Computational prediction of molecular pathogen-host interactions based on dual transcriptome data
 Sylvie Schulze
Sylvie Schulze Jana Schleicher
Jana Schleicher Reinhard Guthke
Reinhard Guthke Jörg Linde
Jörg Linde- Research Group Systems Biology and Bioinformatics, Leibniz-Institute for Natural Product Research and Infection Biology – Hans-Knöll-Institute, Jena, Germany
Organisms constantly interact with other species through physical contact which leads to changes on the molecular level, for example the transcriptome. These changes can be monitored for all genes, with the help of high-throughput experiments such as RNA-seq or microarrays. The adaptation of the gene expression to environmental changes within cells is mediated through complex gene regulatory networks. Often, our knowledge of these networks is incomplete. Network inference predicts gene regulatory interactions based on transcriptome data. An emerging application of high-throughput transcriptome studies are dual transcriptomics experiments. Here, the transcriptome of two or more interacting species is measured simultaneously. Based on a dual RNA-seq data set of murine dendritic cells infected with the fungal pathogen Candida albicans, the software tool NetGenerator was applied to predict an inter-species gene regulatory network. To promote further investigations of molecular inter-species interactions, we recently discussed dual RNA-seq experiments for host-pathogen interactions and extended the applied tool NetGenerator (Schulze et al., 2015). The updated version of NetGenerator makes use of measurement variances in the algorithmic procedure and accepts gene expression time series data with missing values. Additionally, we tested multiple modeling scenarios regarding the stimuli functions of the gene regulatory network. Here, we summarize the work by Schulze et al. (2015) and put it into a broader context. We review various studies making use of the dual transcriptomics approach to investigate the molecular basis of interacting species. Besides the application to host-pathogen interactions, dual transcriptomics data are also utilized to study mutualistic and commensalistic interactions. Furthermore, we give a short introduction into additional approaches for the prediction of gene regulatory networks and discuss their application to dual transcriptomics data. We conclude that the application of network inference on dual-transcriptomics data is a promising approach to predict molecular inter-species interactions.
Introduction
Organisms constantly interact with their abiotic and biotic environment (Koshland, 2002). Generally, biotic interactions are characterized by their effects on the fitness of an organism (Figure 1A): The result of an interaction can be a fitness gain (+), a fitness loss (−), or has no effect on fitness (0). Based on the interaction outcome for the organisms fitness, researchers can classify biotic interactions into competition (−∕−), predator-prey interaction (+∕−), parasite/pathogen-host interaction (+∕−), mutualism (+∕+; including symbiosis), commensalism (+∕0), and amensalism (−∕0) (Begon et al., 2006).
FIGURE 1
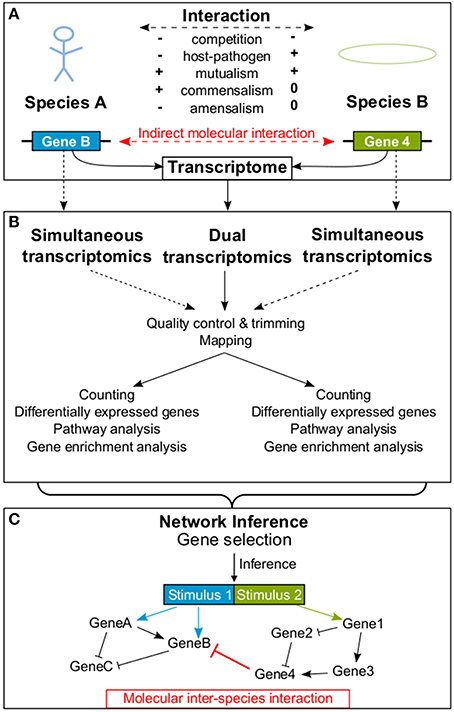
Figure 1. An example is provided how a molecular interaction between two species is detected. Panel (A) provides a schematic overview of possible interactions between organisms of two different species influencing each other's transcriptome. In (B) the processing of dual RNA-seq data extracted simultaneously from both interacting species is shown. In contrast, dotted lines represent simultaneous transcriptomics, where both transcriptomes are analyzed separately. In (C) an exemplary GRN resulting from the inference process is illustrated, including an indirect molecular inter-species interaction between two genes (red bar). In addition, molecular intra-species interactions (black) within each of the two species are shown. Arrowheads indicate activation and bars indicate repression.
Most of these interactions do not only appear on the macroscopic (e.g., organism interaction) or mesoscopic level (e.g., cell-cell interaction), but also affect the molecular level of the cells through molecular mediators. Thus, interactions can be scaled down to changes in gene expression, i.e., to the transcriptome. Biotic interactions change the organisms' environment. These changes, but also the interaction partner themselves, are sensed by receptors on cell surfaces and transmitted into the cell by signaling cascades. Such signals finally (in)activate transcriptional regulators (Groisman and Mouslim, 2006). These regulators alter the expression of their target genes which may in turn regulate other target genes within complex Gene Regulatory Networks (GRNs) (Barabási and Oltvai, 2004; Emmert-Streib et al., 2014). These specific and complex networks determine the cells response to changes caused by a macroscopic interaction. Currently, our knowledge about these complex GRNs is limited, but it would be of great help to understand the molecular basis of biotic interactions taking place on the macroscopic level.
KEY CONCEPT 1. Gene regulatory networks:
A GRN consists of regulatory interactions between genes in order to adjust the mRNA expression levels to an applied stimulus. Commonly, they are visualized as nodes representing genes and edges representing regulatory interactions.
Transcriptomics offers a comprehensive way to study expression changes of all genes of an organism under different conditions (e.g., reviewed by Jenner and Young, 2005; Kammenga et al., 2007; Leroy and Raoult, 2010). Traditionally, microarrays have been applied for transcriptomics. Since the advent of next-generation-sequencing of cDNAs derived from RNA samples (RNA-seq), researchers are able to study transcriptomes with a higher sensitivity and unlimited detection ranges (Mardis, 2008; Wang et al., 2009). One advantage of RNA-seq over microarrays is that RNA-seq offers a species-independent platform which allows for investigations of non-model species.
KEY CONCEPT 2. RNA-seq:
RNA-seq is a next-generation-sequencing technology where gene expression levels are measured based on high-troughput sequencing of RNA molecules.
Given that an interaction between organisms of two different species affects each one's gene expression, it is promising to study the transcriptomes of both species simultaneously in order to understand the molecular basis of the observed interaction. Dual transcriptomics is characterized by simultaneous measurements of the transcriptome from two interacting species where the processing of samples occurs collectively and species specific expression is determined in silico (Westermann et al., 2012). Dual transcriptomics has been successfully applied using both microarrays and RNA-seq and contributed to new knowledge about important biological questions (see Section 2; Table 1). For example, dual transcriptomics data allows to determination of which genes or proteins indirectly or directly interact between two species. Of note, this approach is different to metagenomics, which identifies the entire genome representation of an (environmental) sample; and it is different to comparative transcriptomics, which compares the responses of different species to the same stimulus. Strictly speaking, dual transcriptomics is not the same as simultaneous transcriptomics, where the transcriptome of two interacting species is captured simultaneously but processing is carried out separately for each species/RNA sample (e.g., Oosthuizen et al., 2011; Vojvodic et al., 2015). In what follows, we differentiate between simultaneous and dual transcriptomics, but proposed approaches may work for both.
KEY CONCEPT 3. Dual transcriptomics:
Dual transcriptomics means to simultaneously measure the transcriptome of two interacting species and collectively preprocess samples. Species specific gene expression is determined in silico. Typically, RNA-seq or microarrays are utilized for this purpose.
TABLE 1
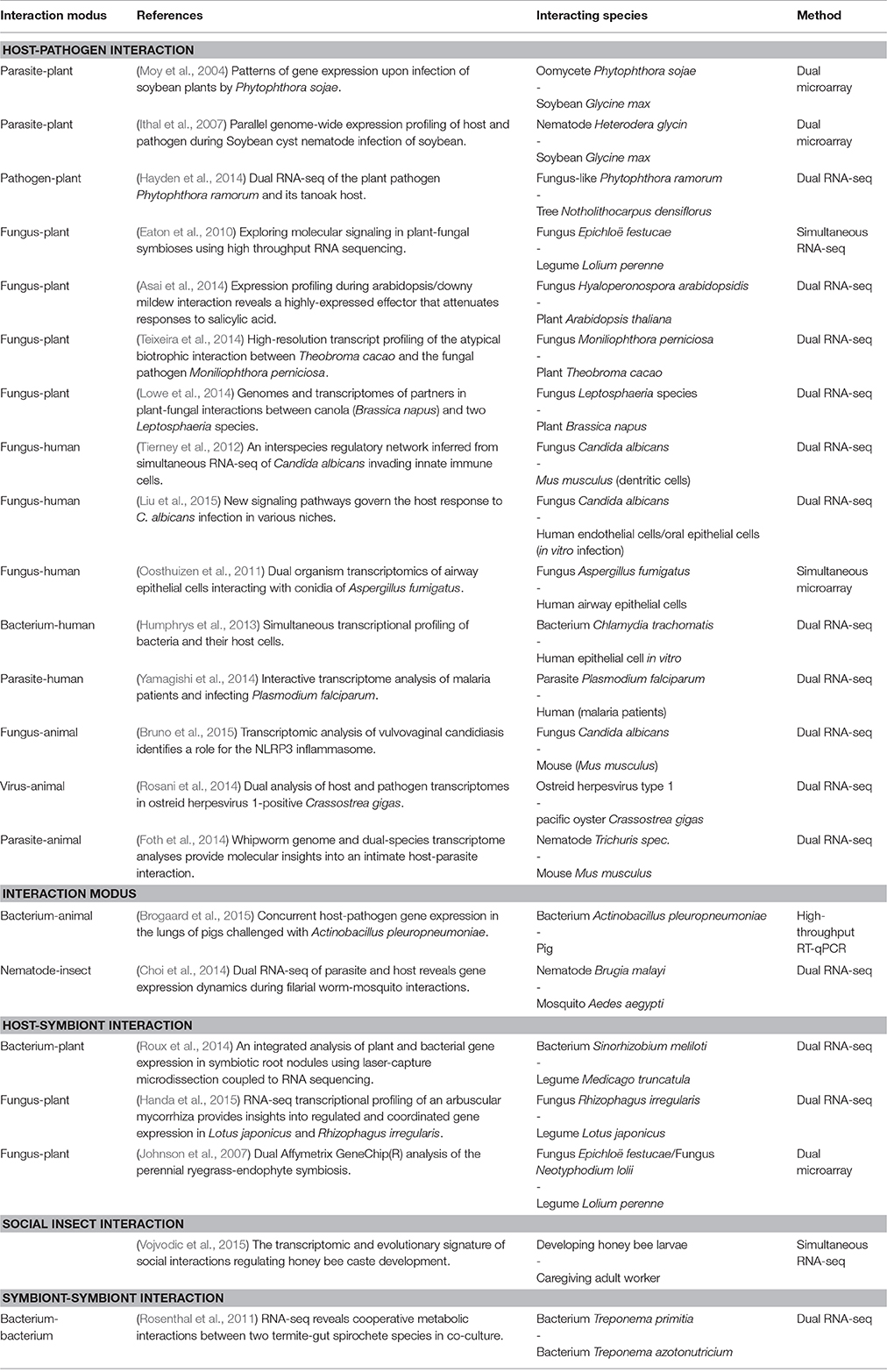
Table 1. Selection of published studies investigating molecular interactions between organisms by applying simultaneous and dual transcriptomics approaches.
Using dual transcriptomics, researchers have identified important genes, pathways and processes during the interaction of interacting species. However, so far only a few studies have elucidated more specifically which gene or pathway in organism A affects which gene or pathway in organism B (Table 1). Moreover, if such information is given it is either based on previous knowledge or was found by small scale experiments. In most studies, underlying transcriptome data were not used to infer molecular interactions between species.
Network Inference (NI) is a Systems Biology approach that predicts molecular regulatory interactions between genes in GRNs from gene expression data. There are different methodological approaches which were successfully applied in a number of studies (reviewed in Hecker et al., 2009b; Emmert-Streib et al., 2014; Linde et al., 2015). Commonly, NI has been applied to predict a GRN in one species. In a pioneering study, we applied NI on dual RNA-seq data of the fungal pathogen Candida albicans interacting with dendritic cells of Mus musculus. We predicted two molecular host-pathogen interactions, which were experimentally validated with help of intracellular staining, cellular binding assays as well as knock-outs of fungal genes and knock-downs of mouse genes followed by rtPCRs (Tierney et al., 2012). To our best knowledge, this is currently the only study which applied NI on dual-transcriptomics data. Guided by this experience, we augmented our NI approach to deal with typical situations for NI based on dual transcriptomics data. In Schulze et al. (2015), we published an improved version of our NI tool NetGenerator (Guthke et al., 2005; Toepfer et al., 2007) —an algorithm to infer GRNs based on time series gene expression data that are simulated by Ordinary Differential Equations (ODEs) and includes prior knowledge (see Section 4). NetGenerator is now capable of dealing with missing data and has more options for handling variance of gene expression data. In addition, we tested the influence of integrating multiple stimuli.
KEY CONCEPT 4. Network inference:
Network inference is a modeling approach to predict gene regulatory networks based on gene expression data.
KEY CONCEPT 5. Dual RNA-seq:
The term dual RNA-seq describes the simultaneous sequencing of mixed RNA-pools originating from two (interacting) species. When a pool of sequenced reads is mapped to genomes, reads are separated and mapped to the genome of the species they originate from.
This Frontiers Focused Review puts the results of Schulze et al. (2015) into a broader context. In addition to dual transcriptome studies dealing with host-pathogen interactions, we review studies from other fields of biology, such as mutualistic interactions. Furthermore, we give an overview of other NI approaches and discuss their possible applications to dual transcriptomics. Finally, we outline the usability of novel extensions proposed in Schulze et al. (2015) and discuss current problems as well as future developments.
Dual Transcriptomics
The term dual RNA-seq describes the transcriptome sequencing of two or more interacting species based on one mixed RNA-pool. This approach of measuring the transcriptome of two different species in one run has multiple advantages over simultaneous transcriptomics. RNA can be extracted directly without separating interacting species. Therewith, measured gene expression data directly reflects changes due to macroscopic interactions and not side effects of separation. Also, sequencing costs are lower because only one library is constructed. In some experimental setups interacting species are separated, their transcriptomes are sequenced in two runs and data processing is carried out separately. We refer to this procedure as simultaneous transcriptomics.
Raw dual RNA-seq data are preprocessed as follows (Figure 1B): Low quality bases of reads are trimmed [e.g., Trimmomatic (Bolger et al., 2014), CutAdapt (Martin, 2011)] and samples are quality controlled [e.g., HTQC (Yang et al., 2013), FastQC (Andrews, 2010)]. When the quality report is checked, researchers need to keep species properties in mind. E.g., the per sequence GC content plot of the FastQC report could show two peaks, if the GC content of investigated species is very different. A crucial step in dual RNA-seq preprocessing is the alignment of reads to their corresponding genomes (“mapping”; reviewed in Engström et al., 2013; Shang et al., 2014). Reads can be mapped consecutively to both genomes, but the order will influence the results since some reads may map to both genomes (depending on their evolutionary distance, sequencing parameters and read length). Alternatively, reads can be mapped in parallel to concatenated genomes. The advantage is, that in case of possible alignments to both genomes, the read mapping tools find the best position. The drawback is, that in case of equally good alignments to both genomes the read will be discarded as multi-mapped read. Also, mapping parameters are crucial, e.g., if intron lengths of studied organisms are very different, a tool non-sensitive to this parameter should be applied. In advance to carrying out experiments, RNA ratios of studied organisms should be determined and reflected by the mapping rates. As a next step, expression values of features (e.g., genes or transcripts) are calculated [e.g., featureCounts (Liao et al., 2014), maxcounts (Finotello et al., 2014)] and testing for differential expression is carried out separately for each species (reviewed in Rapaport et al., 2013; Soneson and Delorenzi, 2013; Zhang et al., 2014).
Sequencing parameters, such as rRNA filtering (filter RNA species of interest) (Westermann et al., 2012), read length (number of sequenced bases of a RNA fragment), single- vs. paired-end libraries (sequence a fragment from one or two ends), sequencing depth (average number how often every base is sequenced), and number of replicates (biological or technical), need to be chosen carefully depending on the species of interest and project aims. For example, the extractable RNA amounts of each species need to be determined and optimized in advance to the sequencing experiment. Therewith, a minimal sequencing depth to achieve sufficient coverage for all species can be calculated. If the species of interest are closely related, researchers might sequence longer paired-end reads to better determine read-genome correspondence and to prevent a large proportion of multi-mapped reads. If researchers aim for a highly reproducible detection of Differentially Expressed Genes (DEGs), Liu et al. (2013) recommended to generate more biological replicates rather than a higher sequencing depth. Nevertheless, if the aim is to detect DEGs with low expression, a high sequencing depth is necessary. Generally, we recommend to generate at least three replicates with a minimum coverage of ~10 fold to detect DEGs.
In dual or simultaneous transcriptome studies based on microarrays, RNA is extracted separately from each species and hybridized to species-specific microarrays. Therefore, preprocessing is not different from standard microarray data preprocessing. Even though, microarray data preprocessing slightly differs for different technological platforms (Gautier et al., 2004; Du et al., 2008), the main steps are (Irizarry et al., 2003): (i) image processing including background substraction, (ii) within array normalization to correct for spatial effects or cross-hybridization on each array, (iii) between array normalization to ensure that expression values have the same empirical distribution across different arrays/slides and (iv) testing for DEGs between conditions. Here, empirical t-statistics combined with a multiple test correction and a fold-change cutoff have emerged as a standard (Smyth, 2005; SEQC/MAQC-III Consortium, 2014).
Dual Transcriptomics Applications
Over the last years, the use of simultaneous transcriptomics to elucidate the molecular interactions between organisms of the same or of different species has gained increasing importance. One major application of simultaneous transcriptomics is the research area of infectious diseases. Different approaches dealing with the simultaneous analysis of expression profiles from two different species, via microarrays or RNA-seq, were published (e.g., Motley et al., 2004; Tierney et al., 2012; Humphrys et al., 2013; Schulze et al., 2015). These methods provide the basis to reveal the complex interplay between invading pathogens and their host. In the following, we briefly review recent simultaneous transcriptomics studies focusing on host-pathogen interactions. In addition, we provide insights to other relevant fields of biological interactions in which simultaneous transcriptomics plays an increasing role (Table 1).
Host-pathogen interactions are relevant in plant ecology due to their consequences for agricultural ecosystems. Eaton et al. (2010) performed high throughput sequencing of both, a plant host and its pathogen to evaluate interactions in a grass-fungal system. The most important fungal genes responsible for the shift of the fungus from a symbiont to a pathogen were revealed. The study data indicates that the protein sakA is important for the switch from a symbiotic to a pathogenic interaction.
On the plant side, changes in the hormone balance and upregulation of the defense response (generally absent in a symbiotic association) were observed to take place during the change of interaction modes. Interestingly, the same grass-fungal system has been investigated in regard to the symbiotic interaction based on a specifically designed dual microarray, which contains probes of two species (Johnson et al., 2007). In both publications, the symbiotic interaction of the fungus Epichloë festucae and the host Lolium perenne is investigated which is characterized by the fungal biosynthesis of secondary metabolites that protect the plant from various biotic and abiotic stresses, while the plant provides nutrients to the fungus and a mechanism of dissemination via seed transmission.
Moy et al. (2004) designed a dual microarray to simultaneously measure gene expression of soybean (Glycine max) and its pathogen Phytophthora sojae on a single array. The authors identified plant genes which are up- or downregulated within 24 h after infection. Analyzing these gene sets, they conclude that during the infection process the pathogen changes from biotrophy to necrotrophy. Similar work in the field of plant-pathogen interaction has been done by Ithal et al. (2007), who investigated the gene expression changes in soybean (G. max) and the soybean cyst nematode based on a dual microarray expression study during the course of infection. In addition, Teixeira et al. (2014) used dual RNA-seq to simultaneously assess the transcriptomes of cacao (Theobroma cacao) and the fungal pathogen Moniliophthora perniciosa, which causes Witches broom disease in its host. The authors found that the pathogen causes a change of the host metabolism to increase nutrition availability. Accordingly, they observed carbon deprivation on the host side and showed that the fungus causes massive metabolic reprogramming in infected shoots.
The fungus C. albicans attracts broad research interest due to its ability to switch from a commensal organism to a pathogen which can cause fatal invasive infections in humans (Cheng et al., 2012). To understand the mechanisms involved in the infection process, predominantly one-sided expression analyses focusing on either the host (e.g., Barker et al., 2005; Kim et al., 2005; Fradin et al., 2007) or the pathogen (e.g., Fradin et al., 2003; Fernández-Arenas et al., 2007; Bruno et al., 2010) were conducted in the past. In recent years, insights in the infection and defense mechanisms were gained by simultaneously measuring the hosts and the fungus gene expression profiles utilizing dual RNA-seq (Tierney et al., 2012; Bruno et al., 2015). In the latest study, Liu et al. (2015) identified several active pathways during C. albicans and host endothelial cell interaction based on dual RNA-seq data. Furthermore, they validated that two of these pathways regulate the uptake of C. albicans by host cells.
Dual transcriptomics was also applied to study host-virus and host-parasite interactions. Dual RNA-seq analysis of pacific oyster (Crassostrea gigas) infected by the ostreid herpesvirus type 1 allowed the exploration of the virus transcriptome and the host innate immune response during the process of infection (Rosani et al., 2014, see also Segarra et al., 2014). Furthermore, to understand the molecular mechanisms of parasitism, a dual RNA-seq approach was used to elucidate the transcriptome of malaria patients and the parasite Plasmodium falciparum based on blood samples (Yamagishi et al., 2014). The authors found characteristic expression changes of human innate immune response pathways involving TLR2 and TICAM2. Moreover, these expression changes correlated with the severity of the malaria infection.
Besides promoting our understanding of host-pathogen interactions, some studies used dual transcriptomics to investigate mutualism between species. Mycorrhiza is the symbiotic association of certain fungal species with plant roots (Brundrett, 2009). Handa et al. (2015) analyzed the gene expression profiles of the legume Lotus japonicus and the mycorrhizal fungus Rhizophagus irregularis during root mycorrhizal development. Some highly co-regulated transcripts encoding membrane traffic-related proteins, transporters and iron transport-related proteins were identified. An expression change of fungal cytochrome P450 was measured and the authors hypothesize, that this might contribute to metabolic pathways required to accommodate roots and soil.
Furthermore, an interesting study of mutualism has been conducted by Rosenthal et al. (2011), who investigated the interplay between two termite gut symbionts (spirochetes) using dual RNA-seq. The authors identified detailed cooperative interaction concerning metabolism during interaction of the symbionts. Another application field is the use of dual RNA-seq in plant-bacteria symbiotic interactions. For example, Roux et al. (2014) applied dual RNA-seq on the model legume Medicago truncatula and its symbiont Sinorhizobium meliloti. Since their expression study was coupled to laser microdissection of nodule regions, authors were able to analyze region sepcific gene expression. The authors found that bacterial transcription factors which control the root apical meristem are also expressed in the nodule meristem. In contrast, plant genes which are higher expressed in nodules than in roots are often associated with regions comprising both plant and bacterial partners.
Recently, simultaneous RNA-seq analysis was applied to elucidate the molecular interaction of social insects. The interplay between developing female honey bee larvae and adult nurse workers was analyzed on the molecular level before and after removal of the queen. By comparing these gene expression profiles, larval and nurse genes associated with caste development were identified (Vojvodic et al., 2015).
This short overview of interaction studies highlights the far-reaching applicability and practicability of dual RNA-seq analysis in the field of biological interactions. Consequently, further development and improvement of suitable methods and approaches, as the prediction of molecular interactions (Schulze et al., 2015), is essential to promote our understanding of the interplay between organisms on the molecular level. This is not only crucial in the research area of infectious diseases, but may also open up research on many symbiotic systems (Eaton et al., 2011) and other biological relevant interaction systems.
Network Inference
Network Inference approaches predict GRNs based on gene expression data. The structure of a GRN is thereby reconstructed from the gene expression data in response to an applied stimulus (reverse engineering).
The input for NI approaches is a gene expression matrix, which contains the expression values (or changes) of genes during treatment with different stimuli and/or over time. The number of included genes depends on the research question and on the applied NI approach. Importantly, the number of possible network structures increases exponentially with the number of included genes (curse of dimensionality). Several strategies are used to overcome the curse of dimensionality. Guided by the observation that biological networks have fewer interactions than expected in random networks, many NI approaches apply the sparseness criterion, i.e., predict the smallest number of interactions needed to fit the measured data. Another property of GRNs which is often applied as a network selection criterion is scale-freeness. This means that the number of interaction partners per gene is power law distributed, i.e., most genes interact with a very low number of genes while a few genes (hubs) have a high number of interaction partners (Barabási and Oltvai, 2004). So called prior knowledge are interactions extracted from literature or additional data sources, such as the occurrence of transcription factor binding sites in promoters. Integration of prior knowledge during NI strongly improves the accuracy of predicted interactions (Hecker et al., 2009a).
The result of a NI can be a correlation network, a Bayesian network or a mathematical model, depending on the underlying NI approach. NI approaches differ in the details of predicted interactions. They may predict either undirected interactions (A and B interact) or directed interactions (A regulates B). Directed interactions may additionally be signed (A induces B, A represses C). It is possible to get a steady or a dynamic network model depending on the NI approach. In dynamic network models, the state at a certain time point depends on its state at previous time points. Dynamic network models can be applied to predict future behavior of a system.
The assessment of NI approaches is difficult as they are often applied to very different research questions and success in one experimental setup does not guarantee success in another one. Since 2006, the “Dialog for Reverse Engineering Assessment and Methodsâ” (DREAM, www.dreamchallenges.org) has launched annual competitions for systems biology methods including NI (Stolovitzky et al., 2007).
In what follows, we give a brief and general description of the most important NI approaches and discuss their advantages and disadvantages. We guide the interested reader to excellent reviews for more comprehensive and detailed overviews of NI approaches (Hecker et al., 2009b; Wu and Chan, 2012; Emmert-Streib et al., 2014; Linde et al., 2015).
Approaches Based on Correlation and Information Theory
One of the most straightforward methods to predict a GRN is to compute the pairwise correlation between each pair of genes. An interaction is predicted if the correlation value is above a user-defined cut-off. This approach is computationally very fast and can be applied to a large number of genes. As the concept of correlation is well-known, results are easy to interpret. However, correlation does not mean causality. For example, consider a transcription factor inducing two target genes. The target genes have a high correlation value but do not interact.
To overcome this problem, approaches based on information theory compute the mutual information based on the pairwise correlation matrix. This term measures the statistical dependency between two random variables, which represent the expression intensities of two genes. Several mutual information based approaches are available (Butte and Kohane, 2000; Basso et al., 2005; Faith et al., 2007; Meyer et al., 2007; Altay and Emmert-Streib, 2010). Typically, these approaches do not integrate prior knowledge, nor do they enforce sparseness or scale-freeness. In general, they infer static undirected networks, but augmentations to generate directed networks exist (Madar et al., 2010).
Bayesian Networks
Another probabilistic approach is Bayesian NI. Here, the expression of each gene is considered to be a random variable which follows a probability distribution. Applying the Bayesian theorem, algorithms sample networks from a prior distribution and the network which best explains the measured data is selected. With help of the prior distribution of networks, prior knowledge can be elegantly integrated. Inferred GRNs are directed and can be static or dynamic (without direct feedback). Bayesian NI approaches (Murphy and Mian, 1999; Hartemink et al., 2001; Rau et al., 2010; Yeung et al., 2011) do not directly apply scale-freeness but this criterion might be included in the prior distribution. A major disadvantage is that accuracy of predicted interactions strongly depends on a relatively high amount of measured expression data.
Linear Regression Based NI
By applying linear regression, the expression of a gene at condition (time point) t is modeled as the weighted sum of the expression of all other genes. Additionally, some approaches include an external stimulus as part of the weighted sum. This represents the change in the environment. The values for the weights are determined by optimization algorithms in order to fit to the measured expression data. Non-zero weights define the network structure, where a positive weight represents an activation and a negative weight a repression. Thus, linear regression results in directed and signed steady networks. Many linear regression approaches apply the sparseness criterion. They predict the GRN which has a minimal (or small) number of interactions but is still able to fit the measured data. Moreover, they softly integrate prior knowledge (e.g., Gardner et al., 2003; Toepfer et al., 2007; Zou and Hastie, 2005; Hecker et al., 2009a; Gustafsson et al., 2005). More recent approaches also predict scale-free GRNs (Hecker et al., 2009a; Gustafsson and Hörnquist, 2010; Altwasser et al., 2012).
Differential Equation Based NI
Systems of ODEs are widely used in physics, chemistry, and biology to describe and model dynamic systems. Yet, ODEs provide an excellent way to mathematically infer GRNs. Here, the expression change of a gene at a time point is modeled as the weighted sum of the expression of all other genes and an external stimuli represents an environmental change. In contrast to steady linear regression models, differential equations model the change of a gene expression, not the gene expression itself.
Differential equation approaches reflect cause-effect relations which can be visualized by directed networks. The environmental change directly leads to fast reacting genes that regulate genes which respond later. Thus, GRNs modeled by ODEs are dynamic with directed and signed (weighted) edges. Similar to linear regression methods, weights are determined by optimization algorithms with the aim to fit measured data. In this NI category, many approaches apply the sparseness criterion and integrate prior knowledge by preferring known interactions during model structure optimization (e.g., Guthke et al., 2005; Greenfield et al., 2013; Zhang et al., 2013). While predicted GRNs are often highly accurate, a drawback is that the approaches are computationally demanding. Thus, they are often applied to a small number of genes which need to be carefully selected (see below).
One approach based on differential equations is NetGenerator (Guthke et al., 2005; Toepfer et al., 2007). The tool applies the sparseness criterion with help of a heuristic search strategy. Furthermore, it softly integrates prior knowledge and has been augmented to predict interactions which are robust against noise in expression data (Linde et al., 2010). NetGenerator was successfully applied to predict GRNs for immune diseases (Guthke et al., 2005), stress adaptation processes of pathogens (Linde et al., 2010, 2012) and rheumatoid arthritis (Kupfer et al., 2014). Since 2013, NetGenerator is able to predict GRNs based on more than one expression data set and more than one stimulus (Weber et al., 2013), which is for example useful for combined drug treatment.
Dual Network Inference
Interacting organisms of two or more species form a complex system. Various direct and indirect interactions take place and trigger multiple responses at different scales. In the following, as an example the complex interactions of the pathogenic fungus C. albicans and the human immune system are outlined in an abstract and simplified way (Figure 2). C. albicans cells can migrate into host tissues where they are exposed to different environmental conditions, such as a change in pH, available nutritions, presence and contact with immune cells. These stimuli are sensed by C. albicans, transmitted through the cell and finally the transcriptional program is changed. Similarly, immune and tissue cells sense the presence of C. albicans and change their transcriptional program. The transcriptional changes of both species can be measured by dual RNA-seq or microarrays. After the processing of transcribed mRNAs, the host adapts to infection. E.g., membrane bound and soluble receptors for pathogen recognition and cell signaling molecules for cell-cell communication are produced. Furthermore, host defense responses are initiated, such as the generation of reactive oxygen species. In turn, the pathogen again changes its transcriptional program to protect itself against host defense mechanisms. GRNs cannot comprehensively describe all molecular mechanisms of such a complex system of interacting species, but predict essential and also indirect interactions. Tierney et al. (2012) predicted inter-species interactions in a system of murine dendritic cells interacting with C. albicans. As outlined before (Figure 2), these predictions are highly indirect but were experimentally validated.
FIGURE 2
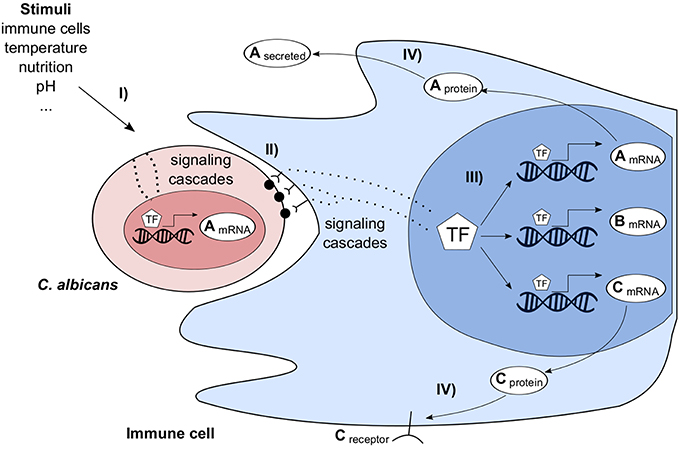
Figure 2. Simplified overview of Candida albicans (red) interacting with an immune cell (blue) and its environment. C. albicans is stimulated by environmental factors (I) leading to a change of its transcriptome. Immune cells recognize the pathogen, e.g., via pattern recognition receptors (II), transmit the signal through the cell and adapt their transcriptional program (III). In turn, this stimulates C. albicans, e.g., by producing cellsurface or extracellular proteins (IV).
Theoretically, all presented NI methods (see Section 4) can be applied to dual transcriptomics data. Some NI approaches additionally allow for prior knowledge as input. Given these inputs, NI approaches may work with dual or simultaneous gene expression data and finally predict a GRN including genes of both interacting species. Molecular interactions can be predicted between genes of one species (molecular intra-species interaction) or between genes of different species (molecular inter-species interaction; Figure 1C). NetGenerator (Guthke et al., 2005; Toepfer et al., 2007) is a tool that requires time series data of DEGs in form of a gene expression matrix, which can consist of genes from two interacting organisms. Relevant genes to be incorporated in the inference have to be selected. A maximum number of 20–30 genes is recommended depending on the number of samples. For example, genes can be selected based on their association to enriched Gene Ontology terms or with the help of expert knowledge.
KEY CONCEPT 6. Molecular intra-species interactions:
Molecular intra-species interactions are gene regulatory interactions predicted between two genes within one species. These predictions can be direct or indirect.
KEY CONCEPT 7. Molecular inter-species interactions:
Molecular inter-species interactions are predictions of a gene from species A interacting with a gene from species B. The predicted inter-species interactions are highly indirect.
To the authors' best knowledge the first practical inter-species application of NI was carried out with NetGenerator based on dual RNA-seq data of M. musculus dendritic cells infected with C. albicans (Tierney et al., 2012). Guided by this experience, we augmented NetGenerator for typical scenaria of dual transcriptomics data (Schulze et al., 2015) which we will introduce in the following.
A change in gene expression is triggered by one or more stimuli, which NetGenerator integrates through one or more time-dependent functions. Such a function is a user-defined input which represents the environmental change over time. If both species respond immediately, one identical stimulus function for both species might be sufficient. It is also possible, that one species responds faster than the other, which could be translated into two or more stimuli functions. NetGenerator was extended to incorporate multiple stimuli by Weber et al. (2013), while making use of multiple stimuli for dual NI was first discussed in Schulze et al. (2015).
NetGenerator was extended in Schulze et al. (2015) to deal with missing data at intermediate time points of time series. For example, this can occur when no gene expression values can be determined due to insufficient coverage or other technical problems. Furthermore, if transcriptome data from two species are combined retrospectively into a dual transcriptomics data set, time points can differ. Internally, NetGenerator handles this problem by interpolating missing data points. NetGenerator does not accept missing values for the first or last time point. In that case, the user has to provide these values, e.g., by setting them to zero or preceding/succeeding values.
Finally, NetGenerator was extended to consider gene expression variances. Biological variance in gene expression data exists for each experimental setup. However, the complex nature of biotic interactions in vivo leads to more variance than in in vitro experiments where under defined conditions only one environmental parameter is changed (e.g., heat shock). For each gene at each time point, a variance is calculated based on replicated measurements. The NI process of NetGenerator is sequential, i.e., it integrates one gene after another. For each gene, an objective function is minimized. In a simplified way, this means that the difference between measured data and simulated time course data should be as small as possible. The extended NetGenerator includes gene expression variances in the objective function. For gene expression values with a large variance, the difference of measured and simulated data is allowed to be larger than for gene expression values with smaller variance.
Issues and Perspectives
Dual transcriptomics paves the way to study the molecular basis of interaction. With the advent of RNA-seq it is now possible to study the transcriptome of non-model species. In this Frontiers Focused Review, we present an overview of dual and simultaneous transcriptomics studies which shows the wide range of possible applications from studying host-pathogen interactions via symbiotic fungal-host interactions to social interactions of insects. Among these studies, there is no example where molecular host-pathogen interactions were solved omics-based by both, dual and simultaneous transcriptomics. The majority of theses studies have identified genes and pathways involved in the interaction process. With dual NI, we present an approach which goes beyond identification of DEGs and uses gene expression data to predict molecular inter-species interactions (Figure 1C; Tierney et al., 2012; Schulze et al., 2015). Hypothetic gene regulatory interactions predicted by NI might be indirect. In fact, genes never directly interact. The most direct interaction, which can be predicted is between a transcription factor coding gene and the transcription factors target gene.
Indirect interactions may represent whole pathways or signaling cascades relevant in the interaction between two species. For a long time, indirect interactions have been regarded as a drawback of NI. In fact, the ability to predict indirect interactions is a big advantage, when NI approaches are applied to dual transcriptomics data. Predicted molecular inter-species interactions are by nature indirect but they are extremely interesting as they indicate which gene from species A influences which gene from species B (e.g., via one or more hidden molecular mediators, receptors, pathways or transcription factors). In future, dual network inference may also be applied to dual transcriptomics experiments where different organisms of the same species interact with each other (e.g., Vojvodic et al., 2015).
As models are always abstractions from a complex reality they do have a number of simplifications/assumptions. We have already discussed, that correlation of two genes does not mean a causal interaction. Other approaches based on regression or differential equations often assume linearity, which means the concentration of an activated gene is a linear function of its activator. However, biochemical kinetics often contain saturation dynamics (e.g., Michaelis Menten kinetics) or synergistic effects (e.g., Hill kinetics). Some tools also allow for non-linear modeling, e.g., NetGenerator (Guthke et al., 2005). This introduces additional parameters that need to be identified and therewith more prior knowledge or experimental data is needed. On the other hand, the assumption of linearity also holds true for wide ranges of biochemical kinetics.
A general assumption in (dual) transcriptomics is that the measured gene expression changes have an influence on the phenotype. However, proteins catalyze most biochemical reactions and shape the structure of a cell. Dual proteomics may pave the way to directly measure proteins. As the overlap between proteomics and transcriptomics differs depending on the experiment (Haider and Pal, 2013), NI approaches combining both -omics approaches are necessary.
Recently, first steps were done toward a systems biology of pathogen-host interactions by combining models of GRNs and signaling networks with models of other levels, in particular by modeling of metabolic and Protein-Protein Interaction (PPI) networks (Durmus et al., 2015; Schleicher et al., 2016). Sometimes, GRN modeling was also supported by prior knowledge retrieved from PPI databases (e.g., Altwasser et al., 2012).
Depending on the number of genes, NI approaches are divided into small scale and large scale NI. Large scale approaches often need a compendium of gene expression data combining different experiments. Thus, predicted interactions are gobal (genome-wide) for the respective interacting species and not specific for a certain condition/treatment. These approaches are useful to identify central genes (hubs) in regulatory networks. Small scale approaches typically focus on certain experimental conditions and are thus useful for dual NI. Even though, statistical (p-values for differential expression) and biological (e.g., member of an interesting biological process) methods for gene selection exist, this process is often subjective. Novel NI approaches need to combine advantages of large scale and small scale methods.
When we study the transcriptome of microorganisms, we need to be aware that we measure a mixture of hundreds or billions of cells, that might even be of different cell types. Expression values are a kind of “average” over the expression of all these different cells which assumes one big identical cell population. In fact, there might be sub-populations during the experiment. Moreover, individual cells may follow a very specific strategy during a biotic interaction. Single-cell RNA-seq allows to measure the transcriptome of each individual (Shapiro et al., 2013; Battle et al., 2014) and will change our understanding of molecular inter-species interactions. While single-cell RNA-seq is already possible for higher organisms, methods are being adapted for small RNA amounts of microorganisms. Single-cell RNA-seq of interacting species will help to identify the molecular basis of two interacting cells. Methods for NI based on such data need to take into account variability between different cells of the same species. In comparison to networks from averaged data, networks of a single cell may help to identify genes and interactions which vary between cells and are connected to a specific phenotype. Such an approach is applicable for individualized medicine (Lu et al., 2014).
This Frontiers Focused Review is mainly dedicated to dual RNA-seq data and modeling of two interacting species such as pathogen-host interaction. RNA-seq opens the door for the investigation of multi-species interaction, in particular the interactions between microorganisms and viruses in oral and gut microbiomes (Bikel et al., 2015). Whereas, metagenomics is focused on the relative abundance of the different species (genomes), the emerging RNA-seq-based metatranscriptomics will provide gene expression data of a biome and, thus, the empirical basis for molecular modeling of multi-species population networks. Examples are metatranscriptomics of the human gut (Franzosa et al., 2014) and its application to the current research on inflammatory bowel disease (Valles-Colomer et al., 2016) and the mixed culture of three bacterial species (Giannoukos et al., 2012). In addition, there are examples of metatranscriptome studies in environmental research, such as to analyse the gene expression and dynamics in the environments of the Pacific (Stewart et al., 2011) or the Amazon River (Satinsky et al., 2014).
Author Contributions
Substantial contributions to the conception and design of the work: all. Drafting the work: SS, JS, RG, JL. Revising the work critically for important intellectual content: all. Final approval of the version to be published: all. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: all.
Funding
SS and JL are supported by the Deutsche Forschungsgemeinschaft (DFG) CRC/Transregio 124 Pathogenic fungi and their human host: Networks of interaction subproject B3 (SS) and subproject INF (JL).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Scott McNamara for revision of English language.
Author Biography

References
Altay, G., and Emmert-Streib, F. (2010). Inferring the conservative causal core of gene regulatory networks. BMC Syst. Biol. 4:132. doi: 10.1186/1752-0509-4-132
Altwasser, R., Linde, J., Buyko, E., Hahn, U., and Guthke, R. (2012). Genome-wide scale-free network inference for Candida albicans. Front. Microbiol. 3:51. doi: 10.3389/fmicb.2012.00051
Andrews, S. (2010). Fastqc: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Asai, S., Rallapalli, G., Piquerez, S. J. M., Caillaud, M.-C., Furzer, O. J., Ishaque, N., et al. (2014). Expression profiling during arabidopsis/downy mildew interaction reveals a highly-expressed effector that attenuates responses to salicylic acid. PLoS Pathog. 10:e1004443. doi: 10.1371/journal.ppat.1004443
Barabási, A.-L., and Oltvai, Z. N. (2004). Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 5, 101–113. doi: 10.1038/nrg1272
Barker, K. S., Liu, T., and Rogers, P. D. (2005). Coculture of THP-1 human mononuclear cells with Candida albicans results in pronounced changes in host gene expression. J. Infect. Dis. 192, 901–912. doi: 10.1086/432487
Basso, K., Margolin, A. A., Stolovitzky, G., Klein, U., Dalla-Favera, R., and Califano, A. (2005). Reverse engineering of regulatory networks in human B cells. Nat. Genet. 37, 382–390. doi: 10.1038/ng1532
Battle, A., Mostafavi, S., Zhu, X., Potash, J. B., Weissman, M. M., McCormick, C., et al. (2014). Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 24, 14–24. doi: 10.1101/gr.155192.113
Begon, M., Townsend, C. R., and Harper, J. L. (2006). Ecology: From individuals to Ecosystems, 4th Edn. Hoboken, NJ: Wiley-Blackwell.
Bikel, S., Valdez-Lara, A., Cornejo-Granados, F., Rico, K., Canizales-Quinteros, S., Soberón, X., et al. (2015). Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: towards a systems-level understanding of human microbiome. Comput. Struct. Biotechnol. J. 13, 390–401. doi: 10.1016/j.csbj.2015.06.001
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brogaard, L., Klitgaard, K., Heegaard, P. M. H., Hansen, M. S., Jensen, T. K., and Skovgaard, K. (2015). Concurrent host-pathogen gene expression in the lungs of pigs challenged with Actinobacillus pleuropneumoniae. BMC Genomics 16:417. doi: 10.1186/s12864-015-1557-6
Brundrett, M. C. (2009). Mycorrhizal associations and other means of nutrition of vascular plants: understanding the global diversity of host plants by resolving conflicting information and developing reliable means of diagnosis. Plant Soil 320, 37–77. doi: 10.1007/s11104-008-9877-9
Bruno, V. M., Shetty, A. C., Yano, J., Fidel, P. L., Noverr, M. C., and Peters, B. M. (2015). Transcriptomic analysis of vulvovaginal candidiasis identifies a role for the NLRP3 inflammasome. mBio 6:e00182-15. doi: 10.1128/mBio.00182-15
Bruno, V. M., Wang, Z., Marjani, S. L., Euskirchen, G. M., Martin, J., Sherlock, G., et al. (2010). Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res. 20, 1451–1458. doi: 10.1101/gr.109553.110
Butte, A. J., and Kohane, I. S. (2000). Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements. Pac. Symp. Biocomput. 5, 418–429.
Cheng, S. C., Joosten, L. A. B., Kullberg, B. J., and Netea, M. G. (2012). Interplay between Candida albicans and the mammalian innate host defense. Infect. Immun. 80, 1304–1313. doi: 10.1128/IAI.06146-11
Choi, Y.-J., Aliota, M. T., Mayhew, G. F., Erickson, S. M., and Christensen, B. M. (2014). Dual RNA-seq of parasite and host reveals gene expression dynamics during filarial worm-mosquito interactions. PLoS Negl. Trop. Dis. 8:e2905. doi: 10.1371/journal.pntd.0002905
Du, P., Kibbe, W. A., and Lin, S. M. (2008). lumi: a pipeline for processing illumina microarray. Bioinformatics 24, 1547–1548. doi: 10.1093/bioinformatics/btn224
Durmus, S., Çakır, T., Özgür, A., and Guthke, R. (2015). A review on computational systems biology of pathogen-host interactions. Front. Microbiol. 6:235. doi: 10.3389/fmicb.2015.00235
Eaton, C. J., Cox, M. P., Ambrose, B., Becker, M., Hesse, U., Schardl, C. L., et al. (2010). Disruption of signaling in a fungal-grass symbiosis leads to pathogenesis. Plant Physiol. 153, 1780–1794. doi: 10.1104/pp.110.158451
Eaton, C. J., Cox, M. P., and Scott, B. (2011). What triggers grass endophytes to switch from mutualism to pathogenism? Plant Sci. 180, 190–195. doi: 10.1016/j.plantsci.2010.10.002
Emmert-Streib, F., Dehmer, M., and Haibe-Kains, B. (2014). Gene regulatory networks and their applications: understanding biological and medical problems in terms of networks. Front. Cell Dev. Biol. 2:38. doi: 10.3389/fcell.2014.00038
Engström, P. G., Steijger, T., Sipos, B., Grant, G. R., Kahles, A., Alioto, T., et al. (2013). Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods 10, 1185–1191. doi: 10.1038/nmeth.2722
Faith, J. J., Hayete, B., Thaden, J. T., Mogno, I., Wierzbowski, J., Cottarel, G., et al. (2007). Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 5:e8. doi: 10.1371/journal.pbio.0050008
Fernández-Arenas, E., Cabezón, V., Bermejo, C., Arroyo, J., Nombela, C., Diez-Orejas, R., et al. (2007). Integrated proteomics and genomics strategies bring new insight into Candida albicans response upon macrophage interaction. Mol. Cell Proteomics 6, 460–478. doi: 10.1074/mcp.M600210-MCP200
Finotello, F., Lavezzo, E., Bianco, L., Barzon, L., Mazzon, P., Fontana, P., et al. (2014). Reducing bias in RNA sequencing data: a novel approach to compute counts. BMC Bioinformatics 15(Suppl. 1):S7. doi: 10.1186/1471-2105-15-S1-S7
Foth, B. J., Tsai, I. J., Reid, A. J., Bancroft, A. J., Nichol, S., Tracey, A., et al. (2014). Whipworm genome and dual-species transcriptome analyses provide molecular insights into an intimate host-parasite interaction. Nat. Genet. 46, 693–700. doi: 10.1038/ng.3010
Fradin, C., Kretschmar, M., Nichterlein, T., Gaillardin, C., D'Enfert, C., and Hube, B. (2003). Stage-specific gene expression of Candida albicans in human blood. Mol. Microbiol. 47, 1523–1543. doi: 10.1046/j.1365-2958.2003.03396.x
Fradin, C., Mavor, A. L., Weindl, G., Schaller, M., Hanke, K., Kaufmann, S. H. E., et al. (2007). The early transcriptional response of human granulocytes to infection with Candida albicans is not essential for killing but reflects cellular communications. Infect. Immun. 75, 1493–1501. doi: 10.1128/IAI.01651-06
Franzosa, E. A., Morgan, X. C., Segata, N., Waldron, L., Reyes, J., Earl, A. M., et al. (2014). Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. U.S.A. 111, E2329–E2338. doi: 10.1073/pnas.1319284111
Gardner, T. S., di Bernardo, D., Lorenz, D., and Collins, J. J. (2003). Inferring genetic networks and identifying compound mode of action via expression profiling. Science 301, 102–105. doi: 10.1126/science.1081900
Gautier, L., Cope, L., Bolstad, B. M., and Irizarry, R. A. (2004). affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315. doi: 10.1093/bioinformatics/btg405
Giannoukos, G., Ciulla, D. M., Huang, K., Haas, B. J., Izard, J., Levin, J. Z., et al. (2012). Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol. 13:r23. doi: 10.1186/gb-2012-13-3-r23
Greenfield, A., Hafemeister, C., and Bonneau, R. (2013). Robust data-driven incorporation of prior knowledge into the inference of dynamic regulatory networks. Bioinformatics 29, 1060–1067. doi: 10.1093/bioinformatics/btt099
Groisman, E. A., and Mouslim, C. (2006). Sensing by bacterial regulatory systems in host and non-host environments. Nat. Rev. Microbiol. 4, 705–709. doi: 10.1038/nrmicro1478
Gustafsson, M., and Hörnquist, M. (2010). Gene expression prediction by soft integration and the Elastic Net – best performance of the DREAM3 gene expression challenge. PLoS ONE 5:e9134. doi: 10.1371/journal.pone.0009134
Gustafsson, M., Hörnquist, M., and Lombardi, A. (2005). Constructing and analyzing a large-scale gene-to-gene regulatory network – Lasso-constrained inference and biological validation. IEEE/ACM Trans. Comput. Biol. Bioinform. 2, 254–261. doi: 10.1109/TCBB.2005.35
Guthke, R., Möller, U., Hoffmann, M., Thies, F., and Töpfer, S. (2005). Dynamic network reconstruction from gene expression data applied to immune response during bacterial infection. Bioinformatics 21, 1626–1634. doi: 10.1093/bioinformatics/bti226
Haider, S., and Pal, R. (2013). Integrated analysis of transcriptomic and proteomic data. Curr. Genomics 14, 91–110. doi: 10.2174/1389202911314020003
Handa, Y., Nishide, H., Takeda, N., Suzuki, Y., Kawaguchi, M., and Saito, K. (2015). RNA-Seq transcriptional profiling of an arbuscular mycorrhiza provides insights into regulated and coordinated gene expression in Lotus japonicus and Rhizophagus irregularis. Plant Cell Physiol. 56, 1490–1511. doi: 10.1093/pcp/pcv071
Hartemink, A. J., Gifford, D. K., Jaakkola, T. S., and Young, R. A. (2001). Using graphical models and genomic expression data to statistically validate models of genetic regulatory networks. Pac. Symp. Biocomput. 422–433.
Hayden, K. J., Garbelotto, M., Knaus, B. J., Cronn, R. C., Rai, H., and Wright, J. W. (2014). Dual RNA-Seq of the plant pathogen Phytophthora ramorum and its tanoak host. Tree Gen. Genomes 10, 489–502. doi: 10.1007/s11295-014-0698-0
Hecker, M., Goertsches, R., Engelmann, R., Thiesen, H.-J., and Guthke, R. (2009a). Integrative modeling of transcriptional regulation in response to antirheumatic therapy. BMC Bioinformatics 10:262. doi: 10.1186/1471-2105-10-262
Hecker, M., Lambeck, S., Toepfer, S., van Someren, E., and Guthke, R. (2009b). Gene regulatory network inference: data integration in dynamic models – a review. Biosystems 96, 86–103. doi: 10.1016/j.biosystems.2008.12.004
Humphrys, M. S., Creasy, T., Sun, Y., Shetty, A. C., Chibucos, M. C., Drabek, E. F., et al. (2013). Simultaneous transcriptional profiling of bacteria and their host cells. PLoS ONE 8:e80597. doi: 10.1371/journal.pone.0080597
Irizarry, R. A., Hobbs, B., Collin, F., Beazer-Barclay, Y. D., Antonellis, K. J., Scherf, U., et al. (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264. doi: 10.1093/biostatistics/4.2.249
Ithal, N., Recknor, J., Nettleton, D., Hearne, L., Maier, T., Baum, T. J., et al. (2007). Parallel genome-wide expression profiling of host and pathogen during soybean cyst nematode infection of soybean. Mol. Plant Microbe Interact. 20, 293–305. doi: 10.1094/MPMI-20-3-0293
Jenner, R. G., and Young, R. A. (2005). Insights into host responses against pathogens from transcriptional profiling. Nat. Rev. Microbiol. 3, 281–294. doi: 10.1038/nrmicro1126
Johnson, L. J., Voisey, R. D., Johnson, A. K., Park, Z. A., Ramakrishna, M., Cao, D., et al. (2007). “Dual Affymetrix GeneChip(R) analysis of the perennial ryegrass-endophyte symbiosis,” in NZ Grassland Association: Endophyte Symposium (Wairakei), 509–513.
Kammenga, J. E., Herman, M. A., Ouborg, N. J., Johnson, L., and Breitling, R. (2007). Microarray challenges in ecology. Trends Ecol. Evol. 22, 273–279. doi: 10.1016/j.tree.2007.01.013
Kim, H. S., Choi, E. H., Khan, J., Roilides, E., Francesconi, A., Kasai, M., et al. (2005). Expression of genes encoding innate host defense molecules in normal human monocytes in response to Candida albicans. Infect. Immun. 73, 3714–3724. doi: 10.1128/IAI.73.6.3714-3724.2005
Koshland, D. E. Jr. (2002). Special essay. The seven pillars of life. Science 295, 2215–2216. doi: 10.1126/science.1068489
Kupfer, P., Huber, R., Weber, M., Vlaic, S., Häupl, T., Koczan, D., et al. (2014). Novel application of multi-stimuli network inference to synovial fibroblasts of rheumatoid arthritis patients. BMC Med. Genomics 7:40. doi: 10.1186/1755-8794-7-40
Leroy, Q., and Raoult, D. (2010). Review of microarray studies for host-intracellular pathogen interactions. J. Microbiol. Methods 81, 81–95. doi: 10.1016/j.mimet.2010.02.008
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Linde, J., Hortschansky, P., Fazius, E., Brakhage, A. A., Guthke, R., and Haas, H. (2012). Regulatory interactions for iron homeostasis in Aspergillus fumigatus inferred by a systems biology approach. BMC Syst. Biol. 6:6. doi: 10.1186/1752-0509-6-6
Linde, J., Schulze, S., Henkel, S., and Guthke, R. (2015). Data- and knowledge-based modeling of gene regulatory networks: an update. EXCLI J. 14, 346–378. doi: 10.17179/excli2015-168
Linde, J., Wilson, D., Hube, B., and Guthke, R. (2010). Regulatory network modelling of iron acquisition by a fungal pathogen in contact with epithelial cells. BMC Syst. Biol. 4:148. doi: 10.1186/1752-0509-4-148
Liu, Y., Shetty, A. C., Schwartz, J. A., Bradford, L. L., Xu, W., Phan, Q. T., et al. (2015). New signaling pathways govern the host response to C. albicans infection in various niches. Genome Res. 25, 679–689. doi: 10.1101/gr.187427.114
Liu, Y., Zhou, J., and White, K. P. (2013). RNA-seq differential expression studies: more sequence or more replication? Bioinformatics 30, 301–304. doi: 10.1093/bioinformatics/btt688
Lowe, R. G. T., Cassin, A., Grandaubert, J., Clark, B. L., de Wouw, A. P. V., Rouxel, T., et al. (2014). Genomes and transcriptomes of partners in plant-fungal-interactions between canola (Brassica napus) and two Leptosphaeria species. PLoS ONE 9:e103098. doi: 10.1371/journal.pone.0103098
Lu, Y.-F., Goldstein, D. B., Angrist, M., and Cavalleri, G. (2014). Personalized medicine and human genetic diversity. Cold Spring Harb. Perspect. Med. 4:a008581. doi: 10.1101/cshperspect.a008581
Madar, A., Greenfield, A., Vanden-Eijnden, E., and Bonneau, R. (2010). DREAM3: network inference using dynamic context likelihood of relatedness and the Inferelator. PLoS ONE 5:e9803. doi: 10.1371/journal.pone.0009803
Mardis, E. R. (2008). Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 9, 387–402. doi: 10.1146/annurev.genom.9.081307.164359
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10. doi: 10.14806/ej.17.1.200
Meyer, P. E., Kontos, K., Lafitte, F., and Bontempi, G. (2007). Information-theoretic inference of large transcriptional regulatory networks. EURASIP J. Bioinform. Syst. Biol. 2007, 1–9. doi: 10.1155/2007/79879
Motley, S. T., Morrow, B. J., Liu, X., Dodge, I. L., Vitiello, A., Ward, C. K., et al. (2004). Simultaneous analysis of host and pathogen interactions during an in vivo infection reveals local induction of host acute phase response proteins, a novel bacterial stress response, and evidence of a host-imposed metal ion limited environment. Cell. Microbiol. 6, 849–865. doi: 10.1111/j.1462-5822.2004.00407.x
Moy, P., Qutob, D., Chapman, B. P., Atkinson, I., and Gijzen, M. (2004). Patterns of gene expression upon infection of soybean plants by Phytophthora sojae. Mol. Plant Microbe Interact. 17, 1051–1062. doi: 10.1094/MPMI.2004.17.10.1051
Murphy, K., and Mian, S. (1999). Modelling Gene Expression Data Using Dynamic Bayesian Networks. Technical Report, University of California at Berkeley.
Oosthuizen, J. L., Gomez, P., Ruan, J., Hackett, T. L., Moore, M. M., Knight, D. A., et al. (2011). Dual organism transcriptomics of airway epithelial cells interacting with conidia of Aspergillus fumigatus. PLoS ONE 6:e20527. doi: 10.1371/journal.pone.0020527
Rapaport, F., Khanin, R., Liang, Y., Pirun, M., Krek, A., Zumbo, P., et al. (2013). Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 14:R95. doi: 10.1186/gb-2013-14-9-r95
Rau, A., Jaffrézic, F., Foulley, J.-L., and Doerge, R. W. (2010). An empirical Bayesian method for estimating biological networks from temporal microarray data. Stat. Appl. Genet. Mol. Biol. 9, 1544–6115. doi: 10.2202/1544-6115.1513
Rosani, U., Varotto, L., Domeneghetti, S., Arcangeli, G., Pallavicini, A., and Venier, P. (2014). Dual analysis of host and pathogen transcriptomes in ostreid herpesvirus 1-positive Crassostrea gigas. Environ. Microbiol. 17, 4200–4212. doi: 10.1111/1462-2920.12706
Rosenthal, A. Z., Matson, E. G., Eldar, A., and Leadbetter, J. R. (2011). RNA-seq reveals cooperative metabolic interactions between two termite-gut spirochete species in co-culture. ISME J. 5, 1133–1142. doi: 10.1038/ismej.2011.3
Roux, B., Rodde, N., Jardinaud, M.-F., Timmers, T., Sauviac, L., Cottret, L., et al. (2014). An integrated analysis of plant and bacterial gene expression in symbiotic root nodules using laser-capture microdissection coupled to RNA sequencing. Plant J. 77, 817–837. doi: 10.1111/tpj.12442
Satinsky, B. M., Crump, B. C., Smith, C. B., Sharma, S., Zielinski, B. L., Doherty, M., et al. (2014). Microspatial gene expression patterns in the amazon river plume. Proc. Natl. Acad. Sci. U.S.A. 111, 11085–11090. doi: 10.1073/pnas.1402782111
Schleicher, J., Conrad, T., Gustafsson, M., Cedersund, G., Guthke, R., and Linde, J. (2016). Facing the challenges of multiscale modelling of bacterial and fungal pathogen-host interactions. Brief Funct. Genomics. doi: 10.1093/bfgp/elv064. [Epub ahead of print].
Schulze, S., Henkel, S. G., Driesch, D., Guthke, R., and Linde, J. (2015). Computational prediction of molecular pathogen-host interactions based on dual transcriptome data. Front. Microbiol. 6:65. doi: 10.3389/fmicb.2015.00065
Segarra, A., Mauduit, F., Faury, N., Trancart, S., Dégremont, L., Tourbiez, D., et al. (2014). Dual transcriptomics of virus-host interactions: comparing two pacific oyster families presenting contrasted susceptibility to ostreid herpesvirus 1. BMC Genomics 15:580. doi: 10.1186/1471-2164-15-580
SEQC/MAQC-III Consortium. (2014). A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat. Biotechnol. 32, 903–914. doi: 10.1038/nbt.2957
Shang, J., Zhu, F., Vongsangnak, W., Tang, Y., Zhang, W., and Shen, B. (2014). Evaluation and comparison of multiple aligners for next-generation sequencing data analysis. BioMed Res. Int. 2014:309650. doi: 10.1155/2014/309650
Shapiro, E., Biezuner, T., and Linnarsson, S. (2013). Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 14, 618–630. doi: 10.1038/nrg3542
Smyth, G. (2005). “limma: Linear models for microarray data,” in Bioinformatics and Computational Biology Solutions Using R and Bioconductor, Stat Biol Health, eds R. Gentleman, V. Carey, W. Huber, R. Irizarry, and S. Dudoit (New York, NY: Springer), 397–420.
Soneson, C., and Delorenzi, M. (2013). A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics 14:91. doi: 10.1186/1471-2105-14-91
Stewart, F. J., Ulloa, O., and DeLong, E. F. (2011). Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ. Microbiol. 14, 23–40. doi: 10.1111/j.1462-2920.2010.02400.x
Stolovitzky, G., Monroe, D., and Califano, A. (2007). Dialogue on reverse-engineering assessment and methods: the dream of high-throughput pathway inference. Ann. N.Y. Acad. Sci. 1115, 1–22. doi: 10.1196/annals.1407.021
Teixeira, P. J. P. L., de Toledo Thomazella, D. P., Reis, O., do Prado, P. F. V., do Rio, M. C. S., Fiorin, G. L., et al. (2014). High-resolution transcript profiling of the atypical biotrophic interaction between Theobroma cacao and the fungal pathogen Moniliophthora perniciosa. Plant Cell 26, 4245–4269. doi: 10.1105/tpc.114.130807
Tierney, L., Linde, J., Müller, S., Brunke, S., Molina, J. C., Hube, B., et al. (2012). An interspecies regulatory network inferred from simultaneous RNA-seq of Candida albicans invading innate immune cells. Front. Microbiol. 3:85. doi: 10.3389/fmicb.2012.00085
Toepfer, S., Guthke, R., Driesch, D., Woetzel, D., and Pfaff, M. (2007). “The NetGenerator algorithm: reconstruction of gene regulatory networks,” in Knowledge Discovery and Emergent Complexity in Bioinformatics, Lecture Notes in Bioinformatics, Vol. 4366 (Ghent; Berlin; Heidelberg: Springer Verlag), 119–130.
Valles-Colomer, M., Darzi, Y., Vieira-Silva, S., Falony, G., Joossens, M., and Raes, J. (2016). Meta-omics in ibd research: applications, challenges and guidelines. J. Crohns Colitis. doi: 10.1093/ecco-jcc/jjw024. [Epub ahead of print].
Vojvodic, S., Johnson, B. R., Harpur, B. A., Kent, C. F., Zayed, A., Anderson, K. E., et al. (2015). The transcriptomic and evolutionary signature of social interactions regulating honey bee caste development. Ecol. Evol. 5, 4795–4807. doi: 10.1002/ece3.1720
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Weber, M., Henkel, S. G., Vlaic, S., Guthke, R., van Zoelen, E. J., and Driesch, D. (2013). Inference of dynamical gene-regulatory networks based on time-resolved multi-stimuli multi-experiment data applying NetGenerator V2.0. BMC Syst. Biol. 7:1. doi: 10.1186/1752-0509-7-1
Westermann, A. J., Gorski, S. A., and Vogel, J. (2012). Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 10, 618–630. doi: 10.1038/nrmicro2852
Wu, M., and Chan, C. (2012). Learning transcriptional regulation on a genome scale: a theoretical analysis based on gene expression data. Brief Bioinform. 13, 150–161. doi: 10.1093/bib/bbr029
Yamagishi, J., Natori, A., Tolba, M., Mongan, A. E., Sugimoto, C., Katayama, T., et al. (2014). Interactive transcriptome analysis of malaria patients and infecting Plasmodium falciparum. Genome Res. 24, 1433–1444. doi: 10.1101/gr.158980.113
Yang, X., Liu, D., Liu, F., Wu, J., Zou, J., Xiao, X., et al. (2013). HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinformatics 14:33. doi: 10.1186/1471-2105-14-33
Yeung, K. Y., Dombek, K. M., Lo, K., Mittler, J. E., Zhu, J., Schadt, E. E., et al. (2011). Construction of regulatory networks using expression time-series data of a genotyped population. Proc. Natl. Acad. Sci. U.S.A 108, 19436–19441. doi: 10.1073/pnas.1116442108
Zhang, X., Liu, K., Liu, Z.-P., Duval, B., Richer, J.-M., Zhao, X.-M., et al. (2013). Narromi: a noise and redundancy reduction technique improves accuracy of gene regulatory network inference. Bioinformatics 29, 106–113. doi: 10.1093/bioinformatics/bts619
Zhang, Z. H., Jhaveri, D. J., Marshall, V. M., Bauer, D. C., Edson, J., Narayanan, R. K., et al. (2014). A comparative study of techniques for differential expression analysis on RNA-Seq data. PLoS ONE 9:e103207. doi: 10.1371/journal.pone.0103207
Keywords: dual transcriptomics, dual RNA-seq, gene regulatory network, molecular inter-species interaction, network inference, host-pathogen interaction
Citation: Schulze S, Schleicher J, Guthke R and Linde J (2016) How to Predict Molecular Interactions between Species? Front. Microbiol. 7:442. doi: 10.3389/fmicb.2016.00442
Received: 09 December 2015; Accepted: 18 March 2016;
Published: 31 March 2016.
Edited by:
Rustam Aminov, Technical University of Denmark, DenmarkReviewed by:
Vitaly V. Ganusov, University of Tennessee, USANichole A. Broderick, University of Connecticut, USA
Paolo Tieri, Consiglio Nazionale delle Ricerche, Italy
Copyright © 2016 Schulze, Schleicher, Guthke and Linde. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: am9lcmcubGluZGVAaGtpLWplbmEuZGU=