 Jialian Xie
Jialian Xie Shaojun Li
Shaojun Li Chenmi Mo1
Chenmi Mo1 Xueqiong Xiao
Xueqiong Xiao- 1Key Laboratory of Plant Pathology of Hubei Province, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, China
- 2State Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
Purpureocillium lilacinum is a promising nematophagous ascomycete able to adapt diverse environments and it is also an opportunistic fungus that infects humans. A microbial inoculant of P. lilacinum has been registered to control plant parasitic nematodes. However, the molecular mechanism of the toxicological processes is still unclear because of the relatively few reports on the subject. In this study, using Illumina paired-end sequencing, the draft genome sequence and the transcriptome of P. lilacinum strain 36-1 infecting nematode-eggs were determined. Whole genome alignment indicated that P. lilacinum 36-1 possessed a more dynamic genome in comparison with P. lilacinum India strain. Moreover, a phylogenetic analysis showed that the P. lilacinum 36-1 had a closer relation to entomophagous fungi. The protein-coding genes in P. lilacinum 36-1 occurred much more frequently than they did in other fungi, which was a result of the depletion of repeat-induced point mutations (RIP). Comparative genome and transcriptome analyses revealed the genes that were involved in pathogenicity, particularly in the recognition, adhesion of nematode-eggs, downstream signal transduction pathways and hydrolase genes. By contrast, certain numbers of cellulose and xylan degradation genes and a lack of polysaccharide lyase genes showed the potential of P. lilacinum 36-1 as an endophyte. Notably, the expression of appressorium-formation and antioxidants-related genes exhibited similar infection patterns in P. lilacinum strain 36-1 to those of the model entomophagous fungi Metarhizium spp. These results uncovered the specific parasitism of P. lilacinum and presented the genes responsible for the infection of nematode-eggs.
Introduction
Parasitic nematodes cause great loss to agriculture, leading to a loss of US$157 billion each year around the world (Abad et al., 2008). For decades, nematode control has largely depended on various chemical nematicides. However, chemical control usually leads to environmental side effects and human health risks (Li et al., 2015). Some bio-control agents such as soil hyphomycetes show great promise. The egg-pathogenic fungus Purpureocillium lilacinum (P. lilacinum), which was previously known as Paecilomyces lilacinus, is one of the most widely reported entophytic filamentous fungi used as a bio-control agent to control root-knot nematodes. One of its strains (PL251) has being registered as a biological nematicide in the USA (Kiewnick et al., 2011). P. lilacinum is frequently isolated from soil, forests, grass, nematodes, and insects (Luangsa-ard et al., 2011). In addition, the fungus has been detected in the rhizosphere of many crops (Lopez et al., 2014). It is also an opportunistic pathogen of immunodeficient humans and other vertebrates (Inglis et al., 2005).
P. lilacinum can grow in a wide range of temperatures with the optimal growth range from 26 to 30°C (Banu et al., 2006). This organism is tolerant to broad range of pH values and can grow on a variety of carbon and nitrogen media (Banu et al., 2006; Sun and Liu, 2006). Moreover, P. lilacinum is well-known for producing all types of proteolytic enzymes and carbohydrate hydrolases (such as serine proteases and chitinases), which can destroy the lipid and chitin layers of nematode eggshell (Wang et al., 2010). Besides, P. lilacinum can also produce various biologically active secondary metabolites including polyketides and non-ribosom-synthesized peptides, such as leucinostatins, which exert a range of biological activities including nematicidal, anti-viral, anti-tumor, and phytotoxicity properties (Mori et al., 1982; Park et al., 2004; Ishiyama et al., 2009).
Gene functional studies of P. lilacinum have identified a few genes that are involved in virulence and fungal development (Wang et al., 2010; Yang et al., 2011). Most studies have focused on practical bio-control effects on crops and medical discoveries in human (Ramam, 2014; López-Medrano et al., 2015). The underlying molecular biological mechanism is still unknown. The genome of a P. lilacinum strain that was isolated from the tannery sludge in India was already sequenced. The report has summarized the fundamental characteristics of the fungus, but the genes related to pathogenesis were not well-explored (Prasad et al., 2015). Here, we sequenced another P. lilacinum strain 36-1, which was specifically isolated from nematode eggs, and performed a comparative analysis with the other sequenced genome of Ascomycetes, especially for nematophagous or entomopathogenic fungi. The comparison revealed a series of gene family expansions and contractions in P. lilacinum 36-1 that could distinguish it from plant pathogens, as well as species-specific genes gain or loss that implemented different pathogenic strategies. Moreover, the transcriptional responses of P. lilacinum strain 36-1 to Meloidogyne incongnita eggs were explored by RNA-seq technique and the genes involved in pathogenicity were demonstrated.
Materials and Methods
Fungus Strains
P. lilacinum strain 36-1 was isolated from the surface of the eggs of Meloidogyne incognita in the field soil from Hubei Province in China (Yang et al., 2015). The fungus were cultured in potato dextrose agar (PDA) medium at 28°C. The conidia of P. lilacinum strain 36-1 were suspended in MM liquid medium, which contained 1g L−1 NH4NO3, 0.5g L−1 KH2PO3, 1.5g L−1 K2HPO3, 1.0 g L−1 NaCl, and MgSO4.7H2O.
Genome Sequencing and Assembly
The genome of P. lilacinum strain 36-1 was shotgun-sequenced with the Illumina next generation sequencing technology. DNA libraries with 500 and 6000 bp insertions were constructed and sequenced with Hiseq2000 at BGI (Shenzhen, China). The genome sequence was assembled using SOAPdenovo 1.05 (Li et al., 2010).
Gene Prediction and Annotations
The gene modes were predicted with a combined method in priority order. Gene structures were generated by Genewise 2.20 (Birney et al., 2004) using protein sequences from Metarhizium anisopliae, Metarhizium acridum, Ophiocordyceps sinensis, Trichoderma virens, and Aspergillus nidulans as references, with the ab initio predictor Augustus 2.6.1 (Keller et al., 2011) and SNAP (Korf, 2004) using annotated information from A. nidulans FGSC A4, GeneMark-ES 2.3e (Borodovsky and Lomsadze, 2011). The final gene annotation set was incorporated and redundancies were removed. Transfer RNAs (tRNAs) were predicted with tRNAscan-SE (Schattner et al., 2005). Secreted proteins were identified with SignalP3.0 using a hidden Markov model (Bendtsen et al., 2004). Transmembrane proteins were predicted with TMHMM 2.0 (Krogh et al., 2001). The subcellular localization of proteins were identified by Targetp1.1 (Emanuelsson et al., 2000). Transposon elements were analyzed with a combined method. RepeatMasker was used to Blast against the Repbase library, and a transposon model was constructed by building an XDFDatabase with P. lilacinum strain 36-1 genome sequences (Saha et al., 2008). Tandem repeat elements were identified by Tandem Repeat Finder (TRF) (Benson, 1999). The RIP (Repeat Induced Point mutation) index was determined with RIPCAL software by referencing them against the non-repetitive control families (Hane and Oliver, 2008). The whole genome alignment between P. lilacinum 36-1 and the India strain was performed using MAUVE aligner version 2.4.0 using progressive algorithm with default parameters prior to reordering the scaffolds by Mauve Contig Mover (MCM) (Darling et al., 2004). Locally collinear blocks (LCB) with lengths of at least 100 bp was displayed by R package genoPlotR (Guy et al., 2011). The genome segment rearrangements and SNPs were identified by reciprocal genomic comparison between P. lilacinum 36-1 and the India strain.
Orthology and Phylogenomic Analysis
Orthologous proteins were identified with ProteinOrtho 5.08 following the default parameters (Lechner et al., 2011). Totally, 19 species from different ecological niches were selected, including the phytopathogenic fungi Fusarium graminearum, Magnaporthe oryzae, Ustilaginoidea virens, Verticillium dahiliae, Rhizoctonia solani, and Ustilago maydis; saprophytes Neurospora crassa, T. reesei, T. virens, and Saccharomyces cerevisiae; human pathogenic fungi A. nidulans, A. fumigatus, and Candida albicans; and nematophagous/entomophagous fungi O. sinensis, Beauveria bassiana, M. acridum, M. anisopliae, Cordyceps militaris, and Pochonia chlamydosporia. In sum, 580 single-copy orthologous proteins were acquired and aligned with muscle 3.8 (Edgar, 2004). The alignment sequences were concatenated and subsequently used for phylogenetic reconstruction by maximum likelihood method with the Dayhoff amino acid substitution model implemented in MEGACC7.0 (Molecular Evolutionary Genetics Analysis Computational Core) (http://www.megasoftware.net/) with 1000 bootstrap replications and complete-deletions for gaps or missing sites.
Protein Family Classification and Evolutionary Analysis
Whole genome protein families were classified by InterProScan analysis (Jones et al., 2014). The peptidase families were identified by BLASTP searching against MEROPS peptidase database release 9.12 with a cutoff e-value of 1e-20 (Rawlings et al., 2014). The transporters were classified on the basis of Transport Classification Database (Saier et al., 2014). Carbohydrate-active enzymes were classified by using hmmer 3.0 (Mistry et al., 2013) to search against a library of catalytic and carbohydrate-binding module enzymes acquired from dbCAN (Yin et al., 2012). G-protein-coupled receptors were selected from the best hits with GPCRDB sequences (Horn et al., 2003) and by confirming that they contained seven transmembrane helices with the amino terminus outside and the carboxyl terminus inside the plasma membrane. Homologs of the Pth11-like GPCRs in Magnaporthales (Kulkarni et al., 2005) were identified by local BLASTP analysis with a cutoff e-value of 1e-10. The divergence time between species was estimated with the Langley-Fitch method with r8s (Marazzi et al., 2012) by calibrating against the reassessed origin of the Ascomycota at 500–650 million years ago (Lücking et al., 2009).
Transcriptome Mapping and Expression Analysis
P. lilacinum strain 36-1 was cultured on potato dextrose agar for 15 days. The conidia were washed out with 10 mL of sterile distilled water and adjusted to a concentration of 1 × 107 cfu mL−1 with MM liquid medium. M. incongnita egg-masses were isolated from tomato roots and sterilized with 1% (v/v) NaClO. The dissociated eggs were suspended in sterile distilled water. The conidia that were precultured in MM liquid media for 24 h were mixed with water or M. incongnita eggs, the concentration of which was adjusted to 2 eggs per microliter. Each treatment was repeated three times. The mixtures were filtered to collect the mycelia after 24 h. The infection process was checked under a light microscope. All the repeat treatments were collected and mixed together following RNA extractions. Total RNAs were extracted according to standard protocol plus an incubation treatment with RNase-free DNase I. mRNAs were purified and then reverse-transcribed into cDNA for library construction. Each library product was sequenced using a library with a read insertion size of 200 bp by using Illumina GA II technology without technology replications. The paired-end reads were filtered as follows: Adaptor sequences were trimmed from the reads with the NGS QC toolkit (ver. 2.3) (Patel and Jain, 2012); and reads with < 80% “Q > 20” bases were removed. We used TopHat2 (ver. 2.1.1) (Trapnell et al., 2009) to map filtered mRNA reads onto the predicted genes model of the genome. The RNA expression count and identification of differentially expressed genes were performed using GFOLD v1.1.1 with a significant cutoff of 0.01 for the fold change. The software GFOLD was especially designed for RNA-Seq reads when no replicate was available, generalizing the reliable fold change by considering the posterior distribution of the log fold change (Feng et al., 2012). To reduce the false positive ratio, we used a stricter expression value (|log2 fold-change| > 2) to select differential expression genes. The gene ontology (GO) annotation of the genome was conducted with ipr2go by a local R script. The GO and KEGG enrichment were analyzed with GOstats in Bioconductor (Falcon and Gentleman, 2007).
Growth Profiles on a Single Carbon Source
The MM medium and agar were mixed together as a basic culture medium in 9 cm Petri dishes. Xylan, cellulose or pectin were used as single carbon source to evaluate the growth abilities of different fungi with diverse carbon sources. Inoculations of P. lilacinum strain 36-1 and other fungi (F. graminearum and T. reesei) were placed on centrals sections of these mediums and cultured at 28°C for 3 days. Three biological replicates were set.
qPCR Analysis
The total RNAs used for RNA-Seq were also employed to analyze the expression levels of the target genes. First, the mRNAs were reverse-transcribed with SuperScript III Reverse Transcriptase (Invitrogen, Shanghai, China) by oligo dT according to the instructions. SsoFast™ EvaGreen Supermix (Bio-Rad, USA) was used to quantify the target genes by putting 5 ng of cDNA products into a 10 μL quantification system. The qPCR cycling parameters were 95°C for 20 s, then 45 cycles of 95°C for 10 s, and 60°C for 20 s. β-Tubulin was used as an endogenous reference gene (Yang et al., 2011). The experiments were conducted in quadruplicate. The 2−ΔΔCt method was used to calculate the transcript abundance. The primers used for qPCR are listed in Table S1.
Results
General Features
The genome of P. lilacinum strain 36-1 was shotgun sequenced up to 75 × coverage using Illumina paired-end sequencing with insertion sizes between 500 and 6000 bp. A draft genome sequence was assembled on the basis of the high-quality reads using the de novo assembler SOAPdenovo (Li et al., 2010). We ultimately obtained a 37.61 Mb genome that was assembled into 240 scaffolds with the N50 size being over 1.3 Mb (Table 1), which was a bit different from the P. lilacinum India strain because of the various sequencing methods and different sources of the fungus (Table 1). The GC contents of the assembled genome was 58.57%, which was the same as the previous sequenced P. lilacinum strain (Table 1). However, P.lilacinum 36-1 seemed to have a more compact genome as indicated by a series of features, such as higher gene density, longer average length of genes and more tRNAs (Table 1). The Whole genome alignment analyses showed large numbers of locally collinear blocks (LCB) between this two strains, but more genome segments rearrangements and SNPs indicated that the genome of P. lilacinum 36-1 was more dynamic than the India strain (Figure S1, Table 1).
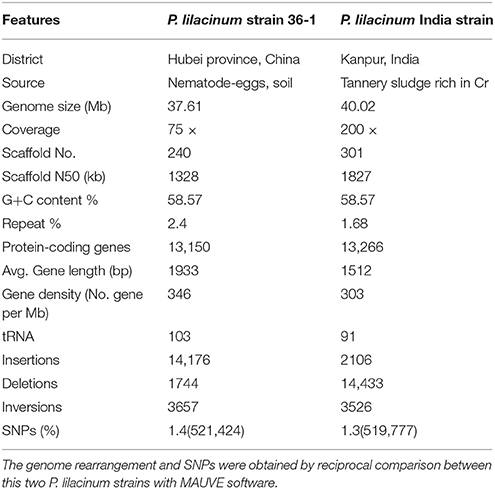
Table 1. Comparison of genome feature between two different strains of P. lilacinum.
We further evaluated the quality of the genome assembly with BWA (Li and Durbin, 2009). As a result, 98.20% of the reads were mapped back to the assembly, which covered over 99.99% of the scaffolds, excluding the gaps. This finding showed that the current assembly covered almost all the genome sites. Moreover, ~97.46% of the assembly was covered by at least 20 × reads, representing the high level of accuracy for assembly at single nucleotide sites (Figure S2). Based on the extensive presence of conserved protein families in eukaryotes, the ratio of core eukaryotic genes (CEGs) was evaluated in the current assembly using CEGMA (Parra et al., 2007). Finally, 242 complete-partial CEGs were found in the current assembly, making up 97.58% the total CEGs. All of these results suggested a largely complete assembly.
The genome of P. lilacinum 36-1 was predicted to have 13150 protein coding genes, which was slightly lower than the number for India strain (Table 1) but higher than other sequenced fungi (Table S2). Relative to fungi in diverse ecological niches, P. lilacinum 36-1 had more line-specific genes with nematophagous/entomophagous fungi (357 genes) than phytopathogenic (158 genes) and saprophytic fungi (54 genes) (Figure 1A). Among the 357 genes, 187 could be annotated in the gene ontology (GO) database (p < 0.05), of which genes with functions of DNA binding, peptidases, kinase activity, oxidoreductase activity, transmembrane transporter activity, and ion binding activity were well-represented. Additionally, P. lilacinum 36-1 shared 4995 homologous genes with other sequenced fungi and had 3172 species-specific genes (Figure 1A). The orphans mapped onto GO database were primarily enriched in terms of metabolic regulation and cellular macromolecule biosynthetic processes (p < 0.05) (Table S3), suggesting a complex of physiological regulation activities in P. lilacinum 36-1.
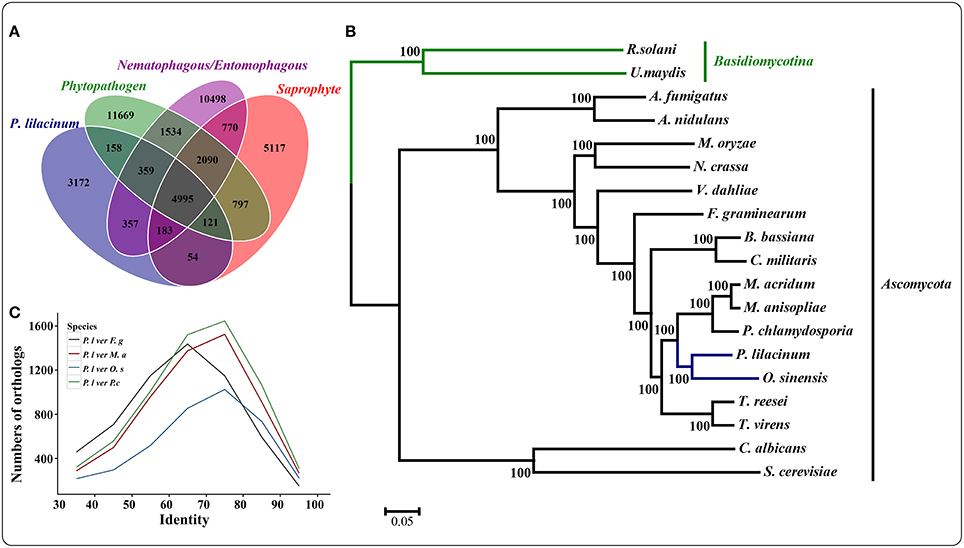
Figure 1. Comparative genomics and evolutionary analysis of P. lilacinum strain 36-1. (A) Numbers of homologous families between different fungi with different lifestyle. Phytopathogenic fungi include Fusarium graminearum, Magnaporthe oryzae, and Ustilaginoidea virens; Saprophytism include Neurospora crassa, Trichoderma reesei, and Aspergillus nidulans. Nematophagous/Entomophagous fungi include Ophiocordyceps sinensis, Beauveria bassiana, Metarhizium acridum, Cordyceps militaris, and Pochonia chlamydosporia; (B) A maximum likelihood phylogenomic tree constructed using the Dayhoff amino acid substitution model showing the evolutionary relationship of P. lilacinum strain 36-1 with other fungi; (C) Numbers of orthologous genes with different levels of protein identity between P. lilacinum strain 36-1 and other fungi. Identity refers to the reciprocal similarity (>30%) of proteins between P. lilacinum and other fungi by BLASTP.
A phylogenetic tree was constructed from 580 single-copy conserved orthologous groups acquired by comparing the protein sequences in 19 species. The results showed that P. lilacinum 36-1 was more closely related to the entomophagous fungus O. sinensis (Figure 1B). According to the Langley-Fitch method, we found that P. lilacinum 36-1 diverged before a split with O. sinensis 74~94 million years ago (MYA) (Figure 1B). However, these two fungi shared far few reciprocal best hit genes than those of the nematode eggs parasite fungus P. chlamydosporia and the entomophagous fungus M. acridum (Figure 1C).
PANTHER (Protein Analysis THrough Evolutionary Relationships, http://pantherdb.org) is widely used for comprehensive protein evolutionary and functional classification (Mi et al., 2016). By using a PANTHER analysis, we compared the profiles for gene expansions or contractions in seven fungi by statistical overrepresentation test with a Bonferroni correction. In comparison with O. sinensis, P. lilacinum 36-1 showed more regulation related genes (P < 0.05), such as enzyme modulators, kinases, membrane traffic proteins, RNA bindings, and transcription factors (Figure 2).
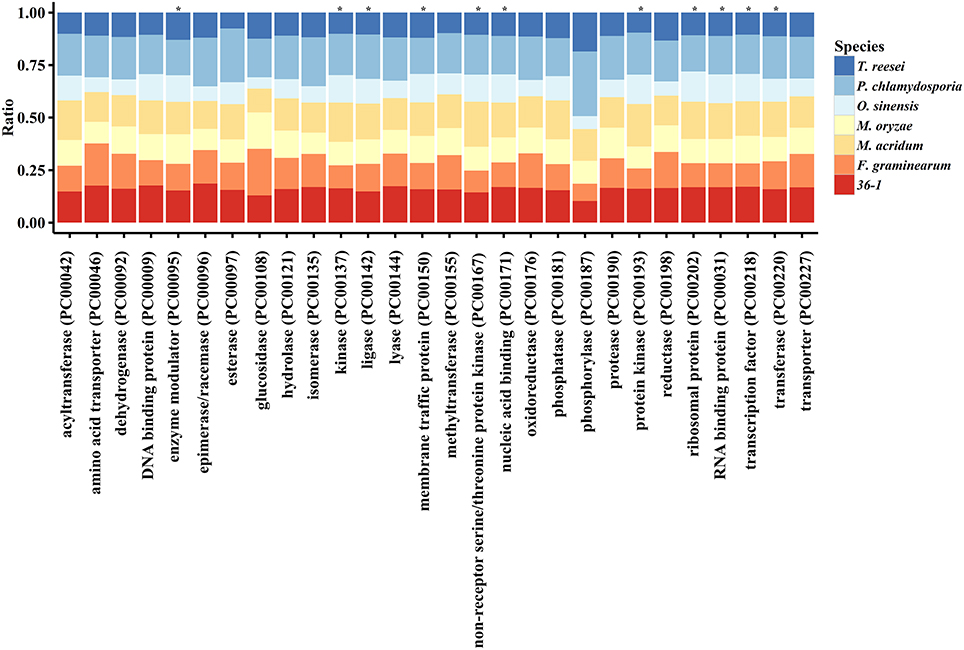
Figure 2. Comparison of gene families between different fungi. *:Indicates significantly expansion of P. lilacinum 36-1 relative to O. sinensis (P < 0.05); Protein class indicates the classification system by PANTHER. Ratio indicates the percentage of protein class in different fungi.
Weak Repeat-Induced Point Mutation (RIP)
The RIP (repeat-induced point mutation) is a genome defense mechanism in fungi that acts against sequence duplication and accumulates G:C to A:T transition mutations (Selker et al., 1987). The RIP index was calculated by using two different indices, namely (CpA+TpG)/(ApC+GpT) and TpA/ApT, which were based on calculations of the dinucleotide frequencies. For the TpA/ApT index, a higher value indicates a stronger RIP, which is the opposite of the (CpA+TpG)/(ApC+GpT) index (Hane and Oliver, 2008). In P. lilacinum 36-1, a low average ratio for the TpA/ApT index (0.78) and a high (CpA+TpG)/(ApC+GpT) index (1.51) was observed, indicating weak RIP performance in P. lilacinum 36-1. The average ratio of di-nucleotide preference in repeat elements also provided evidence for the weak RIP in P. lilacinum 36-1 (Figure S3).
RIP usually occurs during meiosis (Galagan et al., 2003). Two types of compatible MAT genes (MAT1-1 and MAT1-2) are required for sexual development (Kück and Böhm, 2013). The lack of MAT1-2 which contains the HMG_box domain implied the absence of a sexual phase in P. lilacinum 36-1 (Table S4, Figure S4).
RIP impacts on gene duplications, resulting in a decrease in the numbers of gene families (Galagan et al., 2003). A weak RIP could not influence gene expansion in P. lilacinum 36-1. We found 166 pairs of duplicated genes with >90% of nucleotide sequence similarities in P. lilacinum 36-1 in contrast to only 6 pairs in the RIP-effective species N. crassa (Galagan et al., 2003).
Weak Ability to Degrade the Plant Cell Walls
Many plant pathogens require glycoside hydrolases (GHs), carbohydrate esterases (CEs) and polysaccharide lyases (PLs) to degrade the plant cuticle and cell wall. Although P. lilacinum 36-1 could not infect plants, it does have the ability to live in the roots of plants as an endophyte (Lopez et al., 2014). A hierarchical clustering analyses showed that P. lilacinum 36-1 was most related to P. chlamydosporia (Figure 3A), a fungus that acted as an endophyte in monocot and dicot plants.
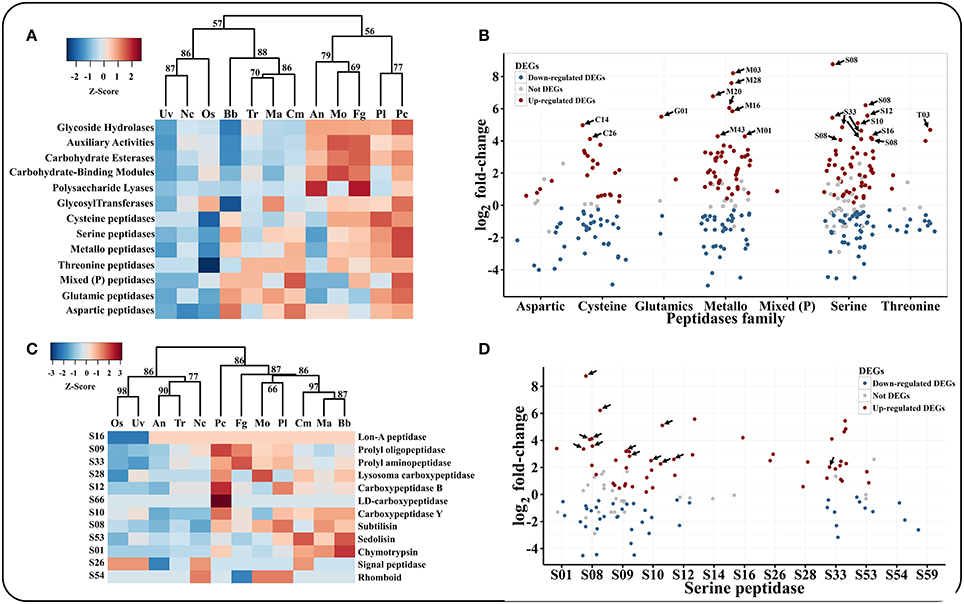
Figure 3. Comparison and expression patterns of CAZYs and peptidase families in P. lilacinum 36-1. (A) Hierarchical clustering of CAZYs and peptidases families in 12 fungi. Uv, U. virens; Nc, N. crassa; Fg, F. graminearum; Os, O. sinensis; Bb, B. bassiana; Tr, T. reesei; Ma, M. acridum; Cm, C. militaris; An, A. nidulans; Mo, M. oryzae; Pl, P. lilacinum; Pc, P. chlamydosporia. Protein families are represented by their family names according to the CAZY and merops database. Approximately unbiased (AU) P-values (%) are computed by 1000 bootstrap resamplings by using the R package pvclust. (B) Expression profile of different peptidase families. Sub-families with high expression values (log2 fold-change > 4) are indicated by arrows. (C) Hierarchical clustering of serine peptidase sub-families in 12 fungi. (D) Expression profile of serine peptidase sub-families. The arrows indicate signal proteins.
Cellulolytic enzymes are used in interactions with plants, especially in plant root colonization by endophytic fungi (Strakowska et al., 2014). P. lilacinum possessed certain numbers of genes that were involved in cellulose and hemicellulose degradation, in particular β-glucosidase (GH3), cellulases (GH5), endo-β-1,4-glucanase (GH12), β-xylosidase (GH43), α-mannosidase (GH47), hydrolyzing lignocellulose (GH35), cellulose-binding domains (family CBM1), and copper-dependent lytic polysaccharide monooxygenases (family AA9) (Table S5). In comparison with plant pathogenic fungi, however, the absence of cellobiohydrolase I (GH6) and cellobiohydrolase II (GH7) in P. lilacinum 36-1 attenuated the capacity for cellulolytic degradation (Table S5). Carbohydrate esterases (CE) cooperate with glycoside hydrolases (GH) in plant polysaccharide degradation to overcome the complexity of the plant cell walls (Biely, 2012). P. lilacinum 36-1 contained more CEs in comparison with the entomophagous fungi, but less than those of cereal pathogens F. graminearum and M. oryzae, such as acetyl xylan esterase (CE1), acetyl xylan esterase (CE2), cutinase (CE5), and arylesterase (CE10) (Table S5), indicating a relatively weaker ability to decompose cell wall. The only exception was that P. lilacinum 36-1 had 3 genes, while M. oryzae did not have any genes and F. graminearum only had one gene of CE7 family (cephalosporin-C deacetylase) (Table S5). Notably, fewer cutinases (CE5) suggested that P. lilacinum 36-1 might not depend on direct penetration into the plant roots by degrading the cuticle. Likewise, P. lilacinum 36-1 contained much less polysaccharide lyase (PL) than plant pathogens, with a lack of pectin lyase (PL1), pectate lyase (PL3) and rhamnogalacturonan lyase (PL4) (Table S5). Therefore, P. lilacinum 36-1 might be unable to degrade pectin effectively during the saprophytic or endogenous phase.
To validate the result discussed above, the capacity of P. lilacinum 36-1 to dissociate components of the cell wall (cellulose, pectin, and xylan) was evaluated in comparison with that of F. graminearum and T. virens. As expected, in comparison with the other two fungi, P. lilacinum 36-1 had a weaker ability to break down the three substances as indicated by the smaller halos observed in the medium by using the substances as the only carbon source (Figure S4). Moreover, P. lilacinum 36-1 grew better in the medium that was supplied with cellulose than that of mediums containing pectin and xylan, which was consistent with the partial cellulose degradation gene family expansion mentioned above.
Serine and Metallo-Peptidases Act as Dominant Factors during Nematode-Eggs Infection
The primary substances of cuticles on nematode eggs are made up of protein-chitin (Wharton, 1980). Thus, nematode eggs pathogens must secrete large amounts of hydrolytic enzymes, such as peptidases or carbohydrate hydrolases, to penetrate the shell and solubilize the host tissues for nutrition (Yang et al., 2007).
P. lilacinum 36-1 possessed 333 genes that encoded peptidases, which were more than those of the other sequenced fungi, with 86 putative secreted proteins (Table S6). The most abundant peptidases families were serine peptidases (121 genes) and metallopeptidases (95 genes), which were much more than those of the plant pathogen, with average numbers of 86 and 78, respectively (Table S6). These two peptidase families were considerably up-regulated during infection (Figure 3B).
Among the serine peptidases, the subtilisin family (S08) had the highest relative expansion with 30 genes in P. lilacinum, a little more than that in P. chlamydosporia (23 genes) and M. acridum (27 genes) (Table S6, Figure 3C). Subtilisin (S08) could be induced by nitrogen sources (including nematode cuticle) and play an important role during the infection of nematophagous fungi (Wang et al., 2010; Zou et al., 2010). Consistent with their important functions, 8 subtilisin coding genes were up-regulated and most of them were putative secreted proteins (Figure 3D). The second common serine peptidases were prolyl oligopeptidase (S09) and prolyl aminopeptidase (S33), which could help P. lilacinum 36-1 to degrade a proline-rich substrate (Table S6). Both of these two gene family had a different expression level when parasitizing nematode-eggs (Figure 3D). Trypsin (S01) is responsible for degrading the chitin-protein layer of the nematode eggshell and affecting the normal embryogenic development of nematodes (Suarez et al., 2004). However, unlike the entomophagous fungi, P. lilacinum 36-1 possessed only 4 genes that coded for trypsin (S01), which was much less than those in B. bassiana (14 genes) and M. acridum (10 genes) (Table S6), and only one of them was up-regulated at a relatively lower level of expression (Figure 3D).
Despite the serine peptidases, the over-representation of metallopeptidases may also function during infection (Table S6). These families such as aminopeptidase (M03), oligopeptidase (M08), and carboxypeptidase (M20) were particularly up-regulated significantly (log2 fold-change > 4) (Figure 3B). In addition, aspartic peptidases which are assist the human pathogen C. albicans by degrading cell surface molecules (Schaller et al., 2005) were also an abundant family in P. lilacinum 36-1 (Table S6). But this family may not be so activate as metallopeptidases, as indicated by their expression profiles (Figure 3B).
Chitinases Are Not the Primary Factors at the Early Stage of Nematode Eggs Infections
Based on the CAZYs database (Carbohydrate-Active enZYmes Database), we found that P. lilacinum 36-1 had a considerable number of carbohydrate-active enzymes. Glycoside hydrolases (GHs), auxiliary activities (AAs) and carbohydrate esterases (CEs) were well-represented in P. lilacinum 36-1 in comparison with entomophagous or saprophytic fungi (Figure 3A).
Chitin is the primary component of nematode eggs. P. lilacinum 36-1 needs to secrete many chitin-degradation genes to digest nematode egg-shells, such as GHs, CBMs, AAs, and CEs. These types of enzymes exhibited clearly different distributions in various fungi (Table S5, Figure 4A). Hierarchical clustering showed an obvious expansion of chitin degradation genes in P. lilacinum 36-1 such as chitinases (GH18), isozymes (GH24, GH25), chitosanase (GH75), chitooligosaccharide oxidase (AA7) and chitin deacetylase (CE4) in P. lilacinum 36-1, suggesting a strong ability to degrade chitin (Figure 4A). During the early stage of nematode eggs infections, these genes were up-regulated at different levels, indicating their importance in nematode eggs-shell degradation (Figure 4B). Moreover, in considering the crucial functions of secretory proteins, we predicted the sub-cellular localizations of these up-regulated genes. Surprisingly, only one chitinase gene belonged to extracellular protein but it had a relatively lower expression value (Figure 4B). By contrast, the secretory proteins CE4, GH75, AA7 and CBM50 (chitin-binding) exhibited a high expression value (Figure 4B). These results suggested that the degradation of chitin in nematode egg-shells might not primarily depend on the chitinase hydrolysis pathway, but on successive processes of chitin deacetylase, chitosanase, and chitooligosaccharide oxidase assisting by chitin binding protein, thus making the egg-shell soluble enough for P. lilacinum 36-1 infection at the early stage (Figure 4C). In particular, the chitin deacetylase in P. lilacinum 36-1 was homologous with an appressorium differentiation related gene (UniPortKB: D1MYV6) in M. oryzae, indicating that the up-regulated chitin degradation genes might be involved in appressorium formation at the early infection stage.
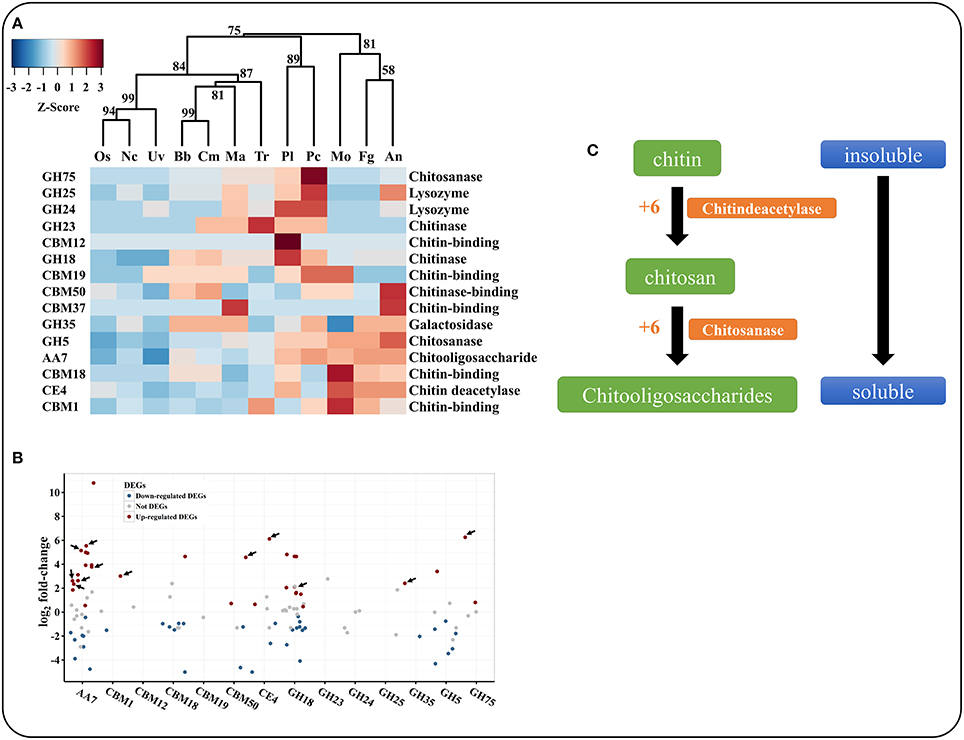
Figure 4. Comparison and expression pattern of chitin degradation genes in P. lilacinum. (A) Hierarchical clustering of chitin degradation families in 12 fungi. Uv, U. virens; Nc, N. crassa; Fg, F. graminearum; Os, O. sinensis; Bb, B. bassiana; Tr, T. reesei; Ma, M. acridum; Cm, C. militaris; An, A. nidulans; Mo, M. oryzae; Pl, P. lilacinum; Pc, P. chlamydosporia. Approximately unbiased (AU) P-values (%) are computed by 1000 bootstrap resamplings by using the R package pvclust. (B) Expression profile of different chitin degradation genes during early stage of P. lilacinum infection nematode-eggs. The arrows indicate signal proteins. (C) The putative pathway of P. lilacinum degradation chitin during infection nematode-eggs. The numbers indicates expression values by transcriptom analysis.
Abundance of Transporters Assist Broad Environmental Adaptation of P. lilacinum 36-1
Membrane transporters are involved in defending against toxic metabolites and in transporting nutrients (Costa et al., 2014). Based on a BLAST search against the TCDB database, we identified 614 transporters in the P. lilacinum 36-1 genome. Interestingly, the transporter content was similar to that of F. graminearum, but much higher than that of entomophagous and saprophytic fungi (Table S7), including the major facilitator superfamily (MFS), amino acid-polyamine-organocation family (APC), drug/metabolite transporter superfamily (DMT), and ATP-binding cassette superfamily (ABC). Considering the closer genetic relationship and the huge differences in inhabited environments between P. lilacinum and O. sinensis, we concluded that the high transporter genes content were the key factors of P. lilacinum 36-1 to adapt diverse environments.
More than 160 transporters were significantly up-regulated (log2 fold-change > 2) when parasitizing nematode-eggs (Table S8). In particular, ~101 genes encoding MFS were up-regulated during nematode eggs infection (Table S8), including the sugar porter (SP) family (2.A.1.1), the anion: cation symporter family (2.A.1.14), drug: H+ Antiporter-1 family (2.A.1.2), and drug: H+ Antiporter-2 family (2.A.1.3), suggesting the strong nutrition-related metabolism of P. lilacinum 36-1 at this stage. Amino acid transporters react at the initial parasitic stage of nematophagous fungi (Rosso et al., 2011). Consistent with their important roles, 16 APC transporters (2.A.3) were significantly up-regulated during P. lilacinum 36-1 infection, especially for arginine permease (2.A.3.10.4) with its eight-fold higher expression level (Table S8). ABC transporters facilitate plant parasitic fungal infection by protecting them from antifungal compounds (Gardiner et al., 2013). Twelve ABC transporter genes were up-regulated, half of which belonged to the pleiotropic drug resistance family (PDR). Moreover, the Pi uptake porter (2.A.1.9.2) and Pi-repressible Pi:Na+ symporter (2.A.20.2.1), in response to the availability of phosphorus under phosphorus-limiting conditions (Versaw, 1995), had remarkable expression (11 and 9-fold, respectively), indicating that P. lilacinum 36-1 might suffer phosphate-restrictive conditions when infecting nematode eggs.
Signal Transduction Genes Involved in Nematode Eggs Infection
G protein–coupled receptors (GPCRs), which are also known as seven-transmembrane domain receptors, are only found in eukaryotes. They constitute a large protein family of receptors that sense molecules outside the cell and activate signal transduction pathways, and ultimately cellular responses (Trzaskowski et al., 2012). In fungi, they are required for pheromone/nutrient sensing and host recognition (Gao et al., 2011). P. lilacinum 36-1 had 70 genes coding for GPCRs in comparison with the insect pathogen M. acridum with 57 genes (Table S9). Two genes encoding STM1, a recognition molecule for nitrogen starvation signals (Chung et al., 2001), were predicted in P. lilacinum 36-1. One of them was up-regulated, which suggested that the fungi may face nitrogen-limited environment during infection, consistent with the insect parasitic process of Metarhizium spp (Gao et al., 2011). In addition, the up-regulation of genes encoding the cAMP receptor indicated that P. lilacinum 36-1 might go through the energy metabolism process. Notably, PTH11 is a pathogenicity gene that was predicted to encode a transmembrane protein and was related to host surface recognition and appressorium formation in Magnaporche oryzae (DeZwaan, 1999). P. lilacinum 36-1 had 53 PTH11-like genes, as many as that of M. anisopliae (Table S9) and 24 of them were up-regulated, indicating their key role during nematode-egg infection (Figure S6).
In fungi, G protein alpha subunits play essential roles during sexual and pathogenic development through the transduction of extracellular signals to affect morphogenetic processes (Tan et al., 2009). Four classes of G protein alpha subunits play distinct roles (Kamato et al., 2015). P. lilacinum 36-1 contained four G-alpha genes. The up-regulated genes PCL_06420 and PCL_11517 showed the best hits (> 35% protein sequence similarity) with MAA_05603 (GPA2) and MAC_04984 (GPA1), both of which were also up-regulated during infection in Metarhizium spp. (Gao et al., 2011), suggesting that a similar signal transduction pattern was involved in the pathogenicity between the egg-parasitic and entomaphagous fungi.
Following extracellular recognition, the downstream kinase pathway was initiated. P. lilacinum 36-1 possessed 139 protein kinases, 18 of which were up-regulated (log2 fold change > 2) during infection (Figure S5). Moreover, the first six highly expressed protein kinase coding genes during this stage were serine/threonine-protein kinases (Figure S5), indicating their potential roles. Like Metarhizium spp., the increased transcript abundance of the G-protein alpha subunit, phosphatidylinositol-specific phospholipase C, calcium/calmodulin-dependent protein kinase, and protein kinase C indicated the mitogen-activated protein kinase (MAPK) pathway was strongly activated in P. lilacinum 36-1 in the early P. lilacinum-eggs interactions (Table S10).
P. Lilacinum 36-1 is Rich in Antibiosis Effector-Like Proteins
Secreted proteins, especially for effectors, play pivotal roles in pathogenicity (Dodds et al., 2009). The P. lilacinum 36-1 genome encoded 879 signal peptides (without transmembrane domains), of which 279 were potential effector-like proteins (length: < 300 bp; cysteine: ≥4) (Table S11). Based on the InterProScan analysis, we compared the numbers of effector-like proteins with the particular function. Despite the hydrolases and signal molecule genes mentioned above, P. lilacinum 36-1 encoded more toxin-like polypeptides than other organisms (Table S11, Figure S7), such as lysozyme, for degrading the bacterial cell walls (Samaranayake et al., 2009) and Kp4, for inhibit cell growth and division by blocking calcium import into mammalian cells (Park et al., 1994) as well as ribonuclease, for inhibiting protein biosynthesis and then resulting in cell death by apoptosis (Lacadena et al., 2007). Both the Kp4 and ribonuclease encoding gene were up-regulated at the early nematode eggs infection (Table S12). In addition, the presence of two aerolysin/hemolysin toxins, bacterial toxins that disrupts human erythrocytes declined (Buckley et al., 1995) (Table S11), which indicated the potential virulence of P. lilacinum 36-1 to humans. Notably, there were eight extracellular membrane proteins with CFEM domains and three of them were up-regulated indicating their importance in the pathogenicity to nematode-eggs (Tables S11, S12).
High-Level Expression of Genes Related to Appressorium Formation during Nematode-Eggs Infection
Like Metarhizium spp., P. lilacinum 36-1 could form appressorium when infecting nematode eggs (Khan et al., 1999). Based on a BLASTP search against appressorium-related genes as retrieved from the UniPort database at an evalue of 1e-05, we found 357 analogous genes in P. lilacinum 36-1, which were more than those in entomophagous fungi, but less than in plant pathogens (Table S13). Curiously, there were little differences among nematophagous fungi, entomophagous fungi, phytopathogens, or saprophytes. All these fungi have most of the appressorium-related genes, except for the Magas2 protein-coding gene (UniPortKB: G3G3V4), which was exclusively present in nematophagous/entomophagous fungi (Table S13). This gene was found to be predominantly expressed in the appressorium-formation stage in M. acridum, but its function was still not well-documented (Zhu et al., unpublished).
During the early infection stage of P. lilacinum 36-1, 68 appressorium formation-related genes were up-regulated (Table S14), suggesting a strong metabolite involved in appressorium-formation. Notably, There were 27 genes having a comparatively high expression level (log2 fold-change >4) (Table S14), which might be used as potential candidate genes in future research.
P. Lilacinum 36-1 Showed Obvious Metabolic Processes Turnover during Nematode Eggs Infections
To identify the genes associated with the infection processes, we used RNA-Seq to compare the gene expression profiles and to identify the difference between non-infection mycelia and mycelia supplied with nematode-eggs as the only nourishment for 24 h. The infections were confirmed under a light microscope (Figures 5A,B). As a result, more than 93% of the reads were mapped to the genome by TopHat2. Relative to the results during growth in water, 2603 genes were significantly up-regulated while 2428 genes were remarkably down-regulated during the parasitism process (GFOLD < 0.01, |log2 fold-change| > 2). And some critical up-regulated genes were confirmed by qPCR (Figure 5C).
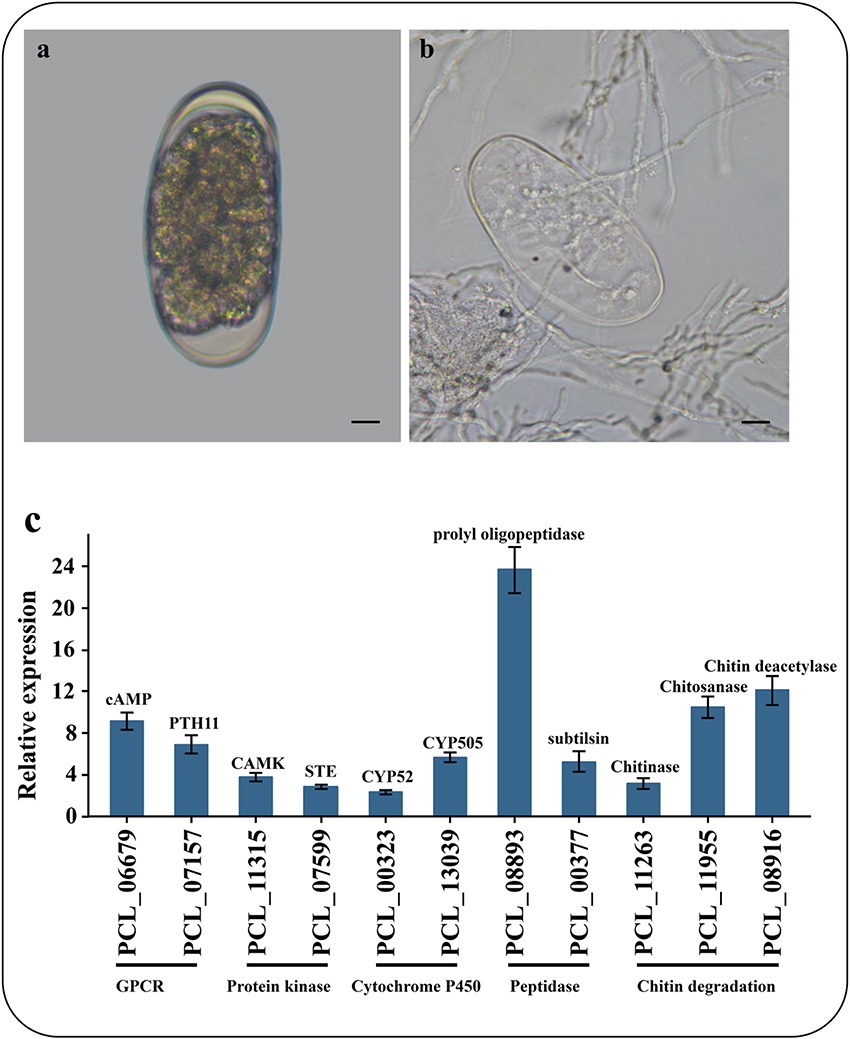
Figure 5. Microscopic observation and qPCR of genes involved in infection. (A) Un-infection nematode-eggs. (B) Infected eggs by P. lilacinum 36-1. Bar indicate 10 μm. (C) qPCR of genes involved in infection. Error bar represent the stand error of averages from four technology replicates.
The GO enrichment results showed that the up-expressed genes related to oxidoreductase activity, structural constitutes of ribosome and transferase activity, as well as transmembrane transporter activity were enriched (P < 0.05; Figure 6A). Consistently, these genes were also significantly enriched in serial energy metabolism-related processes, especially for ribosome biogenesis, oxidative phosphorylation and amino acid metabolism (P < 0.01; Figure 6B). Moreover, the up-regulated genes were also enriched in glutathione metabolism, glycolysis and the pentose phosphate pathway. These results suggested a high turnover of metabolic processes during appressorium formation, which was consistent with the observation in M. oryzae (Oh et al., 2008). A similar situation had also been observed in nematophagous/entomophagous fungi, mycoparasites, or human pathogens (Rosso et al., 2011). In addition, the enrichment of the plant-pathogen interaction pathway (Figure 6B) indicated that P. lilacinum 36-1 might implement a similar strategy as phytopathogens to establish successful infections.
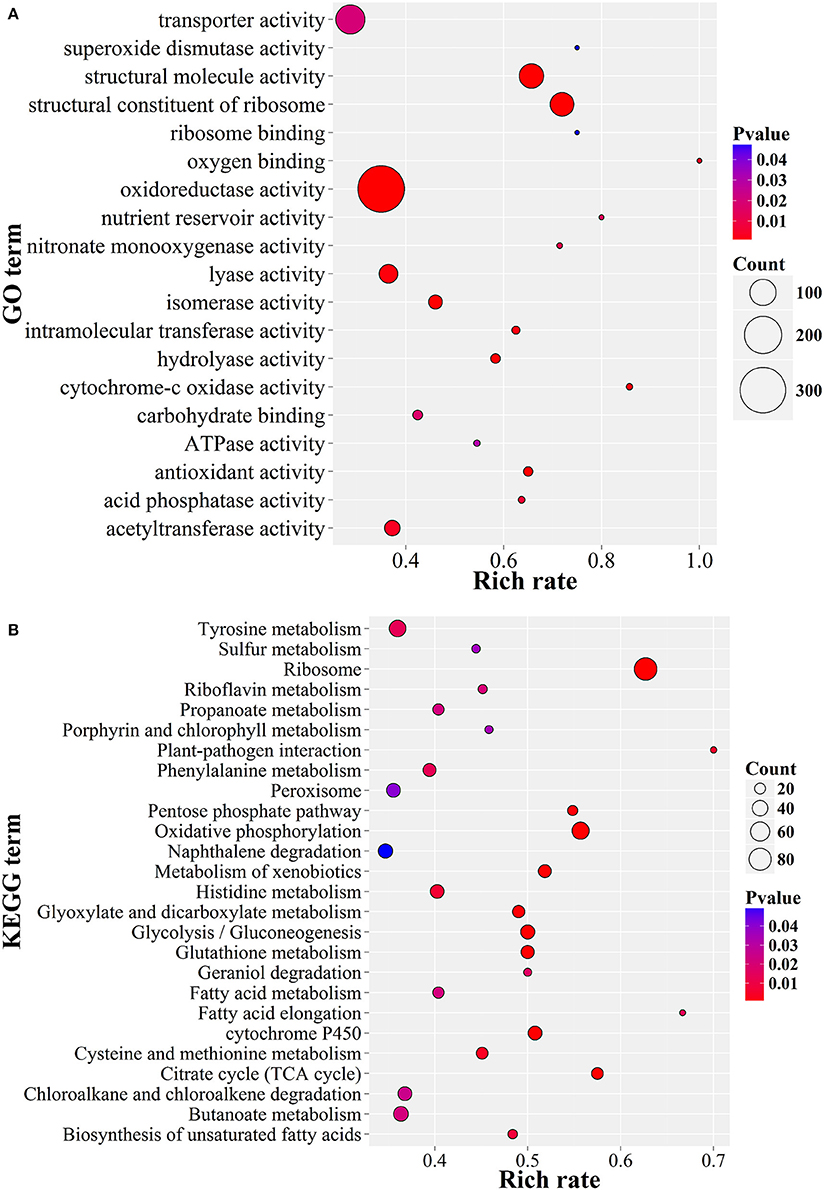
Figure 6. GO and KEGG enrichment of P. lilacinum 36-1 infection nematode-eggs. (A) Gene ontology enrichment of up-regulated genes. (B) KEGG enrichment of up-regulated genes. Count represents the numbers of genes enriched in a particular GO or KEGG term. The bigger the value of rich rate and the smaller the value of P value indicate the degree of enrichment is more significant.
As in Metarhizium spp (Gao et al., 2011), the genes in response to stress were also highly up-expressed, such as superoxide dismutase, HSP20 and HSP30, cytochrome c oxidase, glutathione S-transferase, and superoxide dismutase (Figure 6, Table S15). In addition, Zn (2)-C6 fungal-type DNA-binding and C2H2 transcription factors were also involved in the infection process of P. lilacinum 36-1 (Table S10). Furthermore, gene families such as catalase and superoxide dismutase suppressing the host defense responses and crotonases involved in fatty acid metabolism (Piekarska et al., 2006) were well-explored as virulence factors. These genes were also up-expressed during nematode-eggs infection on the basis of the RNA-Seq analyses (Table S15), all of which could be putative pathogenicity determinants in P. lilacinum 36-1.
Discussion
In this study, we sequenced the genome of a nematode-egg parasitic fungus P. lilacinum 36-1 and further investigated the early responses of the fungus to Meloidogyne incognita eggs. The resultant genome was more compact and dynamic than that of the P. lilacinum strain sequenced before, which was isolated from tannery sludge in India (Prasad et al., 2015).
A phylogenetic analyses showed that P. lilacinum 36-1 was closer to O. sinensis, a strictly obligate parasite (Hu et al., 2013). Moreover, these two fungi shared fewer orthologous genes than those between P. lilacinum 36-1 and P. chlamydosporia, M. acridum, F. graminearum (Figure 1C). In comparison with O. sinensis, P. lilacinum 36-1 contained much more carbohydrate-active enzymes, peptidases, transporters, and signal transduction genes (Figure 2). In considering the huge difference in living environments between P. lilacinum 36-1 and O. sinensis, we speculate that these two fungi may undergo an obvious direction in their evolutionary processes, where P. lilacinum tends to more broad in its habitat scope while O. sinensis is concentrated. The expansion of gene families in P. lilacinum 36-1 may be related to the lower level of repeat-induced point mutations (RIP). P. lilacinum had orthologs (PCL_12430) with the N. crassa RIP defective gene RID (E < 10−10), the only gene known for RIP (Freitag et al., 2002). The retention of this gene in P. lilacinum 36-1 indicates that RIP might play roles at some developmental stages during its evolution even though the genome showed no apparent transitions from C:G to T:A. The RIP has a strong correlation with meiosis during sexual reproduction (Hane and Oliver, 2008). However, the sexual stage of P. lilacinum 36-1 is still unknown. In fact, it is reported that the predominant mode of reproduction in P. lilacinum 36-1 is clonal reproduction in nature (Li et al., 2013). Even so, P. lilacinum 36-1 possessed apparent functional genes whose orthologs in N. crassa and A.nidulans are known to be required for meiosis and sexual development (Table S4), suggesting that P. lilacinum 36-1 may have potential for homothallism or heterothallism under appropriate conditions. The expansion of gene families, lower RIP and loss of sexuality are associated with the transformation from specialization to generalization in Metarhizium spp. (Hu et al., 2014). This mechanism may apply to P. lilacinum 36-1 as indicated by the characteristics of its genome and increased phenotypic flexibility (nematophagous, endogenous, and saprophytic) (Table 2), demonstrating and enlarging the applicability of the theory.
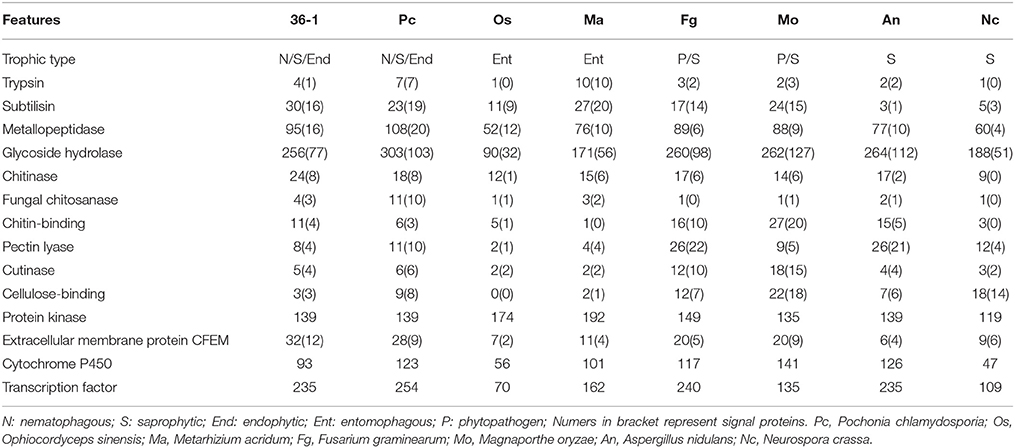
Table 2. Comparison of selected protein families between P. lilacinum 36-1 and other fungi.
The parasitic processes of these fungi include the following successive steps: attraction or recognition, adhesion, penetration, and digestion (Qiaozhen et al., 2009). However, the mechanism through which fungi recognize nematodes and initiate morphological transition remains unknown. Unlike nematode-trapping fungi that sense volatile organic compounds (VOCs) produced by nematodes (Dong and Zhang, 2006), egg-parasitic fungi recognizing nematode-eggs may primarily depend on physical contact due to obstruction of VOCs by egg-shells, requiring signal transduction sensors to perceive environmental cues and thus transfer the signals into intracellular spaces. Extracellular membrane proteins containing an eight-cysteine domain known as CFEM were identified as signal molecules (cell-surface receptors or adhesion molecules) in plant pathogenic fungi (Kulkarni et al., 2005). P. lilacinum 36-1 contained much more CFEM-containing proteins (32) than M. acridum (11) and M. oryzae (20) (Table 2), and one gene had a high expression value of 7.6-fold (Table S15), suggesting the importance of CFEM protein during the recognition of nematode-eggs. Consistent with Metarhizium spp. (Gao et al., 2011), the differential expression of GPCR (PTH11 and STM1) genes may also take part during the recognition process. All these genes could be candidates for further analyses to investigate their function as pathogenicity regulators.
After successful recognition, the nematophagous fungi attach to their hosts via adhesive proteins. Lectins were once considered responsible for adhesive traps in nematode-trapping fungi (Li et al., 2015). Although recent research showed that lectins were not necessary for pathogenicity of nematode-trapping fungi A. oligospora against nematodes (Balogh et al., 2003), the up-regulation of lectin-coding genes when P. lilacinum 36-1 infecting nematode-eggs showed that this protein may be involved in the pathogenicity of egg-parasitic fungi (Table S15). Along with lectins, mad1 is the only gene known to regulate the attachment of the entomopathogenic fungus M. anisopliae to insects (Wang and St. Leger, 2007). In P. lilacinum 36-1, PCL_01111 is homologous to mad1 genes (similarity: 45%; E-value: 2e-149). The up-regulation of PCL_01111 (log2 fold-change: 4.8-fold) suggests that it may mediate the adhesive processes during the early stage of egg-parasitic fungi infection of nematode-eggs. Unexpectedly, the homologous gene PCL_12454 in P. lilacinum 36-1 (protein sequence similarity: 45%; E-value: 2e-53) with mad2, which mediates attachment to plants but not insects in M. anisopliae, was also up-regulated (log2 fold-change: 3-fold). These results reflect the intercommunity and specialization of the adhesion mechanism between egg-parasitic and entomopathogenic fungi.
Egg-parasitic fungi depend on appressoriums, penetration pegs, or lateral mycelial branches to infect nematode eggshells (Li et al., 2015). In comparing P. lilacinum 36-1 with other fungi, we found that most of the appressorium-forming related genes were ubiquitous, even in fungi lacking ability to form appressoriums (Table S13). The conservation of these genes suggests that they are superior regulation mechanisms for mediating appressorium-forming, even though signal transduction genes are included (Table S13). This hypothesis was further supported by the common activation of MAPK and PTH11-like GPCR in Magnaporthe grisea (Gupta and Chattoo, 2007), entomopathogenic (Gao et al., 2011) and egg-parasitic fungus P. lilacinum 36-1.
Because of the chitin and protein components of nematode eggs-shells, extracellular hydrolytic enzymes, such as the chitinases and peptidases in egg-parasitic fungi are required for disintegrating eggshell layers (Yang et al., 2007). Serine peptidases are believed to play important roles in nematode eggs infection (Wang et al., 2010). In comparison with other fungi, P. lilacinum 36-1 contained large numbers of serine peptidases (Figure 3C). Consistent with their vital function, many of them were up-regulated, including S08, S10, S12, S16, and S33 subfamilies (Figure 3D). Moreover, in accordance with a previous report about egg-parasitic fungi Trichoderma harzianum (Szabo et al., 2013), cooperation of different peptidase families was also observed in P. lilacinum 36-1, as indicated by the up-regulation of aspartic peptidase, metalloendopeptidases and the most prominently induced serine peptidases (Figure 3B). Likewise, chitin degradation was also found to have co-expression with peptidases (Yang et al., 2007). To date, most of researches about chitin degradation genes in egg-parasitic fungi focus on the latter infection stages (Dong et al., 2007; Cletus et al., 2013). Particularly, the critical virulence factors chitinase GH18s (Tikhonov et al., 2002) were well-represented (24) and eight of them were predicted to be putatively secreted in P. lilacinum 36-1. However, although many GH18s were up-regulated during nematode-eggs infection, they may be not dominant virulent factors as indicate by only one signal protein which has a relatively lower-level of expression at the same time (Figure 4B). In fact, the high-level expression of chitin deacetylase, chitosanase, and chitooligosaccharide oxidase suggest that these enzymes may play key roles in degrading the chitin of nematode egg-shells during infection assisting by chitin binding related genes (Figure 4C). Some of the genes mentioned above were verified by qPCR (Figure 6B). Notably, chitin deacetylase (CE4) was also found to be involved in appressorium forming, which was consistent with the results mentioned above.
Interestingly, P. lilacinum 36-1 possessed certain numbers of genes related to the degradation of cellulose and xylan, but fewer genes encoding polysaccharide lyases (Table S5). These substances are the primary components of the plant cell walls. This finding suggests that the fungi are able to employ living plant tissues and potential ability to colonize the plant root surface, which is consistent with the frequency isolated of P. lilacinum from the rhizosphere (Lopez et al., 2014). Considering the sedentary plant nematodes and some parasitic migration nematodes infecting the host through roots (Jones et al., 2013), we speculate that there might be a co-evolutionary relationship among P. lilacinum, host plants and nematodes.
Conclusions
In this study, we identified the genome structure, gene contents and transcriptional regulations to nematode eggs of P. lilacinum 36-1. Comparative genomic showed an obvious abundance in peptidases, carbohydrate enzymes, transporters and signal transduction genes with particular functions related to pathogenicity. Furthermore, the transcriptomic analyses indicated that the infection patterns of nematode-eggs parasitic fungi were similar to those of Metarhizium spp. parasitizing insects. The genomic and transcriptomic sequences will facilitate to identifying candidate genes involved in the interactions between nematophagous fungi and nematode-eggs and further to promote practical applications in bio-control.
Nucleotide Sequences Accession Numbers
The whole project sequences were deposited in the DDBJ/EMBL/GenBank repository under BioProject PRJNA281297. The complete genome sequence is available under accession number LCWV00000000. The clean reads have been deposited at NCBI's Sequence Read Archive under accession numbers SRR2536611 and SRR2536612. The Illumina mate-pair reads for RNA-Seq were deposited at the NCBI Sequence Read Archive under accession number SRR2166047.
Author Contributions
YX and GW initiated and designed the research; SL annotated genes, performed protein family analysis, phylogenetic, and genome assembly; JX and CM preformed transcriptome analysis; JX incubated the fungi and nematodes and performed qPCR; JX and SL wrote the paper; DP, GW, and XX reviewed the paper. All the authors read and approved the final manuscript.
Funding
This project was supported by the Special Fund for Agro-scientific Research in the Public Interest 201503114, the Major State Basic Research Development Program (973) 2013CB127504, and Fundamental Research Funds for the Central Universities 2662016PY044.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors appreciate the help of Prof. Chengpeng Bi from the Bioinformatics and Intelligent Computing Lab in Kansas City and Prof. Shiming Liu from Chinese Academy of Agricultural Science in Beijing city for carefully reviewing the paper.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.01084
Abbreviations
RIP: repeat-induced point mutation; ABC: ATP-binding cassette; GH: glycoside hydrolase; GPCR: G-protein coupled receptor; MAPK: mitogen-activated protein kinase; MFS: major facilitator superfamily; GO: gene ontology; KEGG: Kyoto encyclopedia of genes and genomes; qPCR: quantitative real time polymerase chain reaction.
References
Abad, P., Gouzy, J., Aury, J., Castagnone-Sereno, P., Danchin, E. G. J., Deleury, E., et al. (2008). Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat. Biotechnol. 26, 909–915. doi: 10.1038/nbt.1482
Balogh, J., Tunlid, A., and Rosén, S. (2003). Deletion of a lectin gene does not affect the phenotype of the nematode-trapping fungus Arthrobotrys oligospora. Fungal Genet. Biol. 39, 128–135. doi: 10.1016/S1087-1845(03)00023-9
Banu, J. G., Rohini, I., and Gunasekaran, M. (2006). Mass multiplication and formulation of a nematophagous fungus, Paecilomyces lilacinus. Int. J. Nematol. 16, 145–152.
Bendtsen, J. D., Nielsen, H., Von Heijne, G., and Brunak, S. (2004). Improved prediction of signal peptides: signalp 3.0. J. Mol. Biol. 340, 783–795. doi: 10.1016/j.jmb.2004.05.028
Benson, G. (1999). Tandem repeats: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580.
Biely, P. (2012). Microbial carbohydrate esterases deacetylating plant polysaccharides. Biotechnol. Adv. 30, 1575–1588. doi: 10.1016/j.biotechadv.2012.04.010
Birney, E., Clamp, M., and Durbin, R. (2004). Genewise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Borodovsky, M., and Lomsadze, A. (2011). Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr. Protoc. Bioinforma. Chapter 4, Unit 4 6 1–10. doi: 10.1002/0471250953.bi0406s35
Buckley, J. T., Wilmsen, H. U., Lesieur, C., Schulze, A., Pattus, F., Parker, M. W., et al. (1995). Protonation of histidine-132 promotes oligomerization of the channel-forming toxin aerolysin. Biochemistry 34, 16450–16455. doi: 10.1021/bi00050a028
Chung, K. S., Won, M., Lee, S. B., Jang, Y. J., Hoe, K. L., Kim, D. U., et al. (2001). Isolation of a Novel Gene from Schizosaccharomyces pombe: Stm1 encoding a seven-transmembrane loop protein that may couple with the Heterotrimeric Gα2 Protein, Gpa2. J. Biol. Chem. 276, 40190–40201. doi: 10.1074/jbc.M100341200
Cletus, J., Balasubramanian, V., Vashisht, D., and Sakthivel, N. (2013). Transgenic expression of plant chitinases to enhance disease resistance. Biotechnol. Lett. 35, 1719–1732. doi: 10.1007/s10529-013-1269-4
Costa, C., Dias, P. J., Sá-Correia, I., and Teixeira, M. C. (2014). MFS multidrug transporters in pathogenic fungi: do they have real clinical impact? Front. Physiol. 5:197. doi: 10.3389/fphys.2014.00197
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
DeZwaan, T. M. (1999). Magnaporthe grisea Pth11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. Plant Cell Online 11, 2013–2030. doi: 10.1105/tpc.11.10.2013
Dodds, P. N., Rafiqi, M., Gan, P. H. P., Hardham, A. R., Jones, D. A., and Ellis, J. G. (2009). Effectors of biotrophic fungi and oomycetes: pathogenicity factors and triggers of host resistance. New Phytol. 183, 993–1000. doi: 10.1111/j.1469-8137.2009.02922.x
Dong, L. Q., Yang, J. K., and Zhang, K. Q. (2007). Cloning and phylogenetic analysis of the chitinase gene from the facultative pathogen Paecilomyces lilacinus. J. Appl. Microbiol. 103, 2476–2488. doi: 10.1111/j.1365-2672.2007.03514.x
Dong, L. Q., and Zhang, K. Q. (2006). Microbial control of plant-parasitic nematodes: a five-party interaction. Plant Soil 288, 31–45. doi: 10.1007/s11104-006-9009-3
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emanuelsson, O., Nielsen, H., Brunak, S., and von Heijne, G. (2000). Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016. doi: 10.1006/jmbi.2000.3903
Falcon, S., and Gentleman, R. (2007). Using GOstats to test gene lists for GO term association. Bioinformatics 23, 257–258. doi: 10.1093/bioinformatics/btl567
Feng, J., Meyer, C. A., Wang, Q., Liu, J. S., Shirley Liu, X., and Zhang, Y. (2012). GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 28, 2782–2788. doi: 10.1093/bioinformatics/bts515
Freitag, M., Williams, R. L., Kothe, G. O., and Selker, E. U. (2002). A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc. Natl. Acad. Sci. U.S.A. 99, 8802–8807. doi: 10.1073/pnas.132212899
Galagan, J. E., Calvo, S. E., Borkovich, K. A., Selker, E. U., Read, N. D., Jaffe, D., et al. (2003). The genome sequence of the filamentous fungus Neurospora crassa. Nature 422, 859–868. doi: 10.1038/nature01554
Gao, Q., Jin, K., Ying, S.-H., Zhang, Y., Xiao, G., Shang, Y., et al. (2011). Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 7:e1001264. doi: 10.1371/journal.pgen.1001264
Gardiner, D. M., Stephens, A. E., Munn, A. L., and Manners, J. M. (2013). An ABC pleiotropic drug resistance transporter of Fusarium graminearum with a role in crown and root diseases of wheat. FEMS Microbiol. Lett. 348, 36–45. doi: 10.1111/1574-6968.12240
Gupta, A., and Chattoo, B. B. (2007). A novel gene MGA1 is required for appressorium formation in Magnaporthe grisea. Fungal Genet. Biol. 44, 1157–1169. doi: 10.1016/j.fgb.2007.02.014
Guy, L., Kultima, J. R., and Andersson, S. G. E. (2011). GenoPlotR: comparative gene and genome visualization in R. Bioinformatics 26, 2334–2335. doi: 10.1093/bioinformatics/btq413
Hane, J. K., and Oliver, R. P. (2008). RIPCAL: a tool for alignment-based analysis of repeat-induced point mutations in fungal genomic sequences. BMC Bioinformatics 9:478. doi: 10.1186/1471-2105-9-478
Horn, F., Bettler, E., Oliveira, L., Campagne, F., Cohen, F. E., and Vriend, G. (2003). GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res. 31, 294–297. doi: 10.1093/nar/gkg103
Hu, X., Xiao, G., Zheng, P., Shang, Y., Su, Y., Zhang, X., et al. (2014). Trajectory and genomic determinants of fungal-pathogen speciation and host adaptation. Proc. Natl. Acad. Sci. U.S.A. 111, 1–6. doi: 10.1073/pnas.1412662111
Hu, X., Zhang, Y., Xiao, G., Zheng, P., Xia, Y., Zhang, X., et al. (2013). Genome survey uncovers the secrets of sex and lifestyle in caterpillar fungus. Chinese Sci. Bull. 58, 2846–2854. doi: 10.1007/s11434-013-5929-5
Inglis, P. W., Sarmento, R. B. C., Gavião, C. F. C., and Valadares-Inglis, M. C. (2005). DNA fingerprinting of paecilomyces strains of potential use for the biological control of pests. World J. Microbiol. Biotechnol. 21, 1487–1492. doi: 10.1007/s11274-005-7224-3
Ishiyama, A., Otoguro, K., Iwatsuki, M., Namatame, M., Nishihara, A., Nonaka, K., et al. (2009). In vitro and in vivo antitrypanosomal activities of three peptide antibiotics: leucinostatin A and B, alamethicin I and tsushimycin. J. Antibiot. (Tokyo) 62, 343–343. doi: 10.1038/ja.2009.41
Jones, J. T., Haegeman, A., Danchin, E. G. J., Gaur, H. S., Helder, J., Jones, M. G. K., et al. (2013). Top 10 plant-parasitic nematodes in molecular plant pathology. Mol. Plant Pathol. 14, 946–961. doi: 10.1111/mpp.12057
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kamato, D., Thach, L., Bernard, R., Chan, V., Zheng, W., Kaur, H., et al. (2015). Structure, function, pharmacology, and therapeutic potential of the G Protein, Gα/q,11. Front. Cardiovasc. Med. 2:14. doi: 10.3389/fcvm.2015.00014
Keller, O., Kollmar, M., Stanke, M., and Waack, S. (2011). A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763. doi: 10.1093/bioinformatics/btr010
Khan, A., Holland, R., and Williams, K. (1999). Infection of Meloidogyne javanica by Paecilomyces lilacinus. Nematology 1, 131–139. doi: 10.1163/156854199508090
Kiewnick, S., Neumann, S., Sikora, R. a., and Frey, J. E. (2011). Effect of Meloidogyne incognita inoculum density and application rate of Paecilomyces lilacinus strain 251 on biocontrol efficacy and colonization of egg masses analyzed by real-time quantitative PCR. Phytopathology 101, 105–112. doi: 10.1094/PHYTO-03-10-0090
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Kück, U., and Böhm, J. (2013). Mating type genes and cryptic sexuality as tools for genetically manipulating industrial molds. Appl. Microbiol. Biotechnol. 97, 9609–9620. doi: 10.1007/s00253-013-5268-0
Kulkarni, R. D., Thon, M. R., Pan, H., and Dean, R. A (2005). Novel G-protein-coupled receptor-like proteins in the plant pathogenic fungus Magnaporthe grisea. Genome Biol. 6:R24. doi: 10.1186/gb-2005-6-3-r24
Lacadena, J., Álvarez-García, E., Carreras-Sangrà, N., Herrero-Galán, E., Alegre-Cebollada, J., García-Ortega, L., et al. (2007). Fungal ribotoxins: molecular dissection of a family of natural killers. FEMS Microbiol. Rev. 31, 212–237. doi: 10.1111/j.1574-6976.2006.00063.x
Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Lechner, M., Findeiss, S., Steiner, L., Marz, M., Stadler, P. F., and Prohaska, S. J. (2011). Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12:124. doi: 10.1186/1471-2105-12-124
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, J., Li, H., Bi, X., and Zhang, K. Q. (2013). Multiple gene genealogical analyses of a nematophagous fungus Paecilomyces lilacinus from China. J. Microbiol. 51, 423–429. doi: 10.1007/s12275-013-2599-6
Li, J., Zou, C., Xu, J., Ji, X., Niu, X., Yang, J., et al. (2015). Molecular Mechanisms of Nematode-Nematophagous microbe interactions: basis for biological control of plant-parasitic nematodes. Annu. Rev. Phytopathol. 53, 67–95. doi: 10.1146/annurev-phyto-080614-120336
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., et al. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
Lopez, D. C., Zhu-Salzman, K., Ek-Ramos, M. J., and Sword, G. A. (2014). The entomopathogenic fungal endophytes Purpureocillium lilacinum (formerly Paecilomyces lilacinus) and Beauveria bassiana negatively affect cotton aphid reproduction under both greenhouse and field conditions. PLoS ONE 9:e103891. doi: 10.1371/journal.pone.0103891
López-Medrano, R., Pérez Madera, A., and Fuster Foz, C. (2015). [Eye infections caused by Purpureocillium lilacinum: a case report and literature review]. Rev. Iberoam. Micol. 32, 111–114. doi: 10.1016/j.riam.2013.11.004
Luangsa-ard, J., Houbraken, J., van Doorn, T., Hong, S.-B., Borman, A. M., Hywel-Jones, N. L., et al. (2011). Purpureocillium, a new genus for the medically important Paecilomyces lilacinus. FEMS Microbiol. Lett. 321, 141–149. doi: 10.1111/j.1574-6968.2011.02322.x
Lücking, R., Huhndorf, S., Pfister, D. H., Plata, E. R., and Lumbsch, H. T. (2009). Fungi evolved right on track. Mycologia 101, 810–822. doi: 10.3852/09-016
Marazzi, B., Ané, C., Simon, M. F., Delgado-Salinas, A., Luckow, M., and Sanderson, M. J. (2012). Locating evolutionary precursors on a phylogenetic tree. Evolution (N.Y) 66, 3918–3930. doi: 10.1111/j.1558-5646.2012.01720.x
Mi, H., Poudel, S., Muruganujan, A., Casagrande, J. T., and Thomas, P. D. (2016). PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res 44, D336–D342. doi: 10.1093/nar/gkv1194
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., and Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41:e121. doi: 10.1093/nar/gkt263
Mori, Y., Tsuboi, M., Suzuki, M., Fukushima, K., and Arai, T. (1982). Isolation of leucinostatin A and one of its constituents, the new amino acid, 4-methyl-6-(2-oxobutyl)-2-piperidinecarboxylic acid, from Paecilomyces lilacinus A-267. J. Antibiot. (Tokyo) 35, 543–544.
Oh, Y., Donofrio, N., Pan, H., Coughlan, S., Brown, D. E., Meng, S., et al. (2008). Transcriptome analysis reveals new insight into appressorium formation and function in the rice blast fungus Magnaporthe oryzae. Genome Biol. 9:R85. doi: 10.1186/gb-2008-9-5-r85
Park, C. M., Bruenn, J. A., Ganesa, C., Flurkey, W. F., Bozarth, R. F., and Koltin, Y. (1994). Structure and heterologous expression of the Ustilago maydis viral toxin KP4. Mol. Microbiol. 11, 155–164.
Park, J. O., Hargreaves, J. R., McConville, E. J., Stirling, G. R., Ghisalberti, E. L., and Sivasithamparam, K. (2004). Production of leucinostatins and nematicidal activity of Australian isolates of Paecilomyces lilacinus (Thom) Samson. Lett. Appl. Microbiol. 38, 271–276. doi: 10.1111/j.1472-765X.2004.01488.x
Parra, G., Bradnam, K., and Korf, I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. doi: 10.1093/bioinformatics/btm071
Patel, R. K., and Jain, M. (2012). NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7:e30619. doi: 10.1371/journal.pone.0030619
Piekarska, K., Mol, E., van den Berg, M., Hardy, G., van den Burg, J., van Roermund, C., et al. (2006). Peroxisomal fatty acid beta-oxidation is not essential for virulence of Candida albicans. Eukaryot. Cell 5, 1847–1856. doi: 10.1128/EC.00093-06
Prasad, P., Varshney, D., and Adholeya, A. (2015). Whole genome annotation and comparative genomic analyses of bio-control fungus Purpureocillium lilacinum. BMC Genomics 16:1004. doi: 10.1186/s12864-015-2229-2
Qiaozhen, L., Xiaoyang, Z., McIntosh, T., Davis, H., Nemeth, J. F., Pendley, C., et al. (2009). Nematode-trapping fungi eavesdrop on nematode pheromones. Anal. Chem. 81, 8715–8723. doi: 10.1021/ac901991x
Ramam, M. (2014). Cutaneous Paecilomyces lilacinus infections in immunocompromised and immunocompetent patients: retraction. Indian J. Dermatol. Venereol. Leprol. 80, 592. doi: 10.4103/0378-6323.144235
Rawlings, N. D., Waller, M., Barrett, A. J., and Bateman, A. (2014). MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 42, 503–509. doi: 10.1093/nar/gkt953
Rosso, L. C., Finetti-Sialer, M. M., Hirsch, P. R., Ciancio, A., Kerry, B. R., and Clark, I. M. (2011). Transcriptome analysis shows differential gene expression in the saprotrophic to parasitic transition of Pochonia chlamydosporia. Appl. Microbiol. Biotechnol. 90, 1981–1994. doi: 10.1007/s00253-011-3282-7
Saha, S., Bridges, S., Magbanua, Z. V., and Peterson, D. G. (2008). Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 36, 2284–2294. doi: 10.1093/nar/gkn064
Saier, M. H., Reddy, V. S., Tamang, D. G., and Västermark, Å. (2014). The transporter classification database. Nucleic Acids Res. 42, 251–258. doi: 10.1093/nar/gkt1097
Samaranayake, Y. H., Cheung, B. P. K., Parahitiyawa, N., Seneviratne, C. J., Yau, J. Y. Y., Yeung, K. W. S., et al. (2009). Synergistic activity of lysozyme and antifungal agents against Candida albicans biofilms on denture acrylic surfaces. Arch. Oral Biol. 54, 115–126. doi: 10.1016/j.archoralbio.2008.09.015
Schaller, M., Borelli, C., Korting, H. C., and Hube, B. (2005). Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses 48, 365–377. doi: 10.1111/j.1439-0507.2005.01165.x
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Selker, E. U., Cambareri, E. B., Jensen, B. C., and Haack, K. R. (1987). Rearrangement of duplicated DNA in specialized cells of Neurospora. Cell 51, 741–752. doi: 10.1016/0092-8674(87)90097-3
Strakowska, J., Blaszczyk, L., and Chelkowski, J. (2014). The significance of cellulolytic enzymes produced by Trichoderma in opportunistic lifestyle of this fungus. J. Basic Microbiol. 54 (Suppl 1), S2–S13. doi: 10.1002/jobm.201300821
Suarez, B., Rey, M., Castillo, P., Monte, E., and Llobell, A. (2004). Isolation and characterization of PRA1, a trypsin-like protease from the biocontrol agent Trichoderma harzianum CECT 2413 displaying nematicidal activity. Appl. Microbiol. Biotechnol. 65, 46–55. doi: 10.1007/s00253-004-1610-x
Sun, M., and Liu, X. (2006). Carbon requirements of some nematophagous, entomopathogenic and mycoparasitic Hyphomycetes as fungal biocontrol agents. Mycopathologia 161, 295–305. doi: 10.1007/s11046-006-0249-9
Szabo, M., Urban, P., Viranyi, F., Kredics, L., and Fekete, C. (2013). Comparative gene expression profiles of Trichoderma harzianum proteases during in vitro nematode egg-parasitism. Biol. Control 67, 337–343. doi: 10.1016/j.biocontrol.2013.09.002
Tan, K. C., Heazlewood, J. L., Millar, A. H., Oliver, R. P., and Solomon, P. S. (2009). Proteomic identification of extracellular proteins regulated by the Gna1 Gα subunit in Stagonospora nodorum. Mycol. Res. 113, 523–531. doi: 10.1016/j.mycres.2009.01.004
Tikhonov, V. E., Lopez-Llorca, L. V., Salinas, J., and Jansson, H.-B. (2002). Purification and characterization of chitinases from the nematophagous fungi Verticillium chlamydosporium and V. suchlasporium. Fungal Genet. Biol. 35, 67–78. doi: 10.1006/fgbi.2001.1312
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Trzaskowski, B., Latek, D., Yuan, S., Ghoshdastider, U., Debinski, A., and Filipek, S. (2012). Action of Molecular Switches in GPCRs - theoretical and experimental studies. Curr. Med. Chem. 19, 1090–1109. doi: 10.2174/092986712799320556
Versaw, W. K. (1995). A phosphate-repressible, high-affinity phosphate permease is encoded by the pho-5+ gene of Neurospora crassa. Gene 153, 135–139. doi: 10.1016/0378-1119(94)00814-9
Wang, C., and St. Leger, R. J. (2007). The MAD1 adhesin of Metarhizium anisopliae links adhesion with blastospore production and virulence to insects, and the MAD2 adhesin enables attachment to plants. Eukaryot. Cell 6, 808–816. doi: 10.1128/EC.00409-06
Wang, J., Wang, J., Liu, F., and Pan, C. (2010). Enhancing the virulence of Paecilomyces lilacinus against Meloidogyne incognita eggs by overexpression of a serine protease. Biotechnol. Lett. 32, 1159–1166. doi: 10.1007/s10529-010-0278-9
Yang, F., Abdelnabby, H., and Xiao, Y. (2015). The role of a phospholipase (PLD) in virulence of Purpureocillium lilacinum (Paecilomyces lilacinum). Microb. Pathog. 85, 11–20. doi: 10.1016/j.micpath.2015.05.008
Yang, J., Tian, B., Liang, L., and Zhang, K.-Q. (2007). Extracellular enzymes and the pathogenesis of nematophagous fungi. Appl. Microbiol. Biotechnol. 75, 21–31. doi: 10.1007/s00253-007-0881-4
Yang, J., Zhao, X., Liang, L., Xia, Z., Lei, L., Niu, X., et al. (2011). Overexpression of a cuticle-degrading protease Ver112 increases the nematicidal activity of Paecilomyces lilacinus. Appl. Microbiol. Biotechnol. 89, 1895–1903. doi: 10.1007/s00253-010-3012-6
Yin, Y., Mao, X., Yang, J., Chen, X., Mao, F., and Xu, Y. (2012). DbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, 445–451. doi: 10.1093/nar/gks479
Keywords: Purpureocillium lilacinum, parasitism, nematophagous fungi, appressorium, genome, transcriptome
Citation: Xie J, Li S, Mo C, Xiao X, Peng D, Wang G and Xiao Y (2016) Genome and Transcriptome Sequences Reveal the Specific Parasitism of the Nematophagous Purpureocillium lilacinum 36-1. Front. Microbiol. 7:1084. doi: 10.3389/fmicb.2016.01084
Received: 05 February 2016; Accepted: 28 June 2016;
Published: 19 July 2016.
Edited by:
Frederic Lamoth, Lausanne University Hospital, SwitzerlandReviewed by:
Augusto Schrank, Federal University of Rio Grande do Sul, BrazilHolger Heuer, Julius Kühn-Institut, Federal Research Centre for Cultivated Plant, Germany
Copyright © 2016 Xie, Li, Mo, Xiao, Peng, Wang and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaofeng Wang, amtzZ29AbWFpbC5oemF1LmVkdS5jbg==
Yannong Xiao, eGlhb3lhbm5vbmdAbWFpbC5oemF1LmVkdS5jbg==