 Youcef Soufi1
Youcef Soufi1 Boumediene Soufi2*
Boumediene Soufi2*- 1College of Medicine, University of Manitoba, Winnipeg, MB, Canada
- 2Independent Academic Scholar, Magdeburg, Germany
The composition of human skin acts as a natural habitat for various bacterial species that function in a commensal and symbiotic fashion. In a healthy individual, bacterial flora serves to protect the host. Under certain conditions such as minor trauma, impaired host immunity, or environmental factors, the risk of developing skin infections is increased. Although a large majority of bacterial associated skin infections are common, a portion can potentially manifest into clinically significant morbidity. For example, Gram-positive species that typically reside on the skin such as Staphylococcus and Streptococcus can cause numerous epidermal (impetigo, ecthyma) and dermal (cellulitis, necrotizing fasciitis, erysipelas) skin infections. Moreover, the increasing incidence of bacterial antibiotic resistance represents a serious challenge to modern medicine and threatens the health care system. Therefore, it is critical to develop tools and strategies that can allow us to better elucidate the nature and mechanism of bacterial virulence. To this end, mass spectrometry (MS)-based proteomics has been revolutionizing biomedical research, and has positively impacted the microbiology field. Advances in MS technologies have paved the way for numerous bacterial proteomes and their respective post translational modifications (PTMs) to be accurately identified and quantified in a high throughput and robust fashion. This technological platform offers critical information with regards to signal transduction, adherence, and microbial–host interactions associated with bacterial pathogenesis. This mini-review serves to highlight the current progress proteomics has contributed toward the understanding of bacteria that are associated with skin related diseases, infections, and antibiotic resistance.
Introduction
Skin has a primary role to act as a physical barrier in order to protect the body from temperature variations, microorganisms, or toxic substrates. While in utero skin is sterile, after birth it becomes rapidly colonized with numerous microorganisms which aids toward protecting the body (Capone et al., 2011). Bacterially derived dermatologic conditions can range from relatively benign conditions (assuming non-immunocompromised patients) such as cellulitis, erysipelas, folliculitis, or impetigo to serious clinical morbidity, such as necrotizing fasciitis (Stulberg et al., 2002). Underlying medical conditions such as diabetes mellitus or AIDS are examples where commensal bacteria can invade the skin resulting in infections that can manifest from mild to the life threatening (Swartz, 2004).
In recent years there have been growing concerns with respect to the challenges faced in the emergent inability to provide effective therapeutic interventions for patients with pathologies of bacterial etiology, due to increasing rates of antibiotic resistance. Considering the ease of transmission with respect to resistance genes amongst mucosal and epidermal flora, special consideration should be given to dermatologic related bacterial conditions (Espersen, 1998).
Mass spectrometry (MS)-based proteomics has the capability to study proteins and their interactions in order to better understand dysregulations in infection disorders (List et al., 2008), reveal antibiotic resistance mechanisms (Lee et al., 2015) and significant new targets for future drug discovery. Current MS-based proteomics technologies have advanced to the point where they are amenable to any biological system. With regards to bacterial organisms, they are particularly attractive models to apply proteomics based approaches due to their smaller proteomes and modifications compared to eukaryotes allowing for comprehensive proteome coverage (Stekhoven et al., 2014).
Skin Microbiome
The skin is host to many different bacterial species, fungi, viruses and mites. Most of the bacteria fall into four phyla: Actinobacteria, Firmicutes, Bacteroidetes, and Proteobacteria from which Propionibacterium, Corynebacterium, Staphylococcus, Micrococcus, Streptococcus, and Brevibacterium are highly abundant (van Rensburg et al., 2015) Gram-positive bacteria, since Gram-negative members typically cannot cope with the relatively dry environment of healthy human skin (Del Rosso and Leyden, 2007). In certain physiological conditions where the skin becomes moist, some Gram-negative bacteria can colonize such as Acinetobacter sp. (Del Rosso and Leyden, 2007). The diverse community of the skin, known as the skin microbiome, is defined by host physiology, host genotype, immune system, environment, and lifestyle (Grice and Segre, 2011). However, most of these microorganisms are harmless and many bacterial species act in a commensal fashion by which each species benefits through the exchange of nutrients as well as protect the host from pathogens without negatively affecting each other (Mankowska-Wierzbicka et al., 2015). Knowledge toward the bacterial microbiome is mainly derived from the conventional culture-based approaches, although in recent years DNA sequencing technologies combined with bioinformatic analysis and metagenomics approaches have allowed comprehensive examination of microbial communities (Kuczynski et al., 2012), yet uncertainties remain as to what defines a “normal” microbiome (Backhed et al., 2012).
The underlying contribution of the microbiome in the clinical picture of skin disorders is clear from the perspective of antibacterial treatment, however, the molecular dynamics between the microbiome and host remains largely unknown. Disturbances in homeostasis between microbiome and host can manifest into skin disorders such as atopic dermatitis (AD) or psoriasis (Kuo et al., 2013). Both disorders are connected with dysregulation of the skin immune response. However, while AD lesions are characterized by low level, psoriatic lesions are characterized by high level of antimicrobial peptide production (Nomura et al., 2003). Additionally, AD lesions are regularly infected with microbial pathogens.
The bacterial genus of Staphylococcus sp. is composed of approximately 40 different species both commensal (unable to produce virulence factor coagulase) and pathogenic, and play an important role in skin health and pathology (Del Rosso and Leyden, 2007). For example, Staphylococcus aureus is a medically relevant bacterial pathogen that is associated with the increasing rate of antibiotic resistance mechanisms as well as systemic and cutaneous infections (Becker et al., 2007), including AD (Hanifin and Rogge, 1977). However, it was previously thought that S. aureus causes AD alone, a recent study showed that the skin microbiome composition has a temporal change and it depends on disease flares and treatment. In the active state of the disease, the abundance of S. aureus and skin commensal S. epidermidis was increased, while Streptococcus, Propionibacterium, and Corynebacterium were increased following therapy (Kong et al., 2012). Increased abundance of S. epidermidis could reflect a microbial response to overgrowth of S. aureus, since it can selectively inhibit S. aureus growth (Iwase et al., 2010; Zeeuwen et al., 2013). Moreover, a recent study identified the presence of a distinct microbiome capable of direct communication with the host at the sub-epidermal compartments of the skin, an area previously thought to be sterile (Nakatsuji et al., 2013). While these types of studies employing different techniques have characterized the bacterial species that make up the skin microbiome, proteomics is required in order to identify proteins and their respective pathways involved during these interactions, which could directly contribute to the better understanding of the complex interplay between microbiome and host.
Shotgun MS-Based Proteomics
Due to the progress made toward state of the art next generation sequencing methodologies applied to generate fully annotated genomes, the number of fully sequenced microbial genomes has increased dramatically (Ribeiro et al., 2012). This has enabled state of the art MS-based proteomics technologies to rapidly advance in order to study microbial models and their communities in a robust and systematic fashion.
Two-dimensional polyacrylamide gel electrophoresis (2D-PAGE), followed by MS analysis has served as the main proteomics method of choice in the past and has been utilized in many skin associated bacterial pathogen studies (Kohler et al., 2005; Becher et al., 2009; Francois et al., 2014). In this technique, proteins are separated according to their molecular weight and isoelectric point migrating and accumulating as protein spots on the gel matrix. Although this approach can resolve multiples of thousands of proteins, it does carry some disadvantages especially with respect toward the identification of low abundant proteins, proteins with extremely high and low molecular weights, and is rather impractical with respect to large scale site specific analysis of post translational modifications (PTMs) such as phosphorylation.
The inherent limitations associated with 2D-PAGE MS lead to the development of gel-free MS-based approaches better known as “shotgun” proteomics. Advances made in shotgun based proteomics approaches is largely due to technological improvements made in high performance mass spectrometers. Since most biological samples consist of very complex peptide mixtures, mass spectrometers must be capable of ensuring a deep analytical coverage while maintaining a high level of robustness, sensitivity, and measurement accuracy (Mann and Kelleher, 2008). Newer generations of hybrid mass spectrometers such as but not limited to the LTQ-Orbitrap (Hu et al., 2005), LTQ Orbitrap Velos (Olsen et al., 2009), Q-Exactive HF (Scheltema et al., 2014), and Orbitrap Fusion (Erickson et al., 2015) can achieve precise mass accuracy at very high acquisition speeds, and resolution which allows for a complete sampling of complex peptide extracts providing comprehensive proteome coverage. Combined with high performance liquid chromatography (HPLC) technologies, these LC-MS workflows also referred to as gel-free shotgun proteomics allow for the quantification and identification of entire proteomes as well as their protein modifications across different biological samples. An overview of a typical shotgun MS-based experimental workflow is illustrated in Figure 1.
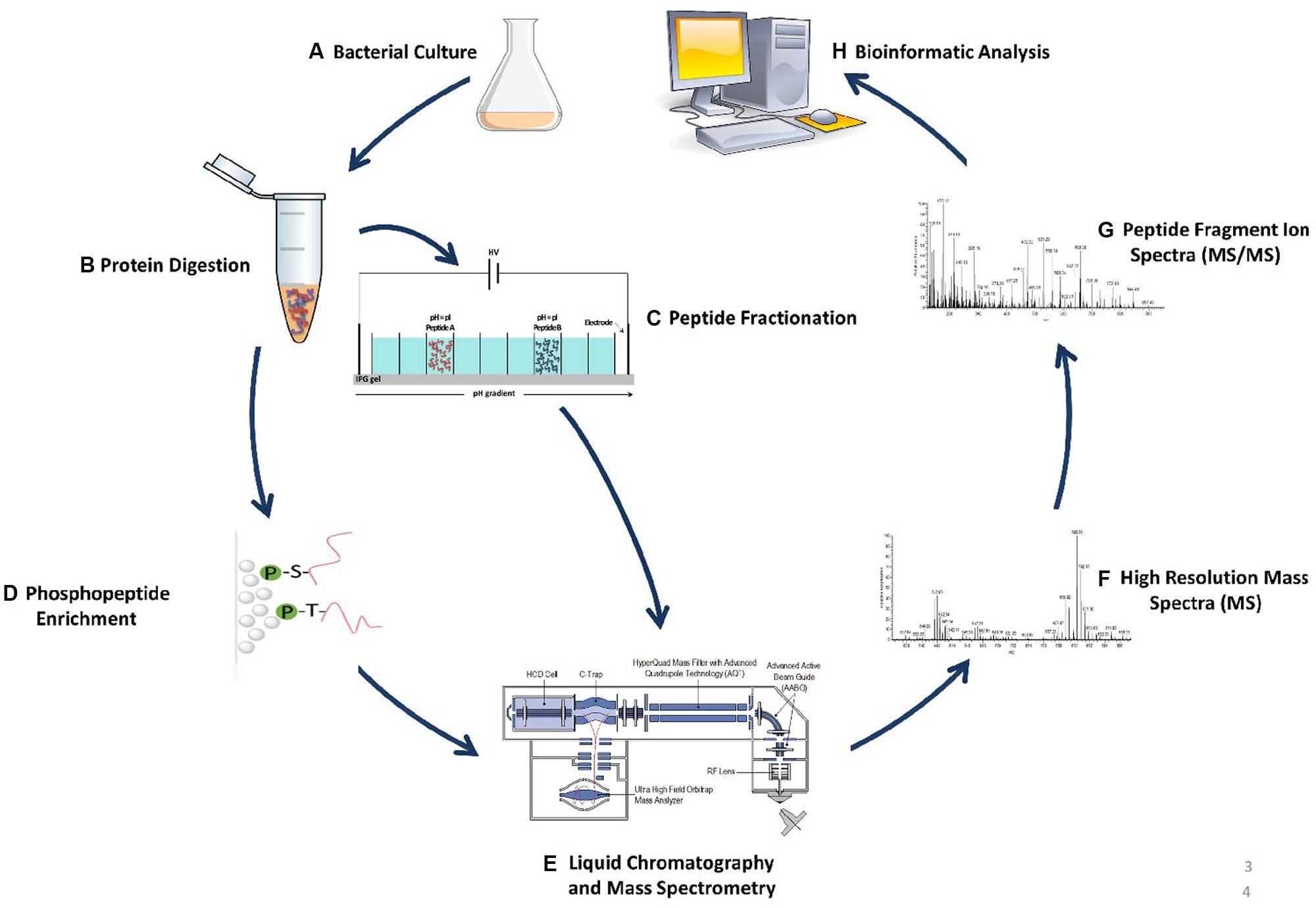
FIGURE 1. Example of shotgun proteomics workflow. This workflow provides a blueprint that can decode protein dynamics from complex crude protein extracts toward elucidation of numerous novel biological functions. (A) Cells are grown in culture or other growth models, and lysed using various procedures. (B) Protein extracts are digested for example by trypsin to generate peptides. (C) Peptides are fractionated in order to decrease peptide complexity which increase both identification and quantification rates upon MS analysis. (D) If PTM analysis is required, various enrichment strategies can be performed such as titanium chromatography in the case of phosphopeptide enrichment. (E) Resulting peptide extracts are separated on nanoflow HPLC and measured on a high resolution mass spectrometer (e.g., Thermo Scientific Q Exactive HF). (F) Relative peptide quantitation (full-scan MS). (G) Peptide identification through sequencing and detection to acquire MS/MS spectra. (H) Data processing utilizing numerous bioinformatic approaches.
For example, a similar approach as described above was applied to the bacterium Propionibacterium acnes (Bek-Thomsen et al., 2014). P. acnes is known to be associated with the inflammatory condition of the sebaceous human follicles known as acne vulgaris (Williams et al., 2012). The authors utilized proteomics in order to identify both human and P. acnes proteins using sebaceous follicular casts. To this end, many proteins involved in wound healing, inflammation, and tissue formation were identified. The most abundant P. acnes proteins were CAMP factors, and surface exposed dermatan sulfate adhesins. This study is the first of its kind in terms of analyzing the proteomes of both humans and bacteria on sebaceous follicular casts and demonstrates the importance of employing shotgun based proteomics workflows (Bek-Thomsen et al., 2014).
A recent study employed a combinatorial approach utilizing 16SrRNA sequencing with ultra-performance liquid chromatography/quadrupole time of flight (UPLC-TOF) MS toward the generation of a 3-dimensional topographical map of microbes and their molecules of origin (peptides, metabolites) distributed on the surface of the human skin (Bouslimani et al., 2015). This information was utilized to identify microbial species present and correlated with the chemical environment of the skin and serves as a powerful approach toward elucidating how the microbiome interacts and subsequently modifies different areas of the human skin in a species specific fashion.
Shotgun MS-Based Quantitative Proteomics
The complete identification of a proteome and its respective PTMs provides a better understanding toward biological function and regulation. However, it is equally important to quantify dynamics of proteins relative to each other under different biological conditions, disease states, and perturbations. This information can be obtained through the use of quantitative proteomics of which numerous approaches have been developed that are compatible with shotgun MS-based proteomics strategies.
One common approach is to introduce a stable non-radioactive isotope label either chemically or metabolically on the peptide or protein level. Through this technique, the relative intensities of peptides are measured, via MS thus identifying and quantifying regulated proteins from different samples through the obtained mass spectra. Examples of such techniques include but they are not limited to Stable Isotope Labeling by Amino acids in Cell culture (SILAC; Ong et al., 2002) or 15N labeling (Hempel et al., 2010; Soufi et al., 2015), both of which have been successfully implemented in a variety of bacteria (Soufi et al., 2010, 2015; Hempel et al., 2011; Soares et al., 2013; Misra et al., 2014; Soufi and Macek, 2014; Boysen et al., 2015). Furthermore, an innovative MS-based technique known as cell type-specific labeling using amino acid precursors (CTAP) has the ability to continuously label the proteome of individual cell types actively growing in a co-culture environment, allowing for the elucidation of the “cell-of-origin” of proteins in multicellular environments (Gauthier et al., 2013). Although this method was demonstrated in eukaryotic models, the application toward bacteria especially those in multispecies environments, such as human microbiomes, biofilms, etc. could significantly contribute toward the better understanding of the dynamics between different bacterial species inhabiting the same environment.
Although metabolic labeling approaches such as SILAC are considered by many to be the most accurate method toward global protein quantitation (Ong et al., 2002), they may be difficult to implement due to technical challenges with incorporation of the chemical or metabolic labeling approach, or challenges posed in complex biological models such as those involving the skin. Therefore, due to rapid advances made in LC-MS, label free quantitation (LFQ) approaches are now possible and routinely performed (Neilson et al., 2011). In this approach, different peptide samples are measured via MS and compared either by the total number of sequenced (MS/MS) spectra or the total extracted ion currents under controlled conditions.
The LFQ methodology was successfully applied to study the role of S. aureus in patients with ectodermal dysplasia and AD (Burian et al., 2015). Patients suffering from AD are highly correlated with the presence of opportunistic S. aureus infections due to having a reduced immune response, and have been shown to contain a lower amount of peptides originating from the natural antimicrobial dermcidin found in sweat leading to decreased levels of antimicrobial activity (Schittek, 2011). The authors proved through proteomics analysis of the secretome of diseased vs. healthy patients that a similar mechanism of reduced sweat derived dermcidin also exists in patients with ectodermal dysplasia, making these individuals highly prone to acquiring S. aureus infections (Burian et al., 2015).
Quantitative proteomics techniques can be utilized to estimate the absolute molar amount or concentration of a particular protein per cell. This information is highly relevant especially within the clinical context (Rodriguez-Suarez and Whetton, 2013). Many variants of this technique exist and all involve spiking in a known concentration of an internal protein standard into the sample, followed by MS analysis, and comparing resulting sample peptide measurements to the internal standard. Techniques include FLEXIQuant (Singh et al., 2009), absolute quantification using protein epitope signature tags (PrEST; Zeiler et al., 2012), intensity based absolute quantification (iBAQ; Schwanhausser et al., 2011), and absolute quantification (AQUA; Gerber et al., 2003). These methods can be applied to bacteria and serve as an important tool toward understanding the underlying mechanisms during bacterial virulence and antibiotic resistance.
Post Translational Modifications (PTMS)
Various PTMs on bacterial proteins such as phosphorylation, acetylation, methylation, and deamidation serve as an efficient means of controlling signal transduction, virulence and regulatory processes. PTMs represent a significant process in the life cycle of bacteria and can modulate key virulence factors and are attractive targets for novel therapies.
Detecting these PTMs in bacteria poses a technical challenge due to the fact that they are difficult to discover as these modifications typically exist at low levels of abundance. To circumvent this issue, specific enrichment strategies targeting certain PTMs can be utilized in order to decrease peptide complexity thereby increasing the likeliness of detection and subsequent characterization. For example, immunoaffinity enrichment is typically employed to select for lysine acetylated peptides (Rardin et al., 2013). Moreover, similar enrichment strategies are employed to capture phosphorylation events on serine, threonine, and tyrosine (S/T/Y) amino acid residues. These PTMs were once thought to exist solely in eukaryotes, however, MS-based proteomics combined with phosphopeptide enrichment strategies such as titanium dioxide chromatography (TiO2; Pinkse et al., 2004), immunoprecipitation (Rush et al., 2005) or immobilized metal ion affinity chromatography (IMAC; Villen and Gygi, 2008) have now established S/T/Y phosphorylation as a frequent and important PTM among different bacterial species (Macek et al., 2007, 2008; Soufi et al., 2008; Lin et al., 2009; Sun et al., 2010; Manteca et al., 2011; Misra et al., 2011; Cousin et al., 2013).
A recent shotgun based high resolution LC-MS/MS approach involving the enrichment of surface proteins using “trypsin shaving” was applied to the opportunistic pathogen S. aureus toward the identification of hydroxymethylation on aspargine and glutamine amino acid residues in an attempt to identify the presence and potential regulatory importance of this PTM on surface proteins which are known to assist in the colonization and invasion of the host cell (Waridel et al., 2012). The authors reported a total of 15 proteins (mostly surface proteins) that contained hydroxymethylation modifications and could play a role in virulence factor modulation.
Pseudomonas aeruginosa, an opportunistic bacterial pathogen, is associated with immunocompromised patients and nosocomial infections. P. aeruginosa infections can enter the body through the skin, and can be life threatening due to their ability to develop antibiotic resistance (Ouidir et al., 2015). A study involving P. aeruginosa, employed an effective combinatorial method of immunoaffinity assays, complex peptide fractionation, and shotgun proteomics toward the identification and characterization of lysine acetylation, a reversible PTM that has recently been implicated as an important regulatory mechanism in many bacteria (Yu et al., 2008; Kim et al., 2013; Zhang et al., 2013; Liao et al., 2014). This approach lead to the identification of 320 acetylated proteins in a wide variety of functional classes. The study also identified novel lysine acetylation events in virulence factors known to assist in host immune response evasion such as chitin binding protein, serine protease, exotoxin A, and hemolysin which potentially implies that lysine acetylation events in P. aeruginosa plays a role in mechanisms involving virulence (Ouidir et al., 2015).
Cysteine phosphorylation in S. aureus was shown to assist in the regulation of bacterial virulence and vancomycin resistance (Sun et al., 2012). Utilizing high resolution MS, the authors elucidated in a site specific fashion, that cysteine phosphorylation events occurred in various proteins many of which are global regulators that control important biological processes (Sun et al., 2012). Moreover, the eukaryotic-like Ser/Thr kinase and phosphatase pair Stk1/Stp1 was found to regulate cysteine phosphorylation in many Gram-positive bacteria providing an important piece of information toward the underlying regulatory mechanism of these events (Sun et al., 2012).
Conclusion and Future Outlook
A wide range of dermatological microbial associated diseases presents current and future challenges to health care providers. While in the last decade metagenomics data provided a higher level of understanding of the microbial skin environment, understanding signal transduction, virulence, regulatory processes, and dynamics between different bacterial species is essential in order to improve the overall standards and quality of patient care and treatment. MS-based proteomics has the capability to provide this knowledge. To this end, a ground-breaking endeavor with proteomics at the forefront is required in order to elucidate the specific mechanisms involved in skin infections, bacterial resistance as well as the complex microbiome and host relationship.
Author Contributions
YS wrote the manuscript. BS assisted with writing of the manuscript and generated the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We would like to thank Katarina Matic, M.D., Ph.D. for critical reading of the manuscript.
References
Backhed, F., Fraser, C. M., Ringel, Y., Sanders, M. E., Sartor, R. B., Sherman, P. M., et al. (2012). Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe 12, 611–622. doi: 10.1016/j.chom.2012.10.012
Becher, D., Hempel, K., Sievers, S., Zuhlke, D., Pane-Farre, J., Otto, A., et al. (2009). A proteomic view of an important human pathogen–towards the quantification of the entire Staphylococcus aureus proteome. PLoS ONE 4:e8176. doi: 10.1371/journal.pone.0008176
Becker, K., Bierbaum, G., Von Eiff, C., Engelmann, S., Gotz, F., Hacker, J., et al. (2007). Understanding the physiology and adaptation of staphylococci: a post-genomic approach. Int. J. Med. Microbiol. 297, 483–501. doi: 10.1016/j.ijmm.2007.04.004
Bek-Thomsen, M., Lomholt, H. B., Scavenius, C., Enghild, J. J., and Bruggemann, H. (2014). Proteome analysis of human sebaceous follicle infundibula extracted from healthy and acne-affected skin. PLoS ONE 9:e107908. doi: 10.1371/journal.pone.0107908
Bouslimani, A., Porto, C., Rath, C. M., Wang, M., Guo, Y., Gonzalez, A., et al. (2015). Molecular cartography of the human skin surface in 3D. Proc. Natl. Acad. Sci. U.S.A. 112, E2120–E2129. doi: 10.1073/pnas.1424409112
Boysen, A., Borch, J., Krogh, T. J., Hjerno, K., and Moller-Jensen, J. (2015). SILAC-based comparative analysis of pathogenic Escherichia coli secretomes. J. Microbiol. Methods 116, 66–79. doi: 10.1016/j.mimet.2015.06.015
Burian, M., Velic, A., Matic, K., Gunther, S., Kraft, B., Gonser, L., et al. (2015). Quantitative proteomics of the human skin secretome reveal a reduction in immune defense mediators in ectodermal dysplasia patients. J. Invest. Dermatol. 135, 759–767. doi: 10.1038/jid.2014.462
Capone, K. A., Dowd, S. E., Stamatas, G. N., and Nikolovski, J. (2011). Diversity of the human skin microbiome early in life. J. Invest. Dermatol. 131, 2026–2032. doi: 10.1038/jid.2011.168
Cousin, C., Derouiche, A., Shi, L., Pagot, Y., Poncet, S., and Mijakovic, I. (2013). Protein-serine/threonine/tyrosine kinases in bacterial signaling and regulation. FEMS Microbiol. Lett. 346, 11–19. doi: 10.1111/1574-6968.12189
Del Rosso, J. Q., and Leyden, J. J. (2007). Status report on antibiotic resistance: implications for the dermatologist. Dermatol. Clin. 25, 127–132. doi: 10.1016/j.det.2007.01.001
Erickson, B. K., Jedrychowski, M. P., Mcalister, G. C., Everley, R. A., Kunz, R., and Gygi, S. P. (2015). Evaluating multiplexed quantitative phosphopeptide analysis on a hybrid quadrupole mass filter/linear ion trap/orbitrap mass spectrometer. Anal. Chem. 87, 1241–1249. doi: 10.1021/ac503934f
Espersen, F. (1998). Resistance to antibiotics used in dermatological practice. Br. J. Dermatol. 139(Suppl. 53), 4–8. doi: 10.1046/j.1365-2133.1998.1390s3004.x
Francois, P., Scherl, A., Hochstrasser, D., and Schrenzel, J. (2014). Proteomic approach to investigate pathogenicity and metabolism of methicillin-resistant Staphylococcus aureus. Methods Mol. Biol. 1085, 231–250. doi: 10.1007/978-1-62703-664-1_14
Gauthier, N. P., Soufi, B., Walkowicz, W. E., Pedicord, V. A., Mavrakis, K. J., Macek, B., et al. (2013). Cell-selective labeling using amino acid precursors for proteomic studies of multicellular environments. Nat. Methods 10, 768–773. doi: 10.1038/nmeth.2529
Gerber, S. A., Rush, J., Stemman, O., Kirschner, M. W., and Gygi, S. P. (2003). Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945. doi: 10.1073/pnas.0832254100
Grice, E. A., and Segre, J. A. (2011). The skin microbiome. Nat. Rev. Microbiol. 9, 244–253. doi: 10.1038/nrmicro2537
Hanifin, J. M., and Rogge, J. L. (1977). Staphylococcal infections in patients with atopic dermatitis. Arch. Dermatol. 113, 1383–1386. doi: 10.1001/archderm.1977.01640100061009
Hempel, K., Herbst, F. A., Moche, M., Hecker, M., and Becher, D. (2011). Quantitative proteomic view on secreted, cell surface-associated, and cytoplasmic proteins of the methicillin-resistant human pathogen Staphylococcus aureus under iron-limited conditions. J. Proteome Res. 10, 1657–1666. doi: 10.1021/pr1009838
Hempel, K., Pane-Farre, J., Otto, A., Sievers, S., Hecker, M., and Becher, D. (2010). Quantitative cell surface proteome profiling for SigB-dependent protein expression in the human pathogen Staphylococcus aureus via biotinylation approach. J. Proteome Res. 9, 1579–1590. doi: 10.1021/pr901143a
Hu, Q., Noll, R. J., Li, H., Makarov, A., Hardman, M., and Graham Cooks, R. (2005). The Orbitrap: a new mass spectrometer. J. Mass Spectrom. 40, 430–443. doi: 10.1002/jms.856
Iwase, T., Uehara, Y., Shinji, H., Tajima, A., Seo, H., Takada, K., et al. (2010). Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465, 346–349. doi: 10.1038/nature09074
Kim, D., Yu, B. J., Kim, J. A., Lee, Y. J., Choi, S. G., Kang, S., et al. (2013). The acetylproteome of gram-positive model bacterium Bacillus subtilis. Proteomics 13, 1726–1736. doi: 10.1002/pmic.201200001
Kohler, C., Wolff, S., Albrecht, D., Fuchs, S., Becher, D., Buttner, K., et al. (2005). Proteome analyses of Staphylococcus aureus in growing and non-growing cells: a physiological approach. Int. J. Med. Microbiol. 295, 547–565. doi: 10.1016/j.ijmm.2005.08.002
Kong, H. H., Oh, J., Deming, C., Conlan, S., Grice, E. A., Beatson, M. A., et al. (2012). Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 22, 850–859. doi: 10.1101/gr.131029.111
Kuczynski, J., Lauber, C. L., Walters, W. A., Parfrey, L. W., Clemente, J. C., Gevers, D., et al. (2012). Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 13, 47–58. doi: 10.1038/nrg3129
Kuo, I. H., Yoshida, T., De Benedetto, A., and Beck, L. A. (2013). The cutaneous innate immune response in patients with atopic dermatitis. J. Allergy Clin. Immunol. 131, 266–278. doi: 10.1016/j.jaci.2012.12.1563
Lee, C. R., Lee, J. H., Park, K. S., Jeong, B. C., and Lee, S. H. (2015). Quantitative proteomic view associated with resistance to clinically important antibiotics in gram-positive bacteria: a systematic review. Front. Microbiol. 6:828. doi: 10.3389/fmicb.2015.00828
Liao, G., Xie, L., Li, X., Cheng, Z., and Xie, J. (2014). Unexpected extensive lysine acetylation in the trump-card antibiotic producer Streptomyces roseosporus revealed by proteome-wide profiling. J. Proteomics 106, 260–269. doi: 10.1016/j.jprot.2014.04.017
Lin, M. H., Hsu, T. L., Lin, S. Y., Pan, Y. J., Jan, J. T., Wang, J. T., et al. (2009). Phosphoproteomics of Klebsiella pneumoniae NTUH-K2044 reveals a tight link between tyrosine phosphorylation and virulence. Mol. Cell. Proteomics 8, 2613–2623. doi: 10.1074/mcp.M900276-MCP200
List, E. O., Berryman, D. E., Bower, B., Sackmann-Sala, L., Gosney, E., Ding, J., et al. (2008). The use of proteomics to study infectious diseases. Infect. Disord. Drug Targets 8, 31–45. doi: 10.2174/187152608784139640
Macek, B., Gnad, F., Soufi, B., Kumar, C., Olsen, J. V., Mijakovic, I., et al. (2008). Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteomics 7, 299–307. doi: 10.1074/mcp.M700311-MCP200
Macek, B., Mijakovic, I., Olsen, J. V., Gnad, F., Kumar, C., Jensen, P. R., et al. (2007). The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Proteomics 6, 697–707. doi: 10.1074/mcp.M600464-MCP200
Mankowska-Wierzbicka, D., Karczewski, J., Dobrowolska-Zachwieja, A., and Adamski, Z. (2015). The microbiome and dermatological diseases. Postepy Hig. Med. Dosw. 69, 978–985.
Mann, M., and Kelleher, N. L. (2008). Precision proteomics: the case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. U.S.A. 105, 18132–18138. doi: 10.1073/pnas.0800788105
Manteca, A., Ye, J., Sanchez, J., and Jensen, O. N. (2011). Phosphoproteome analysis of Streptomyces development reveals extensive protein phosphorylation accompanying bacterial differentiation. J. Proteome Res. 10, 5481–5492. doi: 10.1021/pr200762y
Misra, S. K., Milohanic, E., Ake, F., Mijakovic, I., Deutscher, J., Monnet, V., et al. (2011). Analysis of the serine/threonine/tyrosine phosphoproteome of the pathogenic bacterium Listeria monocytogenes reveals phosphorylated proteins related to virulence. Proteomics 11, 4155–4165. doi: 10.1002/pmic.201100259
Misra, S. K., Moussan Desiree Ake, F., Wu, Z., Milohanic, E., Cao, T. N., Cossart, P., et al. (2014). Quantitative proteome analyses identify PrfA-responsive proteins and phosphoproteins in Listeria monocytogenes. J. Proteome Res. 13, 6046–6057. doi: 10.1021/pr500929u
Nakatsuji, T., Chiang, H. I., Jiang, S. B., Nagarajan, H., Zengler, K., and Gallo, R. L. (2013). The microbiome extends to subepidermal compartments of normal skin. Nat. Commun. 4:1431. doi: 10.1038/ncomms2441
Neilson, K. A., Ali, N. A., Muralidharan, S., Mirzaei, M., Mariani, M., Assadourian, G., et al. (2011). Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553. doi: 10.1002/pmic.201000553
Nomura, I., Gao, B., Boguniewicz, M., Darst, M. A., Travers, J. B., and Leung, D. Y. (2003). Distinct patterns of gene expression in the skin lesions of atopic dermatitis and psoriasis: a gene microarray analysis. J. Allergy Clin. Immunol. 112, 1195–1202. doi: 10.1016/j.jaci.2003.08.049
Olsen, J. V., Schwartz, J. C., Griep-Raming, J., Nielsen, M. L., Damoc, E., Denisov, E., et al. (2009). A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol. Cell. Proteomics 8, 2759–2769. doi: 10.1074/mcp.M900375-MCP200
Ong, S. E., Blagoev, B., Kratchmarova, I., Kristensen, D. B., Steen, H., Pandey, A., et al. (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386. doi: 10.1074/mcp.M200025-MCP200
Ouidir, T., Cosette, P., Jouenne, T., and Hardouin, J. (2015). Proteomic profiling of lysine acetylation in Pseudomonas aeruginosa reveals the diversity of acetylated proteins. Proteomics 15, 2152–2157. doi: 10.1002/pmic.201500056
Pinkse, M. W., Uitto, P. M., Hilhorst, M. J., Ooms, B., and Heck, A. J. (2004). Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal. Chem. 76, 3935–3943. doi: 10.1021/ac0498617
Rardin, M. J., Held, J. M., and Gibson, B. W. (2013). Targeted quantitation of acetylated lysine peptides by selected reaction monitoring mass spectrometry. Methods Mol. Biol. 1077, 121–131. doi: 10.1007/978-1-62703-637-5_8
Ribeiro, F. J., Przybylski, D., Yin, S., Sharpe, T., Gnerre, S., Abouelleil, A., et al. (2012). Finished bacterial genomes from shotgun sequence data. Genome Res. 22, 2270–2277. doi: 10.1101/gr.141515.112
Rodriguez-Suarez, E., and Whetton, A. D. (2013). The application of quantification techniques in proteomics for biomedical research. Mass Spectrom. Rev. 32, 1–26. doi: 10.1002/mas.21347
Rush, J., Moritz, A., Lee, K. A., Guo, A., Goss, V. L., Spek, E. J., et al. (2005). Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101. doi: 10.1038/nbt1046
Scheltema, R. A., Hauschild, J. P., Lange, O., Hornburg, D., Denisov, E., Damoc, E., et al. (2014). The Q exactive HF, a benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteomics 13, 3698–3708. doi: 10.1074/mcp.M114.043489
Schittek, B. (2011). The antimicrobial skin barrier in patients with atopic dermatitis. Curr. Probl. Dermatol. 41, 54–67. doi: 10.1159/000323296
Schwanhausser, B., Busse, D., Li, N., Dittmar, G., Schuchhardt, J., Wolf, J., et al. (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. doi: 10.1038/nature10098
Singh, S., Springer, M., Steen, J., Kirschner, M. W., and Steen, H. (2009). FLEXIQuant: a novel tool for the absolute quantification of proteins, and the simultaneous identification and quantification of potentially modified peptides. J. Proteome Res. 8, 2201–2210. doi: 10.1021/pr800654s
Soares, N. C., Spat, P., Krug, K., and Macek, B. (2013). Global dynamics of the Escherichia coli proteome and phosphoproteome during growth in minimal medium. J. Proteome Res. 12, 2611–2621. doi: 10.1021/pr3011843
Soufi, B., Gnad, F., Jensen, P. R., Petranovic, D., Mann, M., Mijakovic, I., et al. (2008). The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL1403 reveals multiply phosphorylated proteins. Proteomics 8, 3486–3493. doi: 10.1002/pmic.200800069
Soufi, B., Krug, K., Harst, A., and Macek, B. (2015). Characterization of the E. coli proteome and its modifications during growth and ethanol stress. Front. Microbiol. 6:103. doi: 10.3389/fmicb.2015.00103
Soufi, B., Kumar, C., Gnad, F., Mann, M., Mijakovic, I., and Macek, B. (2010). Stable isotope labeling by amino acids in cell culture (SILAC) applied to quantitative proteomics of Bacillus subtilis. J. Proteome Res. 9, 3638–3646. doi: 10.1021/pr100150w
Soufi, B., and Macek, B. (2014). Stable isotope labeling by amino acids applied to bacterial cell culture. Methods Mol. Biol. 1188, 9–22. doi: 10.1007/978-1-4939-1142-4_2
Stekhoven, D. J., Omasits, U., Quebatte, M., Dehio, C., and Ahrens, C. H. (2014). Proteome-wide identification of predominant subcellular protein localizations in a bacterial model organism. J. Proteomics 99, 123–137. doi: 10.1016/j.jprot.2014.01.015
Stulberg, D. L., Penrod, M. A., and Blatny, R. A. (2002). Common bacterial skin infections. Am. Fam. Physician 66, 119–124.
Sun, F., Ding, Y., Ji, Q., Liang, Z., Deng, X., Wong, C. C., et al. (2012). Protein cysteine phosphorylation of SarA/MgrA family transcriptional regulators mediates bacterial virulence and antibiotic resistance. Proc. Natl. Acad. Sci. U.S.A. 109, 15461–15466. doi: 10.1073/pnas.1205952109
Sun, X., Ge, F., Xiao, C. L., Yin, X. F., Ge, R., Zhang, L. H., et al. (2010). Phosphoproteomic analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J. Proteome Res. 9, 275–282. doi: 10.1021/pr900612v
Swartz, M. N. (2004). Clinical practice. Cellulitis. N. Engl. J. Med. 350, 904–912. doi: 10.1056/NEJMcp031807
van Rensburg, J. J., Lin, H., Gao, X., Toh, E., Fortney, K. R., Ellinger, S., et al. (2015). The human skin microbiome associates with the outcome of and is influenced by bacterial infection. MBio 6:e1315-15. doi: 10.1128/mBio.01315-15
Villen, J., and Gygi, S. P. (2008). The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 3, 1630–1638. doi: 10.1038/nprot.2008.150
Waridel, P., Ythier, M., Gfeller, A., Moreillon, P., and Quadroni, M. (2012). Evidence for a new post-translational modification in Staphylococcus aureus: hydroxymethylation of asparagine and glutamine. J. Proteomics 75, 1742–1751. doi: 10.1016/j.jprot.2011.12.014
Williams, H. C., Dellavalle, R. P., and Garner, S. (2012). Acne vulgaris. Lancet 379, 361–372. doi: 10.1016/S0140-6736(11)60321-8
Yu, B. J., Kim, J. A., Moon, J. H., Ryu, S. E., and Pan, J. G. (2008). The diversity of lysine-acetylated proteins in Escherichia coli. J. Microbiol. Biotechnol. 18, 1529–1536.
Zeeuwen, P. L., Kleerebezem, M., Timmerman, H. M., and Schalkwijk, J. (2013). Microbiome and skin diseases. Curr. Opin. Allergy Clin. Immunol. 13, 514–520. doi: 10.1097/ACI.0b013e328364ebeb
Zeiler, M., Straube, W. L., Lundberg, E., Uhlen, M., and Mann, M. (2012). A protein epitope signature tag (PrEST) library allows SILAC-based absolute quantification and multiplexed determination of protein copy numbers in cell lines. Mol. Cell. Proteomics 11, O111009613. doi: 10.1074/mcp.O111.009613
Keywords: mass spectrometry, proteomics, pathogenic bacteria, skin disease, dermatology, bacterial resistance
Citation: Soufi Y and Soufi B (2016) Mass Spectrometry-Based Bacterial Proteomics: Focus on Dermatologic Microbial Pathogens. Front. Microbiol. 7:181. doi: 10.3389/fmicb.2016.00181
Received: 24 October 2015; Accepted: 02 February 2016;
Published: 19 February 2016.
Edited by:
German Bou, Hospital Universitario A Coruña, SpainReviewed by:
Catrin Ffion Williams, Cardiff University, UKLucía Monteoliva, Universidad Complutense de Madrid, Spain
Copyright © 2016 Soufi and Soufi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Boumediene Soufi, Ym91bWVkaWVuZUBnbWFpbC5jb20=