 Bridget Calder
Bridget Calder Claudia Albeldas
Claudia Albeldas Jonathan M. Blackburn
Jonathan M. Blackburn Nelson C. Soares
Nelson C. Soares- Applied and Chemical Proteomics Group, Medical Biochemistry Division, Faculty of Health Sciences, Institute of Infectious Diseases and Molecular Medicine, University of Cape Town, Cape Town, South Africa
Phosphorylation is a post translational modification which can rapidly regulate biochemical pathways by altering protein function, and has been associated with pathogenicity in bacteria. Once engulfed by host macrophages, pathogenic bacteria are exposed to harsh conditions and must respond rapidly in order to survive. The causative agent of TB, Mycobacterium tuberculosis, is unusual amongst the bacteria because it can survive within the host macrophage for decades in a latent state, demonstrating a remarkable capacity to successfully evade the host immune response. This ability may be mediated in part by regulatory mechanisms such as ser/thr/tyr phosphorylation. Mass spectrometry-based proteomics has afforded us the capacity to identify hundreds of phosphorylation sites in the bacterial proteome, allowing for comparative phosphoproteomic studies in the mycobacteria. There remains an urgent need to validate the reported phosphosites, and to elucidate their biological function in the context of pathogenicity. However, given the sheer number of putative phosphorylation events in the mycobacterial proteome, and the technical difficulty of assigning biological function to a phosphorylation event, it will not be trivial to do so. There are currently six published phosphoproteomic investigations of a member of mycobacteria. Here, we combine the datasets from these studies in order to identify commonly detected phosphopeptides and phosphosites in order to present high confidence candidates for further validation. By applying modern mass spectrometry-based techniques to improve our understanding of phosphorylation and other PTMs in pathogenic bacteria, we may identify candidates for therapeutic intervention.
Introduction
Proteins are the bioactive molecule in the cell, and contribute to survival, growth, and reproduction by interacting with each other, and with metabolites, lipids, nucleic acids and carbohydrates, and catalyzing biological reactions (Nørregaard Jensen, 2004). Protein biosynthesis and degradation are tightly regulated by complex biochemical systems in response to the changing needs of the cell. However, there is an additional mechanism which allows cells to respond rapidly and efficiently to the external and internal conditions. Post translational modification (PTM) by selective covalent processing of proteins—by proteolytic cleavage or the addition of a modifying group—can drastically alter the properties of a protein (Mann and Jensen, 2003; Mijakovic, 2010; Stülke, 2010). Post translational modifications add a layer of complexity to both bacterial and eukaryotic mechanisms of adaptation to the surrounding environment.
Modern mass spectrometry (MS) has enabled us to perform high throughput analysis of PTMs. Since modified proteins usually occur at low abundance, an enrichment process is typically carried out for a specific PTM prior to MS analysis (Semanjski and Macek, 2016). This results in increased resolution, sensitivity and fragmentation (Cain et al., 2014) and has contributed to the identification and localization of phosphosites in many species, including pathogenic bacteria. Once thought to be found only in eukaryotes, the discovery of Hanks-type family of kinases in bacteria indicates that a complex bacterial phosphorylation-mediated signaling system exists (Bakal and Davies, 2000). These serine/threonine protein kinases (STPKs) add a phosphate group to a serine/threonine, while BY-kinases add phosphate groups to Tyrosine residues. PTMs in bacteria have been linked to pathogenicity, virulence, resistance and persistence, and are vital for survival (Ge and Shan, 2011; Van Els et al., 2014). MS-based phosphoproteomic analysis has been carried out in a number of bacterial species, including Bacillus subtilis (Macek et al., 2007), Escherichia coli (Macek et al., 2008; Soares et al., 2013), Streptococcus pneumonia (Sun et al., 2009) Listeria monocytogenes (Misra et al., 2011), Acinetobacter baumanii (Soares et al., 2014), and Mycobacterium tuberculosis.
The Role of Phosphorylation in M. tuberculosis
The bacillus M. tuberculosis is the causative agent of tuberculosis (TB), a leading global health crisis which has claimed millions of lives by continuing to evade clinical intervention. This is largely due to its ability to lay dormant for many years in the body, resurfacing if the hosts' immune system becomes compromised (Gengenbacher and Kaufmann, 2012). When the bacilli enter the human lung, they are ingested by alveolar macrophage cells of the human immune system. The macrophages respond by becoming acidic and exposing the pathogen to lytic enzymes, oxygenated lipids, fatty acids, and reactive oxygen and nitrogen intermediates (Schnappinger et al., 2003). In order to survive in the adverse environment of the macrophage, M. tuberculosis needs to react swiftly, which is possible through the regulatory mechanisms afforded by PTMs (Cain et al., 2014). Mycobacterial STPK phosphorylation has been long associated with pathogenicity (Sherman and Grundner, 2014), which has driven efforts to improve our understanding of the role of phosphorylation in M. tuberculosis (Prisic and Husson, 2014). Currently, there are six published manuscripts describing the phosphoproteome of a member of the Mycobacteria—M. tuberculosis H37Rv (Prisic et al., 2010; Kusebauch et al., 2014), M. smegmatis, and M. bovis BCG (Nakedi et al., 2015; Zheng et al., 2015), a clinical isolate of M. tuberculosis Beijing lineage (Fortuin et al., 2015) and a ΔpknE deletion mutant strain of M. tuberculosis (Parandhaman et al., 2014b). While these studies are discussed in more detail below, a summary of the methods used and relevant results for each of them is presented in Table 1. A major difficulty in comparing phosphoproteomic studies is that we cannot compare those phosphosites that were uniquely identified by each study, because it is impossible to determine whether that uniqueness is as a result of biological differences or the stochastic nature of discovery-driven MS-based proteomics. The only available study in the mycobacteria where those uniquely detected phosphosites are comparable is Nakedi et al. (2015), because the study compared two strains under the same experimental conditions. In this case, a relevant conclusion drawn by the authors is that the phosphosite patterns detected in M. smegmatis and M. bovis BCG are often species specific and these phosphorylation events are commonly occurring on entirely different peptides.
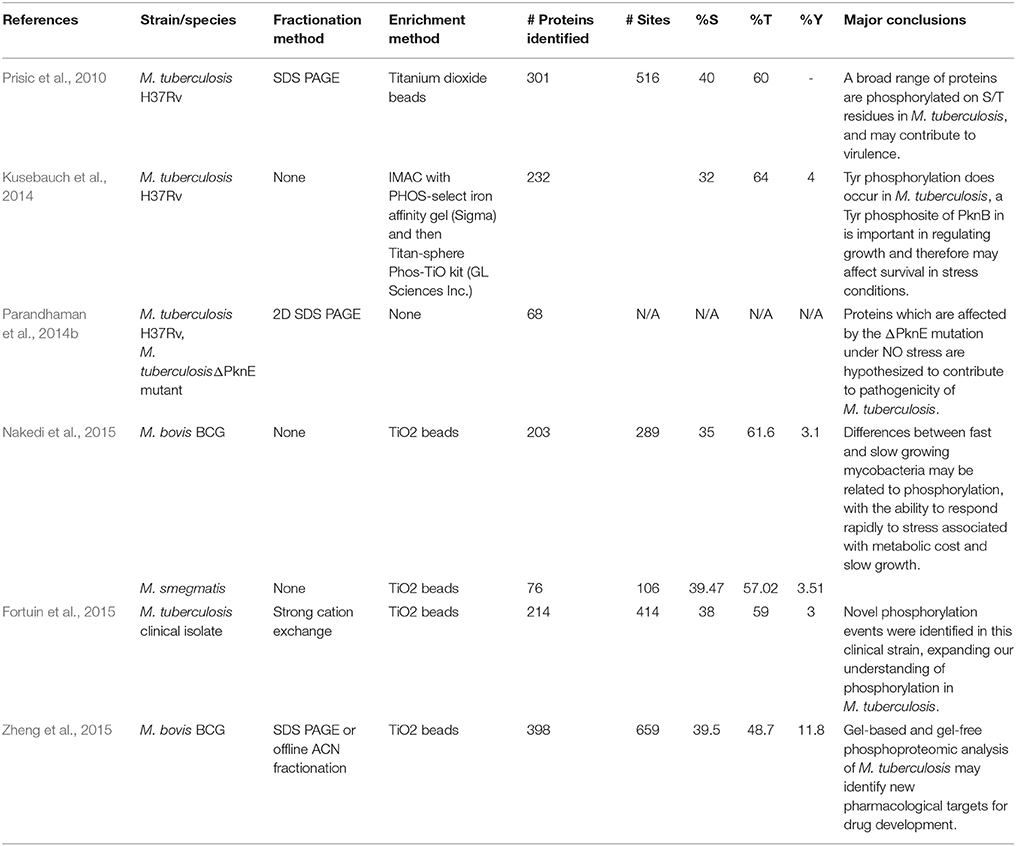
Table 1. An overview of the methods used and results generated in the six currently available published studies investigating phosphorylation in the mycobacteria using mass spectrometry.
The identification of PTMs which contribute to the pathogenicity of M. tuberculosis by enhancing its ability to survive in the macrophage is of great interest to the medical community, as these represent attractive candidates for therapeutic intervention. This review will focus specifically on the use of MS-based techniques which have been used to identify phosphorylated proteins in the mycobacteria, with particular focus on the identification of phosphorylation sites which may contribute to pathogenicity and are conserved across pathogenic strains of mycobacteria.
Comparative Phosphoproteomic Analysis of Mycobacteria
Prisic et al. (2010) were the first to present a global view of the phosphoproteome of M. tuberculosis H37Rv. They used in-gel tryptic digest to proteolytically prepare samples of H37Rv lysate grown under conditions of NO stress, oxidative stress, hypoxia and using glucose, or acetate as a carbon source. A total of 152 samples were analyzed on an LTQ mass spectrometer following enrichment for phosphopeptides using titanium dioxide beads. In this manner, the authors detected a total of 506 phosphosites on 301 proteins and identified a dominant motif for M. tuberculosis STPKs, which was validated using synthetic peptides. This initial investigation reported 40% Ser: 60% Thr phosphorylation, and no Tyr phospho-sites—at the time of publication there was no conclusive molecular evidence for Tyr phosphorylation in M. tuberculosis, although there was a long-established association between Tyr phosphorylation and pathogenicity in other bacteria. (Ilan et al., 1999). Kusebauch et al. (2014) reported Tyr phosphorylation in M. tuberculosis for the first time, after establishing that the known M. tuberculosis STPKs have the capacity to phosphorylate Tyr, and then by carrying out LC MS/MS analysis on M. tuberculosis culture lysate enriched for phosphopeptides. In this manner, they detected 30 high-confidence Tyr phospho-sites on 17 M. tuberculosis proteins, contributing to a Ser: Thr: Tyr ratio of 34:62:4%. Intriguingly, these authors also found an additional 35 Tyr phosphorylation sites in publically accessible MS data from previously published proteomic studies of M. tuberculosis, where the assumption that there was no Tyr phosphorylation in M. tuberculosis had led the authors to overlook them. Subsequent MS based descriptions of the M. tuberculosis phosphoproteome have identified similar proportions of Tyr phosphorylation sites, some of which may be of particular importance in establishing virulence.
Recently, Nakedi et al. (2015) investigated the impact of protein phosphorylation in growth-related functions by measuring differential phosphorylation between two mycobacterial species during exponential growth phase—the fast growing, non-pathogenic, soil dwelling Mycobacterium smegmatis and the slow growing, attenuated strain of Mycobacterium bovis (BCG). BCG had consistently higher phosphorylation levels, with 289 phosphosites on 203 proteins, compared to M. smegmatis with 106 phosphosites found on 76 proteins. The phosphoproteins which were uniquely found in BCG were generally involved in cell growth and stress response. Even under optimal growth conditions, BCG appears to have a high level of phosphorylated stress response proteins which suggests the capability for quick on/off responses to stressors within the host, which ultimately allows the bacteria to respond rapidly and survive more effectively. The potential adaptive advantage for the pathogen may result in a fitness cost of slower growth. In a follow-up phosphoproteomic study, Zheng et al. (2015) found 659 phosphosites on 398 proteins in BCG harvested during stationary phase. The majority (40.1%) of identified phosphoproteins in this case were involved in regulation of metabolism. Again, these findings indicate that phosphorylation plays an important role in the slower metabolism of BCG, which may ultimately increase the capability for persistence within the host, and may be beneficial to the bacteria when faced with drug treatment (Evangelopoulos and McHugh, 2015).
A phosphoproteomic investigation of a hyper-virulent Beijing strain of M. tuberculosis by Fortuin et al. (2015) reported the identification of 414 phosphosites on 214 proteins. Of these, 252 were novel phosphosites which had not been identified in previous phosphoproteome research on the H37Rv strain. Since the capability for complex signaling is directly related to the number of phosphorylation events, it could be inferred that an increasing number of phosphorylation events may play a role in differentiating virulence in different M. tuberculosis strains. This is supported by findings that a highly virulent, drug resistant strain of Acinetobacter baumannii has almost double the number of phosphosites in comparison to the reference strain (Soares et al., 2014). It is also interesting to note that phosphorylation may be linked to drug resistance, which is of concern given the emergence of drug resistant strains of TB (Evangelopoulos and McHugh, 2015). Although the mechanism by which a bacterium might accumulate additional phosphosites is not fully understood, it is of interest that Fortuin et al. (2015) identified phosphorylated forms of 9 out of the 11 STPKs encoded by the M. tuberculosis genome in this hypervirulent Beijing strain (Prisic and Husson, 2014), while Prisic et al. (2010) only identified four in H37Rv.
Although our understanding of the specific activity of Mycobacterial STPKs is largely incomplete, some substrates and their downstream functions have been associated with specific kinases (Supplementary Table 1). While mass spectrometry is capable of identifying hundreds of phospho-substrates, more laborious methods are necessary to associate these substrates with their corresponding kinases. The challenges inherent in mass-spectrometry-based kinase substrate identification are discussed in more detail by Sherman and Grundner (2014). Parandhaman et al. (2014b) made use of a ΔPknE deletion mutant of M. tuberculosis to identify PknE substrates during NO stress in M. tuberculosis, and identified 68 phosphoproteins by combining 2D PAGE MS with phospho-serine and phospho-threonine specific antibodies. The candidate PknE substrates identified in this manner may play a role in dormancy within the macrophage, which may have implications for virulence, once again highlighting the importance of phosphorylation in allowing the bacterium to respond effectively to the host environment. Given this information, and somewhat surprisingly, there is evidence that the PknE gene is non-essential for growth of M. tuberculosis in culture (Sassetti et al., 2003). Indeed, in culture-based models it seems that only three of the STPKs available to M. tuberculosis are individually essential for growth: PknA and PknB (Wehenkel et al., 2008); and PknG, which is essential for survival of M. tuberculosis within the macrophage. Walburger et al. (2004) demonstrated that a strain of M. bovis BCG carrying an inactivated PknG gene showed no differences in growth or cell morphology in liquid medium to the wild type, whereas the mutant was unable to survive when inside a macrophage because the bacteria were no longer able to prevent lysosomal fusion. In many pathogenic bacteria, but particularly in M. tuberculosis, we are increasingly aware of the complexity of the host/pathogen interaction during disease progression, while being limited to culture or animal based models in our attempts to understand it1. The fact that M. tuberculosis has evolved to exist in the intracellular space is highlighted by the metabolic changes which are observed in intracellular M. tuberculosis (Lee et al., 2013), but we have yet to explore the consequences of phosphorylation for M. tuberculosis in vivo. It is conceivable that intracellular STPKs have different activity compared to culture-based systems, and high-throughput phosphoproteomic analysis of intracellular M. tuberculosis would allow us to better understand the role of phosphorylation in the host pathogen interaction.
Identifying Functional Phosphosites for Further Characterization
The aim of discovery-based proteomics has been to catalog as many proteins as possible in a sample, with the intent being to better understand a biological condition or response. However, the volumes of data generated in this manner have not necessarily achieved a deeper understanding of the biological systems in question. This problem is equally as confounding, if not more so, in phosphoproteomic investigations, since it is difficult to attribute biological significance to a discreet phosphorylation event. Many of the detected phosphorylation events reported by these large-scale studies require validation before we can begin to determine their function. To this end, meta-analysis of the available data may provide some insight for future investigations. Although the above-mentioned mycobacterial phosphoproteomic studies were conducted using different MS-based methodologies, and in different strains of mycobacteria, there are discreet phosphorylation events that were commonly observed in several datasets, and are therefore unlikely to be random. A phosphopeptide which is observed in more than one study, or across several mycobacterial strains, may therefore be the best starting point for further investigation in M. tuberculosis. To this end, here we present a compilation of commonly detected phosphopeptides/sites which may thus be of use for future functional phosphoproteomic investigations in the mycobacteria (Supplementary Table 2).
A total of 194 phosphopeptides representing 148 proteins were found to be represented by two or more sets of data according to the available published supplementary information. To facilitate comparison at the protein level, the peptide sequences were matched to H37Rv protein identifiers using the Protein Information Resource (PIR) batch peptide match tool against the Uniprot H37Rv proteome (Chen et al., 2013). GO analysis of the proteins corresponding to these shared phosphopeptides using STRAP 1.5 (Bhatia et al., 2009) revealed that the associated GO terms relate to broad regulatory functions, such as the regulation of cell metabolism, cellular processes, and growth (Figures 1A,B). While this is expected given the established role of phosphorylation in metabolic regulation in bacteria (Kochanowski et al., 2015), this is also potentially noteworthy as we are only beginning to unravel the importance of metabolic regulation for intracellular M. tuberculosis and how this relates to virulence (Eisenreich et al., 2010). Of particular interest are incidences where the more virulent clinical strain is phosphorylated on a different residue compared to the other strains, as in the example of the AEASIETPTPVQSQR peptide of TatA, a Sec-independent protein translocase protein, which was phosphorylated on T7 and/or T9 in M. bovis BCG and M. tuberculosis H37Rv but on S4 in the clinical strain. The functional significance of this difference remains unknown and should be validated and investigated, particularly in light of the contribution of the Tat pathway to virulence in M. tuberculosis (Feltcher et al., 2010). It should be noted that the utility of GO analysis in mycobacteria is limited by the availability of GO annotations and other resources such as KEGG pathway representation, for which coverage is generally poor.
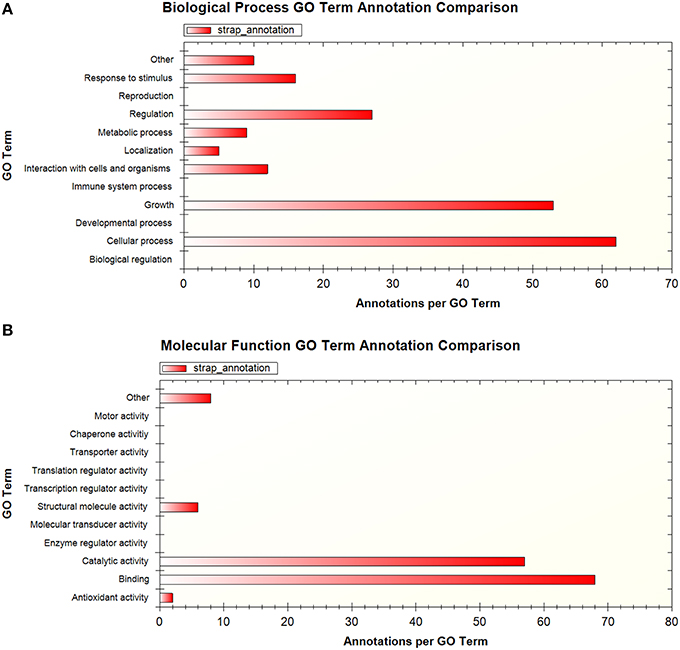
Figure 1. Summary of GO terms associated with proteins that were detected in more than one phosphoproteomic dataset, shown by (A) biological process (B) molecular function. GO analysis performed using STRAP 1.5.
Differential phosphorylation of the STPKs themselves can alter their enzymatic activity, and is another possible mechanism for altered pathogenicity in M. tuberculosis (Chopra et al., 2003; Durán et al., 2005). Supplementary Table 2 highlights that phosphorylation is commonly detected in PknA, B, D, E, G, and H in these mycobacteria; however the differences in localized phosphosites between the virulent and less virulent strains are more pronounced in peptides corresponding to PknA, D, and G. Having already established the importance of PknA (Singh et al., 2006) and G (Walburger et al., 2004) for survival within the macrophage, the biological significance of these differences in phosphorylation and how they contribute virulence should now be ascertained. Forrellad et al. (2013) published a summary of the known virulence factors in the M. tuberculosis complex and their putative contribution to virulence. Cross referencing their table of results to ours identified proteins which are known virulence factors which are also commonly detected in these phosphoproteomic datasets. These proteins and their putative role in virulence in the M. tuberculosis complex are presented in Table 2. Included in these are some previously mentioned STPKs, as well as the proteins KatG, EspR, and IdeR.
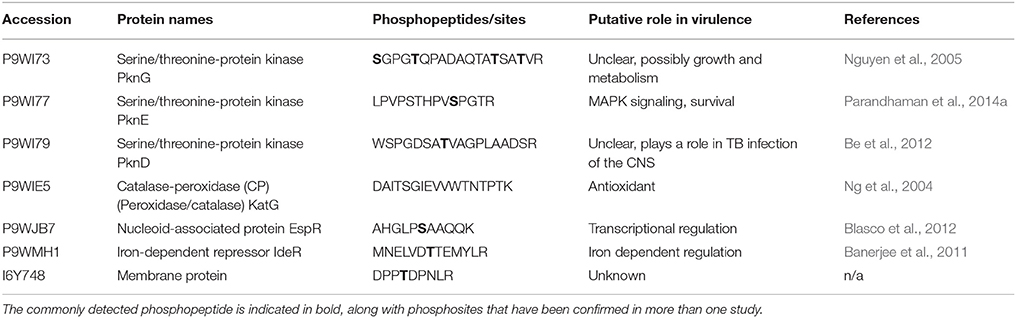
Table 2. Previously reported virulence factors in the M. tuberculosis complex which have been identified in more than one phosphoproteomic dataset, and their putative role in virulence.
Current State of Phosphoproteomics
Currently, the mycobacterial phosphoproteome has been qualitatively described, which has identified many high confidence phosphosites. However, a complete description of the phosphoproteome should include quantitative information for the specific phosphosite in question as well as for the phosphorylated protein. In addition, most of the available phosphoproteomic data is for bacterial culture under a single condition, and yet we recognize that phosphorylation is dynamic and can change rapidly (Macek et al., 2009). In order to address this, it is important to incorporate multiple time points and experimental conditions in future phosphoproteomic investigations. These objectives are specifically challenging for mass spectrometry-based proteomics, and despite promising technological advances the mycobacterial phosphoproteome has not yet been quantitatively assessed (de la Fuente van Bentem et al., 2008). There are many available quantitative proteomic tools which are applicable in bacterial phosphoproteomics, which have been discussed in detail elsewhere (Jers et al., 2008; Macek et al., 2009), however, not all of these are suitable for the mycobacteria. SILAC, for example, has been used successfully to quantify proteins in the bacterium Bacillus subtilis (Ravikumar et al., 2014), but is not currently possible in M. tuberculosis because the available lysine deficient mutants are not viable for SILAC. Alternative quantitative methods include label-free quantitation, which has the benefit of cost effectiveness allowing for multiple time points/conditions to be explored, or dimethyl labeling, which is a promising alternative to iTRAQ and other label-based techniques (Lau et al., 2014). iTRAQ has been successfully used to quantify the phosphorylation of arginine in B. subtilis (Schmidt et al., 2014), and therefore is suitable for measuring S/T/Y phosphorylation in bacterial systems, but is limited by the number of samples that can be analyzed concurrently and the high cost of the iTRAQ reagents. Validation of phosphoproteomic data using targeted proteomics along with specialized analysis software such as Skyline is an exciting prospect, as these powerful tools have the capability to validate specific phosphosites as well as providing quantitative information. The absolute quantitation of modified peptides is possible through a combination of heavy-labeled AQUA peptides and selected reaction monitoring (SRM) MS, although the successful use of this strategy has not yet been reported in bacteria (Kirkpatrick et al., 2005).
Recommendations for Future Research
The field of phosphoproteomics would benefit greatly from an integrated, easily accessible database containing all available information, which would facilitate meta-analysis of PTMs between multiple datasets. Furthermore, such a database for all known PTMs would allow for more in-depth analysis of the cross-talk between bacterial PTMs and how this may relate to pathogenicity, which has been discussed in a review by Soufi et al. (2012). Such a resource could provide a standardized format for reporting PTMs, as well as the opportunity to automatically update database accession numbers for modified proteins—which would in turn facilitate more meaningful comparison between different species or strains. Significant challenges exist prior to the development of such a database, including the lack of standardized reporting in currently published studies. However, the capacity to detect PTMs using MS will only increase with improving technology and the interpretation and management of the data thus generated is therefore crucial if we are to translate this into meaningful clinical applications. Through better understanding of the function of regulatory PTMs in M. tuberculosis, we may reveal the means to control or cure it.
Author Contributions
CA Wrote the original draft of this paper, read, and edited final draft. BC Edited draft submitted by CA, compiled data from phosphoproteomic reviews and performed meta-data analysis, put together final draft of paper. NS Supervised revisions on paper, provided editing support, and directed the topic to be covered by the review. JB Provided supervision and guidance throughout and offered editing support.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
BC and NS thank the South African Medical Research Council for Fellowships. JB acknowledges the South African National Research Foundation for the Research Chair grant. NS acknowledges support in part by the National Research Foundation of South Africa (Grant Numbers 98963 and 95984).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00141
Footnotes
1. ^MISSING:pmid:22320122. MISSING:pmid:22320122. (2016).
References
Bakal, C. J., and Davies, J. E. (2000). No longer an exclusive club: eukaryotic signalling domains in bacteria. Trends Cell Biol. 10, 32–38. doi: 10.1016/S0962-8924(99)01681-5
Banerjee, S., Farhana, A., Ehtesham, N. Z., and Hasnain, S. E. (2011). Iron acquisition, assimilation and regulation in mycobacteria. Infect Genet. Evol. 11, 825–838. doi: 10.1016/j.meegid.2011.02.016
Be, N. A., Bishai, W. R., and Jain, S. K. (2012). Role of Mycobacterium tuberculosis pknD in the pathogenesis of central nervous system tuberculosis. BMC Microbiol. 12:7. doi: 10.1186/1471-2180-12-7
Bhatia, V. N., Perlman, D. H., Costello, C. E., and McComb, M. E. (2009). Software tool for researching annotations of proteins: open-source protein annotation software with data visualization. Anal. Chem. 81, 9819–9823. doi: 10.1021/ac901335x
Blasco, B., Chen, J. M., Hartkoorn, R., Sala, C., Uplekar, S., Rougemont, J., et al. (2012). Virulence regulator EspR of Mycobacterium tuberculosis is a nucleoid-associated protein. PLoS Pathog. 8:e1002621. doi: 10.1371/journal.ppat.1002621
Cain, J. A., Solis, N., and Cordwell, S. J. (2014). Beyond gene expression: the impact of protein post-translational modifications in bacteria. J. Proteomics 97, 265–286. doi: 10.1016/j.jprot.2013.08.012
Chen, C., Li, Z., Huang, H., Suzek, B. E., Wu, C. H., and UniProt Consortium (2013). A fast peptide match service for uniprot knowledgebase. Bioinformatics 29, 2808–2809. doi: 10.1093/bioinformatics/btt484
Chopra, P., Singh, B., Singh, R., Vohra, R., Koul, A., Meena, L. S., et al. (2003). Phosphoprotein phosphatase of Mycobacterium tuberculosis dephosphorylates serine-threonine kinases PknA and PknB. Biochem. Biophys. Res. Commun. 311, 112–120. doi: 10.1016/j.bbrc.2003.09.173
de la Fuente van Bentem, S., Mentzen, W. I., de la Fuente, A., and Hirt, H. (2008). Towards functional phosphoproteomics by mapping differential phosphorylation events in signaling networks. Proteomics 8, 4453–4465. doi: 10.1002/pmic.200800175
Durán, R., Villarino, A., Bellinzoni, M., Wehenkel, A., Fernandez, P., Boitel, B., et al. (2005). Conserved autophosphorylation pattern in activation loops and juxtamembrane regions of Mycobacterium tuberculosis Ser/Thr protein kinases. Biochem. Biophys. Res. Commun. 333, 858–867. doi: 10.1016/j.bbrc.2005.05.173
Eisenreich, W., Dandekar, T., Heesemann, J., and Goebel, W. (2010). Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat. Rev. Microbiol. 8, 401–412. doi: 10.1038/nrmicro2351
Evangelopoulos, D., and McHugh, T. D. (2015). Improving the tuberculosis drug development pipeline. Chem. Biol. Drug Des. 86, 951–960. doi: 10.1111/cbdd.12549
Feltcher, M. E., Sullivan, J. T., and Braunstein, M. (2010). Protein export systems of Mycobacterium tuberculosis: novel targets for drug development? Future Microbiol. 5, 1581–1597. doi: 10.2217/fmb.10.112
Forrellad, M. A., Klepp, L. I., Gioffré, A., Sabio y Garcia, J., Morbidoni, H. R., de la Paz Santangelo, M., et al. (2013). Virulence factors of the Mycobacterium tuberculosis complex. Virulence 4, 3–66. doi: 10.4161/viru.22329
Fortuin, S., Tomazella, G. G., Nagaraj, N., Sampson, S. L., van Pittius, N. C. G., Soares, N. C., et al. (2015). Phosphoproteomics analysis of a clinical Mycobacterium tuberculosis Beijing isolate: expanding the mycobacterial phosphoproteome catalog. Front. Microbiol. 6:6. doi: 10.3389/fmicb.2015.00006
Ge, R., and Shan, W. (2011). Bacterial phosphoproteomic analysis reveals the correlation between protein phosphorylation and bacterial pathogenicity. Genomics. Proteomics Bioinformatics 9, 119–127. doi: 10.1016/S1672-0229(11)60015-6.
Gengenbacher, M., and Kaufmann, S. H. (2012). Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 36, 514–532. doi: 10.1111/j.1574-6976.2012.00331.x
Ilan, O., Bloch, Y., Frankel, G., Ullrich, H., Geider, K., and Rosenshine, I. (1999). Protein tyrosine kinases in bacterial pathogens are associated with virulence and production of exopolysaccharide. EMBO J. 18, 3241–3248. doi: 10.1093/emboj/18.12.3241
Jers, C., Soufi, B., Grangeasse, C., Deutscher, J., and Mijakovic, I. (2008). Phosphoproteomics in bacteria: towards a systemic understanding of bacterial phosphorylation networks. Expert Rev. Proteomics 5, 619–627. doi: 10.1586/14789450.5.4.619
Kirkpatrick, D. S., Gerber, S. A., and Gygi, S. P. (2005). The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods 35, 265–273. doi: 10.1016/j.ymeth.2004.08.018
Kochanowski, K., Sauer, U., and Noor, E. (2015). Posttranslational regulation of microbial metabolism. Curr. Opin. Microbiol. 27, 10–17. doi: 10.1016/j.mib.2015.05.007
Kusebauch, U., Ortega, C., Ollodart, A., Rogers, R. S., Sherman, D. R., Moritz, R. L., et al. (2014). Mycobacterium tuberculosis supports protein tyrosine phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 111, 9265–9270. doi: 10.1073/pnas.1323894111
Lau, H.-T., Suh, H. W., Golkowski, M. G., and Ong, S.-E. (2014). Comparing SILAC- and stable isotope dimethyl-labeling approaches for quantitative proteomics. J. Proteome Res. 13, 4164–4174. doi: 10.1021/pr500630a
Lee, W., VanderVen, B. C., Fahey, R. J., and Russell, D. G. (2013). Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J. Biol. Chem. 288, 6788–6800. doi: 10.1074/jbc.M112.445056
Macek, B., Gnad, F., Soufi, B., Kumar, C., Olsen, J. V., Mijakovic, I., et al. (2008). Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteomics 7, 299–307. doi: 10.1074/mcp.M700311-MCP200
Macek, B., Mann, M., and Olsen, J. V. (2009). Global and site-specific quantitative phosphoproteomics: principles and applications. Annu. Rev. Pharmacol. Toxicol. 49, 199–221. doi: 10.1146/annurev.pharmtox.011008.145606
Macek, B., Mijakovic, I., Olsen, J. V., Gnad, F., Kumar, C., Jensen, P. R., et al. (2007). The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Proteomics 6, 697–707. doi: 10.1074/mcp.M600464-MCP200
Mann, M., and Jensen, O. N. (2003). Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–261. doi: 10.1038/nbt0303-255
Misra, S. K., Milohanic, E., Aké, F., Mijakovic, I., Deutscher, J., Monnet, V., et al. (2011). Analysis of the serine/threonine/tyrosine phosphoproteome of the pathogenic bacterium Listeria monocytogenes reveals phosphorylated proteins related to virulence. Proteomics 11, 4155–4165. doi: 10.1002/pmic.201100259
Nakedi, K. C., Nel, A. J., Garnett, S., Blackburn, J. M., and Soares, N. C. (2015). Comparative Ser/Thr/Tyr phosphoproteomics between two mycobacterial species: the fast growing Mycobacterium smegmatis and the slow growing Mycobacterium bovis BCG. Front. Microbiol. 6:237. doi: 10.3389/fmicb.2015.00237
Ng, V. H., Cox, J. S., Sousa, A. O., MacMicking, J. D., and McKinney, J. D. (2004). Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52, 1291–1302. doi: 10.1111/j.1365-2958.2004.04078.x
Nguyen, L., Walburger, A., Houben, E., Koul, A., Muller, S., Morbitzer, M., et al. (2005). Role of protein kinase G in growth and glutamine metabolism of Mycobacterium bovis BCG. J. Bacteriol. 187, 5852–5856. doi: 10.1128/JB.187.16.5852-5856.2005
Nørregaard Jensen, O. (2004). Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 8, 33–41. doi: 10.1016/j.cbpa.2003.12.009
Parandhaman, D. K., Hanna, L. E., and Narayanan, S. (2014a). PknE, a serine/threonine protein kinase of Mycobacterium tuberculosis initiates survival crosstalk that also impacts HIV coinfection. PLoS ONE 9:e83541. doi: 10.1371/journal.pone.0083541
Parandhaman, D. K., Sharma, P., Bisht, D., and Narayanan, S. (2014b). Proteome and phosphoproteome analysis of the serine/threonine protein kinase E mutant of Mycobacterium tuberculosis. Life Sci. 109, 116–126. doi: 10.1016/j.lfs.2014.06.013
Prisic, S., Dankwa, S., Schwartz, D., Chou, M. F., Locasale, J. W., Kang, C.-M., et al. (2010). Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U.S.A. 107, 7521–7526. doi: 10.1073/pnas.0913482107
Prisic, S., and Husson, R. N. (2014). Mycobacterium tuberculosis serine/threonine protein kinases. Microbiol. Spectr. 2. doi: 10.1128/microbiolspec.MGM2-0006-2013
Ravikumar, V., Shi, L., Krug, K., Derouiche, A., Jers, C., Cousin, C., et al. (2014). Quantitative phosphoproteome analysis of Bacillus subtilis reveals novel substrates of the kinase PrkC and phosphatase PrpC. Mol. Cell. Proteomics 13, 1965–1978. doi: 10.1074/mcp.M113.035949
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84. doi: 10.1046/j.1365-2958.2003.03425.x
Schmidt, A., Trentini, D. B., Spiess, S., Fuhrmann, J., Ammerer, G., Mechtler, K., et al. (2014). Quantitative phosphoproteomics reveals the role of protein arginine phosphorylation in the bacterial stress response. Mol. Cell. Proteomics 13, 537–550. doi: 10.1074/mcp.M113.032292
Schnappinger, D., Ehrt, S., Voskuil, M. I., Liu, Y., Mangan, J. A., Monahan, I. M., et al. (2003). Transcriptional adaptation of Mycobacterium tuberculosis within macrophages insights into the phagosomal environment. J. Exp. Med. 198, 693–704. doi: 10.1084/jem.20030846
Semanjski, M., and Macek, B. (2016). Shotgun proteomics of bacterial pathogens: advances, challenges and clinical implications. Expert Rev. Proteomics 13, 139–156. doi: 10.1586/14789450.2016.1132168
Sherman, D. R., and Grundner, C. (2014). Agents of change-concepts in Mycobacterium tuberculosis Ser/Thr/Tyr phosphosignalling. Mol. Microbiol. 94, 231–241. doi: 10.1111/mmi.12747
Singh, A., Singh, Y., Pine, R., Shi, L., Chandra, R., and Drlica, K. (2006). Protein kinase I of Mycobacterium tuberculosis: cellular localization and expression during infection of macrophage-like cells. Tuberculosis (Edinb) 86, 28–33. doi: 10.1016/j.tube.2005.04.002
Soares, N. C., Spät, P., Krug, K., and and, Macek, B. (2013). Global dynamics of the Escherichia coli proteome and phosphoproteome during growth in minimal medium. J. Proteome Res. 12, 2611–2621. doi: 10.1021/pr3011843
Soares, N. C., Spät, P., Méndez, J. A., Nakedi, K., Aranda, J., and Bou, G. (2014). Ser/Thr/Tyr phosphoproteome characterization of Acinetobacter baumannii: comparison between a reference strain and a highly invasive multidrug-resistant clinical isolate. J. Proteomics 102, 113–124. doi: 10.1016/j.jprot.2014.03.009
Soufi, B., Soares, N. C., Ravikumar, V., and Macek, B. (2012). Proteomics reveals evidence of cross-talk between protein modifications in bacteria: focus on acetylation and phosphorylation. Curr. Opin. Microbiol. 15, 357–363. doi: 10.1016/j.mib.2012.05.003
Stülke, J. (2010). More than just activity control: phosphorylation may control all aspects of a protein's properties. Mol. Microbiol. 77, 273–275. doi: 10.1111/j.1365-2958.2010.07228.x
Sun, X., Ge, F., Xiao, C.-L., Yin, X.-F., Ge, R., Zhang, L.-H., et al. (2009). Phosphoproteomic analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J. Proteome Res. 9, 275–282. doi: 10.1021/pr900612v
Van Els, C. A. C. M., Corbière, V., Smits, K., van Gaans-van den Brink, J. A. M., Poelen, M. C. M., Mascart, F., et al. (2014). Toward Understanding the Essence of Post-Translational Modifications for the Mycobacterium tuberculosis Immunoproteome. Front. Immunol. 5:361. doi: 10.3389/fimmu.2014.00361
Walburger, A., Koul, A., Ferrari, G., Nguyen, L., Prescianotto-Baschong, C., Huygen, K., et al. (2004). Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304, 1800–1804. doi: 10.1126/science.1099384
Wehenkel, A., Bellinzoni, M., Graña, M., Duran, R., Villarino, A., Fernandez, P., et al. (2008). Mycobacterial Ser/Thr protein kinases and phosphatases: physiological roles and therapeutic potential. Biochim. Biophys. Acta 1784, 193–202. doi: 10.1016/j.bbapap.2007.08.006
Keywords: post-translational modification, phosphorylation, mycobacteria, tuberculosis, proteomics, phosphoproteomics, mass spectrometry, virulence
Citation: Calder B, Albeldas C, Blackburn JM and Soares NC (2016) Mass Spectrometry Offers Insight into the Role of Ser/Thr/Tyr Phosphorylation in the Mycobacteria. Front. Microbiol. 7:141. doi: 10.3389/fmicb.2016.00141
Received: 25 October 2015; Accepted: 25 January 2016;
Published: 12 February 2016.
Edited by:
Biswarup Mukhopadhyay, Virginia Tech, USAReviewed by:
Haike Antelmann, Freie Universität Berlin, GermanyIvan Mijakovic, Chalmers University of Technology, Sweden
Copyright © 2016 Calder, Albeldas, Blackburn and Soares. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan M. Blackburn, am9uYXRoYW4uYmxhY2tidXJuQHVjdC5hYy56YQ==;
Nelson C. Soares, bmVsc29uLmRhY3J1enNvYXJlc0B1Y3QuYWMuemE=