
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 06 May 2016
Sec. Terrestrial Microbiology
Volume 7 - 2016 | https://doi.org/10.3389/fmicb.2016.00668
 Kai Xue1,2,3
Kai Xue1,2,3 Jianping Xie2,3,4
Jianping Xie2,3,4 Aifen Zhou2,3
Aifen Zhou2,3 Feifei Liu2,3
Feifei Liu2,3 Dejun Li3
Dejun Li3 Liyou Wu2,3
Liyou Wu2,3 Ye Deng2,3,5
Ye Deng2,3,5 Zhili He2,3
Zhili He2,3 Joy D. Van Nostrand2,3Yiqi Luo3
Joy D. Van Nostrand2,3Yiqi Luo3 Jizhong Zhou1,2,3,6*
Jizhong Zhou1,2,3,6*Soil microbial communities play critical roles in ecosystem functioning and are likely altered by climate warming. However, so far, little is known about effects of warming on microbial functional gene expressions. Here, we applied functional gene array (GeoChip 3.0) to analyze cDNA reversely transcribed from total RNA to assess expressed functional genes in active soil microbial communities after nine years of experimental warming in a tallgrass prairie. Our results showed that warming significantly altered the community wide gene expressions. Specifically, expressed genes for degrading more recalcitrant carbon were stimulated by warming, likely linked to the plant community shift toward more C4 species under warming and to decrease the long-term soil carbon stability. In addition, warming changed expressed genes in labile C degradation and N cycling in different directions (increase and decrease), possibly reflecting the dynamics of labile C and available N pools during sampling. However, the average abundances of expressed genes in phosphorus and sulfur cycling were all increased by warming, implying a stable trend of accelerated P and S processes which might be a mechanism to sustain higher plant growth. Furthermore, the expressed gene composition was closely related to both dynamic (e.g., soil moisture) and stable environmental attributes (e.g., C4 leaf C or N content), indicating that RNA analyses could also capture certain stable trends in the long-term treatment. Overall, this study revealed the importance of elucidating functional gene expressions of soil microbial community in enhancing our understanding of ecosystem responses to warming.
Global climate change resulting from anthropogenic activities (Novotny et al., 2007) has become one of the greatest scientific and political concerns (IPCC, 2007). Near the summit of the Mauna Loa volcano in Hawaii, the daily concentration of atmospheric CO2 has reached a worrisome milestone, 400 ppm, in Monastersky (2013). The yearly average global CO2 concentration is predicted to surpass 400 ppm in only a few years (Monastersky, 2013). The emissions of CO2 and other greenhouse gasses had driven the Earth’s average temperature to increase by 0.74°C in the 20th century, which may continue to increase by 1.1–6.4°C at the end of this century (IPCC, 2007).
Within the global climate change context, the soil microbial community is likely to be influenced by the atmospheric warming. The temperature increase itself affects almost all chemical and biological processes (Shaver et al., 2000) and thus may alter the soil microbial community directly. Meanwhile, warming may influence the soil microbial community via its indirect effects on plant communities (Cheng et al., 2010). For example, plant community shift toward more C4 species was observed in the warming treatment plots (An et al., 2005; Wan et al., 2005; Zhang et al., 2005; Luo et al., 2009). C3 and C4 species differ in their photosynthesis pathways to fix and reduce inorganic CO2 into organic compounds, and C4 species were generally believed to have lower plant tissue quality, e.g., higher lignin content and greater C:N ratio (Kephart and Buxton, 1993). Thus, the above-ground plant community shift under warming may lead to changes in plant-derived soil carbon (C) input that is important substrate for soil microbial community, and hence may alter soil microbial community.
Influences of warming on soil microbial communities have been assessed by various molecular approaches such as phospholipid fatty acid (PLFA) analysis (Zogg et al., 1997; Zhang et al., 2005, 2011; Rinnan et al., 2007; Frey et al., 2008), Biolog (Rinnan et al., 2007; Zhang et al., 2011) or a series of DNA-based methods, including traditional molecular tools [e.g., terminal restriction fragment length polymorphism (T-RFLP), denaturing gradient gel electrophoresis (DGGE), etc.] (Deslippe et al., 2005, 2012; Cookson et al., 2007; Schindlbacher et al., 2011; Sheik et al., 2011; Stockdale et al., 2013) and newly emerged metagenomic technologies (e.g., high throughput sequencing and microarrays; Mackelprang et al., 2011; Sheik et al., 2011; Hayden et al., 2012; Kuffner et al., 2012; Yergeau et al., 2012). Substantial influences of warming on soil microbial communities were found in the majority of these studies, from aspects of fingerprint, taxonomic and phylogenetic compositions. Moreover, crucial roles of soil microbial community in regulating ecosystem C dynamics under warming were discovered by hybridizing DNA from soil microbial community on GeoChip3.0 (Zhou et al., 2012), a functional gene array-based metagenomic tool that can allow to assess genes in a highly comprehensive and standardized manner.
Due to the importance of microbial communities in mediating soil biogeochemical processes (van Bruggen and Semenov, 2000; Jackson et al., 2003; Xue et al., 2011), influences of warming on microbial community activities may potentially affect the biogeochemical cycling and ecosystem functioning. However, DNA-based technologies are hard to directly reflect the actual activity of active microbial populations (McGrath et al., 2010; Simon and Daniel, 2011; Franzosa et al., 2015). In contrast, RNA-based approaches can provide better insights into functional attributes of active populations in microbial community (McGrath et al., 2010; Simon and Daniel, 2011; Takasaki et al., 2013). RNA-based approaches have already been applied in global climate change studies, e.g., discovering the microbial response to ocean acidification due to increased atmospheric CO2 concentration (Gilbert et al., 2008) and some investigations in peat soils (Tveit et al., 2013; Tuorto et al., 2014).
By using DNA-based GeoChip3.0, we discovered crucial roles of soil microbial community in regulating ecosystem C dynamics under warming through three mechanisms: shifts in the functional gene composition of soil microbial community, enriched genes for labile C but not recalcitrant C degradation, and enriched genes for nutrient cycling (Zhou et al., 2012). To better understand functional attributes of active populations in microbial community under warming, we used RNA-based GeoChip 3.0 in this study to assess effects of warming on functional gene expressions of soil microbial community in the same experimental site after 9 years of warming (targeting 2°C above ambient temperature) at the Great Plain Apiaries in Oklahoma, USA. GeoChip 3.0 contains approximately 28, 000 probes covering more than 57,000 gene variants from 292 functional gene families involved in C, nitrogen (N), phosphorus (P) and sulfur (S) cycles and other processes (He et al., 2010). We hypothesized that expressions of key functional genes of soil microbial community involved in important biochemistry processes would be stimulated by warming, and these changes would be closely linked to the dynamics of environmental attributes. Our results showed that expressions of genes involved in recalcitrant C degradation, P and S cycling were stimulated by warming, but both increased and decreased gene expressions were observed for labile C degradation and N cycling. Moreover, the expressed gene composition was closely related to both dynamic (e.g., soil moisture) and stable environmental attributes (e.g., C4 leaf C or N content).
This study was conducted at the Kessler Farm Field Laboratory (KFFL) located in the Great Plain Apiaries in McClain County, Oklahoma, USA (34°58′54′′N, 97°31′14′′W; Luo et al., 2001). The grassland is dominated by C4 grasses (Andropogon gerardii, Sorghastrum nutans, Schizachyrium scoparium, Panicum virgatum, and Eragrostis spp.), C3 forbs (Ambrosia psilostachya and Xanthocephalum texanum), and C3 annual grass (Bromus japonicus; Zhang et al., 2005; Sherry et al., 2008). Based on Oklahoma Climatological Survey, the mean annual temperature and precipitation in this site was 16.3°C and 967 mm, respectively. The soil is silt loam (36% sand, 55% silt, and 10% clay in the top 15 cm) and part of Nash–Lucien complex, typically with high fertility, neutral pH, high available water capacity and a deep moderately penetrable root zone (USDA, 1963).
The experiment was established in November 1999 with a blocked split-plot design, in which warming is a primary factor nested by clipping (annual removal of above-ground biomass). In this study, we focus on examining the effects of warming on expressions of functional genes in soil microbial community and thus only samples from unclipped sub-plots were collected. There were six replicates for each treatment. In 1 m × 1 m subplots, the warming treatment (targeting +2°C) was created by suspending infrared radiators (Kalglo Electronics, Bethlehem, Pennsylvania) 1.5 m above the ground; while dummy infrared radiators were used in control plots to exclude the shading effect of the device itself. Soil samples were collected from the top 15 cm layer in October 2008. In each subplot, four soil cores (2.5 cm diameter × 15 cm deep) were taken, likely including both rhizosphere soil and bulk soil, and then composited to one sample. Like many long term experimental sites, destructive sampling is restricted. Thus, only one-time point can be allowed for sampling. All samples were sieved by 2 mm sieves and stored at -80°C before analyses.
The measurements or calculations for environmental attributes in 2008, including soil temperature, NH4+, NO3- and total N contents, as well as heterotrophic respiration and autotrophic respiration, were described in detail in Supplementary Materials and Methods. Other environmental attributes used for CCA and Mantel test (see Statistical Analysis) were from previous studies to investigate treatment effects (Niu et al., 2010; Cheng et al., 2011; Xu et al., 2012a,b, 2014, 2015; Zhou et al., 2012), all collected in 2008 (Supplementary Table S1).
Total DNA and RNA from the soil microbial community were extracted from 5 g soil according to the method developed previously (Hurt et al., 2001). Total RNA was isolated and purified with Qiagen RNeasy®Mini Kit (Qiagen, Germantown, MD, USA). RNA quantity and quality were assessed based on the ratios of 260/280 nm and 260/230 nm absorbance by a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA), as well as agarose gel image.
Total RNA (500 ng) was amplified with modified protocols described previously (Gao et al., 2007) since limited amount of total RNA was obtained from soil samples. Briefly, first strand cDNA was synthesized with random primer T7N6 bearing a T7 promoter and six random oligonucleotides and reverse transcriptase superscript III. The product was used for the second strand cDNA synthesis with a cocktail containing Escherichia coli DNA polymerase I, RNaseH, and DNA ligase. After the synthesis of second strand cDNA, T4 DNA polymerase was added to polish the end. The double strand DNA was then purified with phenol:chloroform:isoamyl alcohol and in vitro transcribed with MegaScript T7 kit (Life technologies, Grand Island, NY, USA). The amplified RNA was purified with Qiagen RNeasy Mini Kit (Qiagen, Germantown, MD, USA) and eluted in RNase free H2O. Amplified RNA was labeled with fluorescent dye Cy5 via reverse transcription reaction. The 10 μg amplified RNA was mixed with 3.3 μl random primers (3.0 μg/μl; Life Technologies, Grand Island, NY, USA) and adjusted the volume to 10 μl with water. After 10 min of denaturation at 70°C and immediate chilling on ice, the labeling mix containing 6 μl 5X first strand buffer, 3 μl 0.1 M DTT, 1.5 μl dNTP mix (10 mM dA/G/CTP and 0.5 mM dTTP), 1 μl RNase inhibitor and 1 μl Cy5-dUTP was added and incubated in the dark at room temperature for 10 min. Then 1 μl Superscript reverse transcriptase II was added to the mixture and incubated at 42°C for 2 h. Labeled products were purified with QIAquick PCR purification kit (Qiagen, Germantown, MD, USA) and vacuum dried.
GeoChip 3.0 was used in this study as described previously (Wu et al., 2006; Xue et al., 2013). The Cy5-labeled cDNA was re-suspended in 50 μl hybridization solution [40% formamide, 5x SSC, 5 μg of unlabeled herring sperm DNA (Promega, Madison, WI, USA), and 0.1% SDS] and 2 μl universal standard DNA (0.2 pmol μl-1) labeled with the fluorescent dye Cy-3 (Liang et al., 2010), denatured at 95°C for 3 min and maintained at 65°C until loaded onto microarray slides. Arrays were hybridized on a MAUI Hybridization Station (Roche, South San Francisco, CA, USA) for 10 h at 42°C. After washing and drying, the microarrays were scanned using a ScanArray Express Microarray Scanner (Perkin Elmer, Boston, MA, USA) at 633 nm with a laser power of 90% and a photomultiplier tube (PMT) gain of 75%. The obtained images were analyzed by ImaGene version 6.0 (Biodiscovery, El Segundo, CA, USA) to determine the intensity of each spot.
Raw data from ImaGene were submitted to the Microarray Data Manager System1 and analyzed by the following steps: (i) spots (probes) flagged as 1 or 3 by Imagene and with a signal to noise ratio (SNR) less than 3.0 were removed as poor-quality spots; (ii) Cy5 intensities of functional genes were normalized by a two-step method based on mean Cy3 intensities of universal standards in each sub-grid within a slide and in each slide among samples; (iii) outliers were defined as probe intensities out of the 95% confident interval for each probe in control or warming treatment and removed until no outliers were identified; (iv) genes with less than 0.34 time of the final positive probes or two probes were removed; (v) probes that appeared in less than two of six replicates in each treatment were removed. After these steps, relative abundances were calculated through dividing signal intensities for individual probes by the sum of intensities for all probes detected in each sample. Then relative abundances were multiplied by the mean value of intensity sums over all samples. A natural logarithm transformation (x+1) was performed for each amplified relative abundance plus 1.
Detrended correspondence analysis (DCA), three non-parametric tests (multiple response permutation procedure, MRPP; permutational multivariate analysis of variance, ADONIS; analysis of similarity, ANOSIM), Mantel test and canonical correlation analysis (CCA) were performed by R version 2.9.1 (The R Foundation for Statistical Computing, Vienna, Austria). The three non-parametric tests were based on Bray–Curtis, Horn, and Euclidean dissimilarity indexes; whilst mantel test were based on Euclidean dissimilarity. To construct a CCA model, 11 environmental attributes were selected based on their biological importance and p-values of single-factor CCA models, including soil temperature, soil moisture, total soil organic C (TOC), the proportion of soil C derived from C4 plant species (F-C4), soil 15N content, C3 species above-ground biomass, C4 species above-ground biomass, C4 leaf C content, C4 leaf C:N ratio, C3 leaf C content, and below net primary productivity (BNPP). Then the automatic forward selection by permutation tests was applied in the stepwise analysis to choose constrained variables in the final CCA model, combined with the variance inflation factor (VIF) criterion (VIF < 20).
Treatment differences between warming and control for expressed gene abundances were analyzed by two-tailed paired t-test by R version 2.9.1 (The R Foundation for Statistical Computing, Vienna, Austria). The significance was defined as p ≤ 0.05, while marginally significance was defined as 0.05 < p ≤ 0.10.
The GeoChip analysis was performed with cDNA that was reversely transcribed from total RNA to investigate functional gene expressions. A total of 2,347 probes (∼10% of the probes on the arrays) were detected in all samples, of which 1,733 and 1,947 were in control and warming treatments, respectively. In this way, there were a big portion, 1333 probes (56.8%), were overlapped between two treatments. Meanwhile, among all 2,347 detected probes, 400 (17.0%) and 414 (26.2%) were unique to control and warming treatments, respectively. The composition of expressed genes differed significantly (p ≤ 0.005) between control and warming, as revealed by all three non-parametric dissimilarity tests (MRPP, ADONIS, and ANOSIM) based on Bray–Curtis dissimilarity (Table 1). Consistently, DCA profile also illustrated that the warming samples were separated clearly from control samples (Figure 1).
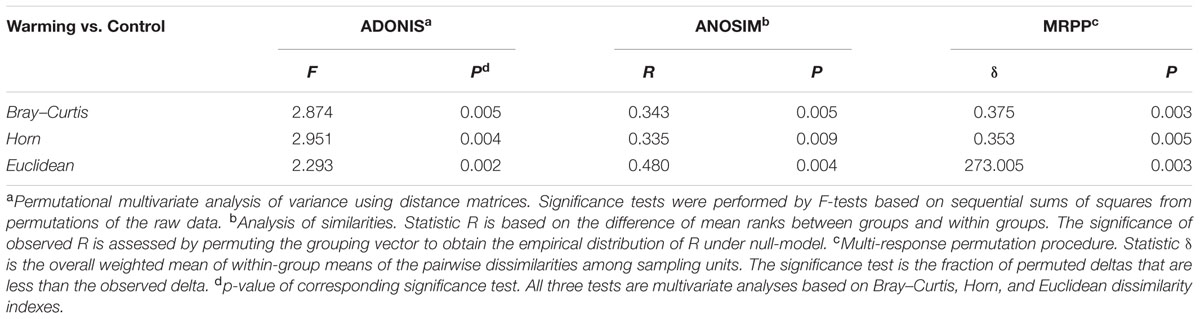
TABLE 1. Non-parametric analyses to test dissimilarity of communities between warming and control.
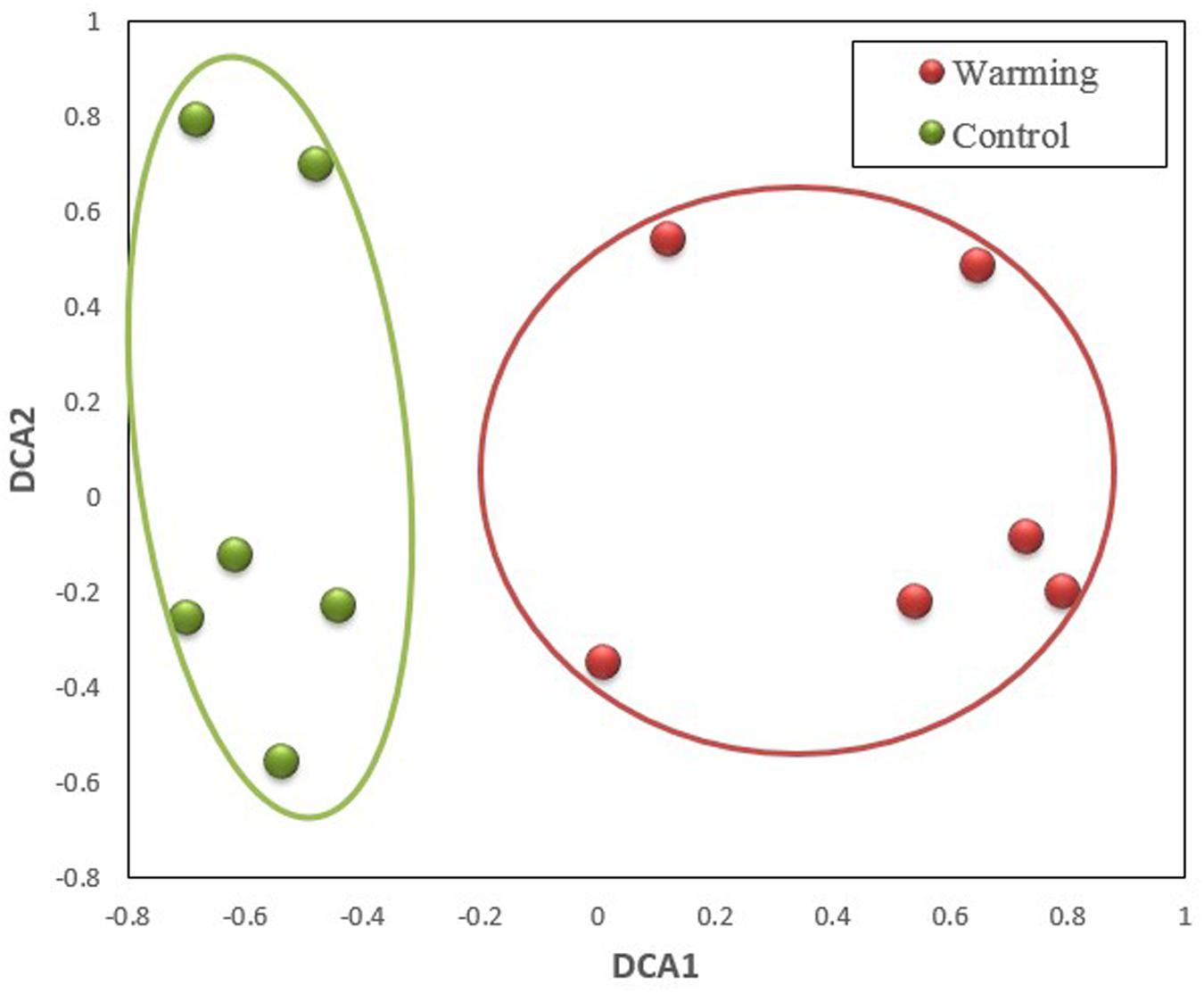
FIGURE 1. Detrended correspondence analysis (DCA) of microbial community composition in control and warming treatments based on expressed functional genes.
Expressed genes involved in biochemical cycles of C/N/P/S are of our particular interests. A total of 26 expressed genes encoding enzymes for degrading C components were detected. Among them, 34.6% (9 of 26) had significantly or marginally significantly higher abundances in warming than control samples (Figure 2), including not only labile, but also recalcitrant C decomposition genes, e.g., vanA encoding vanillate O-demethylase oxygenase (p = 0.02) for degrading aromatic compounds; glx encoding glyoxal oxidase (p = 0.07) and mnp encoding manganese peroxidase (p = 0.03) for lignin degradation. Meanwhile, abundances of three expressed genes were decreased by warming significantly, all involved in labile C degradation, including those encoding mannanase (p = 0.03) and xylanase (p = 0.02) for hemicellulose degradation, and acetylglucosaminidase (p = 0.04) for chitin degradation. The rest of C degradation genes did not differ significantly by warming.
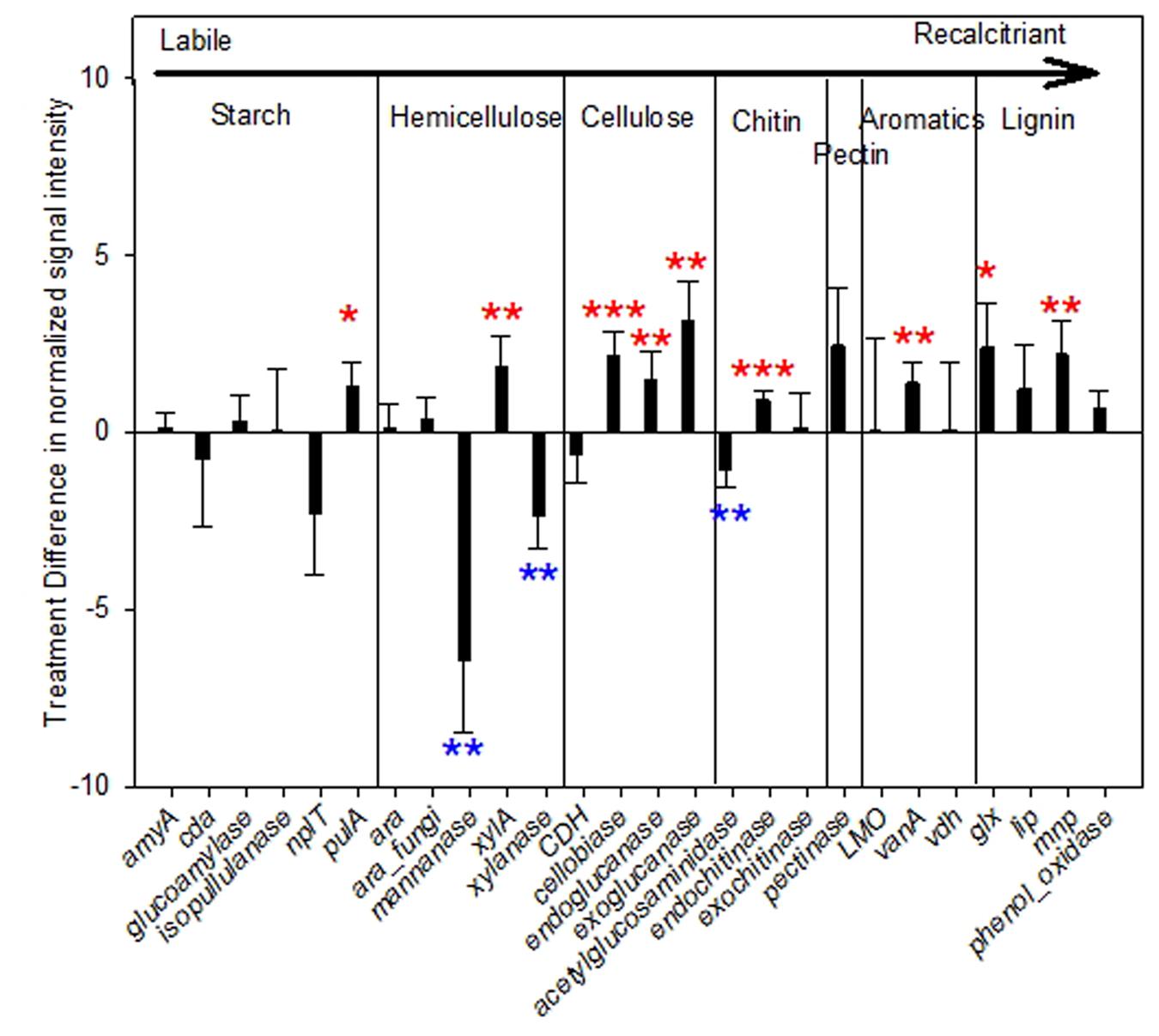
FIGURE 2. Difference of normalized signal intensity between warming and control for expressed functional genes involved in carbon (C) degradation. The complexity of carbon is presented in order from labile to recalcitrant from left to right. Error bars represent standard error. Significance according to two tailed paired t-test were labeled by ∗∗∗p ≤ 0.01, ∗∗p ≤ 0.05, ∗p ≤ 0.10; while red and blue colors represent increases and decreases caused by warming.
There were 13 expressed genes detected in N cycling. Among them, 30.7% (4 of 13) were stimulated significantly or marginally significantly by warming (Figure 3). On the contrary, warming also decreased abundances of three expressed genes significantly or marginally significantly, including hzo encoding hydrazine oxidase (p = 0.03) for anammox, nirK encoding copper-containing nitrite reductase (p = 0.06) for denitrification, and amoA encoding ammonia monooxygenase (p = 0.06) for nitrification. Abundances of the rest of expressed N genes did not differ between control and warming, including those for ammonification, assimilatory N reduction and N fixation processes.
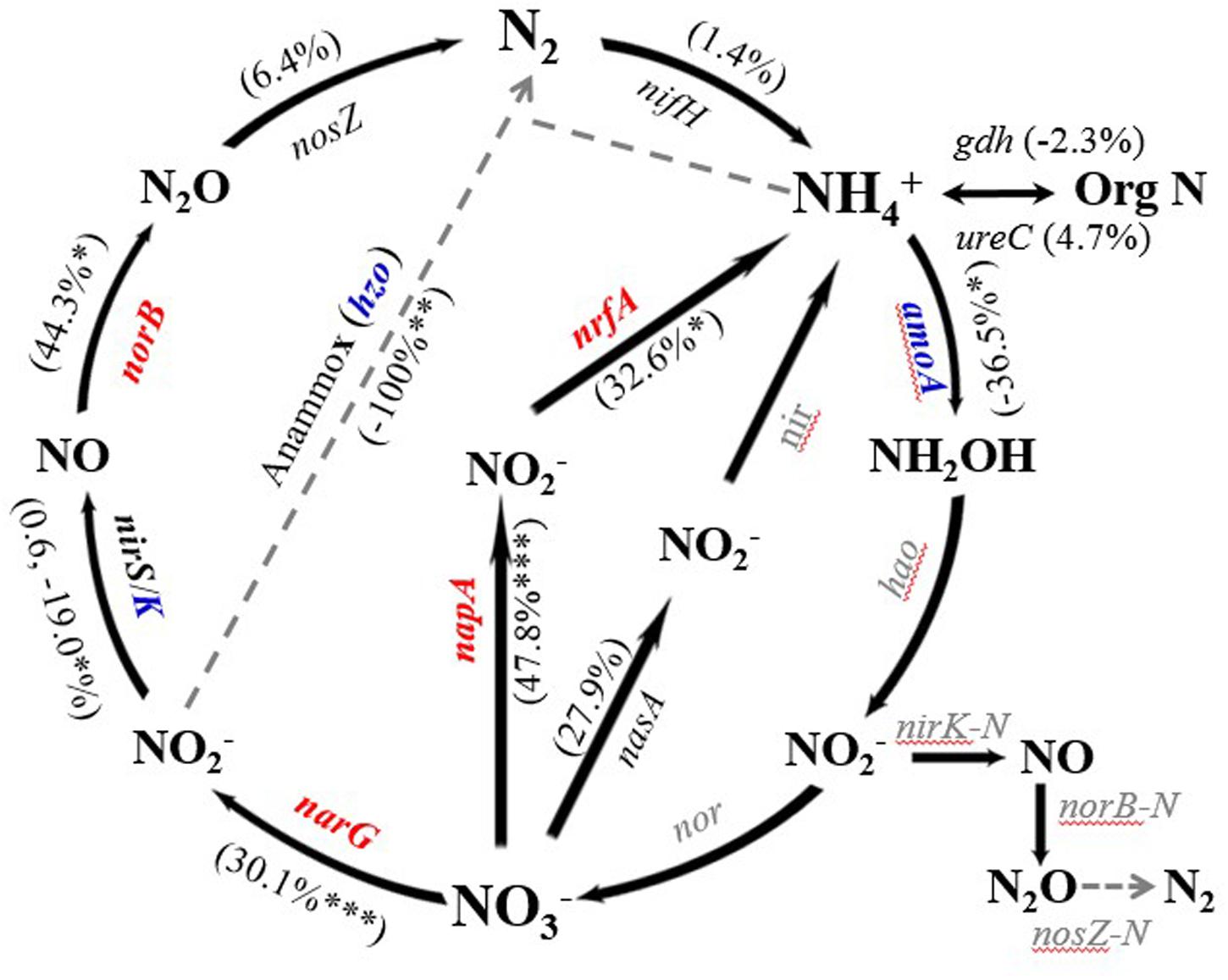
FIGURE 3. Percentage change of normalized signal intensity from expressed functional genes involved in nitrogen (N) cycling to warming. Significance according to two tailed paired t-test were labeled by ∗∗∗p ≤ 0.01, ∗∗p ≤ 0.05, ∗p ≤ 0.10. Red and blue colors represent increases and decreases caused by warming; while gray color means that the expressed genes were not present on the version of GeoChip used, or were undetected.
Different from C and N cycles, all expressed genes involved in P and S cycles were stimulated by warming on average (Figure 4). Among them, changes in ppx encoding exopolyphosphatase (p = 0.02) and the gene encoding phytase (p = 0.08) for P utilization, dsrA encoding the large subunit of sulfite reductase (p = 0.02) for sulfite reduction, and sox for sulfur oxidation (p < 0.001) were significant or marginally significant.
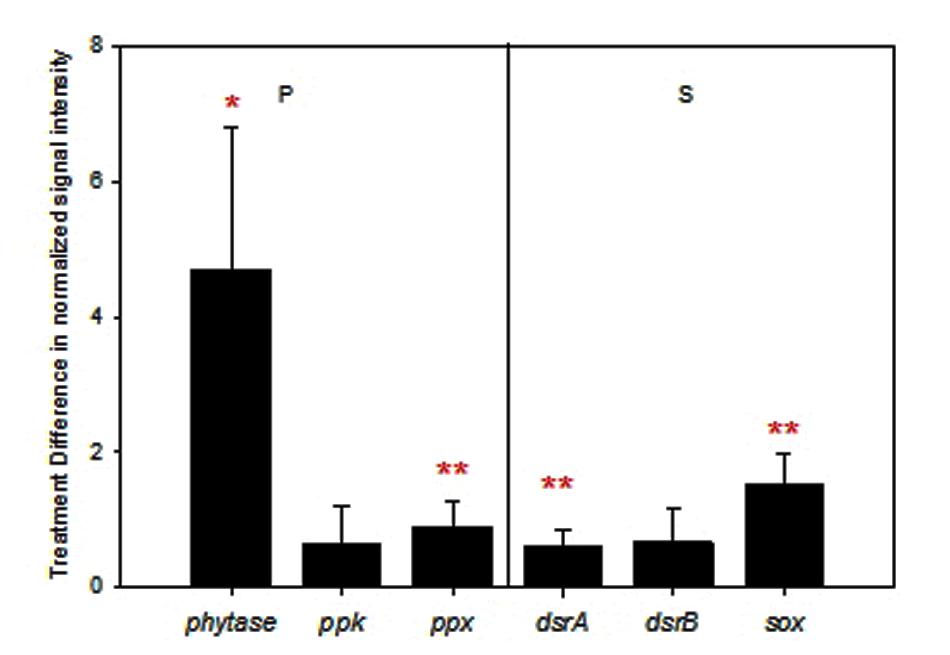
FIGURE 4. Difference of normalized signal intensity between warming and control for expressed functional genes involved in phosphorus (P) and sulfur (S) cycling. Error bars represent standard error. Significance according to two tailed paired t-test were labeled by ∗∗∗p ≤ 0.01, ∗∗p ≤ 0.05, ∗p ≤ 0.10; while red and blue colors represent increases and decreases caused by warming.
Canonical correlation analysis was performed to investigate the relationship between environmental attributes and the structure of expressed functional genes in microbial communities. Four attributes were chosen in the final CCA model (F = 1.91, p = 0.005), including soil temperature (F = 1.76, p = 0.015 for partial CCA), soil moisture (F = 1.67, p = 0.039 for partial CCA), C4 leaf C:N (F = 1.82, p = 0.055 for partial CCA) and BNPP (F = 1.38, p = 0.130 for partial CCA). In the CCA profile (Figure 5), warming samples separated clearly from control samples along the first canonical axis (CCA1), explaining 24.8% of the total variation in expressed gene composition. Projections of environmental attributes by CCA revealed that warming samples were positively correlated with soil temperature, BNPP and C4 leaf C:N, but negatively correlated with soil moisture. These projections were consistent with observations in these attributes when investigating treatment effects (Niu et al., 2010; Xu et al., 2012a, 2014). Moreover, variation portioning analysis (VPA) was performed based on partial CCA and found that each single factor explained 9.4–12.4% of the total variance in expressed functional gene structure; while their interaction explained 6.9% in total, leaving 47.8% as unexplained (Figure 5).
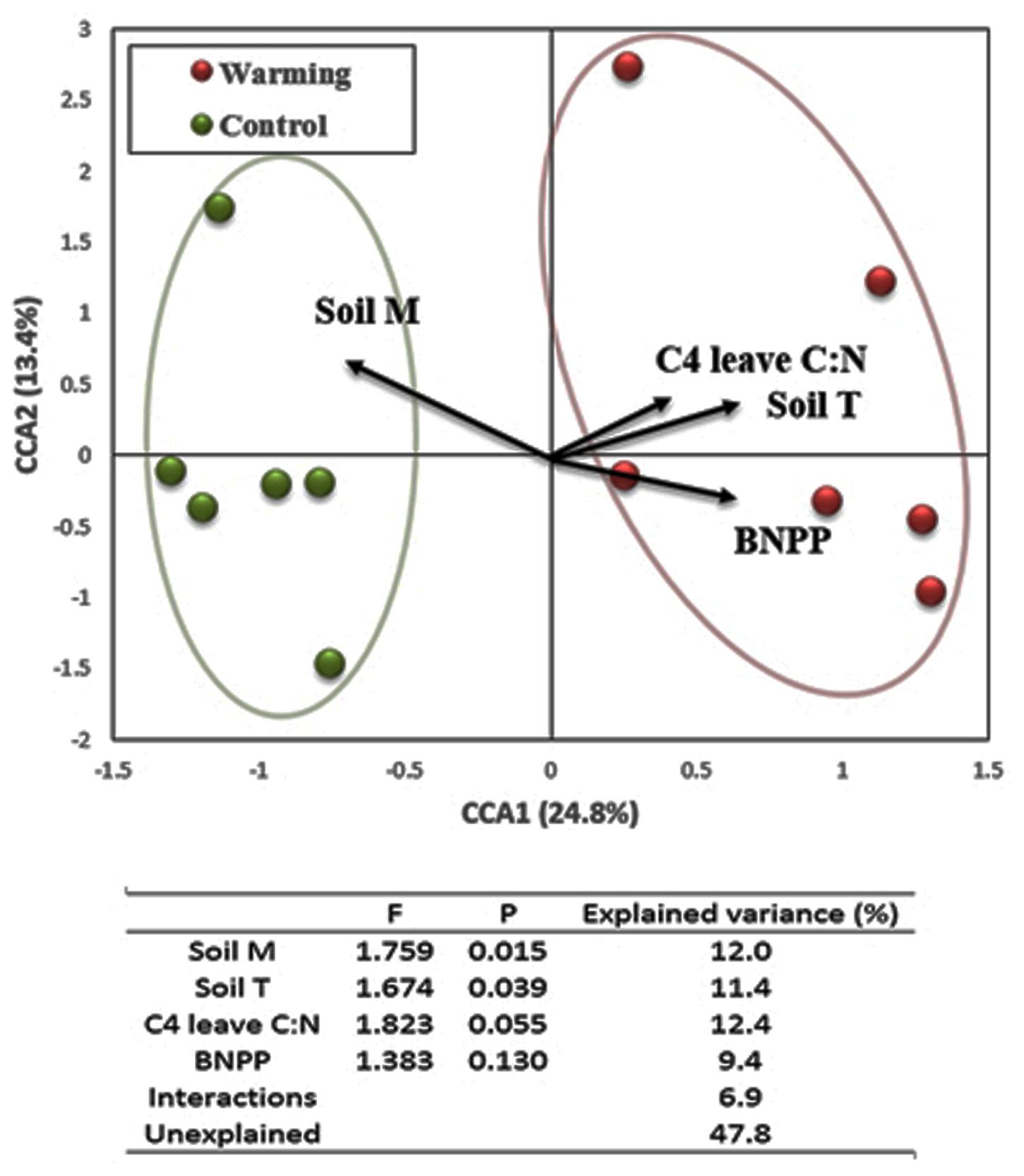
FIGURE 5. Canonical correspondence analysis (CCA) of microbial community composition based on expressed functional genes with selected environmental variables. The table presents results from partial CCA and variation partitioning analysis (VPA). The selected environmental variables include soil moisture (Soil M), soil temperature (Soil T), leaf C:N ratio in C4 species (C4 leaf C:N), and below-ground net primary productivity (BNPP).
To further examine the linkage between environmental attributes and certain groups of expressed genes, Mantel test was performed (Table 2). The structure of all expressed C degradation genes was correlated significantly (p ≤ 0.05) with soil moisture, TOC, above-ground net primary productivity (ANPP) and C4 species leaf C content. Other than these variables, the proportion of recalcitrant C pool in TOC, C4 AGB and BNPP were correlated significantly (p < 0.05) with expressed genes for decomposing recalcitrant C components only (e.g., aromatic compounds). In N cycling, surprisingly, no significant correlation was observed between any expressed gene groups and soil NH4+, NO3- or total N content (Table 2). Rather, the structure of all expressed N genes was correlated significantly with C4 species leaf N content (p = 0.03) and its C:N ratio (p = 0.02). Moreover, expressed assimilatory N reduction genes were correlated with TOC significantly (p = 0.04); while expressed ammonification genes were correlated significantly with soil 13C content (p = 0.02) and the proportion of soil C derived from C4 species (p = 0.03).
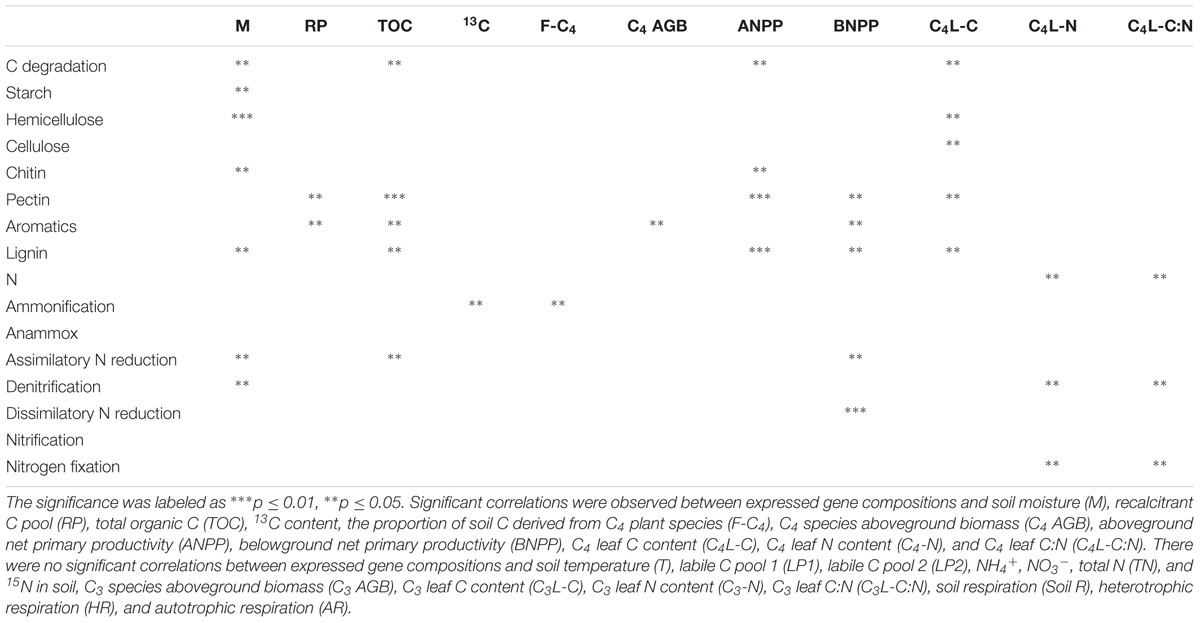
TABLE 2. Mantel tests between composition of expressed gene catalogs involved in C degradation/N processes and environmental variables.
One of the biggest scientific challenges of the 21st century is to better understand biological mechanisms regulating ecosystem responses to climate change and feedbacks that can amplify or dampen climate change (Heimann and Reichstein, 2008). Within this context, an understanding of soil microbial community is critical to our ability to assess terrestrial ecosystem responses and feedbacks (Bardgett et al., 2008). In the previous study conducted in the same experimental site, by using DNA-based GeoChip analysis, shifted functional gene composition of soil microbial community was observed under a long-term warming treatment (Zhou et al., 2012). In this study, by using RNA-based GeoChip analysis, we also found that the compositions of expressed functional genes in soil microbial community were altered significantly under warming. This finding well agrees with the general notion of shifted microbial community composition under warming observed in either the same (Zhang et al., 2005; Sheik et al., 2011) or different experiment sites (Zogg et al., 1997; Deslippe et al., 2005, 2012; Cookson et al., 2007; Rinnan et al., 2007; Frey et al., 2008; Mackelprang et al., 2011; Sheik et al., 2011; Zhang et al., 2011; Hayden et al., 2012; Kuffner et al., 2012; Yergeau et al., 2012; Stockdale et al., 2013), mainly tested by DNA-based methods.
In this experimental site, alterations in soil microclimate (e.g., soil temperature) were observed under warming (Xu et al., 2012b). The quantity of soil C input from plant that provides substrates for microorganisms likely increased under warming due to the total AGB increase. However, its quality possibly decreased as the proportion of soil C derived from C4 species increased with warming (Zhou et al., 2012), reflecting the plant community shift toward more C4 species with generally higher lignin content (Kephart and Buxton, 1993) and observed greater C:N ratio in plant tissues (Niu et al., 2010). These changes involved in soil physicochemical properties could be linked to the composition shift of expressed functional genes in soil microbial community. Close relationship between soil physicochemical properties and soil microbial community are widely recognized (Fierer et al., 2009; Peralta et al., 2013). Consistently, the CCA and VPA analyses revealed that the model consisting of soil temperature, soil moisture, leaf C:N ratio in C4 species and BNPP explained 52.2% of total variance in the structure of all expressed functional genes significantly (p = 0.005).
Different results in characterizing microbial communities by using DNA and RNA-based methods were observed in this and many other studies (Frias-Lopez et al., 2008; Gilbert et al., 2008; Baldrian et al., 2012; DeAngelis and Firestone, 2012). For example, in the previous study, unchanged recalcitrant C degradation were discovered under warming based on DNA-GeoChip analysis (Zhou et al., 2012), though the plant community shift towards more C4 species had long been observed in the field (An et al., 2005; Wan et al., 2005; Zhang et al., 2005; Luo et al., 2009). In this study, expressed genes for degrading more recalcitrant C components (i.e., aromatic components and lignin) increased with warming. Moreover, none of the functional genes involved in C degradation or N cycling decreased with warming based on the DNA-GeoChip analysis in the previous study (Zhou et al., 2012), while different changing directions (increase and decrease) were observed for expressed genes involved in labile C degradation and N cycling in this study. These phenomena were likely due to the fact that DNA and RNA-based methods characterize microbial communities in different aspects. RNA-based analyses only detect instantaneously expressed genes from metabolically active populations in the microbial community (Dell’Anno et al., 1998; Mills et al., 2005); while DNA-based surveys cannot discern active or inactive members if microbial populations do not change (Dell’Anno et al., 1998; Bodrossy et al., 2006). Also, RNA-based analyses would be influenced greatly by environmental conditions when sampling, while DNA-based analyses could include “historical” information (Anderson and Parkin, 2007) from inactive (e.g., dormant cells and spores) and dead cells (Dell’Anno et al., 1998; Bodrossy et al., 2006).
Stimulated gene expressions for degrading more recalcitrant C components under warming were likely linked to the quality decrease in soil C input due to the plant community shift toward more C4 species. Consistently, significant correlations were observed between expressed genes for degrading more recalcitrant C (e.g., aromatic components) and the measured soil recalcitrant C pool. In this experimental site, the AGB increase likely brought more labile C components into the soil and the microorganisms with higher capacity of degrading recalcitrant C components may not gain fitness in the community structure in the long-term as reflected by the DNA analysis. However, the metabolic state of those microorganisms could be possibly changed as reflected by corresponding expressed gene transcripts. Disproportional expressions of genes to the abundances of their originated cells were also observed by Baldrian et al. (2012) who found that some of the most abundant expressed cbhI genes for cellulose hydrolysis were from low abundance fungi species. Microorganisms have the ability to mediate their metabolic states in different niches, which are governed by the principles of redox reactions and chemical thermodynamics (Merlo et al., 2011). It has been reported that the soil substrate availability (poor or rich in organic C) could affect the metabolic enthalpy change of microbial activity (Barros et al., 2000). The stimulated expressions of genes involved in more recalcitrant C degradation may decrease the long-term soil C stability and trigger a positive feedback to climate warming.
Different changing directions (increase and decrease) for expressed genes involved in labile C degradation and N cycling may partially or completely reflect the dynamics of labile C components and available N at the time of sampling, which may mask the general trends of warming influences as reflected by the DNA analysis. Both labile C and available N pools have short mean residence time (MRT) in soils. The MRT of active fraction of soil organic C (e.g., plant residues and metabolites from soil biota) is only in months (Paul et al., 2001). Though the MRT of plant N in soils is in years (Silla and Escudero, 2004; Finzi et al., 2007), the available N (i.e., NH4+ or NO3-) could be transferred or taken up by plants and microorganisms in a much shorter time, like days or even hours (Inselsbacher et al., 2010). These facts imply that these pools, especially the available N, may be quite dynamic in the field, thereby likely resulting in different changing directions (increase and decrease) of corresponding gene transcripts.
Soil moisture, a dynamic variable in the field along time, was important in shaping expressed functional genes according to its significant correlations with the structures of C degradation or N genes. Interestingly, these expressed genes were also correlated with a few more “stable” variables. For example, the structure of all expressed C degradation genes were correlated with the TOC or C content in C4 species leafs; while the structure of all expressed N genes were correlated with the N content or C:N ratio in C4 species leafs. Linkages of RNA-based community composition with both dynamic and stable environmental attributes imply that information obtained from RNA analyses was still capable of capturing some stable trends caused by the long-term treatment, though DNA analyses would be better for this purpose. It may depend on both the strength of treatment effects and the dynamics of changes.
P and S are critical nutrient elements for plant growth (Eaton, 1950; Stewart et al., 1966; Fredeen et al., 1989). In P and S cycling, the average abundances of expressed genes were all higher under warming, suggesting a stable trend of accelerated nutrient cycling processes. This phenomenon was possibly related to the enhanced substrate input from plants, as well as promoted uptake from biological communities. The photosynthetic P use efficiency was suggested to be similar in C3 and C4 species (Halsted and Lynch, 1996). The P demand could be higher along with the stimulated growth of C4 species under warming. The S assimilatory pathway in plants is known to be well-coordinated with the N assimilatory pathway (Reuveny et al., 1980; Koprivova et al., 2000; Prosser et al., 2001; Kopriva and Koprivova, 2005) and the plant N use efficiency could be limited by the deficiency of soil S supply (Salvagiotti et al., 2009). In this field site, the N use efficiency was higher in C4 species and increased by warming (Brown, 1978; Oaks, 1994; An et al., 2005), which may demand higher S supply. In this way, if the accelerated P and S cycling mediated by microorganisms led to higher supplies of P and S, it was likely to serve as a key mechanism to sustain higher plant growth in the long-term warming treatment.
In summary, substantial changes of expressed functional genes in soil microbial communities were observed in this experimental site after long-term warming treatment. In the previous study, shifts in the functional gene composition of soil microbial community, enriched genes for labile C but not recalcitrant C degradation, and enriched genes for nutrient cycling were discovered under warming based on DNA-GeoChip analysis (Zhou et al., 2012). Findings from this study based on RNA-GeoChip analysis were illustrated in a conceptual model (Figure 6). Warming stimulated the growth of C4 species with higher C:N ratio and possibly greater lignin content. Thus, the quality of soil C input from plant decreased under warming, likely linked to stimulated gene expressions for degrading more recalcitrant C. Moreover, consistent trends of stimulated expressions were observed for genes in phosphorus and sulfur cycling, but not in labile carbon degradation and nitrogen cycling. These results suggest that the RNA-based GeoChip analysis is a robust tool to analyze soil microbial community in global climate change studies, though its combination with DNA-based analysis is needed as the dynamics of gene expression at the time of sampling may mask the general trends that can be captured by the DNA analysis. The stimulated genes expressions for degrading recalcitrant C may reveal changes in metabolic states of soil microorganisms with pathways of degrading recalcitrant C, likely to decrease the long-term soil C stability and trigger a positive feedback to climate warming. However, the accelerated P and S cycling processes implied by strong stimulating impacts of warming on P and S gene expressions may serve as a key negative feedback mechanism to sustain higher plant growth under warming. Though further studies are still needed to evaluate the weight of each mechanism in determining the whole direction of ecosystem feedback, our findings elucidate the importance of soil microbial community in regulating ecosystem responses to climate warming.
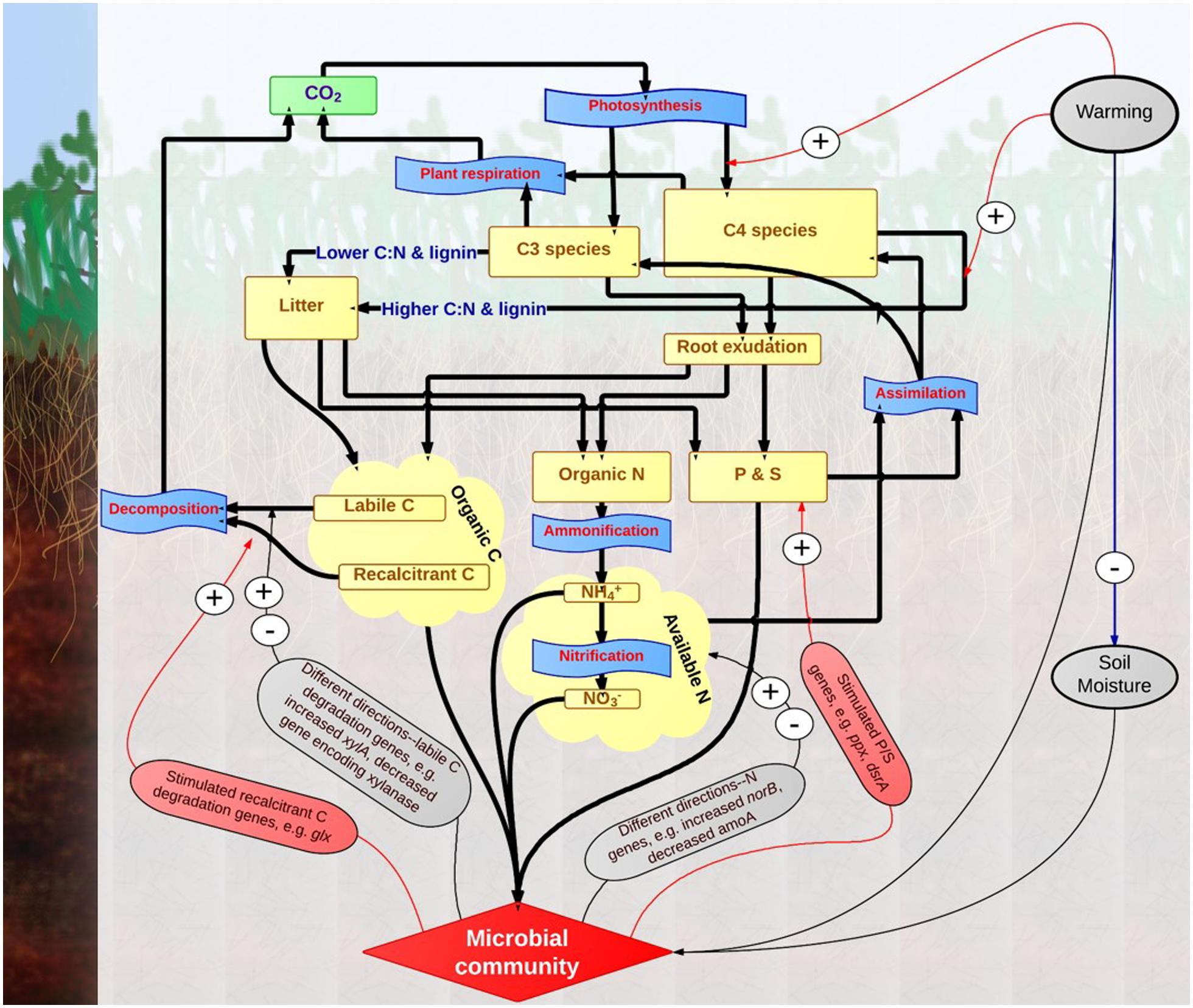
FIGURE 6. Conceptual model of warming impacts on grassland ecosystem processes based on results from this study. Greenhouse gas pool of CO2 is represented by square frames in green color, material pools are represented by square frames in yellow color, and biological processes are represented by punched tape frames in blue color. Material flows are represented by thicker rows in black color. Impacts of environmental attributes (e.g., soil temperature) and microbial community are represented by narrow rows in red (increase), blue (decrease) or black (different directions) colors.
All authors contributed intellectual input and assistance to this study and manuscript preparation. JZ and YL developed the original concepts. KX, JX, AZ, FL, DL, LW, YD, ZH, and JN contributed reagents, experiment conduction, data collection, and data analysis. Specifically, JX, AZ, KX, and FL handled all soils processing and subsampling for microbial analysis. JX and AZ did GeoChip hybridization. KX, YD, LW, ZH, and JN performed data analysis. KX wrote the manuscript with help from JZ, YL, FL, LW, ZH, and JN. All authors involved in revising this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the United States Department of Energy, Biological Systems Research on the Role of Microbial Communities in Carbon Cycling Program (DE-SC0004601), and Oklahoma Bioenergy Center (OBC).
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00668
An, Y. A., Wan, S. Q., Zhou, X. H., Subedar, A. A., Wallace, L. L., and Luo, Y. Q. (2005). Plant nitrogen concentration, use efficiency, and contents in a tallgrass prairie ecosystem under experimental warming. Global Change Biol. 11, 1733–1744. doi: 10.1111/j.1365-2486.2005.01030.x
Anderson, I. C., and Parkin, P. I. (2007). Detection of active soil fungi by RT-PCR amplification of precursor rRNA molecules. J. Microbiol. Methods 68, 248–253. doi: 10.1016/j.mimet.2006.08.005
Baldrian, P., Kolarik, M., Stursova, M., Kopecky, J., Valaskova, V., Vetrovsky, T., et al. (2012). Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 6, 248–258. doi: 10.1038/ismej.2011.95
Bardgett, R. D., Freeman, C., and Ostle, N. J. (2008). Microbial contributions to climate change through carbon cycle feedbacks. ISME J. 2, 805–814. doi: 10.1038/ismej.2008.58
Barros, N., Feijóo, S., Fernández, S., Simoni, J. A., and Airoldi, C. (2000). Application of the metabolic enthalpy change in studies of soil microbial activity. Thermochim. Acta. 356, 1–7. doi: 10.1016/S0040-6031(00)00495-0
Bodrossy, L., Stralis-Pavese, N., Konrad-Koszler, M., Weilharter, A., Reichenauer, T. G., Schofer, D., et al. (2006). mRNA-based parallel detection of active methanotroph populations by use of a diagnostic microarray. Appl. Environ. Microbiol. 72, 1672–1676. doi: 10.1128/Aem.72.2.1672-1676.2006
Brown, R. H. (1978). A Difference in N Use Efficiency in C3 and C4 Plants and its Implications in Adaptation and Evolution. Crop Sci. 18, 93–98. doi: 10.2135/cropsci1978.0011183X001800010025x
Cheng, X., Luo, Y., Xu, X., Sherry, R., and Zhang, Q. (2011). Soil organic matter dynamics in a North America tallgrass prairie after 9 yr of experimental warming. Biogeosciences 8, 1487–1498. doi: 10.5194/bg-8-1487-2011
Cheng, X. L., Luo, Y. Q., Su, B., Zhou, X. H., Niu, S. L., Sherry, R., et al. (2010). Experimental warming and clipping altered litter carbon and nitrogen dynamics in a tallgrass prairie. Agric. Ecosyst. Environ. 138, 206–213. doi: 10.1016/j.agee.2010.04.019
Cookson, W. R., Osman, M., Marschner, P., Abaye, D. A., Clark, I., Murphy, D. V., et al. (2007). Controls on soil nitrogen cycling and microbial community composition across land use and incubation temperature. Soil Biol. Biochem. 39, 744–756. doi: 10.1016/j.soilbio.2006.09.022
DeAngelis, K. M., and Firestone, M. K. (2012). Phylogenetic clustering of soil microbial communities by 16S rRNA but not 16S rRNA genes. Appl. Environ. Microbiol. 78, 2459–2461. doi: 10.1128/AEM.07547-11
Dell’Anno, A., Fabiano, M., Duineveld, G. C. A., Kok, A., and Danovaro, R. (1998). Nucleic acid (DNA, RNA) quantification and RNA/DNA ratio determination in marine sediments: Comparison of spectrophotometric, fluorometric, and high-performance liquid chromatography methods and estimation of detrital DNA. Appl. Environ. Microbiol. 64, 3238–3245.
Deslippe, J. R., Egger, K. N., and Henry, G. H. R. (2005). Impacts of warming and fertilization on nitrogen-fixing microbial communities in the Canadian High Arctic. FEMS Microbiol. Ecol. 53, 41–50. doi: 10.1016/j.femsec.2004.12.002
Deslippe, J. R., Hartmann, M., Simard, S. W., and Mohn, W. W. (2012). Long-term warming alters the composition of Arctic soil microbial communities. FEMS Microbiol. Ecol. 82, 303–315. doi: 10.1111/j.1574-6941.2012.01350.x
Eaton, S. V. (1950). Effects of Phosphorus Deficiency on Growth and Metabolism of Soybean - Contributions from the Hull-Botanical-Laboratory-613. Bot. Gazette 111, 426–436. doi: 10.1086/335613
Fierer, N., Strickland, M. S., Liptzin, D., Bradford, M. A., and Cleveland, C. C. (2009). Global patterns in belowground communities. Ecol. Lett. 12, 1238–1249. doi: 10.1111/j.1461-0248.2009.01360.x
Finzi, A. C., Norby, R. J., Calfapietra, C., Gallet-Budynek, A., Gielen, B., Holmes, W. E., et al. (2007). Increases in nitrogen uptake rather than nitrogen-use efficiency support higher rates of temperate forest productivity under elevated CO2. Proc. Natl. Acad. Sci.U.S.A. 104, 14014–14019. doi: 10.1073/pnas.0706518104
Franzosa, E. A., Hsu, T., Sirota-Madi, A., Shafquat, A., Abu-Ali, G., Morgan, X. C., et al. (2015). Sequencing and beyond: integrating molecular ’omics’ for microbial community profiling. Nat. Rev. Microbiol. 13, 360–372. doi: 10.1038/nrmicro3451
Fredeen, A. L., Rao, I. M., and Terry, N. (1989). Influence of Phosphorus-Nutrition on Growth and Carbon Partitioning in Glycine-Max. Plant Physiol. 89, 225–230. doi: 10.1104/Pp.89.1.225
Frey, S. D., Drijber, R., Smith, H., and Melillo, J. (2008). Microbial biomass, functional capacity, and community structure after 12 years of soil warming. Soil Biol. Biochem. 40, 2904–2907. doi: 10.1016/j.soilbio.2008.07.020
Frias-Lopez, J., Shi, Y., Tyson, G. W., Coleman, M. L., Schuster, S. C., Chisholm, S. W., et al. (2008). Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U.S.A. 105, 3805–3810. doi: 10.1073/pnas.0708897105
Gao, H. C., Yang, Z. M. K., Gentry, T. J., Wu, L. Y., Schadt, C. W., and Zhou, J. Z. (2007). Microarray-based analysis of microbial community RNAs by whole-community RNA amplification. Appl. Environ. Microbiol. 73, 563–571. doi: 10.1128/AEM.01771-06
Gilbert, J. A., Field, D., Huang, Y., Edwards, R., Li, W. Z., Gilna, P., et al. (2008). Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS ONE 3:e3042. doi: 10.1371/Journal.Pone.0003042
Halsted, M., and Lynch, J. (1996). Phosphorus responses of C-3 and C-4 species. J. Exp. Bot. 47, 497–505. doi: 10.1093/Jxb/47.4.497
Hayden, H. L., Mele, P. M., Bougoure, D. S., Allan, C. Y., Norng, S., Piceno, Y. M., et al. (2012). Changes in the microbial community structure of bacteria, archaea and fungi in response to elevated CO2 and warming in an Australian native grassland soil. Environ. Microbiol. 14, 3081–3096. doi: 10.1111/j.1462-2920.2012.02855.x
He, Z., Deng, Y., Van Nostrand, J. D., Tu, Q., Xu, M., Hemme, C. L., et al. (2010). GeoChip 3.0 as a high throughput tool for analyzing microbial community structure, composition, and functional activity. ISME J. 4, 1167–1179. doi: 10.1038/ismej.2010.46
Heimann, M., and Reichstein, M. (2008). Terrestrial ecosystem carbon dynamics and climate feedbacks. Nature 451, 289–292. doi: 10.1038/Nature06591
Hurt, R. A., Qiu, X. Y., Wu, L. Y., Roh, Y., Palumbo, A. V., Tiedje, J. M., et al. (2001). Simultaneous recovery of RNA and DNA from soils and sediments. Appl. Environ. Microbiol. 67, 4495–4503. doi: 10.1128/AEM.67.10.4495-4503.2001
Inselsbacher, E., Hinko-Najera Umana, N., Stange, F. C., Gorfer, M., Schuller, E., Ripka, K., et al. (2010). Short-term competition between crop plants and soil microbes for inorganic N fertilizer. Soil Biol. Biochem. 42, 360–372. doi: 10.1016/j.soilbio.2009.11.019
IPCC. (2007). Climate Change 2007: Synthesis Report. Contribution of Working Groups I, II and III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Geneva: Intergovernmental Panel on Climate Change.
Jackson, L. E., Calderon, F. J., Steenwerth, K. L., Scow, K. M., and Rolston, D. E. (2003). Responses of soil microbial processes and community structure to tillage events and implications for soil quality. Geoderma 114, 305–317. doi: 10.1016/S0016-7061(03)00046-6
Kephart, K. D., and Buxton, D. R. (1993). Forage quality responses of C3 and C4 perennial grasses to shade. Crop Sci. 33, 831–837. doi: 10.2135/cropsci1993.0011183X003300040040x
Kopriva, S., and Koprivova, A. (2005). Sulfate assimilation and glutathione synthesis in C-4 plants. Photosynth. Res. 86, 363–372. doi: 10.1007/s11120-005-3482-z
Koprivova, A., Suter, M., den Camp, R., Brunold, C., and Kopriva, S. (2000). Regulation of sulfate assimilation by nitrogen in Arabidopsis. Plant Physiol. 122, 737–746. doi: 10.1104/Pp.122.3.737
Kuffner, M., Hai, B., Rattei, T., Melodelima, C., Schloter, M., Zechmeister-Boltenstern, S., et al. (2012). Effects of season and experimental warming on the bacterial community in a temperate mountain forest soil assessed by 16S rRNA gene pyrosequencing. FEMS Microbiol. Ecol. 82, 551–562. doi: 10.1111/j.1574-6941.2012.01420.x
Liang, Y. T., He, Z. L., Wu, L. Y., Deng, Y., Li, G. H., and Zhou, J. Z. (2010). Development of a common oligonucleotide reference standard for microarray data normalization and comparison across different microbial communities. Appl. Environ. Microbiol. 76, 1088–1094. doi: 10.1128/AEM.02749-09
Luo, Y. Q., Sherry, R., Zhou, X. H., and Wan, S. Q. (2009). Terrestrial carbon-cycle feedback to climate warming: experimental evidence on plant regulation and impacts of biofuel feedstock harvest. Global Change Biol. Bioenergy 1, 62–74. doi: 10.1111/j.1757-1707.2008.01005.x
Luo, Y. Q., Wan, S. Q., Hui, D. F., and Wallace, L. L. (2001). Acclimatization of soil respiration to warming in a tall grass prairie. Nature 413, 622–625. doi: 10.1038/35098065
Mackelprang, R., Waldrop, M. P., DeAngelis, K. M., David, M. M., Chavarria, K. L., Blazewicz, S. J., et al. (2011). Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480, 368–371. doi: 10.1038/Nature10576
McGrath, K. C., Mondav, R., Sintrajaya, R., Slattery, B., Schmidt, S., and Schenk, P. M. (2010). Development of an environmental functional gene microarray for soil microbial communities. Appl. Environ. Microbiol. 76, 7161–7170. doi: 10.1128/AEM.03108-09
Merlo, M. E., Jankevics, A., Takano, E., and Breitling, R. (2011). Exploring the metabolic state of microorganisms using metabolomics. Bioanalysis 3, 2443–2458. doi: 10.4155/bio.11.248
Mills, H. J., Martinez, R. J., Story, S., and Sobecky, P. A. (2005). Characterization of microbial community structure in Gulf of Mexico gas hydrates: comparative analysis of DNA- and RNA-derived clone libraries. Appl. Environ. Microbiol. 71, 3235–3247. doi: 10.1128/Aem.71.6.3235-3247.2005
Monastersky, R. (2013). Global carbon dioxide levels near worrisome milestone. Nature 497, 13–14. doi: 10.1038/497013a
Niu, S. L., Sherry, R. A., Zhou, X. H., Wan, S. Q., and Luo, Y. Q. (2010). Nitrogen regulation of the climate-carbon feedback: evidence from a long-term global change experiment. Ecology 91, 3261–3273. doi: 10.1890/09-1634.1
Novotny, A. M., Schade, J. D., Hobbie, S. E., Kay, A. D., Kyle, M., Reich, P. B., et al. (2007). Stoichiometric response of nitrogen-fixing and non-fixing dicots to manipulations of CO2, nitrogen, and diversity. Oecologia 151, 687–696. doi: 10.1007/s00442-006-0599-5
Oaks, A. (1994). Efficiency of Nitrogen-Utilization in C-3 and C-4 Cereals. Plant Physiol. 106, 407–414.
Paul, E. A., Collins, H. P., and Leavitt, S. W. (2001). Dynamics of resistant soil carbon of midwestern agricultural soils measured by naturally occurring C-14 abundance. Geoderma 104, 239–256. doi: 10.1016/S0016-7061(01)00083-0
Peralta, R. M., Ahn, C., and Gillevet, P. M. (2013). Characterization of soil bacterial community structure and physicochemical properties in created and natural wetlands. Sci. Total Environ. 443, 725–732. doi: 10.1016/j.scitotenv.2012.11.052
Prosser, I. M., Purves, J. V., Saker, L. R., and Clarkson, D. T. (2001). Rapid disruption of nitrogen metabolism and nitrate transport in spinach plants deprived of sulphate. J. Exp. Bot. 52, 113–121. doi: 10.1093/jexbot/52.354.113
Reuveny, Z., Dougall, D. K., and Trinity, P. M. (1980). Regulatory coupling of nitrate and sulfate assimilation pathways in cultured tobacco cells. Proc. Natl. Acad. Sci. U.S.A. 77, 6670–6672. doi: 10.1073/pnas.77.11.6670
Rinnan, R., Michelsen, A., Baath, E., and Jonasson, S. (2007). Fifteen years of climate change manipulations alter soil microbial communities in a subarctic heath ecosystem. Global Change Biol. 13, 28–39. doi: 10.1111/j.1365-2486.2006.01263.x
Salvagiotti, F., Castellarin, J. M., Miralles, D. J., and Pedrol, H. M. (2009). Sulfur fertilization improves nitrogen use efficiency in wheat by increasing nitrogen uptake. Field Crops Res. 113, 170–177. doi: 10.1016/j.fcr.2009.05.003
Schindlbacher, A., Rodler, A., Kuffner, M., Kitzler, B., Sessitsch, A., and Zechmeister-Boltenstern, S. (2011). Experimental warming effects on the microbial community of a temperate mountain forest soil. Soil Biol. Biochem. 43, 1417–1425. doi: 10.1016/j.soilbio.2011.03.005
Shaver, G. R., Canadell, J., Chapin, F. S., Gurevitch, J., Harte, J., Henry, G., et al. (2000). Global warming and terrestrial ecosystems: a conceptual framework for analysis. Bioscience 50, 871–882. doi: 10.1641/0006-3568(2000)050[0871:GWATEA]2.0.CO;2
Sheik, C. S., Beasley, W. H., Elshahed, M. S., Zhou, X. H., Luo, Y. Q., and Krumholz, L. R. (2011). Effect of warming and drought on grassland microbial communities. ISME J. 5, 1692–1700. doi: 10.1038/ismej.2011.32
Sherry, R. A., Weng, E. S., Arnone, J. A., Johnson, D. W., Schimel, D. S., Verburg, P. S., et al. (2008). Lagged effects of experimental warming and doubled precipitation on annual and seasonal aboveground biomass production in a tallgrass prairie. Global Change Biol. 14, 2923–2936. doi: 10.1111/j.1365-2486.2008.01703.x
Silla, F., and Escudero, A. (2004). Nitrogen-use efficiency: trade-offs between N productivity and mean residence time at organ, plant and population levels. Funct. Ecol. 18, 511–521. doi: 10.1111/j.0269-8463.2004.00872.x
Simon, C., and Daniel, R. (2011). Metagenomic analyses: past and future trends. Appl. Environ. Microbiol. 77, 1153–1161. doi: 10.1128/AEM.02345-10
Stewart, B. A., Porter, L. K., and Viets, F. G. (1966). Effect of sulfur content of straws on rates of decomposition and plant growth. Soil Sci. Soc. Am. Proc. 30:355. doi: 10.2136/sssaj1966.03615995003000030017x
Stockdale, E. A., Banning, N. C., and Murphy, D. V. (2013). Rhizosphere effects on functional stability of microbial communities in conventional and organic soils following elevated temperature treatment. Soil Biol. Biochem. 57, 56–59. doi: 10.1016/j.soilbio.2012.08.020
Takasaki, K., Miura, T., Kanno, M., Tamaki, H., Hanada, S., Kamagata, Y., et al. (2013). Discovery of glycoside hydrolase enzymes in an avicel-adapted forest soil fungal community by a metatranscriptomic approach. PLoS ONE 8:e55485. doi: 10.1371/journal.pone.0055485
Tuorto, S. J., Darias, P., McGuinness, L. R., Panikov, N., Zhang, T. J., Haggblom, M. M., et al. (2014). Bacterial genome replication at subzero temperatures in permafrost. ISME J. 8, 139–149. doi: 10.1038/ismej.2013.140
Tveit, A., Schwacke, R., Svenning, M. M., and Urich, T. (2013). Organic carbon transformations in high-Arctic peat soils: key functions and microorganisms. ISME J. 7, 299–311. doi: 10.1038/ismej.2012.99
USDA (1963). National Cooperative Soil Survey, Soil Survey of McClain Country. Stillwater: Agricultural Experimental Station.
van Bruggen, A. H. C., and Semenov, A. M. (2000). In search of biological indicators for soil health and disease suppression. Appl. Soil Ecol. 15, 13–24. doi: 10.1016/S0929-1393(00)00068-8
Wan, S. Q., Hui, D. F., Wallace, L., and Luo, Y. Q. (2005). Direct and indirect effects of experimental warming on ecosystem carbon processes in a tallgrass prairie. Global Biogeochem. Cycles 19:GB2014. doi: 10.1029/2004gb002315
Wu, L. Y., Liu, X., Schadt, C. W., and Zhou, J. Z. (2006). Microarray-based analysis of subnanogram quantities of microbial community DNAs by using whole-community genome amplification. Appl. Environ. Microbiol. 72, 4931–4941. doi: 10.1128/AEM.02738-05
Xu, X., Luo, Y. Q., Shi, Z., Zhou, X. H., and Li, D. J. (2014). Consistent proportional increments in responses of belowground net primary productivity to long-term warming and clipping at various soil depths in a tall-grass prairie. Oecologia 174, 1045–1054. doi: 10.1007/s00442-013-2828-z
Xu, X., Niu, S. L., Sherry, R. A., Zhou, X. H., Zhou, J. Z., and Luo, Y. Q. (2012a). Interannual variability in responses of belowground NPP and NPP partitioning to long-term warming and clipping in a tallgrass prairie. Global Change Biol. 18, 1648–1656. doi: 10.1111/j.1365-2486.2012.02651.x
Xu, X., Sherry, R. A., Niu, S. L., Zhou, J. Z., and Luo, Y. Q. (2012b). Long-term experimental warming decreased labile soil organic carbon in a tallgrass prairie. Plant Soil 361, 307–365. doi: 10.1007/s11104-012-1265-9
Xu, X., Shi, Z., Li, D. J., Zhou, X. H., Sherry, R. A., and Luo, Y. Q. (2015). Plant community structure regulates responses of prairie soil respiration to decadal experimental warming. Global Change Biol. 21, 3846–3853. doi: 10.1111/gcb.12940
Xue, K., Serohijos, R. C., Devare, M., and Thies, J. E. (2011). Decomposition rates and residue-colonizing microbial communities of Bacillus thuringiensis insecticidal protein Cry3Bb-expressing (Bt) and non-Bt corn hybrids in the field. Appl. Environ. Microbiol. 77, 839–846. doi: 10.1128/AEM.01954-10
Xue, K., Wu, L. Y., Deng, Y., He, Z. L., Van Nostrand, J., Robertson, P. G., et al. (2013). Functional gene differences in soil microbial communities from conventional, low-input, and organic farmlands. Appl. Environ. Microbiol. 79, 1284–1292. doi: 10.1128/Aem.03393–3312
Yergeau, E., Bokhorst, S., Kang, S., Zhou, J. Z., Greer, C. W., Aerts, R., et al. (2012). Shifts in soil microorganisms in response to warming are consistent across a range of Antarctic environments. ISME J. 6, 692–702. doi: 10.1038/ismej.2011.124
Zhang, N. L., Xia, J. Y., Yu, X. J., Ma, K. P., and Wan, S. Q. (2011). Soil microbial community changes and their linkages with ecosystem carbon exchange under asymmetrically diurnal warming. Soil Biol. Biochem. 43, 2053–2059. doi: 10.1016/j.soilbio.2011.06.001
Zhang, W., Parker, K. M., Luo, Y., Wan, S., Wallace, L. L., and Hu, S. (2005). Soil microbial responses to experimental warming and clipping in a tallgrass prairie. Global Change Biol. 11, 266–277. doi: 10.1111/j.1365-2486.2005.00902.x
Zhou, J. Z., Xue, K., Xie, J. P., Deng, Y., Wu, L. Y., Cheng, X. H., et al. (2012). Microbial mediation of carbon-cycle feedbacks to climate warming. Nat. Clim. Change 2, 106–110. doi: 10.1038/Nclimate1331
Keywords: RNA, functional gene expression, global climate change, warming, GeoChip
Citation: Xue K, Xie J, Zhou A, Liu F, Li D, Wu L, Deng Y, He Z, Van Nostrand JD, Luo Y and Zhou J (2016) Warming Alters Expressions of Microbial Functional Genes Important to Ecosystem Functioning. Front. Microbiol. 7:668. doi: 10.3389/fmicb.2016.00668
Received: 04 November 2015; Accepted: 21 April 2016;
Published: 06 May 2016.
Edited by:
Steve Brian Pointing, Auckland University of Technology, New ZealandReviewed by:
Sascha M. B. Krause, University of Washington, USACopyright © 2016 Xue, Xie, Zhou, Liu, Li, Wu, Deng, He, Van Nostrand, Luo and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jizhong Zhou, anpob3VAb3UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.