 Aaron Lerner
Aaron Lerner Rustam Aminov
Rustam Aminov Torsten Matthias2
Torsten Matthias2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Microbiol., 05 February 2016
Sec. Microbial Immunology
Volume 7 - 2016 | https://doi.org/10.3389/fmicb.2016.00084
This article is part of the Research TopicMicrobial and environmental factors in autoimmune and inflammatory diseasesView all 18 articles
The gut ecosystem with myriads of microorganisms and the high concentration of immune system cells can be considered as a separate organ on its own. The balanced interaction between the host and microbial cells has been shaped during the long co-evolutionary process. In dysbiotic conditions, however, this balance is compromised and results in abnormal interaction between the host and microbiota. It is hypothesize here that the changed spectrum of microbial enzymes involved in post-translational modification of proteins (PTMP) may contribute to the aberrant modification of host proteins thus generating autoimmune responses by the host, resulting in autoimmune diseases.
Intricate host–microbe symbiotic relationships in the human gut have evolved during the long-term coevolution between the two. It resulted in fine-tuned inter kingdom molecular adaptations that benefit both sides (Donia and Fischbach, 2015). In particular, the commensal microbiota benefits from the continuous food supply, exogenous and endogenous, and constant physico-chemical conditions. Advantages for the host organism include metabolic, structural, protective, and other beneficial functions exerted by the commensal microbiota. The importance of commensal microbiota for the proper development and functioning of the host immune system is also well-recognized (Paun and Danska, 2015).
Our knowledge on the topic expands continuously. The complex microbiota of our gastrointestinal tract consists of at least 1000 bacterial species, the majority of which belong to the Firmicutes and Bacteroidetes phyla (Qin et al., 2010). The microbiota composition and function is very dynamic and multiple environmental factors affect its quantity, quality and functionality. Age, mode of delivery, infant feeding practices, the use of drugs (antibiotics), diet, industrial food additives, epidemiology, climate, eco-catastrophes, migrations, and many more are the influential factors (Lerner, 2011; Chassaing et al., 2015; Lerner and Matthias, 2015a,b,c).
It is not our intention to cover all the aforementioned factors that may affect host-microbe interaction. Here, we will provide an update on the dysbiotic situations, where the normal composition of the commensal is compromised, which may result in pathologies such as autoimmune diseases. More specifically, we will be focusing on the microbial enzymatic machinery that is involved in post-translational modification of proteins (PTMP). We hypothesize that the enzymes produced by the dysbiotic microbial community may process luminal proteins differently than that of the normal community. The abnormal PTMP may produce neo-epitopes that are autoimmunogenic and may induce systemic autoimmune responses resulting in autoimmune diseases.
Presently, it is apparent that the microbiota and its products have a profound effect on the development and maintenance of the immune system. Germ-free animals, for example, have an impaired immune system that can be functionally restored after the inoculation of commensal bacteria (MacPherson et al., 2001, 2002; Mazmanian et al., 2005). The extent of dependency of the immune system on commensals may even suggest the commensalocentric view. At the same time, not all commensals are alike. The dysbiotic populations, with no identifiable pathogens, can still confer the susceptibility to immune-mediated diseases (Paun and Danska, 2015). Our focus here on autoimmune diseases and several mechanisms of the microbial involvement in the promotion of autoimmunity have been suggested (Chervonsky, 2013). The first one is the molecular mimicry, where microbial peptides are identical, or similar enough to self-peptides. Second, it could be bystander activation during infection, with the induction of costimulation and cytokine production by APCs, which, at the same time, may presents self-antigens. The third suggested mechanism is the “amplification of autoimmunity by cytokines” elicited by microbial activation of professional APCs and the innate lymphoid cells to produce proinflammatory cytokines by T cells. And the fourth suggestion is the involvement of the whole human intestinal system, including dysbiotic community, exogenous enzymes produced by the dysbiotic populations, and the corresponding PTMP activity that generates neo-epitopes.
The multiple animal models of human autoimmune diseases (AD) suggest the direct involvement of commensal microbiota in disease development. Under the germ-free conditions no disease is developing in the animal models of IBD, rheumatoid arthritis and multiple sclerosis, supporting the notion of “no bugs, no disease,” while in some others they are only attenuated (Wu and Wu, 2012). In some models of the human ADs, causality is strengthened by the reintroduction of specific microorganisms restoring the disease severity.
Some members of the gut microbiota have been linked to ADs. Changing a single bacterial species and/or the entire commensal community can alter the outcome of a specific AD due to the imbalance of pathological/protective immune responses (Wu and Wu, 2012). The summary of specific microbial species in relation to defined animal models of ADs and their functions, in relation to disease progression, is shown in Table 1.
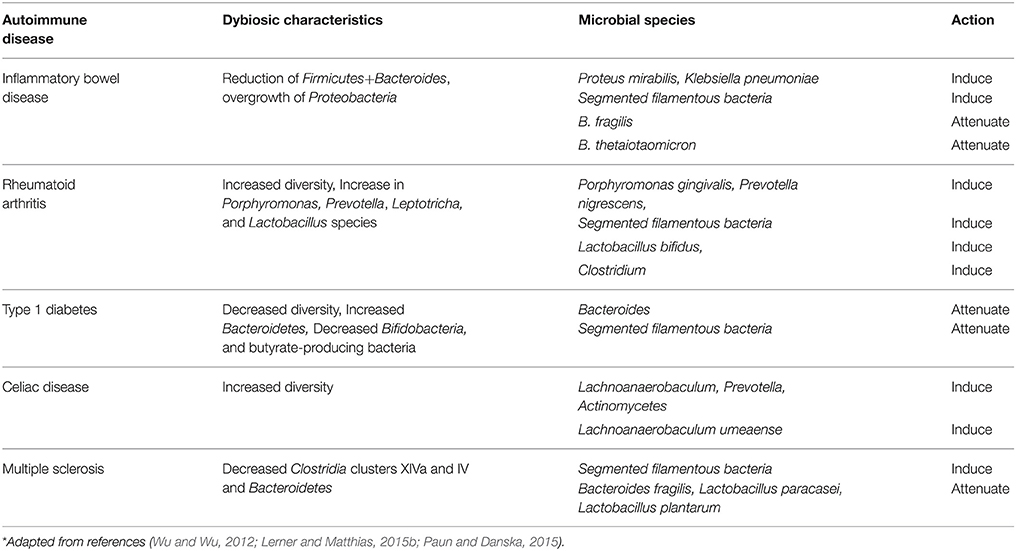
Table 1. Autoimmune disease and associated dysbiotic characteristics*.
Endogenous and microbial enzymes have the capacity of intestinal enzymatic neo-antigen generation by PTMP. The corresponding modifications taking place in the intestine include peptides crosslinking, de/amination/deamidation, de/phosphorylation, a/deacetylation, de/tyrosination, de/glutamylation, de/glycylation, ubiquitination, palmitoylation, glycosylation, galactosylation, arginylation, methylation, citrullination, sumoylation, and carbamylation. For example, human endogenous intestinal enzymes and microbial transglutaminases (tTg, mTg) induce multiple neo-epitopes on the Tg-peptide docked complex or citrullination by peptidylarginine deiminase resulting in the formation of autoantibodies in celiac disease and rheumatoid arthritis, respectively. A similar phenomenon is observed in other ADs. Bacterial species, their corresponding enzymes capable of PTM of host proteins and potential involvement in disease are listed in Table 2.
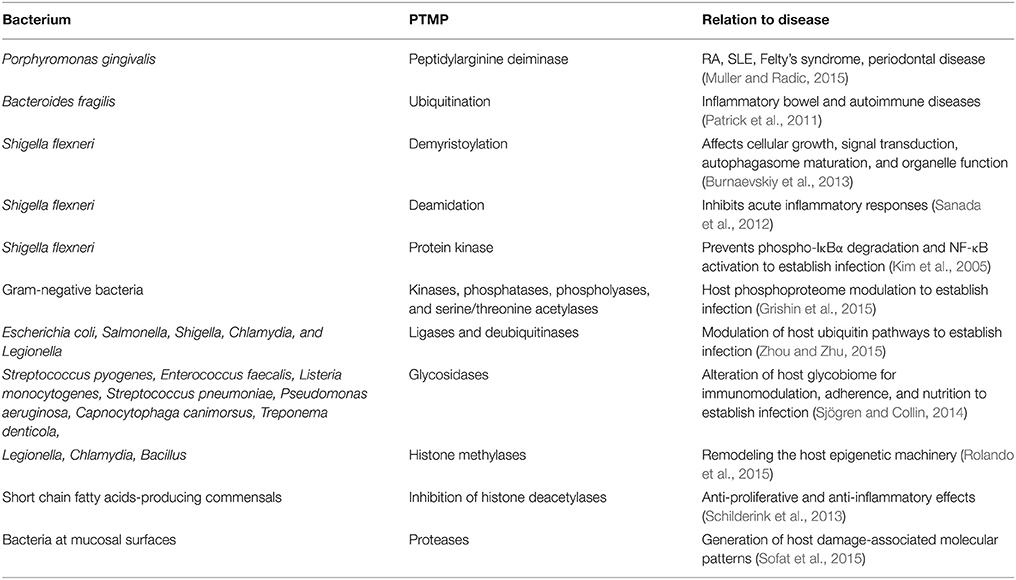
Table 2. Bacterial enzymes capable of post-translational modification of host proteins and potential involvement in disease.
Bacteria possess an amazing capacity for adaptation and survival strategies, including differential expression of transcriptome and proteome, variations in growth physiology, and in developmental behavior. PTMP contribute substantially to this adaptability and bacterial cell cycle regulation (Grageasse et al., 2015). On the other hand, the microbial PTMP has a paramount significance to the host. Their enzymatic apparatus is capable to transform naïve/self or non-self-peptides to autoimmunogenic ones. Several examples are worth mentioning.
A well-described PTMP is the citrullination, where an arginine residue in a protein is converted to a citrulline residue. The host enzyme is activated during apoptosis, autophagy, and NETosis processes, which are well-known as being implicated in autoimmunity (Valesini et al., 2015). In fact, some anti-citrullinated protein antibodies may serve as good diagnostic markers for autoimmune diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and Felty's syndrome (Muller and Radic, 2015). The citrullinated proteins have also been detected in polymyositis and IBD. Some bacteria such as Porphyromonas gingivalis possess the corresponding enzymes, peptidylarginine deiminases, which may citrullinate human proteins (Wegner et al., 2010). The authors suggested that P. gingivalis-mediated citrullination of bacterial and host proteins may lead to the generation of antigens driving the autoimmune response in RA. Experimental verification of the specificity and activity of peptidylarginine deiminase from P. gingivalis revealed that it is primarily cell surface associated, heat stable, and display the optimal activity under alkaline conditions characteristic for the inflamed environment (Abdullah et al., 2013). Moreover, the enzyme has very broad substrate specificity and modifies arginine residues in all positions of all proteins tested. Thus, its presence in inflamed tissue may foster autoimmune reactions by altering host epitopes. In experimental models of periodontal disease and arthritis it has been found that peptidylarginine deiminase from P. gingivalis is a key contributor to the pathogenesis of these diseases (Gully et al., 2014). In human subjects, there is a strong association between the presence of P. gingivalis peptidylarginine deiminase and diseases such as RA and periodontitis (Laugisch et al., 2015; Shimada et al., 2015). In both diseases, the microbial enzyme convert arginine to a citrulline residue, thus creating a neo-antigen.
More recently, an additional PTMP activity, lysine acetylation, has been detected in P. gingivalis in addition to its citrullination capacity (Butler et al., 2015). A unique homolog of the eukaryotic ubiquitin has been found in the predominant bacterium of the human gut, Bacteroides fragilis (Patrick et al., 2011). In eukaryotes post-translational regulation by ubiquitination plays an important role in regulation of intracellular proteolysis and modification of protein function. The ubiquitination process is also central for immune surveillance and response to invading pathogens. Its presence in a predominant human gut bacterium may have important implications for our understanding of AD development.
One of the factors contributing to autoimmune progression is the cellular environment that may change PTMP (Cañas et al., 2015). PTMP of histones alter the chromatin architecture thus generating “open” and “closed” states, and these structural changes can modulate gene expression under specific conditions. While methylation and acetylation are the best-characterized histone PTMP, citrullination by the protein arginine deiminases represents another important player in this process (Slade et al., 2014). Juxtaposition of citrullinated histones with infectious pathogens and complement and immune complexes may compromise the tolerance to nuclear autoantigens and promote autoimmunity (Muller and Radic, 2015). Like a double-edged sword, histones can be post-translationally modified also by Tg cross-linking, a well-described PTMP (Lerner and Matthias, 2015c). Likewise, microbial transglutaminase, a member of the Tg family, is known for its pivotal function in bacterial survival (Lerner and Matthias, 2015c). Bacterial glyosidases are involved in PTMP and many key proteins of the immune system are glycosylated (Sjögren and Collin, 2014). The glycosylation sites of IgE, IgM, IgD, IgE, IgA, and IgG are functionally important, and they are responsible for the well-documented association between alterations of the serum glycome and autoimmunity. The altered glycan theory of autoimmunity has been recently suggested (Maverakis et al., 2015). It implies that each AD has a unique glycan signature characterized by the site-specific relative abundances of individual glycan structures on immune cells and extracellular proteins. This especially concerns the site-specific glycosylation patterns of different immunoglobulin classes and subclasses (Maverakis et al., 2015).
A well-characterized example of PTMP is the tTg in celiac disease that will be discussed in the next section.
This topic still needs to be revealed in greater details, but there are some hints for at least two ADs. In celiac disease, the autoantigen is tTg, capable of deamidating, or transamidating gliadin (Reif and Lerner, 2004; Lerner et al., 2015a). This PTMP occurs below the epithelium, resulting in neo-epitopes of gliadin docked on the tTg, inducing anti-tTg, or anti neo-epitope tTg autoantibodies. These are the well-known serological markers of celiac disease (Lerner, 2014; Lerner et al., 2015b). More recently, a family member of tTg, the microbial Tg that is heavily used in the food industry, has been shown as a potent inducer of specific antibodies in celiac disease patients (Lerner and Matthias, 2015d). Interestingly, the same food additive has been suggested as a new environmental trigger and potential inducer of celiac disease (Lerner and Matthias, 2015a,c).
A number of PTMPs are relevant to IBD. These pathways include phosphorylation, neddylation hydroxylation, and cleavage of cytokine precursor forms by the inflammasome. The interplay of these rapid response mechanisms enables rapid adaptation to incoming inflammatory signals. Cytokine induced barrier breakdown allows for bacterial translocation to the basal aspect of intestinal epithelial cells. Bacterial antigens and endogenous danger signals are recognized by the adaptive and innate immune system, triggering a variety of reactions including apoptosis, increased cytokine release, loss of tight junctional proteins, and barrier breakdown (Ehrentraut and Colgan, 2012).
In rheumatoid arthritis, citrullination is a major post-translational modification of arginine, which converts naïve peptides into the immunogenic neo-epitopes. This PTMP constitutes the basis for the specific prediction of disease activity due to the production of anti-citrullinated protein antibodies (Lerner and Matthias, 2015b). It has been suggested that infectious agents that release toxins such as lipopolysaccharides at mucosal surfaces may trigger the inflammatory response with a potential to cause citrullination of various proteins such as fibronectin, fibrinogen, and collagen (Sofat et al., 2015).
Theoretically, it can be assumed that PTMP may take place in the lumen, on the intestinal, or buccal mucosal surfaces, in the interepithelial spaces, or below the epithelium. There are more questions, however, than definitive answers.
We hypothesize here that the PTM enzymes of dysbiotic gut microbiota behave like a Trojan horse. They are essential for the microbial growth and survival in the gut, but are detrimental to the human host. This enzymatic machinery is capable of PTMP, turning naïve peptides to immunogenic ones by generating, or exposing neo-epitopes, thus compromising tolerance and inducing autoimmunity. Peptides crosslinking, de/amination/deamidation, de/phosphorylation, a/deacetylation, de/tyrosination, de/glutamylation, de/glycylation, ubiquitination, palmitoylation, glycosylation, galactosylation, arginylation, methylation, citrullination, sumoylation and carbamylation are some examples for PTMP taking place in the intestine. The corresponding microbial enzymes that encounter the closely related host substrates under the optimal local conditions, act as post-translational modifiers of the host's peptides in the initiation, progression and maintenance of human systemic ADs.
There are a number of factors, genetic and environmental, that have been identified as conducive to dysbiotic conditions in humans. We hypothesize here that the spectrum and activities of enzymes, which are normally involved in PTMP, become biased in the dysbiotic microbial community. The examples are given demonstrating how these activities may affect post-translational modification of host proteins thus generating new aberrant epitopes. These epitopes may generate host autoimmune responses and trigger autoimmune diseases. Much less is known, however, how to convert the “pathobiota” back to the “normobiota” to restore the balanced host-microbe interaction with a normal PTMP pattern. Answering this question will be the basis for the development of efficient therapeutic strategies to prevent autoimmune diseases.
AL and RA designed, performed literature search and wrote the manuscript; TM designed, constructed table, edited and wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdullah, S. N., Farmer, E. A., Spargo, L., Logan, R., and Gully, N. (2013). Porphyromonas gingivalis peptidylarginine deiminase substrate specificity. Anaerobe 23, 102–108. doi: 10.1016/j.anaerobe.2013.07.001
Burnaevskiy, N., Fox, T. G., Plymire, D. A., Ertelt, J. M., Weigele, B. A., Selyunin, A. S., et al. (2013). Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature 496, 106–109. doi: 10.1038/nature12004
Butler, C. A., Veith, P. D., Nieto, M. F., Dashper, S. G., and Reynolds, E. C. (2015). Lysine acetylation is a common post-translational modification of key metabolic pathway enzymes of the anaerobe Porphyromonas gingivalis. J. Proteomics 128, 352–364. doi: 10.1016/j.jprot.2015.08.015
Cañas, C. A., Cañas, F., Bonilla-Abadía, F., Ospina, F. E., and Tobón, G. J. (2015). Epigenetics changes associated to environmental triggers in autoimmunity. Autoimmunity. doi: 10.3109/08916934.2015.1086996. [Epub ahead of print].
Chassaing, B., Koren, O., Goodrich, J. K., Poole, A. C., Srinivasan, S., Ley, R. E., et al. (2015). Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519, 92–96. doi: 10.1038/nature14232
Chervonsky, A. V. (2013). Microbiota and autoimmunity. Cold Spring Harb. Perspect. Biol. 5:a007294. doi: 10.1101/cshperspect.a007294
Donia, M. S., and Fischbach, M. A. (2015). Small molecules from the human microbiota. Science 349, 1254766. doi: 10.1126/science.1254766
Ehrentraut, S. F., and Colgan, S. P. (2012). Implications of protein post-translational modifications in IBD. Inflamm. Bowel Dis. 18, 1378–1388. doi: 10.1002/ibd.22859
Grageasse, C., Stülke, J., and Mijakovic, I. (2015). Regulatory potential of post-translational modifications in bacteria. Front. Microbiol. 6:500. doi: 10.3389/fmicb.2015.00500
Grishin, A. M., Beyrakhova, K. A., and Cygler, M. (2015). Structural insight into effector proteins of Gram-negative bacterial pathogens that modulate the phosphoproteome of their host. Protein Sci. 24, 604–620. doi: 10.1002/pro.2636
Gully, N., Bright, R., Marino, V., Marchant, C., Cantley, M., Haynes, D., et al. (2014). Porphyromonas gingivalis peptidylarginine deiminase, a key contributor in the pathogenesis of experimental periodontal disease and experimental arthritis. PLoS ONE 9:e100838. doi: 10.1371/journal.pone.0100838
Kim, D. W., Lenzen, G., Page, A. L., Legrain, P., Sansonetti, P. J., and Parsot, C. (2005). The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. 102, 14046–14051. doi: 10.1073/pnas.0504466102
Laugisch, O., Wong, A., Sroka, A., Kantyka, T., Koziel, J., Neuhaus, K., et al. (2015). Citrullination in the periodontium - a possible link between periodontitis and rheumatoid arthritis. Clin. Oral Investig. doi: 10.1007/s00784-015-1556-7. [Epub ahead of print].
Lerner, A. (2011). The last two millennias echo-catastrophes are the driving forces for the potential genetic advantage mechanisms in celiac disease. Med. Hypotheses 77, 773–776. doi: 10.1016/j.mehy.2011.07.034
Lerner, A. (2014). Serological diagnosis of celiac disease –moving beyond the tip of the iceberg. Int. J. Celiac Dis. 2, 64–66. doi: 10.12691/ijcd-2-2-8
Lerner, A., and Matthias, T. (2015a). Changes in intestinal tight junction permeability associated with industrial food additives explain the rising incidence of autoimmune disease. Autoimmun. Rev. 14, 479–489. doi: 10.1016/j.autrev.2015.01.009
Lerner, A., and Matthias, T. (2015b). Rheumatoid arthritis-celiac disease relationship: joints get that gut feeling. Autoimm. Rev. 14, 1038–1047. doi: 10.1016/j.autrev.2015.07.007
Lerner, A., and Matthias, T. (2015c). Possible association between celiac disease and bacterial transglutaminase in food processing: a hypothesis. Nutr. Rev. 73, 544–552. doi: 10.1093/nutrit/nuv011
Lerner, A., and Matthias, T. (2015d). “Microbial transglutaminase is a potential environmental inducer of celiac disease,” in Autoantigens, Autoantibodies, Autoimmunity, Vol. 10, eds K Conrad, E. K. L. Chan, L. E. C. Andrade, G. Steiner, G. J. M. Pruijn, and Y Shoenfeld (Lengerich: Pabst Science Publishers), 227–233.
Lerner, A., Neidhöfer, S., Jeremias, P., and Matthias, T. (2015b). “The diversities between the neo-epitope and the IgA- tissue transglutaminase autoantibodies in celiac disease,” in Autoantigens, Autoantibodies, Autoimmunity, Vol. 10, eds K Conrad, E. K. L. Chan, L. E. C. Andrade, G. Steiner, G. J. M. Pruijn, and Y Shoenfeld (Lengerich: Pabst Science Publishers), 220–226.
Lerner, A., Neidhöfe, S., and Torsten Matthias, T. (2015a). Transglutaminase 2 and anti transglutaminase 2 autoantibodies in celiac disease and beyond. Part B: anti-transglutaminase 2 autoantibodies: friends or enemies. Immunome Res. 11:100. doi: 10.4172/1745-7580.10000100
MacPherson, A. J., Hunziker, L., McCoy, K., and Lamarre, A. (2001). IgA responses in the intestinal mucosa against pathogenic and non-pathogenic microorganisms. Microbes Infect. 3, 1021–1035. doi: 10.1016/S1286-4579(01)01460-5
MacPherson, A. J., Martinic, M. M., and Harris, N. (2002). The functions of mucosal T cells in containing the indigenous commensal flora of the intestine. Cell. Mol. Life Sci. 59, 2088–2096. doi: 10.1007/s000180200009
Maverakis, E., Kim, K., Shimoda, M., Gershwin, M. E., Patel, F., Wilken, R., et al. (2015). Glycans in the immune system and the Altered Glycan Theory of Autoimmunity: a critical review. J. Autoimmun. 57, 1–13. doi: 10.1016/j.jaut.2014.12.002
Mazmanian, S. K., Liu, C. H., Tzianabos, A. O., and Kasper, D. L. (2005). An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118. doi: 10.1016/j.cell.2005.05.007
Muller, S., and Radic, M. (2015). Citrullinated Autoantigens: from diagnostic markers to pathogenetic mechanisms. Clin. Rev. Allergy Immunol. 49, 232–239. doi: 10.1007/s12016-014-8459-2
Patrick, S., Jobling, K. L., O'Connor, D., Thacker, Z., Dryden, D. T., and Blakely, G. W. (2011). A unique homologue of the eukaryotic protein-modifier ubiquitin present in the bacterium Bacteroides fragilis, a predominant resident of the human gastrointestinal tract. Microbiology 157, 3071–3078. doi: 10.1099/mic.0.049940-0
Paun, A., and Danska, J. S. (2015). Immuno-ecology: how the microbiome regulates tolerance and autoimmunity. Curr. Opin. Immunol. 37, 34–39. doi: 10.1016/j.coi.2015.09.004
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). MetaHIT Consortium A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
Reif, S., and Lerner, A. (2004). Tissue transglutaminase – the key player in celiac disease: a review. Autoimmun. Rev. 3, 40–45. doi: 10.1016/S1568-9972(03)00065-X
Rolando, M., Gomez-Valero, L., and Buchrieser, C. (2015). Bacterial remodelling of the host epigenome: functional role and evolution of effectors methylating host histones. Cell. Microbiol. 17, 1098–1107. doi: 10.1111/cmi.12463
Sanada, T., Kim, M., Mimuro, H., Suzuki, M., Ogawa, M., Oyama, A., et al. (2012). The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature 483, 623–626. doi: 10.1038/nature10894
Schilderink, R., Verseijden, C., and de Jonge, W. J. (2013). Dietary inhibitors of histone deacetylases in intestinal immunity and homeostasis. Front. Immunol. 4:226. doi: 10.3389/fimmu.2013.00226
Shimada, A., Kobayashi, T., Ito, S., Okada, M., Murasawa, A., Nakazono, K., et al. (2015). Expression of anti-Porphyromonas gingivalis peptidylarginine deiminase immunoglobulin G and peptidylarginine deiminase-4 in patients with rheumatoid arthritis and periodontitis. J. Periodontal Res. 51, 103–111. doi: 10.1111/jre.12288
Sjögren, J., and Collin, M. (2014). Bacterial glycosidases in pathogenesis and glycoengineering. Future Microbiol. 9, 1039–1051. doi: 10.2217/fmb.14.71
Slade, D. J., Horibata, S., Coonrod, S. A., and Thompson, P. R. (2014). A novel role for protein arginine deiminase 4 in pluripotency: the emerging role of citrullinated histone H1 in cellular programming. Bioessays 36, 736–740. doi: 10.1002/bies.201400057
Sofat, N., Wait, R., Robertson, S. D., Baines, D. L., and Baker, E. H. (2015). Interaction between extracellular matrix molecules and microbial pathogens: evidence for the missing link in autoimmunity with rheumatoid arthritis as a disease model. Front. Microbiol. 5:783. doi: 10.3389/fmicb.2014.00783
Valesini, G., Gerardi, M. C., Iannuccelli, C., Pacucci, V. A., Pendolino, M., and Shoenfeld, Y. (2015). Citrullination and autoimmunity. Autoimmun. Rev. 14, 490–497. doi: 10.1016/j.autrev.2015.01.013
Wegner, N., Wait, R., Sroka, A., Eick, S., Nguyen, K. A., Lundberg, K., et al. (2010). Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 62, 2662–2672. doi: 10.1002/art.27552
Wu, H. J., and Wu, E. (2012). The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 3, 4–14. doi: 10.4161/gmic.19320
Keywords: microbiome, dysbiosis, intestine, post-translational modification, autoimmune disease
Citation: Lerner A, Aminov R and Matthias T (2016) Dysbiosis May Trigger Autoimmune Diseases via Inappropriate Post-Translational Modification of Host Proteins. Front. Microbiol. 7:84. doi: 10.3389/fmicb.2016.00084
Received: 20 November 2015; Accepted: 18 January 2016;
Published: 05 February 2016.
Edited by:
Linda F. Van Dyk, University of Colorado Denver Anschutz Medical Campus, USAReviewed by:
Dina Weilhammer, Lawrence Livermore National Lab, USACopyright © 2016 Lerner, Aminov and Matthias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaron Lerner, YWFyb25sZXJuZXIxOTQ4QGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.