 Nargis Khan
Nargis Khan Aurobind Vidyarthi1
Aurobind Vidyarthi1 Javed N. Agrewala
Javed N. Agrewala- 1Council of Scientific and Industrial Research – Institute of Microbial Technology, Chandigarh, India
- 2Department of Cytology and Gynecologic Pathology, Postgraduate Institute of Medical Education and Research, Chandigarh, India
T cells play a cardinal role in imparting protection against Mycobacterium tuberculosis (Mtb). However, ample time is required before T-cells are able to evoke efficient effector responses in the lung, where the mycobacterium inflicts disease. This delay in T cells priming, which is termed as lag phase, provides sufficient time for Mtb to replicate and establish itself within the host. In contrast, innate immunity efficiently curb the growth of Mtb during initial phase of infection through several mechanisms. Pathogen recognition by innate cells rapidly triggers a cascade of events, such as apoptosis, autophagy, inflammasome formation and nitric oxide production to kill intracellular pathogens. Furthermore, bactericidal mechanisms such as autophagy and apoptosis, augment the antigen processing and presentation, thereby contributing substantially to the induction of adaptive immunity. This manuscript highlights the role of innate immune mechanisms in restricting the survival of Mtb during lag phase. Finally, this article provides new insight for designing immuno-therapies by targeting innate immune mechanisms to achieve optimum immune response to cure TB.
Introduction
Tuberculosis continues to affect public health worldwide. About one third of the global population is infected with Mtb but only 3–10% succumb to disease (Barry et al., 2009; Ottenhoff and Kaufmann, 2012). Therefore, greater than 90% of infected population remains asymptomatic, which determines the intricate balance between host immunity and Mtb. Understanding the immune response of these asymptomatic individuals can be highly informative and will provide potentially new pathways for the development of anti-TB drugs and vaccines.
Over the past several decades, research related to defense against Mtb was largely focused on the T cells because of their remarkable ability to generate Mtb specific immunity, followed by an enduring memory response to counter subsequent infections (Stenger and Modlin, 1999). Undoubtedly, T cells play a crucial role in protection against Mtb. However, recent information has chiseled the long belief that T cells are the sentinels in Mtb protection, in part due to the substantial lag period between infection and the establishment of specific T-cell responses (Shaler et al., 2012). Recruitment of DCs to the site of infection, followed by their Mtb acquisition and transportation toward draining lymph nodes to prime naïve T cells takes 9–11 days after the invasion of the pathogen. Hence, T cell activation occurs after considerable time of infection. This delay between the onset of infection and generation of specific effector T cells provides enough time for Mtb to establish an infection (Figure 1). Once established, Mtb ultimately hampers the antigen processing and priming of naïve T cells (Roberts and Robinson, 2014). Eventually, obstructs the generation and propagation of anti-Mtb T cell responses. However, despite the lag phase in T-cell responses, >90% of infected individuals are asymptomatic, raising the possibility of the involvement of other factors in controlling TB.
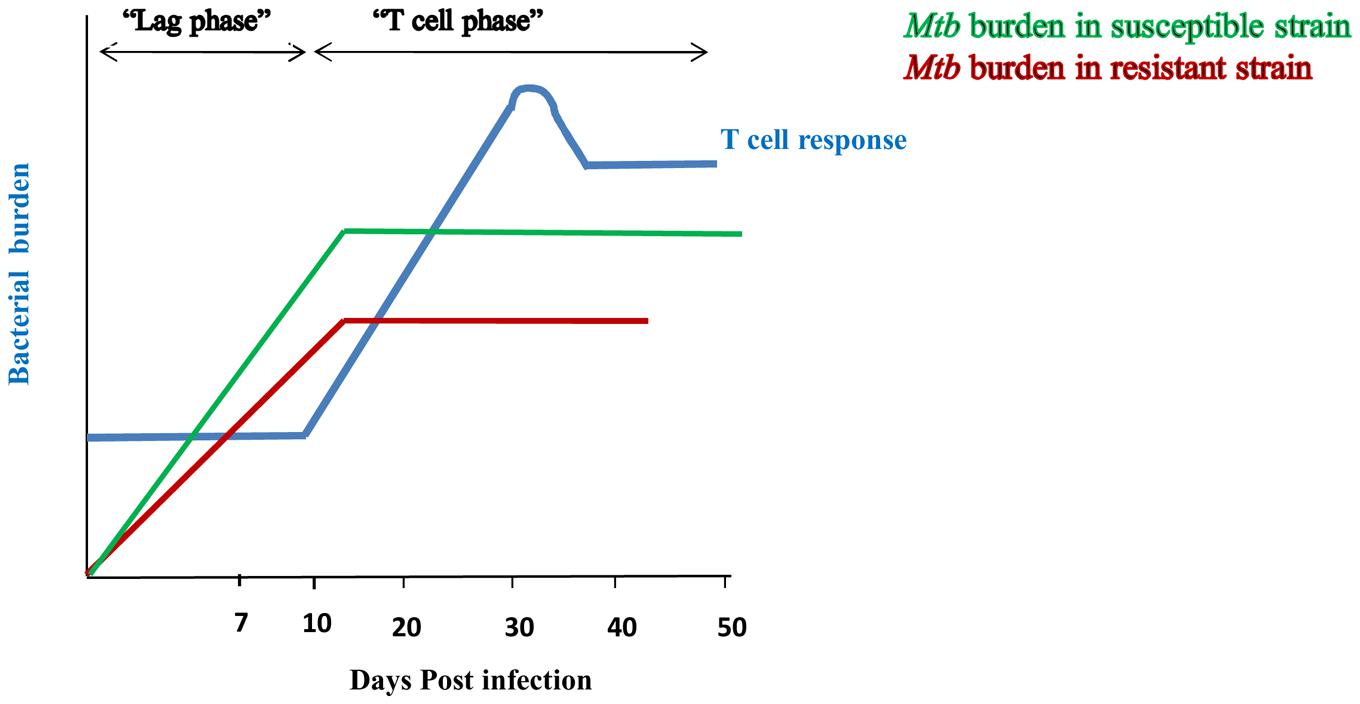
FIGURE 1. Innate immunity restricts the bacterial burden during lag phase of T cell response. Initiation of T cell response occurs after 9–11 days of Mtb infection and peaks at 20–25 days (-). Delay in the duration for the generation of effective T cell response is considered as its “lag phase”. Susceptible strain (-) of rabbit shows high bacterial burden during lag phase of T cell response; whereas resistant strain (-) signifies lesser bacterial burden. However, after initiation of T cell response, both the strains restrict Mtb growth. It indicates that lesser bacterial burden during lag phase of T cell response in resistant strain of rabbit is due to involvement of innate immunity.
Lurie’s fundamental studies with resistant and susceptible inbred rabbits proved that the innate response effectively controls the growth of Mtb at early times of infection. After 7 days of inhalation of tubercle bacilli, lungs of susceptible animals showed 20–30-fold more bacteria than resistant strain (Lurie, 1964; Arthur et al., 1994) (van Crevel et al., 2002) (Figure 1). Protection during the initial phase of infection clearly indicates that T cells are not at the forefront in controlling the infection, but, rather that components of the innate immune system play a pivotal role in generating efficient immunity against Mtb. Therefore, it is important to dissect the mechanisms responsible for curbing the growth of Mtb during the lag phase of T cell response. Understanding these mechanisms could pave ways in designing novel therapeutic strategies and vaccines to enhance the immune response more efficiently against Mtb.
Why Focus More on Innate Immunity?
In the past, the role of innate immunity was ignored in inducing a protective response against Mtb. Recent reports show that the function of innate immunity is even more effective than T cell response against Mtb (Fremond et al., 2004; Nicolle et al., 2004). Mice with defective MyD88 signaling show optimal T cell response, yet there is no significant reduction in the lung bacterial burden of Mtb challenged mice, compared to wild type (Nicolle et al., 2004). In another study it was shown that MyD88 knockout mice show interferon-(IFN)-γ production in response to mycobacterial antigens but Mtb infection became lethal within 4 weeks of post infection with 2 log10 higher CFUs in the lung (Fremond et al., 2004). TLR-2 deficient or TLR-9 KO mice show high levels of IFN-γ and tumor necrosis factor (TNF)-α, with high infiltration of CD4 T cells and CD8 T cells in lungs but these mice succumb to Mtb infection (Carlos et al., 2009). These reports indicate that innate immunity has a more profound role than to simply assist adaptive immunity. Moreover, an optimal acquired immune response is not sufficient to compensate for defective innate immunity. Collectively, these studies suggest that it is important to dissect out the functions assisted by innate immunity to induce a protective response against Mtb.
Innate Mechanisms Act and Foster T Cells to React against Mtb
Based on the recent studies, innate immunity has gained much more impetus due to its profound role in early control of Mtb infection and in sustaining the T cell response (Sia et al., 2015). These innate mechanisms represent the target to explore in designing new strategies to control Mtb. Taking into consideration all these facts, herein we compile the contribution of the bactericidal mechanisms: autophagy, apoptosis, inflammasome formation, and nitric oxide (NO) production in limiting the growth of Mtb (Table 1). Additionally, we will discuss how innate signaling delivered through pattern recognition receptors (PRRs) such as toll like receptors (TLRs), nucleotide binding oligomerization domain like receptors (NLRs) can augment these mechanisms.

TABLE 1. The role of innate immune mechanisms in restricting the survival of Mtb during lag phase.
Autophagy
Autophagy is evolved as a stress response that endows cells with a capability to adjust their biomass and turn over constituents at the time of starvation. It targets the cytoplasmic material, including macromolecules, organelles, and cells undergoing unscheduled apoptosis to lysosomes for degradation, thus periodically cleaning their interiors. Furthermore, autophagy has crucial roles in various biological processes, which include aging, development, degenerative diseases, and cancer (Huang and Brumell, 2014; Jiang and Mizushima, 2014). In addition, it also helps in elimination of pathogens, which exploit the cytosolic compartment for their regular life cycle, or those that are evolved with the capability to arrest phago-lysosome biogenesis (Flannagan et al., 2009). Autophagy is initiated with the sequestering of pathogens intracellularly to form a double membrane envelope that is known as autophagosome. Autophagosome fuses with lysosomes to form autolysosome to degrade pathogens. Thereby, autophagy facilitates the trafficking of mycobacteria to the lysosome for degradation (Gutierrez et al., 2004; Deretic et al., 2009). Similar results are reported in the case of BCG. In addition, autophagy also transports a large proportion of ubiquitinated proteins to lysosomes and augments the bactericidal capacity of lysosomal fraction (Alonso et al., 2007). Mtb escapes the immune mechanism by neutralizing the acidification of phagosomes (Deretic et al., 2006; Russell, 2011). Autophagy overcomes this Mtb evasion strategy by targeting phagosomes containing bacterium to lysosomes (Jo, 2013). Thus, autophagy provides an additional barrier to neutralize an attempt made by the mycobacterium to manipulate phagosomes maturation.
Noteworthy, autophagy processes bacteria for degradation within early hours of infection, as evidenced by conversion of autophagy marker LC3-I to LC3-II in Mtb infected DCs and Mφs (Khan et al., 2016). This experiment categorically indicates that autophagy guards the host against Mtb during the initial phase of infection. Importantly, animals with defective autophagy showed increase in the bacterial burden in lungs of Mtb challenged animals despite of predominance of Th17 immunity (Castillo et al., 2012). Furthermore, these animals showed remarkable gross tubercle lesions, in contrast to the smaller infected foci in the lungs of control animals. It signifies that autophagy also aid in preventing excessive inflammatory reactions in the host.
Currently, the only available vaccine for TB is BCG. Nonetheless, BCG has failed to reduce the global TB burden. Interestingly, one of the factors associated with the failure of BCG in TB-endemic areas is BCG ability to hamper the fusion of phagosome with lysosomes (Soualhine et al., 2007; Sun et al., 2007; Gowthaman et al., 2012). This interference in the antigen processing and presentation to T cells, results in defective T cell response. Since autophagy can overcome the problem of phago-lysosome biogenesis, targeting autophagy can substantially contribute to improve efficacy of BCG vaccine (Jagannath et al., 2009). It has been shown in a very elegant study that mice immunized with rapamycin-treated DCs infected with BCG showed enhanced Th1 cells mediated protection when challenged with virulent Mtb. Rapamycin induced enhancement in antigen presentation was attenuated when autophagy was suppressed by 3-methyladenine or by small interfering RNA against beclin-1 (Jagannath et al., 2009). Targeting autophagy may open new avenues in boostering the efficacy of BCG or immune response against Mtb. Dissecting the host factors that regulates autophagy can help in restricting the growth of Mtb and simultaneously improving the processing and presentation of antigen and enhancing T cell immunity.
Autophagy can be induced due to starvation, treatment with IFN-γ or rapamycin (Bento et al., 2015). Additionally, triggering through PRRs has direct correlation with the induction of autophagy. Signaling through TLR-4, TLR-3, TLR-7 and NOD-2 receptor can potently induce autophagy (Delgado et al., 2008; Delgado and Deretic, 2009; Cooney et al., 2010; Yuk et al., 2012). TLR-7 triggering enables Mφs to reduce the survival of Mtb via induction of autophagy (Delgado et al., 2008). It was further supported through suppression of autophagy by knocking down beclin and Atg5 through siRNA. Innate triggering through NOD-1 and NOD-2 enhances autophagy induction and reduces the bacterial burden within 6 h of infection (Travassos et al., 2010). Murine immunity related to guanosine tri-phosphate induces autophagy and generate large autolysosomal organelles, as a mechanism for the elimination of intracellular Mtb. Furthermore, human Irgm1 ortholog (IRGM) augments autophagy and reduces intracellular bacillary load (Singh et al., 2006). Many evidences have been documented to show the potential for autophagy based therapies to target Mtb (Bento et al., 2015). Similarly, exploring innate receptors to augment autophagy could also be one of the strategies to boost the host immunity against Mtb. Moreover, it would be beneficial in overcoming the failure associated with BCG vaccine.
Reactive Nitrogen and Oxygen Intermediates
Nitric oxide and reactive nitrogen intermediates (RNI) are considered potent antimicrobial agents acquired by innate cells. Mφs are the major producer of NO. It is released by inducible nitric oxide synthase (iNOs), a heme-protein that catalyzes the oxidation of L-arginine to NO and citrulline. NO production is a critical defense mechanism in determining the outcome of TB infection, since it reduces the survival of Mtb (Rich et al., 1997). This information was corroborated with the result observed in mouse model where abrogation of iNOs activity produces dramatic increase in microbial burden (MacMicking et al., 1997). Further, disruption of iNOs gene expression results in a high rate of Mtb dissemination and mortality. Stimulation of innate molecules, such as TLRs or NODs trigger the expression of iNOs, which ultimately kills Mtb, as evidenced by colony forming units (CFU) assay (Chan et al., 2001). In the mouse model of TB, NO secretion is well known to be an antimicrobial defense mechanism. However, its role in humans is still controversial. Forthcoming evidences indicate that human Mφs and alveolar epithelial cells upon infection with Mtb secrete NO to inhibit the intracellular growth of Mtb. Additionally, iNOs and markers associated with NO are highly expressed in the Mφs obtained from broncho-alveolar lavage of TB patients and not healthy individuals (Nicholson et al., 1996). Interestingly, patients infected with multidrug resistant (MDR) strain of Mtb produce less NO (Sharma et al., 2004). More startling observation came from the report that chemotherapy appears to cure TB in immune-competent mice but fails to do so in NOS2-deficient animals (Nathan and Shiloh, 2000). It concludes that bactericidal drug uses NO pathway for efficient killing of Mtb (Ciccone et al., 2003).
IFN-γ induces the production of NO (Flesch and Kaufmann, 1991). After several days of Mtb infection, T cells produce IFN-γ. However, production and action of NO is observed within 3 h and it persists for few days in the circulation (Akaki et al., 1997; Rich et al., 1997). Early production of NO signifies that IFN-γ, which is a potential stimulator for NO release, is not being released by T cells, but instead by the cells of innate immunity such as natural killer (NK) cells and γδ T cells (Sada-Ovalle et al., 2008). It suggests that innate immune cells are responsible for early release of NO to restrict the growth of Mtb during the initial period of infection. NO is also reported to regulate the synthesis and release of several pro-inflammatory cytokines including IL-1β, TNF-α, and IL-8, which subsequently affect the production of NO in the feedback loop (Kuo et al., 2000). Microbicidal activity of Mφs is also associated with reactive oxygen intermediates (ROI). However, their role in constraining the growth of Mtb is not highly significant (Chan et al., 1992; Akaki et al., 1997; Lamichhane, 2011). Probable reason documented is that although ROI appear immediately upon Mtb infection but Mφs cease to produce it within 2 h of infection. Further, ROI also have shorter half-lives.
In essence, the NO kills Mtb and as well as augments host immunity against the pathogen is well documented in the literature (Chan et al., 2001). It is important to mention that TLRs signaling contributes substantially in release of NO (West et al., 2011). 1, 25-dihydroxyvitamin D is a potent inducer of NO and suppresses the growth of Mtb (Rockett et al., 1998). TLRs triggering enhances the bactericidal activity of Mφs by upregulating the expression of the vitamin D receptor and by inducing the enzyme that catalyzes the conversion of 25-dihydroxyvitamin D3 to active 1, 25-dihydroxyvitamin D leading to the induction of antimicrobial peptide cathelicidin (Liu et al., 2006). Hence, it may be considered as a critical molecule in designing new therapeutic strategies to treat TB. TLR ligands are known to induce NO production in antigen presenting cells (APCs) and NO restricts the growth of Mtb (Khan et al., 2015).
Apoptosis
Apoptosis is commonly known as ‘programmed cell death (PCD)’. It is a phenomenon that occurs when a cell committing suicide confines its cytoplasmic content within membrane bound vesicles named as apoptotic bodies. These membrane bound vesicles express molecules known as ‘eat me or find me’ signals. ‘Eat me’ signals help in the recognition of these unwanted moieties by phagocytic cells (Behar et al., 2011). Furthermore, phagocytic cells remove them through a mechanism known as efferocytosis; the process known to engulf and remove apoptotic cells. Failure in efferocytosis results in the disintegration of apoptotic bodies and release of intracellular contents. This causes inflammation that is known as secondary necrosis (Martin et al., 2012). Importantly, apoptosis makes a crucial contribution to the host immune response and determines the outcome of infection. It abolishes the protected intracellular niche favoring the replication of Mtb, thus forcing the bacteria to search for a new habitat. The caspase family of serine proteases are the central molecules responsible for the execution of apoptosis. Apoptosis is classically induced by three pathways. First is through ligation or oligomerization of tumor necrosis factor receptor (TNFR) family. Ligation of cell surface receptor such as TNFR or Fas, results in the subsequent activation of caspases and the induction of apoptotic vesicles. Intrinsic apoptosis occurs in response to oxidative stress, nutrient starvation or intracellular stress, which changes the mitochondrial membrane permeability. It results in the translocation of cytochrome c from mitochondria to the cytoplasm, leading to the activation of caspases. The third pathway is mediated by granzyme B released from cytolytic T cells and NK cells.
Mycobacterium tuberculosis induces apoptosis through the classical extrinsic pathway. Encounter of Mtb with innate cells such as DCs and Mφs induces the release of TNF-α and triggers apoptosis. Apoptosis limits the replication of Mtb by sequestering bacilli in apoptotic vesicles and by activating nearby uninfected Mφs. This phenomenon has been demonstrated through a classical experiment in which uninfected autologous Mφs were cultured with apoptotic or necrotic or non-apoptotic infected Mφs. Interestingly, significant inhibition in the growth of Mtb was seen when apoptotic cells were cultured with uninfected Mφs. In coculture experiments, elimination of Mtb was anticipated through efferocytosis. Later, antimicrobial effect enacted by naïve Mφs was shown to be contact independent (Hartman and Kornfeld, 2011). Interleukin-1 signaling in naïve Mφs mediates the cross-talk with infected-Mφs. It exhibits NO-dependent antimicrobial activity against bacilli in autolysosomes of heavily infected Mφs (Hartman and Kornfeld, 2011). Noteworthy, the discrepancy occurs in the induction of apoptosis by avirulent versus virulent Mtb (Chen et al., 2006). Multiple reports indicate that virulent Mtb induces necrosis to avoid host defensive strategies, whereas attenuated strain is associated with apoptosis (Chen et al., 2006; Divangahi et al., 2009). Despite comparable amount of TNF-α, cells infected with avirulent strain are more susceptible to apoptosis. It was revealed that difference in level of apoptosis between Mtb strains is due to an evasion strategy used by the virulent strain of Mtb. Cells infected with virulent Mtb secrete more IL-10, which induces the release of TNFR-2. Soluble TNFR-2 forms a complex with TNF-α and downregulates the TNF-α induced apoptosis (Balcewicz-Sablinska et al., 1998). Furthermore, it has been demonstrated by Annexin V binding and intracellular caspase staining that early secretory antigen target (ESAT)-6 of Mtb induces apoptosis in human Mφs (Choi et al., 2010). Additionally, the expression profile of apoptotic genes shows up-regulation of anti-apoptotic genes in virulent Mtb infected Mφs.
In addition to the restriction of the Mtb growth during early phase of infection, apoptosis has a considerable role in the induction of the acquired cellular immune response (Winau et al., 2006). The role of both CD4 T cells and CD8 T cells are well documented in immunity against Mtb. However, the mechanism underlying the presentation of antigens to CD8 T cells in context with MHC-I molecules remains enigmatic. Recently, it has been shown that apoptosis of infected Mφs facilitates the release of mycobacterial antigens in apoptotic vesicles, thereby allowing their access to bystander APCs to present antigen to CD8 T cells. Inhibition of apoptotic blebbing using caspase inhibitors, hampers the CD8 T cell response (Winau et al., 2006). Therefore, it may be concluded that triggering of apoptosis can efficiently control the Mtb growth at early time points; and at later stages it potentially contributes in the generation of antigen specific CD8 T cells.
Neutrophils are important cells of innate immunity. They play a significant role in imparting protection to Mtb (Andersson et al., 2014). These are the first cells to be recruited at the site of infection. Neutrophils phagocytose Mtb. Furthermore, Mtb infected neutrophils undergo apoptosis and are phagocytosed by Mφs. These Mφs then release TNF-α to form granulomas and control acute Mtb infection (Perskvist et al., 2002). Further, inhibition of apoptosis in neutrophils delays the priming of CD4 T cells. Hence it implies that apoptosis plays a decisive role in controlling Mtb infection by activating innate as well as adaptive immunity. TLRs induced apoptosis such as TLR-3, 4 has been explored for cancer therapy (Salaun et al., 2007). Interestingly, TLRs show enough potential for triggering apoptosis in Mtb infected cells. LpqH, a 19 and 38 kDa lipoprotein of Mtb induces the Mφ cell death in TLR-2 dependent manner (Ciaramella et al., 2000; Sanchez et al., 2012). 38 kDa lipoprotein of Mtb elicits the TNF-α release in TLR-2 dependent manner and induces apoptosis in infected Mφs (Sanchez et al., 2009). Apoptosis has been shown to improve the efficacy of BCG vaccine. Deletion of the secA2 gene of Mtb, which encodes a component of a virulence-associated bacterial protein, triggers the apoptosis of infected cells and enhances the priming of antigen specific CD8 T cells. Vaccination with secA2 deleted Mtb mutant induces better protection than BCG against Mtb (Boom, 2007; Hinchey et al., 2007). rBCG strain that secretes listeriolysin of Lysteria monocytogens induces more efficacious protection than BCG against Mtb by facilitating the cross priming by inducing apoptosis (Grode et al., 2005). This evidence indicates that targeting apoptosis could be one of the potential strategies to prevent TB.
Although apoptosis is the well-studied PCD, but it is not the only mechanism responsible for this process. A new form of non-apoptotic PCD has been termed as paraptosis. Insulin like growth factor I receptor has been identified as a molecule involved in inducing paroptosis. It is characterized by cytoplasmic vacuolation, along with mitochondrial swelling, lack of apoptotic morphology, caspase activation and inhibition by caspase inhibitors (Sperandio et al., 2004). A few reports suggest that this form of cell death is driven by an alternative caspase-9 activity that is Apaf-1-independent (Sperandio et al., 2000). Since, paraptosis follows the pathway different from apoptosis, it could be a novel therapeutic target to kill pathogens that inhibits apoptosis. Little is known about the effect of paraptosis on the immune system and moieties involved in it. A few apoptotic inducers have been shown to elicit paraptosis (Amarante-Mendes et al., 1998). Nothing is known about its role in TB. In the future, it may be an interesting line of investigation to understand the contribution of paraptosis in limiting the Mtb growth.
Inflammasome
Mycobacterium tuberculosis activates the cascade of events mediating the release of an array of pro-inflammatory cytokines such as IL-6, IL-12, and TNF-α that play a defensive role in eliciting innate immunity (Cooper et al., 2011). Similarly, IL-1β and IL-18 have an influential role in imparting protection to Mtb. IL-18 enhances the production of IFN-γ and its abrogation results in less IFN-γ release and impaired NK cell function (Kawakami et al., 2000). Simultaneously, IL-1R1-deficient mice show 2-log increase in bacterial load in the lung and necrotic pneumonia within 4 weeks of Mtb exposure. It is notable to mention that cell mediated immunity (CMI), which is considered the hallmark of protection against Mtb is not sufficient in restricting bacterial burden in IL-1R deficient mice, despite efficient pulmonary CD4 T cell and CD8 T cell responses (Fremond et al., 2007).
Unlike other proinflammatory cytokines, IL-1β and IL-18 are synthesized as precursors known as pro-IL-1β and pro-IL-18 (Sansonetti et al., 2000). Multiple signaling pathways triggered through TLRs and cytokines result in the transcription of pro-IL-1β and pro-IL-18. However, their maturation requires processing by active caspases. Distinct caspases regulate the apoptosis and maturation of IL-1β and IL-18. Caspase-1 regulates the maturation of IL-1β and IL-18 (Dinarello, 2006). Importantly, release of IL-1β and IL-18 is highly regulated phenomenon, which is dependent on the activation of caspase-1 and its homolog by multimeric protein complex termed as inflammasomes (Vladimer et al., 2013). These complexes are critical in the proteolytic processing of pro-IL-1β and pro-IL-18 into their active form (Netea et al., 2010; Briken et al., 2013). The inflammasome is classically composed of NOD like receptors (NLRs), the adaptor molecule PYCARD/ASC, and pro-caspase-1, which when proteolyzed to caspase-1 provides the enzymatic activity of the inflammasome. Pro-caspase-1 forms the core of the inflammasome. However, the constitution of NLRs within the inflammasome varies according to the type of pathogen involved. The NLR family members NALP3, NAIP5, or IPAF and the adaptor apoptosis speck-like protein (ASC) are involved in caspase-1 activation. Inflammasome plays an important role in host defense against Mtb, since mice deficient in IL-1 receptor (IL-1RI), IL-1β or IL-18 are more susceptible to infection with Mtb. Furthermore, a defect in ASC adaptor protein shows the exacerbation of disease without restricting the Mtb growth. Early secreted antigenic target protein 6 kilodalton secretion system (ESX)-1 encoded in RD-1 region of Mtb promotes the release of IL-1β by inflammasome activation. ESX-1 mediated inflammasome formation depends on host NLRP-3 and ASC protein (Mishra et al., 2010). RD-1 deficient Mtb fails to induce a strong activation of caspase-1 resulting in inefficient secretion of IL-1β and IL-18. This observation signifies that the failure of BCG to mount optimal protection against Mtb is due to absence of RD-1 dependent induction of IL-1β and IL-18 (Kurenuma et al., 2009). Interestingly, treatment with exogenous IL-18 reduces the bacterial load in mice. Recently, viral and bacterial RNA have been shown to trigger NLRP3 and activate inflammasome (Mitoma et al., 2013). It suggests that prophylactic strategies employing recombinant BCG expressing innate ligands, which are efficient in inducing inflammasome formation, can boost its protective efficacy against TB. Mtb genes Rv0198c (zmp1), plays a critical role in preventing caspase-1-dependent activation and secretion of IL-1β. zmp1-deleted Mtb triggered activation of the inflammasome, resulting in increased release of IL-1β, enhanced maturation of Mtb containing phagosomes, improved mycobacterial clearance by macrophages, and reduction in bacterial load in the lungs of Mtb aerosol-infected mice (Master et al., 2008). Zmp1 is an important virulence determinant and represents a potentially useful drug target. Furthermore, it has been shown that binding of vitamin D induces IL-1β secretion and prevent infection. This information supports the idea of exploiting vitamin D in clinical trials against Mtb (Verway et al., 2013).
Inflammasomes are also reported to play an important part in amplifying the adaptive immune response. Importantly, inflammasome processed IL-1β promotes the differentiation of naïve CD4 T cells to Th17 subtype. It synergizes with IL-6 and promotes Th17 cell development via up-regulation of key cytokine IL-17, transcription factors, IRF4 and RORγt. Furthermore, IL-1β can coordinate with IL-6 and IL-23 in the absence of TGF-β signaling to induce pathogenic Th17 cells (Ghoreschi et al., 2010). In addition to Th1 cells, Th17 cells also play a cardinal role in generating anti-Mtb response. IL-17 induces the expression of chemokines that results in the recruitment of various cells to the site of infection. Furthermore, memory Th17 cells promote rapid migration of Th1 cells by enhancing the expression of chemokines (Khader and Cooper, 2008). Toll-like receptors and NOD-2 expressed on APCs are responsible for the induction and release of cytokines like IL-6, TGF-β, and IL-12 that are responsible for the differentiation of Th17 cells and Th1 cells, respectively (Khan et al., 2016). Hence immunotherapies involving agonists of innate immunity can be explored in the generation of protective immunity against Mtb (Chodisetti et al., 2015). The above showcased points indicate that innate immunity efficiently controls the Mtb growth during early phase of infection. Moreover, it creates a platform for adaptive immunity.
Conclusion
Continuous efforts are undertaken to generate an effective vaccine against TB. However, a possible candidate that can achieve the WHO-STOP-TB program has not yet been formulated. Eleven candidate vaccines are currently in clinical trials. Failure of BCG to protect against Mtb warrants a serious attempt to reinvigorate BCG potency for inducing optimal immune response (Singh et al., 2010; Gowthaman et al., 2011, 2012).
Recently, innate immunity has emerged as a cornerstone in limiting the growth of Mtb (Fremond et al., 2004; Nicolle et al., 2004; Carlos et al., 2009). Innate immunity not only initiates series of events to assist adaptive immunity but also restricts the growth of TB bacilli at the initial phase of infection. Nonetheless, failure of innate killing mechanisms results in unobstructed growth of Mtb and provides enough opportunity for the pathogen to breach the barrier of the immune system. It indicates that targeting innate immunity is a judicious approach to consider, while designing vaccines or therapeutics. Inefficiency of innate immunity provides an opportunity for unimpeded Mtb growth. Later, the Mtb conquers the adaptive immunity. Adequate innate immunity is capable of restricting the growth of Mtb during the “lag phase” of T cell response. Limiting the growth of Mtb during the initial phase of infection provides enough time for T cells to reach the site of infection and curtail Mtb replication. However, impaired innate immunity is incompetent in curbing the proliferation of Mtb. It results in unhindered growth of Mtb, which ultimately interferes in the activation of adaptive immunity. Biological therapies involving innate ligands for TLRs and NLRs will benefit the quest for novel treatment modalities for TB. We speculate that mycobacterial vaccines engineered with ligands for PRRs may enhance the potency of innate immunity to limit the Mtb growth and sustain the adaptive arm of immunity.
Author Contributions
Conception and design of the work: JA, NK. Drafting of manuscript: JA, NK, AV, SJ.
Funding
This study is supported by the Council of Scientific and Industrial Research (CSIR), New Delhi, India.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful to M. Amir and Drs. Jeffery Downey, Atul K. Verma, Gurpreet Kaur for English editing of the manuscript.
Abbreviations
BCG, Bacillus Calmette–Guerin; DCs, dendritic cells; Mtb, Mycobacterium tuberculosis; PCD, programmed cell death. TB, tuberculosis.
References
Akaki, T., Sato, K., Shimizu, T., Sano, C., Kajitani, H., Dekio, S., et al. (1997). Effector molecules in expression of the antimicrobial activity of macrophages against Mycobacterium avium complex: roles of reactive nitrogen intermediates, reactive oxygen intermediates, and free fatty acids. J. Leukoc. Biol. 62, 795–804.
Alonso, S., Pethe, K., Russell, D. G., and Purdy, G. E. (2007). Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl. Acad. Sci. U.S.A. 104, 6031–6036. doi: 10.1073/pnas.0700036104
Amarante-Mendes, G. P., Finucane, D. M., Martin, S. J., Cotter, T. G., Salvesen, G. S., and Green, D. R. (1998). Anti-apoptotic oncogenes prevent caspase-dependent and independent commitment for cell death. Cell Death Differ. 5, 298–306. doi: 10.1038/sj.cdd.4400354
Andersson, H., Andersson, B., Eklund, D., Ngoh, E., Persson, A., Svensson, K., et al. (2014). Apoptotic neutrophils augment the inflammatory response to Mycobacterium tuberculosis infection in human macrophages. PLoS ONE 9:e101514. doi: 10.1371/journal.pone.0101514
Arthur, M., Dannenberg, J., and Rook, G. A. W. (1994). “Pathogenesis of pulmonary tuberculosis: an interplay of tissue-damaging and macrophage-activating immune responses—dual mechanisms that control bacillary multiplication,” in Tuberculosis: Pathogenesis, Protection, and Control, ed. B. R. Bloom (Washington, DC: American Society for Microbiology), 459–483.
Balcewicz-Sablinska, M. K., Keane, J., Kornfeld, H., and Remold, H. G. (1998). Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J. Immunol. 161, 2636–2641.
Barry, C. E. III, Boshoff, H. I., Dartois, V., Dick, T., Ehrt, S., Flynn, J., et al. (2009). The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7, 845–855. doi: 10.1038/nrmicro2236
Behar, S. M., Martin, C. J., Booty, M. G., Nishimura, T., Zhao, X., Gan, H. X., et al. (2011). Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 4, 279–287. doi: 10.1038/mi.2011.3
Bento, C. F., Empadinhas, N., and Mendes, V. (2015). Autophagy in the fight against tuberculosis. DNA Cell Biol. 34, 228–242. doi: 10.1089/dna.2014.2745
Boom, W. H. (2007). New TB vaccines: is there a requirement for CD8 T cells? J. Clin. Invest. 117, 2092–2094. doi: 10.1172/JCI32933
Briken, V., Ahlbrand, S. E., and Shah, S. (2013). Mycobacterium tuberculosis and the host cell inflammasome: a complex relationship. Front. Cell. Infect. Microbiol. 3:62. doi: 10.3389/fcimb.2013.00062
Carlos, D., Frantz, F. G., Souza-Junior, D. A., Jamur, M. C., Oliver, C., Ramos, S. G., et al. (2009). TLR2-dependent mast cell activation contributes to the control of Mycobacterium tuberculosis infection. Microbes Infect. 11, 770–778. doi: 10.1016/j.micinf.2009.04.025
Castillo, E. F., Dekonenko, A., Arko-Mensah, J., Mandell, M. A., Dupont, N., Jiang, S., et al. (2012). Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. U.S.A. 109, E3168–E3176. doi: 10.1073/pnas.1210500109
Chan, E. D., Chan, J., and Schluger, N. W. (2001). What is the role of nitric oxide in murine and human host defense against tuberculosis? Current knowledge. Am. J. Respir. Cell Mol. Biol. 25, 606–612. doi: 10.1165/ajrcmb.25.5.4487
Chan, J., Xing, Y., Magliozzo, R. S., and Bloom, B. R. (1992). Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J. Exp. Med. 175, 1111–1122. doi: 10.1084/jem.175.4.1111
Chen, M., Gan, H., and Remold, H. G. (2006). A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol. 176, 3707–3716. doi: 10.4049/jimmunol.176.6.3707
Chodisetti, S. B., Gowthaman, U., Rai, P. K., Vidyarthi, A., Khan, N., and Agrewala, J. N. (2015). Triggering through Toll-like receptor 2 limits chronically stimulated T-helper type 1 cells from undergoing exhaustion. J. Infect. Dis. 211, 486–496. doi: 10.1093/infdis/jiu472
Choi, H. H., Shin, D. M., Kang, G., Kim, K. H., Park, J. B., Hur, G. M., et al. (2010). Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett. 584, 2445–2454. doi: 10.1016/j.febslet.2010.04.050
Ciaramella, A., Martino, A., Cicconi, R., Colizzi, V., and Fraziano, M. (2000). Mycobacterial 19-kDa lipoprotein mediates Mycobacterium tuberculosis-induced apoptosis in monocytes/macrophages at early stages of infection. Cell Death Differ. 7, 1270–1272. doi: 10.1038/sj.cdd.4400761
Ciccone, R., Mariani, F., Cavone, A., Persichini, T., Venturini, G., Ongini, E., et al. (2003). Inhibitory effect of NO-releasing ciprofloxacin (NCX 976) on Mycobacterium tuberculosis survival. Antimicrob. Agents Chemother. 47, 2299–2302. doi: 10.1128/AAC.47.7.2299-2302.2003
Cooney, R., Baker, J., Brain, O., Danis, B., Pichulik, T., Allan, P., et al. (2010). NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97. doi: 10.1038/nm.2069
Cooper, A. M., Mayer-Barber, K. D., and Sher, A. (2011). Role of innate cytokines in mycobacterial infection. Mucosal Immunol. 4, 252–260. doi: 10.1038/mi.2011.13
Delgado, M. A., and Deretic, V. (2009). Toll-like receptors in control of immunological autophagy. Cell Death Differ. 16, 976–983. doi: 10.1038/cdd.2009.40
Delgado, M. A., Elmaoued, R. A., Davis, A. S., Kyei, G., and Deretic, V. (2008). Toll-like receptors control autophagy. EMBO J. 27, 1110–1121. doi: 10.1038/emboj.2008.31
Deretic, V., Delgado, M., Vergne, I., Master, S., De Haro, S., Ponpuak, M., et al. (2009). Autophagy in immunity against Mycobacterium tuberculosis: a model system to dissect immunological roles of autophagy. Curr. Top. Microbiol. Immunol. 335, 169–188. doi: 10.1007/978-3-642-00302-8_8
Deretic, V., Singh, S., Master, S., Harris, J., Roberts, E., Kyei, G., et al. (2006). Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell. Microbiol. 8, 719–727. doi: 10.1111/j.1462-5822.2006.00705.x
Dinarello, C. A. (2006). Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am. J. Clin. Nutr. 83, 447S–455S.
Divangahi, M., Chen, M., Gan, H., Desjardins, D., Hickman, T. T., Lee, D. M., et al. (2009). Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 10, 899–906. doi: 10.1038/ni.1758
Flannagan, R. S., Cosio, G., and Grinstein, S. (2009). Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 7, 355–366. doi: 10.1038/nrmicro2128
Flesch, I. E., and Kaufmann, S. H. (1991). Mechanisms involved in mycobacterial growth inhibition by gamma interferon-activated bone marrow macrophages: role of reactive nitrogen intermediates. Infect. Immun. 59, 3213–3218.
Fremond, C. M., Togbe, D., Doz, E., Rose, S., Vasseur, V., Maillet, I., et al. (2007). IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J. Immunol. 179, 1178–1189. doi: 10.4049/jimmunol.179.2.1178
Fremond, C. M., Yeremeev, V., Nicolle, D. M., Jacobs, M., Quesniaux, V. F., and Ryffel, B. (2004). Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J. Clin. Invest. 114, 1790–1799. doi: 10.1172/JCI200421027
Ghoreschi, K., Laurence, A., Yang, X. P., Tato, C. M., Mcgeachy, M. J., Konkel, J. E., et al. (2010). Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467, 967–971. doi: 10.1038/nature09447
Gowthaman, U., Rai, P. K., Khan, N., Jackson, D. C., and Agrewala, J. N. (2012). Lipidated promiscuous peptides vaccine for tuberculosis-endemic regions. Trends Mol. Med. 18, 607–614. doi: 10.1016/j.molmed.2012.07.008
Gowthaman, U., Singh, V., Zeng, W., Jain, S., Siddiqui, K. F., Chodisetti, S. B., et al. (2011). Promiscuous peptide of 16 kDa antigen linked to Pam2Cys protects against Mycobacterium tuberculosis by evoking enduring memory T-cell response. J. Infect. Dis. 204, 1328–1338. doi: 10.1093/infdis/jir548
Grode, L., Seiler, P., Baumann, S., Hess, J., Brinkmann, V., Nasser Eddine, A., et al. (2005). Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guerin mutants that secrete listeriolysin. J. Clin. Invest. 115, 2472–2479. doi: 10.1172/JCI24617
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G. A., Colombo, M. I., and Deretic, V. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766. doi: 10.1016/j.cell.2004.11.038
Hartman, M. L., and Kornfeld, H. (2011). Interactions between naive and infected macrophages reduce Mycobacterium tuberculosis viability. PLoS ONE 6:e27972. doi: 10.1371/journal.pone.0027972
Hinchey, J., Lee, S., Jeon, B. Y., Basaraba, R. J., Venkataswamy, M. M., Chen, B., et al. (2007). Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J. Clin. Invest. 117, 2279–2288. doi: 10.1172/JCI31947
Huang, J., and Brumell, J. H. (2014). Bacteria-autophagy interplay: a battle for survival. Nat. Rev. Microbiol. 12, 101–114. doi: 10.1038/nrmicro3160
Jagannath, C., Lindsey, D. R., Dhandayuthapani, S., Xu, Y., Hunter, R. L. Jr., and Eissa, N. T. (2009). Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat. Med. 15, 267–276. doi: 10.1038/nm.1928
Jiang, P., and Mizushima, N. (2014). Autophagy and human diseases. Cell Res. 24, 69–79. doi: 10.1038/cr.2013.161
Jo, E. K. (2013). Autophagy as an innate defense against mycobacteria. Pathog. Dis. 67, 108–118. doi: 10.1111/2049-632X.12023
Kawakami, K., Koguchi, Y., Qureshi, M. H., Miyazato, A., Yara, S., Kinjo, Y., et al. (2000). IL-18 contributes to host resistance against infection with Cryptococcus neoformans in mice with defective IL-12 synthesis through induction of IFN-gamma production by NK cells. J. Immunol. 165, 941–947. doi: 10.4049/jimmunol.165.2.941
Khader, S. A., and Cooper, A. M. (2008). IL-23 and IL-17 in tuberculosis. Cytokine 41, 79–83. doi: 10.1016/j.cyto.2007.11.022
Khan, N., Pahari, S., Vidyarthi, A., Aqdas, M., and Agrewala, J. N. (2015). NOD-2 and TLR-4 signaling reinforces the efficacy of dendritic cells and reduces the dose of TB drugs against Mycobacterium tuberculosis. J. Innate Immun. doi: 10.1159/000439591 [Epub ahead of print].
Khan, N., Vidyarthi, A., Pahari, S., Negi, S., Aqdas, M., Nadeem, S., et al. (2016). Signaling through NOD-2 and TLR-4 bolsters the T cell priming capability of dendritic cells by inducing autophagy. Sci. Rep. 6, 19084. doi: 10.1038/srep19084
Kuo, H. P., Wang, C. H., Huang, K. S., Lin, H. C., Yu, C. T., Liu, C. Y., et al. (2000). Nitric oxide modulates interleukin-1beta and tumor necrosis factor-alpha synthesis by alveolar macrophages in pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 161, 192–199. doi: 10.1164/ajrccm.161.1.9902113
Kurenuma, T., Kawamura, I., Hara, H., Uchiyama, R., Daim, S., Dewamitta, S. R., et al. (2009). The RD1 locus in the Mycobacterium tuberculosis genome contributes to activation of caspase-1 via induction of potassium ion efflux in infected macrophages. Infect. Immun. 77, 3992–4001. doi: 10.1128/IAI.00015-09
Lamichhane, G. (2011). Mycobacterium tuberculosis response to stress from reactive oxygen and nitrogen species. Front. Microbiol. 2:176. doi: 10.3389/fmicb.2011.00176
Liu, P. T., Stenger, S., Li, H., Wenzel, L., Tan, B. H., Krutzik, S. R., et al. (2006). Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311, 1770–1773. doi: 10.1126/science.1123933
Lurie, M. B. (1964). Resistance to tuberculosis: Experimental Studies in Native and Acquired Defensive Mechanisms. Cambridge, MA: Harvard University Press.
MacMicking, J. D., North, R. J., Lacourse, R., Mudgett, J. S., Shah, S. K., and Nathan, C. F. (1997). Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 94, 5243–5248. doi: 10.1073/pnas.94.10.5243
Martin, C. J., Booty, M. G., Rosebrock, T. R., Nunes-Alves, C., Desjardins, D. M., Keren, I., et al. (2012). Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 12, 289–300. doi: 10.1016/j.chom.2012.06.010
Master, S. S., Rampini, S. K., Davis, A. S., Keller, C., Ehlers, S., Springer, B., et al. (2008). Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 3, 224–232. doi: 10.1016/j.chom.2008.03.003
Mishra, B. B., Moura-Alves, P., Sonawane, A., Hacohen, N., Griffiths, G., Moita, L. F., et al. (2010). Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell. Microbiol. 12, 1046–1063. doi: 10.1111/j.1462-5822.2010.01450.x
Mitoma, H., Hanabuchi, S., Kim, T., Bao, M., Zhang, Z., Sugimoto, N., et al. (2013). The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity 39, 123–135. doi: 10.1016/j.immuni.2013.07.001
Nathan, C., and Shiloh, M. U. (2000). Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U.S.A. 97, 8841–8848. doi: 10.1073/pnas.97.16.8841
Netea, M. G., Simon, A., Van De Veerdonk, F., Kullberg, B. J., Van Der Meer, J. W., and Joosten, L. A. (2010). IL-1beta processing in host defense: beyond the inflammasomes. PLoS Pathog. 6:e1000661. doi: 10.1371/journal.ppat.1000661
Nicholson, S., Bonecini-Almeida Mda, G., Lapa, E., Silva, J. R., Nathan, C., Xie, Q. W., et al. (1996). Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J. Exp. Med. 183, 2293–2302. doi: 10.1084/jem.183.5.2293
Nicolle, D. M., Pichon, X., Bouchot, A., Maillet, I., Erard, F., Akira, S., et al. (2004). Chronic pneumonia despite adaptive immune response to Mycobacterium bovis BCG in MyD88-deficient mice. Lab. Invest. 84, 1305–1321. doi: 10.1038/labinvest.3700149
Ottenhoff, T. H., and Kaufmann, S. H. (2012). Vaccines against tuberculosis: where are we and where do we need to go? PLoS Pathog. 8:e1002607. doi: 10.1371/journal.ppat.1002607
Perskvist, N., Long, M., Stendahl, O., and Zheng, L. (2002). Mycobacterium tuberculosis promotes apoptosis in human neutrophils by activating caspase-3 and altering expression of Bax/Bcl-xL via an oxygen-dependent pathway. J. Immunol. 168, 6358–6365. doi: 10.4049/jimmunol.168.12.6358
Rich, E. A., Torres, M., Sada, E., Finegan, C. K., Hamilton, B. D., and Toossi, Z. (1997). Mycobacterium tuberculosis (MTB)-stimulated production of nitric oxide by human alveolar macrophages and relationship of nitric oxide production to growth inhibition of MTB. Tuber. Lung Dis. 78, 247–255. doi: 10.1016/S0962-8479(97)90005-8
Roberts, L. L., and Robinson, C. M. (2014). Mycobacterium tuberculosis infection of human dendritic cells decreases integrin expression, adhesion and migration to chemokines. Immunology 141, 39–51. doi: 10.1111/imm.12164
Rockett, K. A., Brookes, R., Udalova, I., Vidal, V., Hill, A. V., and Kwiatkowski, D. (1998). 1,25-Dihydroxyvitamin D3 induces nitric oxide synthase and suppresses growth of Mycobacterium tuberculosis in a human macrophage-like cell line. Infect. Immun. 66, 5314–5321.
Russell, D. G. (2011). Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol. Rev. 240, 252–268. doi: 10.1111/j.1600-065X.2010.00984.x
Sada-Ovalle, I., Chiba, A., Gonzales, A., Brenner, M. B., and Behar, S. M. (2008).Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog. 4:e1000239. doi: 10.1371/journal.ppat.1000239
Salaun, B., Romero, P., and Lebecque, S. (2007). Toll-like receptors’ two-edged sword: when immunity meets apoptosis. Eur. J. Immunol. 37, 3311–3318. doi: 10.1002/eji.200737744
Sanchez, A., Espinosa, P., Esparza, M. A., Colon, M., Bernal, G., and Mancilla, R. (2009). Mycobacterium tuberculosis 38-kDa lipoprotein is apoptogenic for human monocyte-derived macrophages. Scand. J. Immunol. 69, 20–28. doi: 10.1111/j.1365-3083.2008.02193.x
Sanchez, A., Espinosa, P., Garcia, T., and Mancilla, R. (2012). The 19 kDa Mycobacterium tuberculosis lipoprotein (LpqH) induces macrophage apoptosis through extrinsic and intrinsic pathways: a role for the mitochondrial apoptosis-inducing factor. Clin. Dev. Immunol. 2012, 950503. doi: 10.1155/2012/950503
Sansonetti, P. J., Phalipon, A., Arondel, J., Thirumalai, K., Banerjee, S., Akira, S., et al. (2000). Caspase-1 activation of IL-1beta and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity 12, 581–590. doi: 10.1016/S1074-7613(00)80209-5
Shaler, C. R., Horvath, C., Lai, R., and Xing, Z. (2012). Understanding delayed T-cell priming, lung recruitment, and airway luminal T-cell responses in host defense against pulmonary tuberculosis. Clin. Dev. Immunol. 2012, 628293. doi: 10.1155/2012/628293
Sharma, S., Sharma, M., Roy, S., Kumar, P., and Bose, M. (2004). Mycobacterium tuberculosis induces high production of nitric oxide in coordination with production of tumour necrosis factor-alpha in patients with fresh active tuberculosis but not in MDR tuberculosis. Immunol. Cell Biol. 82, 377–382. doi: 10.1111/j.0818-9641.2004.01245.x
Sia, J. K., Georgieva, M., and Rengarajan, J. (2015). Innate immune defenses in human tuberculosis: an overview of the interactions between Mycobacterium tuberculosis and innate immune cells. J. Immunol. Res. 2015, 747543. doi: 10.1155/2015/747543
Singh, S. B., Davis, A. S., Taylor, G. A., and Deretic, V. (2006). Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313, 1438–1441. doi: 10.1126/science.1129577
Singh, V., Gowthaman, U., Jain, S., Parihar, P., Banskar, S., Gupta, P., et al. (2010). Coadministration of interleukins 7 and 15 with bacille Calmette-Guerin mounts enduring T cell memory response against Mycobacterium tuberculosis. J. Infect. Dis. 202, 480–489. doi: 10.1086/653827
Soualhine, H., Deghmane, A. E., Sun, J., Mak, K., Talal, A., Av-Gay, Y., et al. (2007). Mycobacterium bovis bacillus Calmette-Guerin secreting active cathepsin S stimulates expression of mature MHC class II molecules and antigen presentation in human macrophages. J. Immunol. 179, 5137–5145. doi: 10.4049/jimmunol.179.8.5137
Sperandio, S., De Belle, I., and Bredesen, D. E. (2000). An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. U.S.A. 97, 14376–14381. doi: 10.1073/pnas.97.26.14376
Sperandio, S., Poksay, K., De Belle, I., Lafuente, M. J., Liu, B., Nasir, J., et al. (2004). Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 11, 1066–1075. doi: 10.1038/sj.cdd.4401465
Stenger, S., and Modlin, R. L. (1999). T cell mediated immunity to Mycobacterium tuberculosis. Curr. Opin. Microbiol. 2, 89–93. doi: 10.1016/S1369-5274(99)80015-0
Sun, J., Deghmane, A. E., Soualhine, H., Hong, T., Bucci, C., Solodkin, A., et al. (2007). Mycobacterium bovis BCG disrupts the interaction of Rab7 with RILP contributing to inhibition of phagosome maturation. J. Leukoc. Biol. 82, 1437–1445. doi: 10.1189/jlb.10.1189
Travassos, L. H., Carneiro, L. A., Ramjeet, M., Hussey, S., Kim, Y. G., Magalhaes, J. G., et al. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62. doi: 10.1038/ni.1823
van Crevel, R., Ottenhoff, T. H., and Van Der Meer, J. W. (2002). Innate immunity to Mycobacterium tuberculosis. Clin. Microbiol. Rev. 15, 294–309. doi: 10.1128/CMR.15.2.294-309.2002
Verway, M., Bouttier, M., Wang, T. T., Carrier, M., Calderon, M., An, B. S., et al. (2013). Vitamin D induces interleukin-1beta expression: paracrine macrophage epithelial signaling controls M. tuberculosis infection. PLoS Pathog. 9:e1003407. doi: 10.1371/journal.ppat.1003407
Vladimer, G. I., Marty-Roix, R., Ghosh, S., Weng, D., and Lien, E. (2013). Inflammasomes and host defenses against bacterial infections. Curr. Opin. Microbiol. 16, 23–31. doi: 10.1016/j.mib.2012.11.008
West, A. P., Brodsky, I. E., Rahner, C., Woo, D. K., Erdjument-Bromage, H., Tempst, P., et al. (2011). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480. doi: 10.1038/nature09973
Winau, F., Weber, S., Sad, S., De Diego, J., Hoops, S. L., Breiden, B., et al. (2006). Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 24, 105–117. doi: 10.1016/j.immuni.2005.12.001
Keywords: Mycobacterium tuberculosis, innate immunity, apoptosis, autophagy, nitric oxide, inflammasome
Citation: Khan N, Vidyarthi A, Javed S and Agrewala JN (2016) Innate Immunity Holding the Flanks until Reinforced by Adaptive Immunity against Mycobacterium tuberculosis Infection. Front. Microbiol. 7:328. doi: 10.3389/fmicb.2016.00328
Received: 28 September 2015; Accepted: 01 March 2016;
Published: 14 March 2016.
Edited by:
Tao Dong, Oxford University, UKReviewed by:
António Gil Castro, University of Minho, PortugalAmedeo Amedei, University of Florence, Italy
Paras Jain, Albert Einstein College of Medicine, USA
Ana M. S. Guimaraes, University of Sao Paulo, Brazil
Copyright © 2016 Khan, Vidyarthi, Javed and Agrewala. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nargis Khan, nargisrauf@gmail.com; Javed N. Agrewala, javed@imtech.res.in