Virus Res. 83, 73. doi: 10.1016/B978-0-12-394438-2.00003-7
Molton, J. S., Tambyah, P. A., Ang, B. S. P., Ling, M. L., and Fisher, D. A. (2013). The global spread of healthcare-associated multidrug-resistant bacteria: a perspective from Asia. Clin. Infect. Dis. 56, 1310–1318. doi: 10.1093/cid/cit020
Mullard, A. (2014). New drugs cost US$2.6 billion to develop. Nat. Rev. Drug Discov. 13, 877. doi: 10.1038/nrd4507
Nakonieczna, A., Cooper, C. J., and Gryko, R. (2015). Bacteriophages and bacteriophage-derived endolysins as potential therapeutics to combat Gram-positive spore forming bacteria. J. Appl. Microbiol. 119, 620–631. doi: 10.1111/jam.12881
Nathan, C., and Cars, O. (2014). Antibiotic resistance- problems, progress and prospects. N. Engl. J. Med. 37, 1761–1763. doi: 10.1056/NEJMp1408040
Nilsson, A. S. (2014). Phage therapy—constraints and possibilities. Ups. J. Med. Sci. 119, 192–198. doi: 10.3109/03009734.2014.902878
Oliveira, H., Boas, D. V., Mesnage, S., Kluskens, L. D., Lavigne, R., Sillankorva, S., et al. (2016). Structural and enzymatic characterization of ABgp46, a novel phage endolysin with broad anti-Gram-negative bacterial activity. Front. Microbiol. 7:208. doi: 10.3389/fmicb.2016.00208
Oliveira, H., Sillankorva, S., Merabishvili, M., Kluskens, L. D., and Azeredo, J. (2015). Unexploited opportunities for phage therapy. Front. Pharmacol. 6:180. doi: 10.3389/fphar.2015.00180
Parracho, H. M., Burrowes, B. H., Enright, M. C., McConville, M. L., and Harper, D. R. (2012). The role of regulated clinical trials in the development of bacteriophage therapeutics. J. Mol. Genet. Med. 6, 279–286. doi: 10.4172/1747-0862.1000050
Pelfrene, E., Willebrand, E., Sanches, A. C., Sebris, Z., and Cavaleri, M. (2016). Bacteriophage therapy: a regulatory perspective. J. Antimicrob. Chemother. 71, 2071–2074. doi: 10.1093/jac/dkw083
Piddock, L. J. V. (2012). The crisis of no new antibiotics-what is the way forward? Lancet Infect. Dis. 12, 249–253. doi: 10.1016/S1473-3099(11)70316-4
Pirnay, J. P., De Vos, D., Verbeken, G., Merabishvili, M., Chanishvili, N., Vaneechoutte, M., et al. (2011). The phage therapy paradigm: prêt-à-porter or sur-mesure? Pharm. Res. 28, 934–937. doi: 10.1007/s11095-010-0313-5
Rhoads, D. D., Wolcott, R. D., Kuskowski, M. A., Wolcott, B. M., Ward, L. S., and Sulakvelidze, A. (2009). Bacteriophage therapy of venous leg ulcers in humans: results of a phase I safety trial. J. Wound Care 18, 237–243. doi: 10.12968/jowc.2009.18.6.42801
Rose, T., Verbeken, G., De Vos, D., Merabishvili, M., Vaneechoutte, M., Lavigne, R., et al. (2014). Experimental phage therapy of burn wound infection: difficult first steps. Int. J. Burns Trauma 4, 66–73.
Saier, M. H., and Reddy, B. L. (2015). Holins in bacteria, eukaryotes, and archaea: multifunctional xenologues with potential biotechnological and biomedical applications. J. Bacteriol. 197, 7–17. doi: 10.1128/JB.02046-14
Salem, M., Virtanen, S., Korkeala, H., and Skurnik, M. (2015). Isolation and characterization of Yersinia-specific bacteriophages from pig stools in Finland. J. Appl. Microbiol. 118, 599–608. doi: 10.1111/jam.12722
Sarker, S. A., McCallin, S., Barretto, C., Berger, B., Pittet, A. C., Sultana, S., et al. (2012). Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 434, 222–232. doi: 10.1016/j.virol.2012.09.002
Sarker, S. A., Sultana, S., Reuteler, G., Moine, D., Descomebes, P., Charton, F., et al. (2016). Oral phage therapy of acute bacterial diarrhea with two coliphage preparations: a randomized trial in children from Bangladesh. EBio. Med. 4, 124–137. doi: 10.1016/j.ebiom.2015.12.023
Sauder, A. B., Quinn, M. R., Brouillette, A., Caruso, S., Cresawn, S., Erill, I., et al. (2016). Genomic characterization and comparison of seven Myoviridae bacteriophage infecting Bacillus thuringiensis. Virology 489, 243–251. doi: 10.1016/j.virol.2015.12.012
Saussereau, E., Vachier, I., Chiron, R., Godbert, B., Sermet, I., Dufour, N., et al. (2014). Effectiveness of bacteriophages in the sputum of cystic fibrosis patients. Clin. Microbiol. Infect. 20, O983–O990. doi: 10.1111/1469-0691.12712
Scannell, J. W., Blanckley, A., Boldon, H., and Warrington, B. (2012). Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Disc. 11, 191–200. doi: 10.1038/nrd3681
Schmerer, M., Molineux, I. J., and Bull, J. J. (2014). Synergy as a rationale for phage therapy using phage cocktails. PeerJ 2:e590. doi: 10.7717/peerj.590
Smith, H. O., Hutchison, C. A., Pfannkoch, C., and Venter, J. C. (2003). Generating a synthetic genome by whole genome assembly: φX174 bacteriophage from synthetic oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 100, 15440–15445. doi: 10.1073/pnas.2237126100
Sonderholm, J. (2009). Wild-card patent extensions as a means to incentive research and development of antibiotics. J. Law Med. Ethics 37, 240. doi: 10.1111/j.1748-720X.2009.00368.x
Valera, M. C., Cardoso, F. G. D. R., Maekawa, L. E., Camargo, C. H. R., de Oliveira, L. D., and Carvalho, C. A. T. (2015). In vitro antimicrobial and anti-endotoxin action of Zingiber Officinale as auxiliary chemical and medicament combined to calcium hydroxide and chlorhexidine. Acta Odontol. Scand. 73, 556–561. doi: 10.3109/00016357.2014.949846
Van Boeckel, T. P., Gandra, S., Ashok, A., Caudron, Q., Grenfell, B. T., Levin, S. A., et al. (2014). Global antibiotic consumption 2000 to 2010: an analysis of national pharmaceutical sales data. Lancet Infect. Dis. 14, 742–750. doi: 10.1016/S1473-3099(14)70780-7
Verbeken, G., Pirnay, J. P., De Vos, D., Jennes, S., Zizi, M., Lavigne, R., et al. (2012). Optimizing the European regulatory framework for sustainable bacteriophage therapy in human medicine. Arch. Immunol. Ther. Exp. (Warsz.) 60, 161–172. doi: 10.1007/s00005-012-0175-0
Verbeken, G., Pirnay, J. P., De Vos, D., Jennes, S., Zizi, M., Lavigne, R., et al. (2014). Call for a dedicated European legal framework for bacteriophage therapy. Arch. Immunol. Ther. Exp. (Warsz.) 62, 117–129. doi: 10.1007/s00005-014-0269-y
Verduri, A., Luppi, F., D'Amico, R., Balduzzi, S., Vicini, R., Liverani, A., et al. (2015). Antibiotic treatment of severe exacerbations of chronic obstructive pulmonary disease with procalcitonin: a randomized noninferiority trial. PLoS ONE 10:e0118241. doi: 10.1371/journal.pone.0118241
Whitfield, K., Huemer, K. H., Winter, D., Thirstrup, S., Libersa, C., Barraud, B., et al. (2010). Compassionate use of interventions: results of a European Clinical Research Infrastructures Network (ECRIN) survey of ten European countries. Trials 11, 104. doi: 10.1186/1745-6215-11-104
Wise, J. (2015). Report calls for $2bn global fund to kickstart antibiotic development. BMJ 350:h2635 doi: 10.1136/bmj.h2635
Young, R., and Gill, J. J. (2015). Phage therapy redux-What is to be done? Science. 350, 1163–1164. doi: 10.1126/science.aad6791
Keywords: bacteriophage, phage therapy, adaptive pathways, alternative licensing, pharmaceutical regulation
Citation: Cooper CJ, Khan Mirzaei M and Nilsson AS (2016) Adapting Drug Approval Pathways for Bacteriophage-Based Therapeutics. Front. Microbiol. 7:1209. doi: 10.3389/fmicb.2016.01209
Received: 27 May 2016; Accepted: 20 July 2016;
Published: 03 August 2016.
Edited by:
Peter Mullany, University College London, UKReviewed by:
Eric Christopher Keen, Washington University in St. Louis, USACopyright © 2016 Cooper, Khan Mirzaei and Nilsson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Callum J. Cooper, Y2FsbHVtLmNvb3BlckBzdS5zZQ==
Anders S. Nilsson, YW5kZXJzLnMubmlsc3NvbkBzdS5zZQ==
†Present Address: Mohammadali Khan Mirzaei, Department of Microbiology and Immunology, McGill University, Montreal, QC, Canada
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.