 Runyu Yuan1,2,3
Runyu Yuan1,2,3 Zheng Wang4
Zheng Wang4 Yinfeng Kang3Jie Wu1,2Lirong Zou1,2Lijun Liang1,2Yingchao Song1,2Xin Zhang1,2Hanzhong Ni1,2Jinyan Lin1,2Changwen Ke1,2*
Yinfeng Kang3Jie Wu1,2Lirong Zou1,2Lijun Liang1,2Yingchao Song1,2Xin Zhang1,2Hanzhong Ni1,2Jinyan Lin1,2Changwen Ke1,2*- 1Key Laboratory for Repository and Application of Pathogenic Microbiology, Research Center for Pathogens Detection Technology of Emerging Infectious Diseases, Guangdong Provincial Center for Disease Control and Prevention, Guangzhou, China
- 2WHO Collaborating Centre for Surveillance, Research and Training of Emerging Infectious Disease, Guangzhou, China
- 3Key Laboratory of Zoonosis Prevention and Control of Guangdong, College of Veterinary Medicine, South China Agricultural University, Guangzhou, China
- 4School of Public Health, Sun Yat-Sen University, Guangzhou, China
First identified in May 2014 in China's Sichuan Province, initial cases of H5N6 avian influenza virus (AIV) infection in humans raised great concerns about the virus's prevalence, origin, and development. To evaluate both AIV contamination in live poultry markets (LPMs) and the risk of AIV infection in humans, we have conducted surveillance of LPMs in Guangdong Province since 2013 as part of environmental sampling programs. With environmental samples associated with these LPMs, we performed genetic and phylogenetic analyses of 10 H5N6 AIVs isolated from different cities of Guangdong Province from different years. Results revealed that the H5N6 viruses were reassortants with hemagglutinin (HA) genes derived from clade 2.3.4.4 of H5-subtype AIV, yet neuraminidase (NA) genes derived from H6N6 AIV. Unlike the other seven H5N6 viruses isolated in first 7 months of 2014, all of which shared remarkable sequence similarity with the H5N1 AIV in all internal genes, the PB2 genes of GZ693, GZ670, and ZS558 more closely related to H6N6 AIV and the PB1 gene of GZ693 to the H3-subtype AIV. Phylogenetic analyses revealed that the environmental H5N6 AIV related closely to human H5N6 AIVs isolated in Guangdong. These results thus suggest that continued reassortment has enabled the emergence of a novel H5N6 virus in Guangdong, as well as highlight the potential risk of highly pathogenic H5N6 AIVs in the province.
Introduction
Depending on their pathotypes, avian influenza viruses (AIVs) have inherently different pathogeneses in the infection and distribution of lesions. According to pathotype, AIVs can divide into non-pathogenic AIVs (NPAIV), low pathogenic AIVs (LPAIV), and highly pathogenic AIVs (HPAIV). In birds, NPAIVs (e.g., H6N6) usually present no clinical symptoms; by contrast, LPAIVs (e.g., H9N2) can cause mild respiratory or gastrointestinal infection, and HPAIVs (e.g., H5N1 and H7N2) can induce systemic, multi-organ infection, as well as high morbidity, and mortality (Swayne and Halvorson, 2003; Yuan et al., 2014).
A continual threat to animal and human health, HPAIVs have caused infections and deaths in not only countless birds, but also many humans. In 1997, the H5N1 HPAIV, the internal genes of which derived from the H6N1 NPAIV, infected 18 people in Hong Kong, six of whom died from the infection (Claas et al., 1998; Subbarao et al., 1998). Furthermore, since 2003, the H5N1 HPAIV has caused outbreaks both in birds and humans in more than 60 countries, including China (Yuan et al., 2014; WHO, 2015a). Recently, H5-subtype HPAIV s—that is, variants of different NA subtypes—have also caused outbreaks in poultry in China (i.e., subtypes H5N1, H5N2, H5N5, H5N6, and H5N8), as well as in South Korea (i.e., subtype H5N8), Japan (i.e., subtype H5N8), Laos (i.e., subtype H5N8), and Vietnam (i.e., subtypes H5N1 and H5N6; WHO, 2014d; OIE, 2015). In March 2014, an outbreak of H5N6 HPAIV in poultry was reported in Laos and, that April, in Vietnam (Wong et al., 2015). Genetic studies have shown that the H5N6 virus has exchanged genes from the H5N1 and H6N6 AIVs that circulate widely in ducks (Shen et al., 2015). Although little is known about the potential of these novel viruses to infect humans, a few isolated cases have been detected. On May 6, 2014, one such case of H5N6 infection in China's Sichuan Province was fatal (CDC China, 2014; WHO, 2014c), and later that year, another severe case of infection occurred in Guangdong Province in December (WHO, 2014a). As of February 2016, nine cases of H5N6 AIVs infection in humans have been confirmed in China, six of them in Guangdong Province (WHO, 2015b,c, 2016a,b,c).
Since 2013, several surveillance systems for pandemic preparedness have been established in China, including those at live poultry markets (LPM) and sentinel hospitals. These surveillance systems have played a vital role in the early detection of warning signs of AIV infection in humans. During our study's surveillance period, we isolated 10 H5N6 AIVs in environmental samples from LPMs in Guangdong Province, and to better understand their genetic diversity and evolution, we analyzed their related epidemiological and sequence data.
Materials and Methods
Ethics Statement
This research was reviewed and approved by the South China Agricultural University Experimental Animal Welfare Ethics Committee (permit no. 2014-11).
Sample Collection
Beginning on April 16, 2013, in order to better monitor LPMs for AIV contamination and assess the risk of AIV infection in humans, environmental sampling programs were implemented in Guangdong Province. Environmental samples were taken from poultry excrement, epilator swabs, and sewage swabs—the latter two from drains in meat preparation areas or around cages—whereas chopping swab samples were gathered randomly from butcher boards or knives at LPMs each week.
Virus Isolation
Samples were first tested for influenza A by using real-time polymerase chain reactions (qPCR) in the laboratories of the district's Centers for Disease Control and Prevention (CDC). Positive influenza A samples were probed to detect subtypes H5, H7, and H9 by using qPCR in local CDC laboratories, and results were later verified by Guangdong's CDC. H5-positive samples were further analyzed by using qPCR to detect the presence of the N6 gene. All qPCR-detected primers and probes were provided by the Chinese CDC. Samples positive with H5N6 subtypes were purified and propagated in 10-d embryonated chicken eggs free of specific pathogens and stored at −70°C until used. Subtypes of the viruses were further identified by hemagglutination (HA) inhibition assay. All experiments were carried out in animal biosafety level 3 facilities.
Genomic Sequencing
Viral RNA was first extracted from allantoic fluid by using an RNA extraction kit (QIAamp Viral RNA Mini Kit, Qiagen, Hilden, Germany). Reverse transcription and polymerase chain reaction (PCR) amplification of all eight gene segments used pre-amplification reagents (PathAmp™ FluA, Life Technologies, Guilford, Connecticut, USA). PCR products were purified and quantified with a purification kit (AmpureXP, Beckman Coulter, Porterville, CA, USA) according to the manufacturer's instructions. The full genomes of the viruses were sequenced with a sequencing kit (Ion PGM Sequencing 200 Kit version 2, Life Technologies), specifically with the kit's Ion 316 Chip V2 and according to the manufacturer's instructions.
Sequence Analysis
To align and analyze the sequences, multiple sequences of the representative AIVs were downloaded from GenBank databases (Li et al., 2010; Yuan et al., 2014, 2016). Full-length gene sequences were implemented and edited with Lasergene 7.1 (DNASTAR, Madison, Wisconsin, USA). A neighbor-joining algorithm and maximum-likelihood trees model were estimated for all eight genes—namely, HA, NA, PB2, PB1, PA, NP, M, and NS—by using genetic analysis software [Molecular Evolutionary Genetics Analysis (MEGA) version 6.06] with 1000 bootstrap trials. Branches with bootstrap values exceeding 50% were grouped together in the trees.
Nucleotide sequences obtained in our study, all listed by their accession numbers, are currently available from GenBank (Table 1).
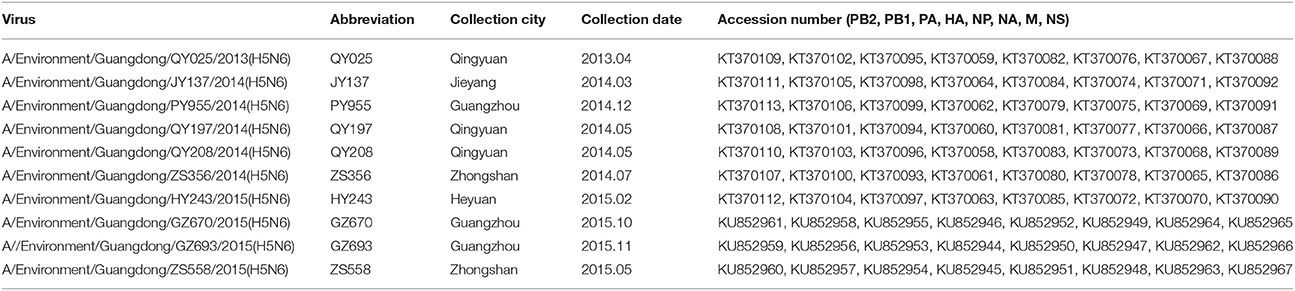
Table 1. Isolation of H5N6-subtype avian influenza viruses from live poultry markets in Guangdong, 2013–2015.
Results
Prevalence of the H5-Subtype AIV in LPMs
From April 2013 to December 2015, a total of 32,452 fecal and swabs were collected from LPMs in 21 cities in Guangdong Province (Table 2). Among all of the samples, 6865 (21.2%) were positive for influenza A, 14.6% of which with the H5 subtype. The H5N1 subtype was the most prevalent among the H5 subtypes, followed by H5N6; also observed were H5N2, H5N3, H5N4, H5N5, H5N7, H5N8, and H5N9. During the same period, we selected 10 H5N6 subtypes among the 66 H5N6-positive samples in different cities of Guangdong Province in different years to analyze the evolution of the subtype (Table 1).
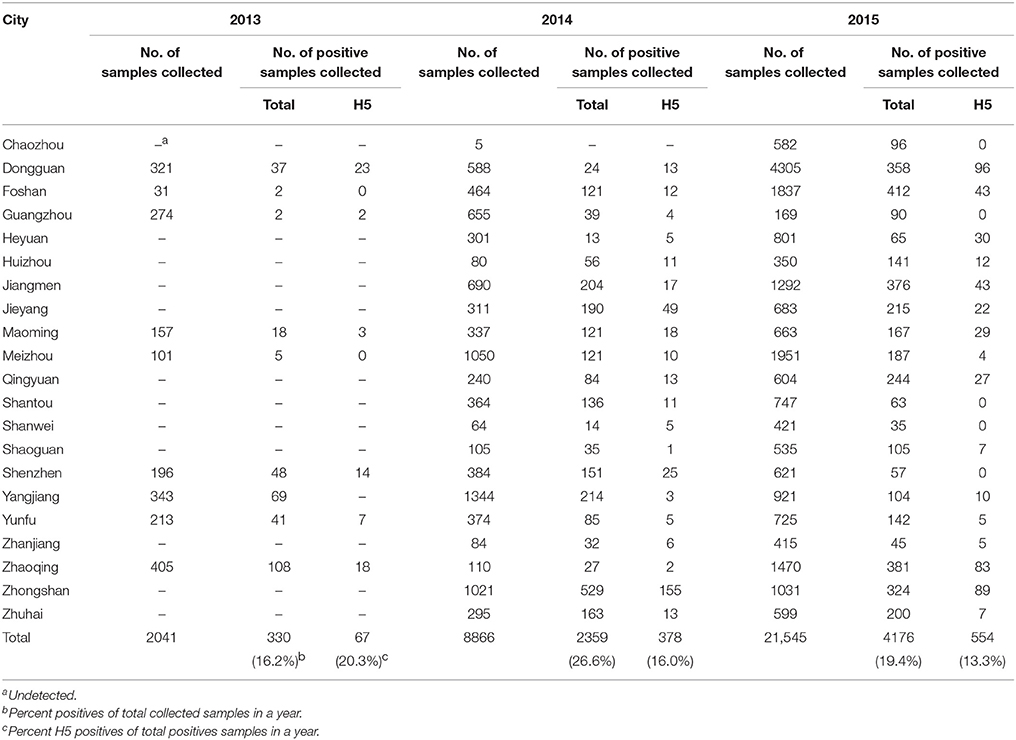
Table 2. Environment surveillance of H5-subtype avian influenza viruses in Guangdong, 2013–2015.
Phylogenetic Analysis of Surface Genes
The genomes of the 10 H5N6 AIVs isolated from environmental samples were sequenced by using a next-generation sequencer (Ion PGM, Life Technologies). The complete genomes of the 10 samples were compared with nucleotide sequences of some viruses in GenBank databases.
Phylogenetic analyses demonstrated the origin and evolution of H5N6 AIVs in China. As results of the phylogenetic analysis of H5 and related viruses show, the HA gene of all 10 viruses clustered into clade 2.3.4.4 (Figure 1A) and thus related more closely to the H5N2 HPAIV, A/chicken/Zhejiang/727159/2014(H5N2), which circulates in Zhejiang Province (Figures 1A, 2). In addition, QY025, QY197, QY208, GZ670, and ZS558 shared 98.5–99.9% highest nucleotide similarity with A/chicken/Dongguan/2690/2013(H5N6) (GD-H5N6), JY137, PY955, and ZS356 shared 99.1–99.6% highest nucleotide similarity with A/chicken/Shenzhen/1395/2013(H5N6) (GD-H5N6), and HY243 shared 99.1% highest nucleotide similarity with JX-H5N6. More singularly, GZ693 shared 98.7% highest nucleotide similarity with A/Guangdong/ZQ874/2015(H5N6) (ZQ874), which was found to have recently infected human in Zhaoqing, Guangdong Province.
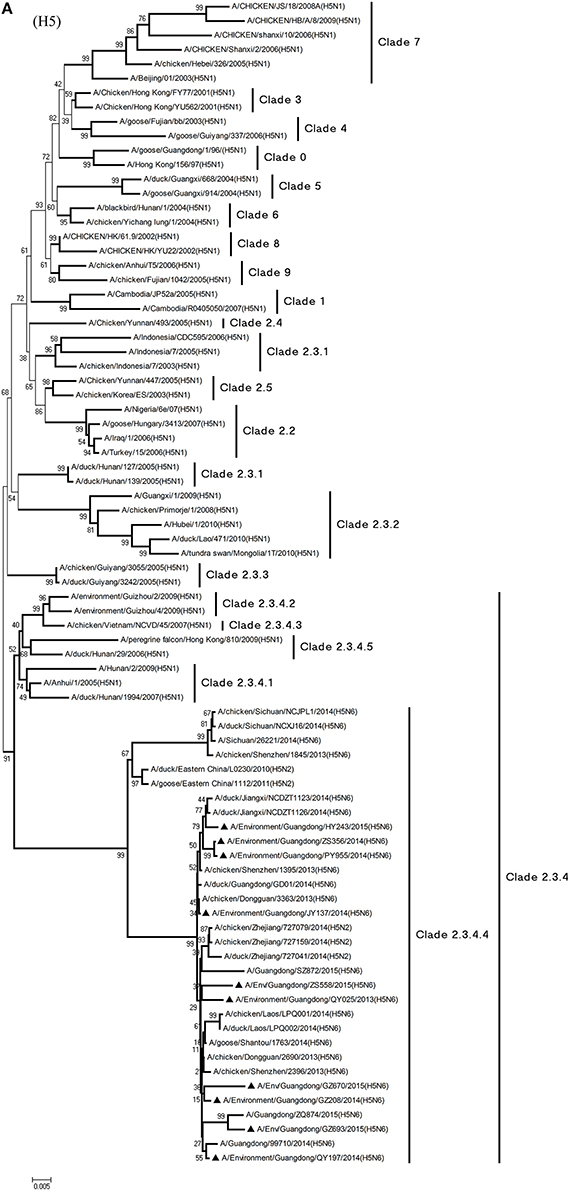
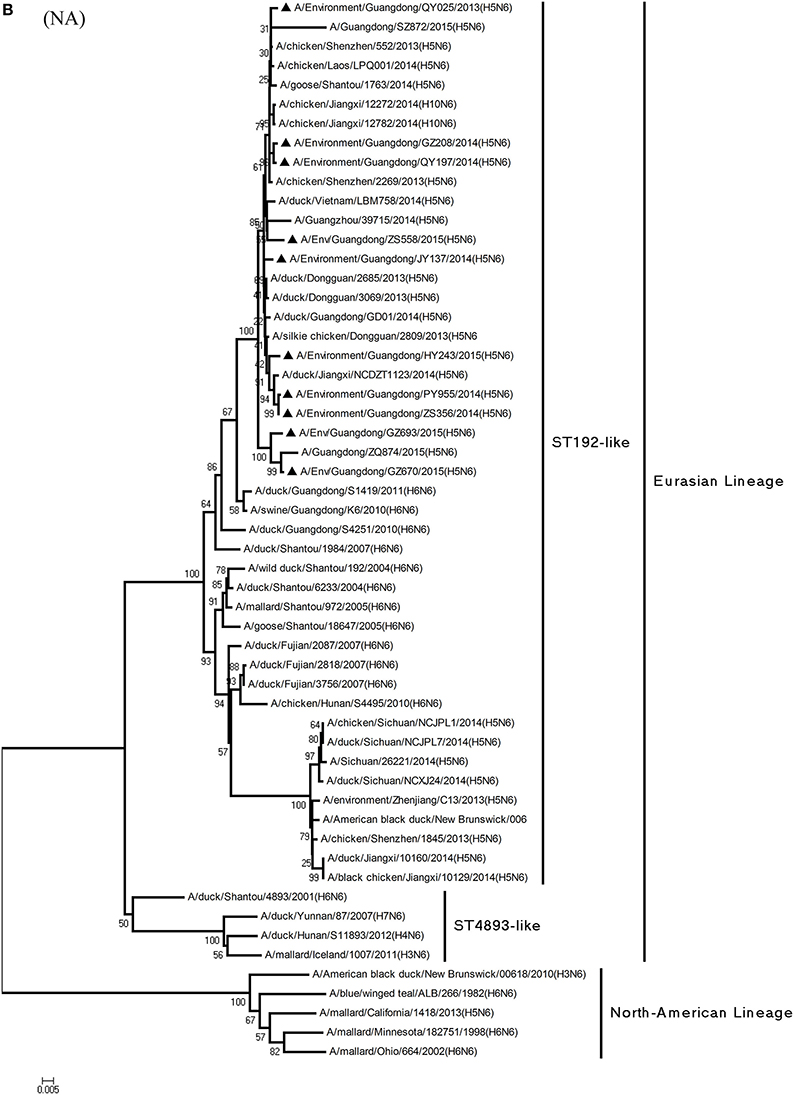
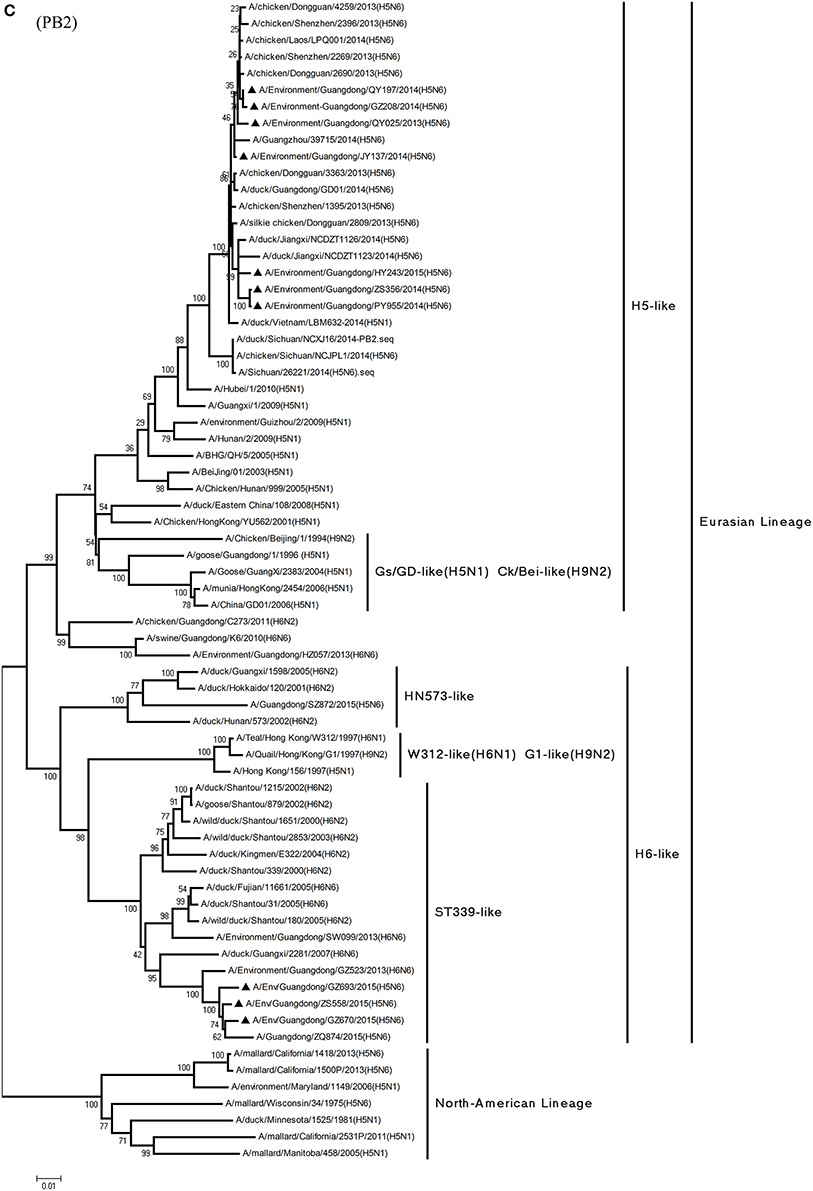
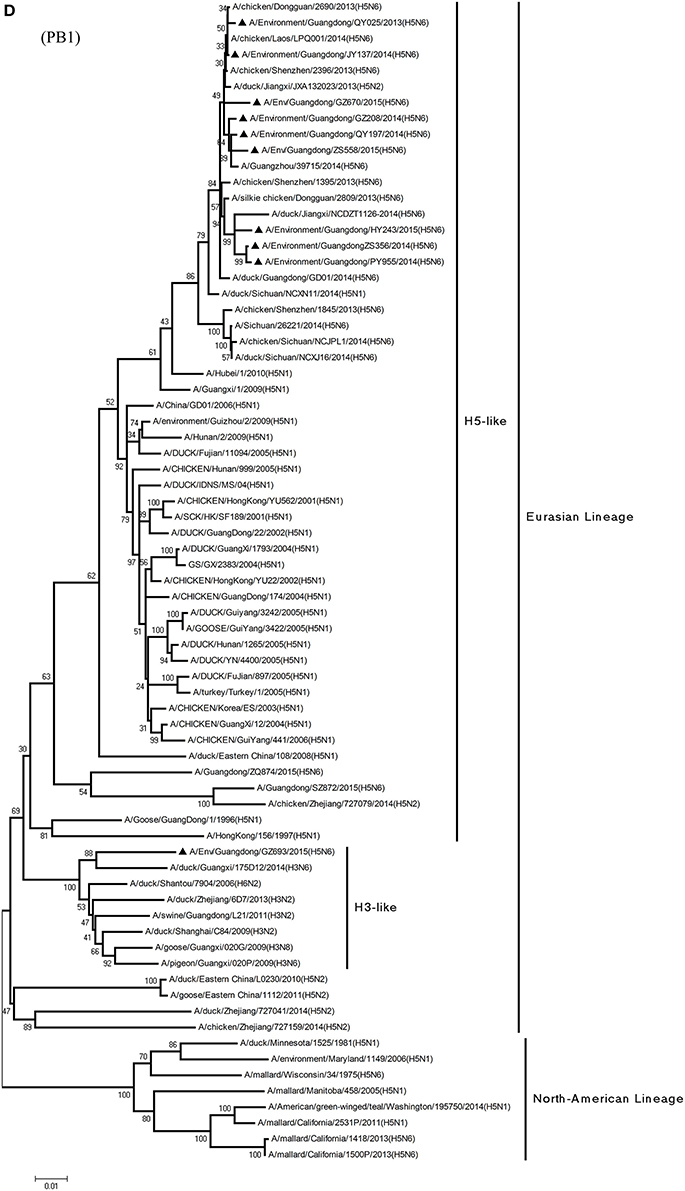
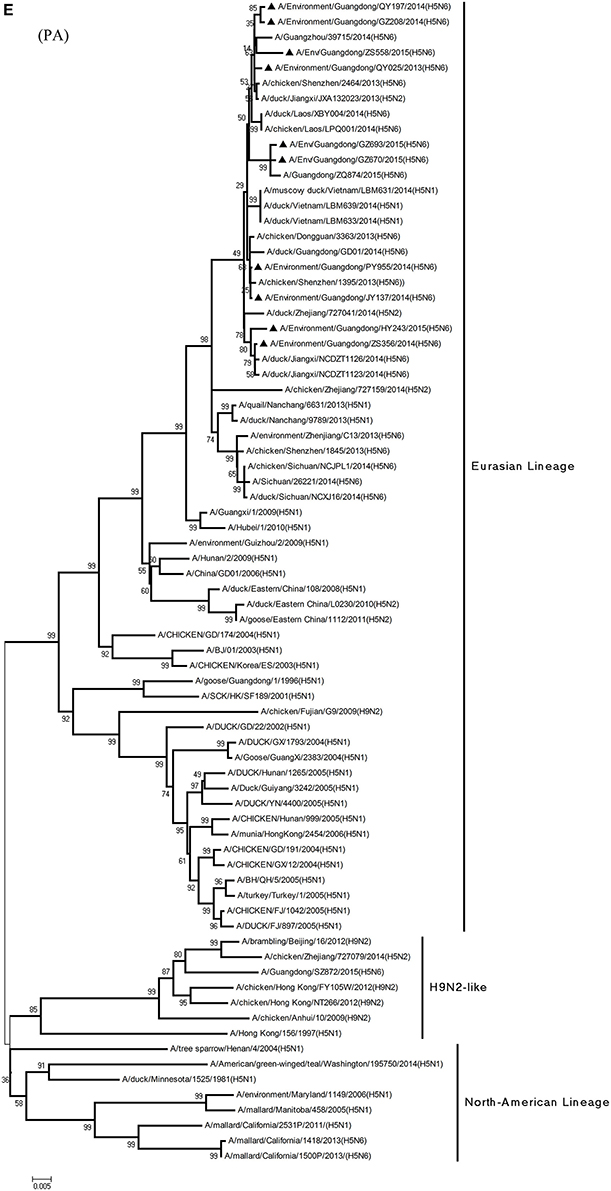
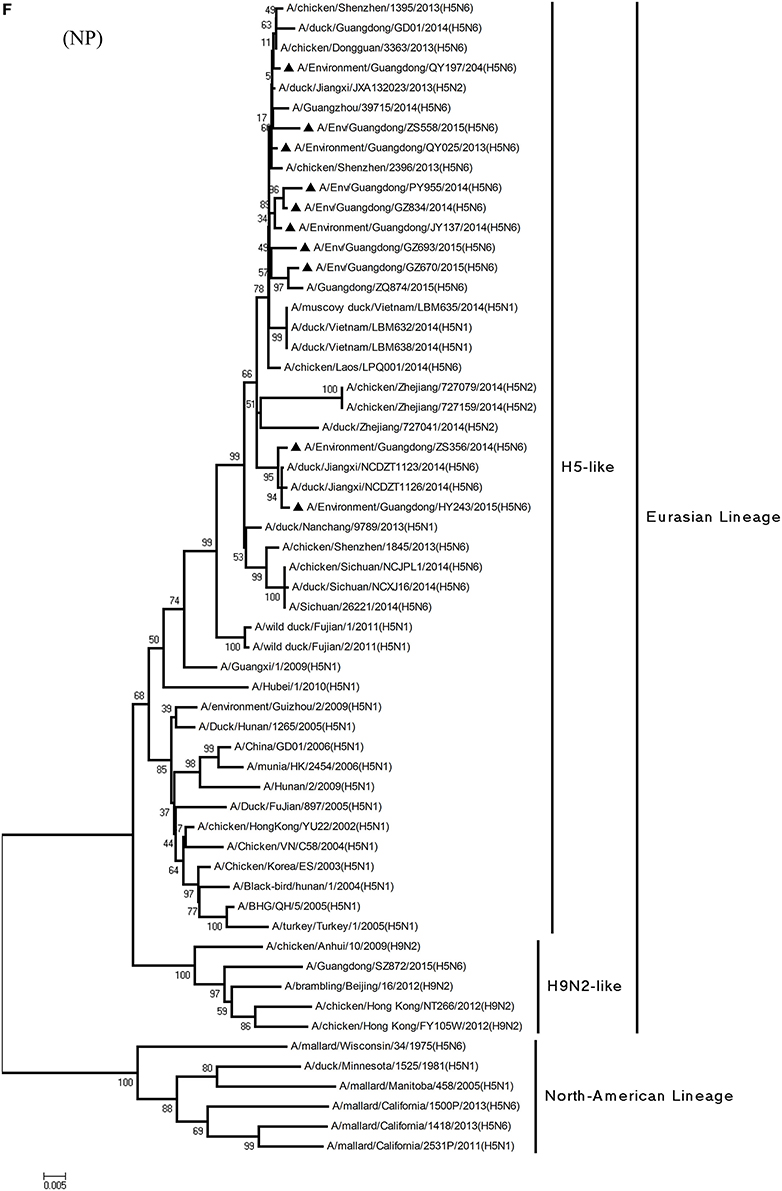
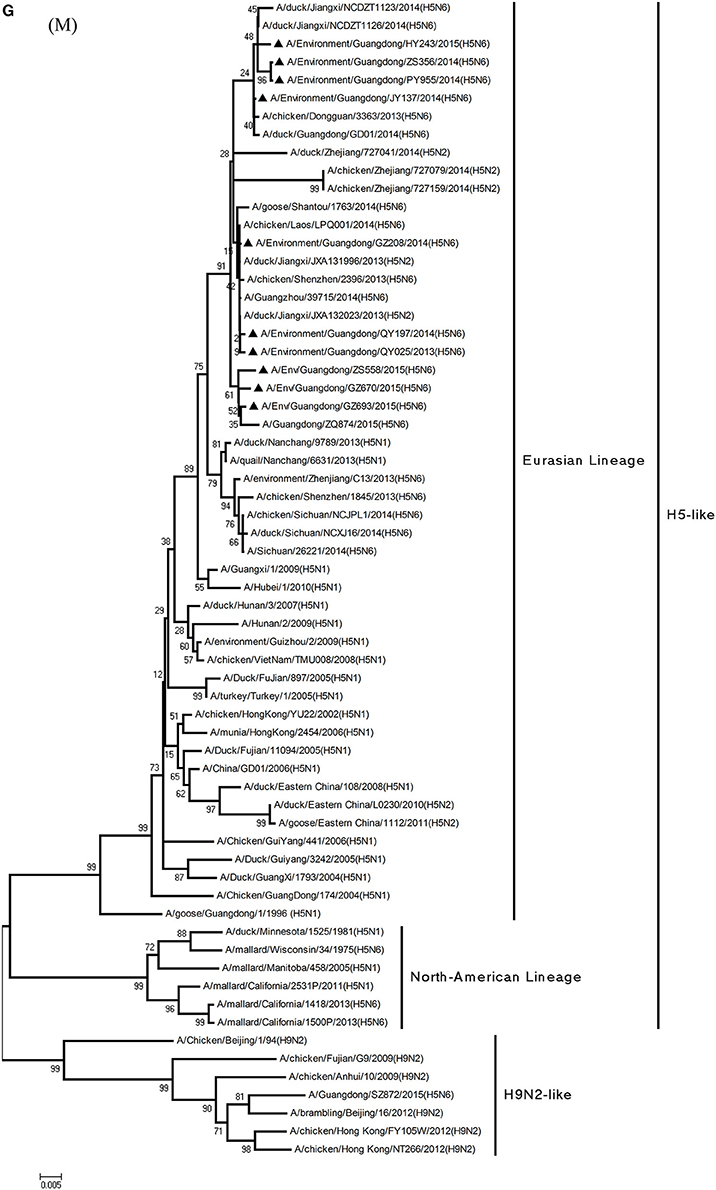
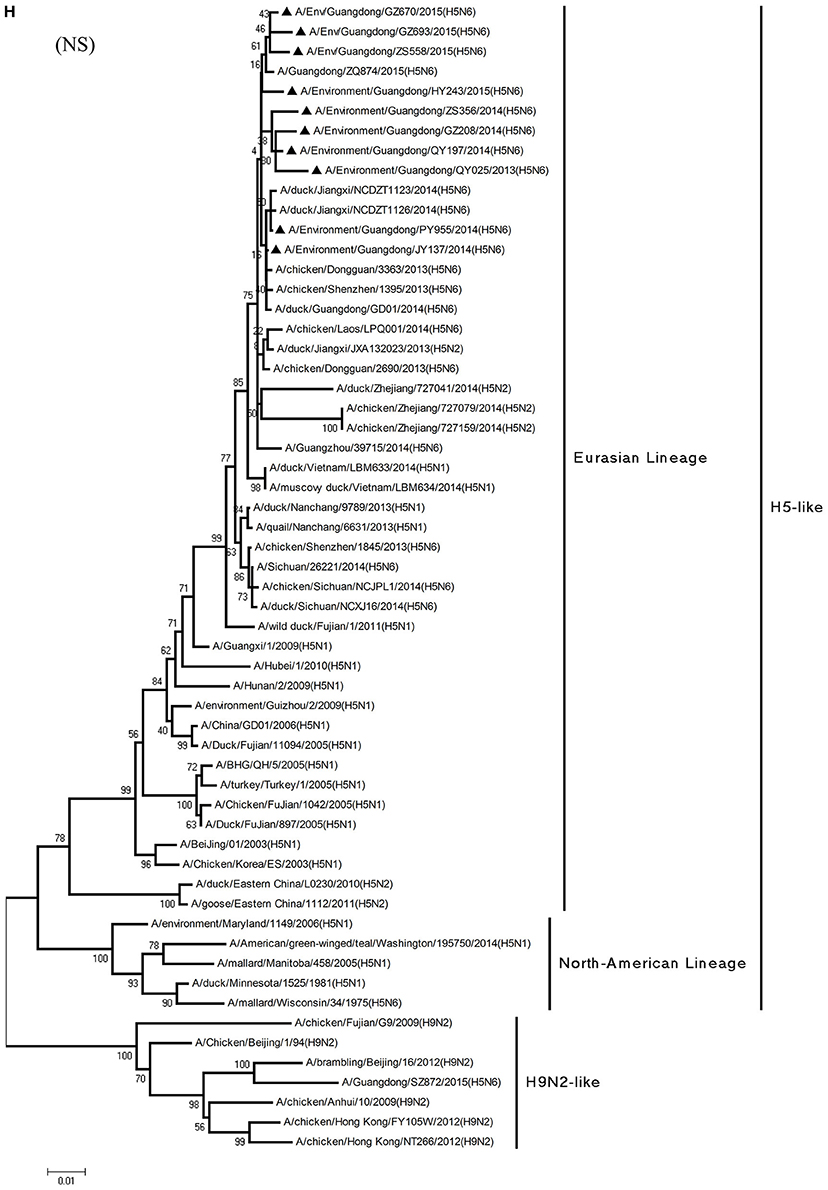
Figure 1. Phylogenetic analyses of the open reading frames of H5N6-subtype avian influenza viruses. Viruses highlighted with black triangles (▴) were characterized in the present study. The tree was constructed using the neighbor-joining of Molecular Evolutionary Genetics Analysis 6.06, with 1000 bootstrap trials to ensure confidence in the groupings.
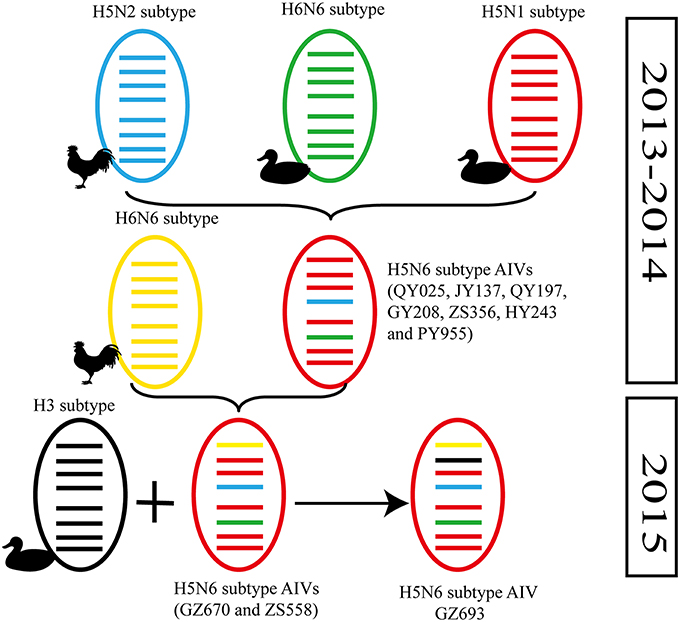
Figure 2. Possible genetic contributions of known donor AIVs to the novel H5N6 avian influenza virus in Southern China. From top to bottom, the eight genes in each schematic virus particle are the PB2, PB1, PA, HA, NP, NA, M, and NS genes. Genes of the same lineage appear in the same color.
Phylogenetic analysis of the N6-NA gene indicated that it likely originated in H6N6 AIVs found in domestic ducks in southern China (Huang et al., 2012). According to their geographical location, the HA genes of H6-subtype AIV can be identified as either of Eurasian or North American lineage. As Figure 1B shows, those of Eurasian lineage can be divided into two groups: Group 1 (ST192-like) and Group 2 (ST4893-like) (Huang et al., 2012). All 10 viruses were of Group 1 of Eurasian lineage, represented by A/wild duck/Shantou/192/2004 (H6N6) (Figures 1B, 2). QY025, QY197, and QY208 shared 97.2–98.8% nucleotide similarity with A/chicken/Shenzhen/552/2013(H5N6) (GD-H5N6), JY137, ZS356, HY243, and ZS558 shared 96.5–99.6% highest nucleotide similarity with A/chicken/Dongguan/2685/2013(H5N6) (GD-H5N6), PY955 shared 99.6% highest nucleotide similarity with JX-H5N6, and GZ670 and GZ693 shared 96.0–96.9% highest nucleotide similarity with ZQ874 isolated from a patient in Guangdong. In all, these results suggest that the surface genes of the 10 reassortant viruses derived from H5 and H6 AIV subtypes circulating in poultry in China.
Phylogenetic Analysis of Internal Genes
Phylogenetic analyses of internal genes showed that the PB2, PB1, PA, M, and NS genes of all 10 viruses were of Eurasian lineage (Figures 1C–H). H5N6 AIVs did not cluster with H5N1 AIVs, but formed an independent lineage (Figure 1).
In the PB2 gene, QY025, QY197, QY208, JY137, and ZS356 shared 99.1–99.9% highest nucleotide similarity with GD-H5N6, whereas ZS356, PY955 and HY243 shared more than 99.4% highest nucleotide similarity with JX-H5N6. GZ670, GZ693, and ZS558 originated from H6-subtype AIVs and were ST339-like viruses, represented by A/wild duck/Shantou/339/2000 (H6N2) (Figure 1C).
From the PB1 gene, PY955 and HY243 shared 99.0–99.1% highest nucleotide similarity with DG-H5N6, whereas the other seven viruses shared more than 98.8% highest nucleotide similarity with Vietnam-H5N6. Meanwhile, the PB1 gene of GZ693 shared highest nucleotide similarity with H3-subtype AIVs.
Regarding the PA gene, ZS356, and HY243 shared 99.7–99.3% highest nucleotide similarity with JX-H5N6, whereas the other eight viruses shared more than 99.3% highest nucleotide similarity with GD-H5N6. As for the NP gene, PY955, ZS356, and HY243 shared more than 99.7% highest nucleotide similarity with JX-H5N6, though the other seven viruses shared 99.4–99.8% highest nucleotide similarity with GD-H5N6, and concerning the M gene, all 10 viruses shared 99.6–100% highest nucleotide similarity with JX-H5N6. Lastly, regarding the NS gene, PY955 and JY137 shared 97.6–99.9% highest nucleotide similarity with JX-H5N6, whereas the other eight viruses shared 98.7–99.4% highest nucleotide similarity with GD-H5N6.
In particular, the 10 environmental viruses shared more than 96.0% high nucleotide similarity with the H5N6 AIVs isolated from patients in Guangdong. Phylogenetic analysis demonstrated that the internal genes of seven AIVs isolated within the first 7 months of 2014 related more closely to H5N1 HPAIVs circulating in poultry in China. By contrast, GZ670, GZ693, and ZS558 isolated in 2015 diverged from previously sequenced H5N6 AIVs and related more closely to H6N2 AIVs in the PB2 gene (Figure 2).
Molecular Characterization
The HA gene of all 10 H5N6 AIVs showed the HPAIV amino acid sequence RERRRKR↓G at the cleavage site of HA1 and HA2. Amino acid residues Q226 and G228, according to H3 numbering, occurred in the receptor-binding pocket of HA1, thus indicating that the viruses preferred to bind to the AIV receptor (Ha et al., 2001). Each of the 10 AIVs had six potential N-linked glycosylation sites at HA1 (26 or 27, 39, 181, 209, and 302) and two in HA2 (499 and 558). However, ZS558 revealed A254T mutation in an extra potential glycosylation site, whereas GZ693 exhibited six potential N-linked glycosylation sites in HA1 (i.e., at positions 27, 39, 180, 208, 230, and 301) and two in HA2 (i.e., at positions 498 and 557).
The NA proteins of JY137 and PY955 exhibited 12 amino acid deletion residues (i.e., at positions 59–70) in the neck, which could boost its virulence in mammals (Matsuoka et al., 2009). The key antiviral neuraminidase inhibitor drugs sites of the NA and M genes, such as position H275 of the NA gene (NA of GS/GD number) and position S31 of the M gene, showed no mutations (Scholtissek et al., 1998; Suzuki et al., 2003).
The PB2 gene of the 10 isolated viruses was E at position 627 and D at position 701, which indicates that all isolated viruses derived from avian sources (Li et al., 2005). At the same time, all environmental viruses were M at position 317 of the PB1 protein, which implies that they are hardly either pathogenic or non-pathogenic to mice (Katz et al., 2000). The AIVs could suppress a host's antiviral defenses relative to the antiviral effects of cytokines such as interferon. All viruses had P42S and D92E mutations in the NS1 protein, which suggests that they could enhance resistance to cytokines (Jiao et al., 2008; Qi et al., 2009).
Discussion
At present, H5N1 AIVs have become endemic in waterfowl and domestic poultry in China, Southeast Asia, North America, and Africa, where they have evolved into multiple phylogenetic lineages (WHO/OIE/FAO, 2012). The regular transmission of H5N1 HPAIVs among waterfowl and domestic poultry has facilitated genetic diversity among circulating clades in poultry in China (Duan et al., 2008; Vijaykrishna et al., 2008). In particular, the AIVs of clades 2.3.2, 2.3.4, and 7.2 have cocirculated predominantly in domestic poultry and waterfowl in China continuously since 2007 (Smith et al., 2009; Jiang et al., 2010; Li et al., 2010). At the same time, evolutionary clades such as 2.3.4.5 and 2.3.4.6—recently redefined as clade 2.3.4.4—have been reported (Gu et al., 2013). Moreover, H5-subtype AIVs from clade 2.3.4.4 appear to be gradually replacing AIVs from clade 2.3.4.2, especially in waterfowl. In March 2014, an emergent H5N6 AIV caused an outbreak in poultry in Laos (Wong et al., 2015), and later, a flock of ducks was infected with H5N6 AIVs in Guangdong Province (Shen et al., 2015). Genetic analysis suggested that subtype H5N6 AIVs originated from clade 2.3.2.1b and variant clade 2.3.4 in H5N1 AIVs (Shen et al., 2015; Wong et al., 2015). As phylogenic analysis shows, of the 10 H5N6 AIVs isolated as part of LPM surveillance during 2013–2015, all environmental samples belonged to novel clade 2.3.4.4 and probably evolved to form a new subcluster, unlike those of H5N6 s previously identified in Sichuan Province.
Alongside HA evolution, the NA gene of H5N1 AIV has frequently reassorted with other subtypes of AIVs circulating in poultry (Zhao et al., 2008; Neumann et al., 2010). The new reassortments, including H5N3, H5N6, and H5N8, together with H7N9 and H9N2, are currently cocirculating in domestic poultry and waterfowl worldwide. In our study, H5N6 AIVs were natural recombinants, the NA gene of which derived from H6N6 AIVs circulating broadly in ducks in southern China. Within the first 7 months of 2014, internal genes of H5N6 reassortants were derived from the genetic backbone of the H5N1 subtype (Wu et al., 2015). Interestingly, for H5N6 viruses isolated after 2015, we noted the divergence of three H5N6 reassortants—namely, GZ670, GZ693, and ZS558—isolated after 2015 (Figure 2). The PB2 genes of GZ670, GZ693, and ZS558 were not grouped into the same clusters as other reported H5N6 viruses, but within the same clusters as H6N2 AIVs. Furthermore, the PB1 gene of GZ693 was clustered as a H3-subtype AIV. These results indicate that H5N6 AIV is constantly evolving, and as such, novel AIVs possessing H5- and H6-derived internal genes and other AIVs possessing specific mammal-derived mutations could enhance virulence and transmissibility in humans.
After December 2014, the first H5N6 AIV infections in humans in Guangdong Province seemed to an appeared to stop. From December 2015 to January 2016, however, five H5N6 AIV infections in humans were reported in Guangdong Province (WHO, 2014b, 2016a,b,c). Consistent with the evolution of H5N6 AIVs isolated from LPMs, the sequences of H5N6 AIVs isolated from patients are constantly evolving. The whole gene sequences of the first human H5N6 AIV were similar to those of the H5N6 AIVs isolated in early 2015 in LPMs in Guangdong Province. Meanwhile, the whole gene sequences of the other four human H5N6 AIVs were consistent with those of H5N6 AIVs isolated from LPMs in late 2015. Molecular characterization and phylogenetic analysis exhibited a highly close genetic relationship between the viruses isolated from humans and LPMs, thereby suggesting that infection in humans might be caused by the LPM environment.
LPMs have been deemed potential hotbeds for infection with H5N1 and H7N9 AIVs in humans (Wan et al., 2011; Shi et al., 2013). Some human–human transmission of AIVs (e.g., H5N1 and H7N9) has been reported (Wang et al., 2008; Qi et al., 2013), and as of February 2016, nine confirmed human infections with subtype H5N6 had occurred in China's Sichuan, Guangdong, and Yunnan Provinces (CDC China, 2014; WHO, 2015b,c, 2016a,b,c). In particular, the patient infected with H5N6 AIV in Guangzhou had visited an LPM before the onset of illness and could have acquired the infection there (Yang et al., 2015). The other patient infected with H5N6 AIV and who died in Sichuan Province was a merchant at a local LPM. Moreover, the other seven cases of infection had visited LPMs in the past. Perhaps above all, we isolated 10 H5N6 AIVs in LPMs, which indicates that LPMs are potential sources of AIV infection in humans.
In conclusion, we analyzed the evolution of H5N6 samples isolated from LPM environments. Epidemiological and experimental data suggest that the H5N6 subtype currently has a limited capacity for chicken–human or environment–human transmission. LPMs can provide sufficient opportunities for close contact among waterfowl, domestic poultry, mammals, and humans, as well as potential AIV infection, which in turn results in the emergence of novel AIVs. Large-scale surveillance of LPMs therefore continues to be essential to identifying novel reassortants and sequence mutations among existing AIV subtypes.
Author Contributions
Conceived and designed the experiments: RY, CK. Performed the experiments: RY, ZW, JW, LL. Analyzed the data: RY. Contributed reagents/materials/analysis tools: RY, LZ, YS, HN, JL, XZ, CK. Wrote the paper: RY, YK.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the Research Project of H7N9 Influenza of Guangdong (2014; No. 1046), the Scientific and Technological Research of Prevention and Control of H7N9 Subtype Avian Influenza Virus (20140224), the Science and Technology Planning Project of Guangzhou City, China (Grant Number. 2014J4100091, 2013J4200020), Science and Technology Planning Project of Guangdong Province (Grant Number. 2013B020307006), and the Medical Scientific Research Foundation of Guangdong Province, China (Grant Number. A2012078).
References
CDC China (2014). Avian Influenza A(H5N6) Virus was Detected in a Respiratory Tract Sample from a Patient Who Died of Severe Pneumonia in Sichuan. Available online at: http://www.chinacdc.cn/mtdx/crbxx/201405/t20140507_96416.htm
Claas, E. C., Osterhaus, A. D., van Beek, R., De Jong, J. C., Rimmelzwaan, G. F., Senne, D. A., et al. (1998). Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet 351, 472–477. doi: 10.1016/S0140-6736(97)11212-0
Duan, L., Bahl, J., Smith, G. J., Wang, J., Vijaykrishna, D., Zhang, L. J., et al. (2008). The development and genetic diversity of H5N1 influenza virus in China, 1996-2006. Virology 380, 243–254. doi: 10.1016/j.virol.2008.07.038
Gu, M., Zhao, G., Zhao, K., Zhong, L., Huang, J., Wan, H., et al. (2013). Novel variants of clade 2.3.4 highly pathogenic avian influenza A(H5N1) viruses, China. Emerg. Infect. Dis. 19, 2021–2024. doi: 10.3201/eid1912.130340
Ha, Y., Stevens, D. J., Skehel, J. J., and Wiley, D. C. (2001). X-ray structures of H5 avian and H9 swine influenza virus hemagglutinins bound to avian and human receptor analogs. Proc. Natl. Acad. Sci. U.S.A. 98, 11181–11186. doi: 10.1073/pnas.201401198
Huang, K., Zhu, H., Fan, X., Wang, J., Cheung, C. L., Duan, L., et al. (2012). Establishment and lineage replacement of H6 influenza viruses in domestic ducks in southern China. J. Virol. 86, 6075–6083. doi: 10.1128/JVI.06389-11
Jiang, W. M., Liu, S., Chen, J., Hou, G. Y., Li, J. P., Cao, Y. F., et al. (2010). Molecular epidemiological surveys of H5 subtype highly pathogenic avian influenza viruses in poultry in China during 2007-2009. J. Gen. Virol. 91(Pt 10), 2491–2496. doi: 10.1099/vir.0.023168-0
Jiao, P., Tian, G., Li, Y., Deng, G., Jiang, Y., Liu, C., et al. (2008). A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J. Virol. 82, 1146–1154. doi: 10.1128/JVI.01698-07
Katz, J. M., Lu, X., Tumpey, T. M., Smith, C. B., Shaw, M. W., and Subbarao, K. (2000). Molecular correlates of influenza A H5N1 virus pathogenesis in mice. J. Virol. 74, 10807–10810. doi: 10.1128/JVI.74.22.10807-10810.2000
Li, Y., Shi, J., Zhong, G., Deng, G., Tian, G., Ge, J., et al. (2010). Continued evolution of H5N1 influenza viruses in wild birds, domestic poultry, and humans in China from 2004 to 2009. J. Virol. 84, 8389–8397. doi: 10.1128/JVI.00413-10
Li, Z., Chen, H., Jiao, P., Deng, G., Tian, G., Li, Y., et al. (2005). Molecular basis of replication of duck H5N1 influenza viruses in a mammalian mouse model. J. Virol. 79, 12058–12064. doi: 10.1128/JVI.79.18.12058-12064.2005
Matsuoka, Y., Swayne, D. E., Thomas, C., Rameix-Welti, M. A., Naffakh, N., Warnes, C., et al. (2009). Neuraminidase stalk length and additional glycosylation of the hemagglutinin influence the virulence of influenza H5N1 viruses for mice. J. Virol. 83, 4704–4708. doi: 10.1128/JVI.01987-08
Neumann, G., Green, M. A., and Macken, C. A. (2010). Evolution of highly pathogenic avian H5N1 influenza viruses and the emergence of dominant variants. J. Gen. Virol. 91(Pt 8), 1984–1995. doi: 10.1099/vir.0.020750-0
OIE (2015). Update on Highly Pathogenic Avian Influenza in Animals (Type H5 and H7). Available online at: http://www.oie.int/en/animal-health-in-the-world/update-on-avian-influenza/2015/
Qi, X., Li, X., Rider, P., Fan, W., Gu, H., Xu, L., et al. (2009). Molecular characterization of highly pathogenic H5N1 avian influenza A viruses isolated from raccoon dogs in China. PLoS ONE 4:e4682. doi: 10.1371/journal.pone.0004682
Qi, X., Qian, Y.-H., Bao, C.-J., Guo, X.-L., Cui, L.-B., Tang, F.-Y., et al. (2013). Probable person to person transmission of novel avian influenza A (H7N9) virus in Eastern China, 2013: epidemiological investigation. BMJ 347:f4752. doi: 10.1136/bmj.f4752
Scholtissek, C., Quack, G., Klenk, H. D., and Webster, R. G. (1998). How to overcome resistance of influenza A viruses against adamantane derivatives. Antiviral Res. 37, 83–95. doi: 10.1016/S0166-3542(97)00061-2
Shen, H., Wu, B., Chen, Y., Bi, Y., and Xie, Q. (2015). Influenza A(H5N6) virus reassortant, Southern China, 2014. Emerg. Infect. Dis. 21, 1261–1262. doi: 10.3201/eid2107.140838
Shi, J., Deng, G., Liu, P., Zhou, J., Guan, L., Li, W., et al. (2013). Isolation and characterization of H7N9 viruses from live poultry markets—implication of the source of current H7N9 infection in humans. Chin. Sci. Bull. 58, 1857–1863. doi: 10.1007/s11434-013-5873-4
Smith, G. J., Vijaykrishna, D., Ellis, T. M., Dyrting, K. C., Leung, Y. H., Bahl, J., et al. (2009). Characterization of avian influenza viruses A (H5N1) from wild birds, Hong Kong, 2004-2008. Emerg. Infect. Dis. 15, 402–407. doi: 10.3201/eid1503.081190
Subbarao, K., Klimov, A., Katz, J., Regnery, H., Lim, W., Hall, H., et al. (1998). Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science 279, 393–396. doi: 10.1126/science.279.5349.393
Suzuki, H., Saito, R., Masuda, H., Oshitani, H., Sato, M., and Sato, I. (2003). Emergence of amantadine-resistant influenza A viruses: epidemiological study. J. Infect. Chemother. 9, 195–200. doi: 10.1007/s10156-003-0262-6
Swayne, D. E., and Halvorson, D. A. (2003). “Influenza,” in Diseases of Poultry, eds Y. M. Saif, H. J. Barnes, J. R. Glisson, A. M. Fadly, L. R. McDougald, and D. E. Swayne (Ames, IA: Iowa State University Press), 135–160.
Vijaykrishna, D., Bahl, J., Riley, S., Duan, L., Zhang, J. X., Chen, H., et al. (2008). Evolutionary dynamics and emergence of panzootic H5N1 influenza viruses. PLoS Pathog. 4:e1000161. doi: 10.1371/journal.ppat.1000161
Wan, X. F., Dong, L., Lan, Y., Long, L. P., Xu, C., Zou, S., et al. (2011). Indications that live poultry markets are a major source of human H5N1 influenza virus infection in China. J. Virol. 85, 13432–13438. doi: 10.1128/JVI.05266-11
Wang, H., Feng, Z., Shu, Y., Yu, H., Zhou, L., Zu, R., et al. (2008). Probable limited person-to-person transmission of highly pathogenic avian influenza A (H5N1) virus in China. Lancet 371, 1427–1434. doi: 10.1016/S0140-6736(08)60493-6
WHO/OIE/FAO (2012). Continued evolution of highly pathogenic avian influenza A (H5N1): updated nomenclature. Influenza Other Respir. Viruses 6, 1–5. doi: 10.1111/j.1750-2659.2011.00298.x
WHO (2014a). Human Infection with Avian Influenza A(H5N6) Virus – China. Available online at: http://www.who.int/csr/don/28-december-2014-avian-influenza/en/
WHO (2014b). Human Infection with Avian Influenza A(H5N6) Virus – China. Available online at: http://www.who.int/csr/don/28-december-2014-avian-influenza/en/
WHO (2014c). Influenza at the Human-Animal Interface (HAI). Available online at: http://www.who.int/influenza/human_animal_interface/en/
WHO (2014d). WHO China Statement on H5N6. Available online at: http://www.wpro.who.int/china/mediacentre/releases/2014/20140507/en/
WHO (2015a). Cumulative Number of Confirmed Human Cases for Avian Influenza A(H5N1) Reported to WHO, 2003-2015. Available online at: http://www.who.int/influenza/human_animal_interface/EN_GIP_20150717cu mulativeNumberH5N1cases.pdf?ua=1
WHO (2015b). Human Infection with Avian Influenza A(H5N6) Virus – China. Available online at: http://www.who. int/csr/don/12-february-2015-avian-influenza/en/
WHO (2015c). Human Infection with Avian Influenza A(H5N6) Virus – China. Available online at: http://www.who.int/csr/don/14-july-2015-avian-influenza/en/
WHO (2016a). Human Infection with Avian Influenza A(H5N6) Virus – China. Available online at: http://www.who.int/csr/don/4-january-2016-avian-influenza-china/en/
WHO (2016b). Human Infection with Avian Influenza A(H5N6) Virus – China. Available online at: http://www.who.int/csr/don/11-january-2016-avian-influenza-china/en/
WHO (2016c). Human Infection with Avian Influenza A(H5N6) Virus – China. Available online at: http://www.who.int/csr/don/26-january-2016-avian-influenza-china/en/
Wong, F. Y., Phommachanh, P., Kalpravidh, W., Chanthavisouk, C., Gilbert, J., Bingham, J., et al. (2015). Reassortant highly pathogenic influenza A(H5N6) virus in Laos. Emerg. Infect. Dis. 21, 511–516. doi: 10.3201/eid2103.141488
Wu, H., Lu, R., Peng, X., Xu, L., Cheng, L., Lu, X., et al. (2015). Novel reassortant highly pathogenic H5N6 avian influenza viruses in poultry in China. Infect. Genet. Evol. 31, 64–67. doi: 10.1016/j.meegid.2015.01.019
Yang, Z. F., Mok, C. K., Peiris, J. S., and Zhong, N. S. (2015). Human infection with a novel avian Influenza A(H5N6) virus. N. Engl. J. Med. 373, 487–489. doi: 10.1056/NEJMc1502983
Yuan, R., Cui, J., Zhang, S., Cao, L., Liu, X., Kang, Y., et al. (2014). Pathogenicity and transmission of H5N1 avian influenza viruses in different birds. Vet. Microbiol. 168, 50–59. doi: 10.1016/j.vetmic.2013.10.013
Yuan, R., Zou, L., Kang, Y., Wu, J., Zeng, X., Lu, J., et al. (2016). Reassortment of avian influenza A/H6N6 viruses from live poultry markets in Guangdong, China. Front. Microbiol. 7:65. doi: 10.3389/fmicb.2016.00065
Keywords: reassortant, highly pathogenicity, avian influenza virus, H5N6, live poultry market
Citation: Yuan R, Wang Z, Kang Y, Wu J, Zou L, Liang L, Song Y, Zhang X, Ni H, Lin J and Ke C (2016) Continuing Reassortant of H5N6 Subtype Highly Pathogenic Avian Influenza Virus in Guangdong. Front. Microbiol. 7:520. doi: 10.3389/fmicb.2016.00520
Received: 04 February 2016; Accepted: 29 March 2016;
Published: 13 April 2016.
Edited by:
Akio Adachi, Tokushima University Graduate School, JapanReviewed by:
Julie McAuley, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, AustraliaRam P. Kamal, Battelle, USA
Karoline Bragstad, The Norwegian Institute of Public Health, Norway
Copyright © 2016 Yuan, Wang, Kang, Wu, Zou, Liang, Song, Zhang, Ni, Lin and Ke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changwen Ke, kecw1965@aliyun.com