 Heini Kutvonen1†
Heini Kutvonen1† Pauliina Rajala
Pauliina Rajala Malin Bomberg
Malin Bomberg- 1Material Recycling and Geotechnology, VTT Technical Research Centre of Finland, Espoo, Finland
- 2Materials Performance, VTT Technical Research Centre of Finland, Espoo, Finland
We investigated the N-utilizing bacterial community in anoxic brackish groundwater of the low and intermediate level nuclear waste repository cave in Olkiluoto, Finland, at 100 m depth using 15N-based stable isotope probing (SIP) and enrichment with 14∕15N-ammonium or 14∕15N-nitrate complemented with methane. Twenty-eight days of incubation at 12°C increased the concentration of bacterial 16S rRNA and nitrate reductase (narG) gene copies in the substrate amended microcosms simultaneously with a radical drop in the overall bacterial diversity and OTU richness. Hydrogenophaga/Malikia were enriched in all substrate amended microcosms and Methylobacter in the ammonium and ammonium+methane supplemented microcosms. Sulfuricurvum was especially abundant in the nitrate+methane treatment and the unamended incubation control. Membrane-bound nitrate reductase genes (narG) from Polarimonas sp. were detected in the original groundwater, while Burkholderia, Methylibium, and Pseudomonas narG genes were enriched due to substrate supplements. Identified amoA genes belonged to Nitrosomonas sp. 15N-SIP revealed that Burkholderiales and Rhizobiales clades belonging to the minority groups in the original groundwater used 15N from ammonium and nitrate as N source indicating an important ecological function of these bacteria, despite their low number, in the groundwater N cycle in Olkiluoto bedrock system.
Introduction
Nitrogen (N) is one of the basic elements of all life forms and is essential for the production of amino and nucleic acids (Bothe et al., 2007). N depletion may be a growth-limiting factor in many environments, such as deep bedrock groundwater. However, use of explosives in e.g., construction into bedrock environments and mining may increase the concentration of N compounds in these environments, due to under detonated explosives (Beller et al., 2004). Use of pesticides, such as atrazine, has also been shown to be a source of N pollution in karst aquifers that are in connection to agricultural sites (Iker et al., 2010). Despite the generally low metabolic activity of deep groundwater microorganisms (e.g., D'Hondt et al., 2002; Rajala et al., 2015a) these N compounds may affect the activity and growth of deep bedrock microbial communities.
Nitrate is generally present only at low concentrations in pristine anoxic crystalline bedrock groundwater because it has been used as an alternative electron acceptor to oxygen and reduced to N2 in anaerobic redox processes (Bothe et al., 2007; Posiva, 2009). Dissimilatory nitrate reduction is a process where the oxidative force of nitrate is used as energy source in the oxidation of organic substrates by denitrifying bacteria. Denitrifying bacteria are mostly heterotrophic and often facultatively anaerobic with the ability to switch between oxygen and nitrate respiration depending on the environmental conditions (Luque-Almagro et al., 2011). Nitrate-reducing bacteria (NRB) have been shown by MPN enrichment studies to colonize groundwater in Olkiluoto, Finland at least to a depth of 300 m (Pedersen, 2008). The same study showed that the distribution and abundance of NRB correlated with that of the cultivable heterotrophic aerobic bacteria, indicating that nitrate as an optional terminal electron acceptor if oxygen is not available. However, depending on depth of the groundwater and location of the study site, denitrification and nitrate respiration in deep crystalline bedrock groundwater have been predicted to be low based on studies focusing on detection of essential genes for denitrification in deep crystalline bedrock environments (Nyyssönen et al., 2014; Purkamo et al., 2015). Nevertheless, NRB otherwise below the detection limit have been shown to significantly increase the transcription of narG genes in response to increased concentration of methane together with sulfate under N2 atmosphere in deep groundwater (Rajala et al., 2015a). In addition, it has been shown that certain ε-proteobacterial lineages couple reduction of nitrate to simultaneous oxidation of sulfide (reviewed in Campbell et al., 2006).
Ammonia/ammonium is an essential nutrient and energy source in both oceanic and terrestrial hydrothermal environments, where genes involved in ammonia oxidation have been found (Wang et al., 2009). However, little is known about the microbial community metabolizing ammonia or ammonium in pristine deep and cold crystalline bedrock groundwater. In the nitrification process ammonia is oxidized via nitrite to nitrate by the ammonia monooxygenase (AMO) enzyme. In pristine groundwater the concentration of ammonia is low. However, ammonia may be produced during fermentation of biomass, such as dead biofilm and ancient organic material trapped in the rock. In addition, under-detonated explosives may serve as an ultimate source for the production of ammonia by the DNRA process. Nevertheless, ammonia-oxidizing bacteria (AOB) have been found to be scarce in Fennoscandian Shield groundwater (Purkamo et al., 2015).
In addition to functioning as energy sources both nitrate and ammonia are N sources for microorganisms. Nyyssönen et al. (2014) showed by analysing metagenomic data from the Outokumpu deep borehole, that ammonia was the main source of N in the deep groundwater.
The N cycle in deep crystalline bedrock groundwater environments have not yet been extensively studied. In karst-bedrock environment the aerobic and anaerobic oxidation of ammonia has been shown to be slow (Ray et al., 2010) and the microbial communities changed as a result of N contamination by atrazine (Iker et al., 2010). In deep geological repositories for spent nuclear fuel, introduced N compounds may have great implications for the long-term safety of the stored nuclear waste. In Finland, low and intermediate level radioactive waste is stored in a repository at 100 m depth in the bedrock of Olkiluoto Island since 1992. This waste consists of metallic and organic materials from maintenance and decommissioning activities in the nuclear power plant. N compounds, such as ammonium and nitrate may increase microbial activity in this environment, which may cause microbially induced corrosion of the steel and cause release of radionuclides (Rajala et al., 2015b). Here we aimed to gain more insight to both the energy-producing and assimilatory aspects of the N cycle, which could affect the stability of the low and intermediate level radioactive waste repository. We focused especially on nitrate and ammonium in the deep groundwater, since these compounds may be introduced in to the groundwater system at the repository site through construction of the repository. In microcosm experiments we followed the consumption on these N compounds as well as the change in the general bacterial as well as NRB and AOB communities influenced by these compounds. In addition, we aimed to identify the nitrate and ammonium assimilating bacterial community by Stable Isotope Probing (SIP) with 15N labeled substrates.
Materials and Methods
Chemistry
The chemical composition of groundwater in the beginning of the experiment was analyzed at the ALS Finland. Dissolved metals were analyzed by ICP-OES and N compounds spectrophotometrically according to the CSN ISO 11732 and CSN ISO 13395. pH, redox-potential, oxygen content and temperature of the water were measured inside an anaerobic glove bag with a HACH Sension 156 m (Hach Lange Gmbh, Germany) and electrical conductivity with a CDM92 conductivity meter (Radiometer-analytical, France) on site.
Experimental Setup
Groundwater samples were collected in May 2013 from drill hole VLJ-KR9 in the low and intermediate level nuclear waste repository tunnel situated in the crystalline bedrock at the site of the Olkiluoto nuclear power plant, Finland. The drill hole VLJ-KR9 was drilled in 1995 in to the wall of the VLJ-tunnel at approximately 95 m depth from the ground surface. The total length of the drill hole is 20.04 m and it bears slightly downwards reaching a depth of 95.249 m from the ground surface. The first 66 mm of the drill hole is cased and the opening is sealed with a welded flange plug equipped with a pressure sensor and tap for sampling. The drill hole is regularly purged. Before the sampling a sterile, gas tight polyacetate tube was connected to the sampling tap of the borehole and groundwater was run for 30 min before the start of the sample collection in order to remove excess air from the tube.
Altogether 30 groundwater samples of 2.4 L each were collected in sterile borosilicate glass bottles previously rendered anaerobic under constant N2 flow. The groundwater was introduced to the bottles through the sterile polyacetate tube directly from the borehole located in the wall of the tunnel. In order to reduce exposure to oxygen in the microcosm bottles a volume of 50% overflow was allowed before each bottle was sealed with a tight-fitting butyl rubber septum and open-top screw-cap. The water samples were transported in a cooling-box with ice to the laboratory within 1d of sampling. The sample bottles were divided into 10 treatment groups with three replicates per treatment. The treatments were; (1) baseline sample, biomass was captured on 0.2 μm pore-size cellulose acetate membranes (Corning, MA, USA) by filtering 2.4 L groundwater on-site immediately after sampling and immediately frozen on dry ice, (2) Na15NO3 addition, (3) 15NH4Cl addition, (4) Na15NO3+CH4addition, (5)15NH4Cl+CH4addition, (6) Na14NO3addition, (7)14NH4Cl addition, (8) Na14NO3+CH4addition, (9)14NH4Cl+CH4 addition, and (10) no substrate additions (incubation control). N substrates and methane were added at the beginning of the experiment and again after 14 days of incubation. The N substrates (sodium nitrate and ammonium chloride, atom 98% 15N, Sigma-Aldrich) were sterilized using a 0.2 μm pore size syringe filter and rendered anaerobic by sterile N2 gas flow for 30 min. Five milliliters N substrates (62.5 mM stock solution) were added to a final concentration of 0.13 mM. The N substrates and 5 mL sterile methane (treatments 4, 5, 8, and 9), i.e., approximately 2 mL L−1 sample water, were added aseptically to the 2.4 L microcosms through the butyl rubber stoppers using a sterile syringe and needle. The microcosms were incubated at a temperature of +12°C, which was as close to that of the drill hole at 95 m depth from ground surface as possible for 28 d.
The biomass of each sample was collected by vacuum suction on 0.2 μm-pore-size cellulose acetate membranes (Corning, MA, USA) at the end of the incubation period. The filter membranes were cut out of the filter funnels using sterile scalpels, inserted into sterile 50 mL screw cap tubes (Corning, MA, USA) and frozen at −80°C until use.
The consumption or production of nitrate and ammonium was monitored by subsamples retrieved at the beginning of the experiment before and after addition of substrates, after 14 days incubation before and after addition of substrates and at the end of the incubation period. At each sampling time, 5 mL subsamples were retrieved using a sterile syringe and needle pushed through the butyl rubber septum of each bottle. The concentration of nitrate and ammonium of each subsample was measured spectrophotometrically using the Hach–Lange DR 2800 spectrophotometer (Hach–Lange, UK) and the LCK304 kit for ammonium and LCK339 kit for nitrate according to the manufacturer's instructions. The consumption and production of nitrate and ammonium was determined by comparing the concentration of N compounds after 14 and 28d of incubation to the concentrations immediately after substrate addition. The balance of N substrates in the different incubations was normalized to relative abundances of the measured N compounds in each different incubation type for better comparison between different treatments.
DNA Extraction
Filters with the collected biomass were carefully thawed on ice. Microbial DNA was extracted using the Metagenomic isolation kit for water (Epicentre, USA). The DNA was extracted according to the manufacturer's protocol and eluted in 50 μL elution buffer.
Quantification of the Number of Bacteria and Nitrate Reducers by qPCR
The bacterial population in the original groundwater and in the water samples after incubation with or without substrates was determined by 16S rRNA gene targeted qPCR using universal bacterial 16S rRNA gene-targeting primers fD1 (Weisburg et al., 1991) and P2 (Muyzer et al., 1993). In addition, the concentration of narG genes of the nitrate reducers was tested with primers narG1960m2f/narG2050M2F (López-Gutiérrez et al., 2004).
The qPCR reactions were performed in 10 μL reaction volumes using the KAPA 2 × Syrb® FAST qPCR-kit on a LightCycler480 qPCR machine (Roche Applied Science, Germany). Each reaction contained 2.5 μM of relevant forward and reverse primer and 1 μL DNA extract. Each reaction was run in triplicate and no-template control reactions were used to determine background fluorescence in the reactions.
The qPCR conditions consisted of an initial denaturation at 95°C for 10 min followed by 45 amplification cycles of 15 s at 95°C, 30 s at 55°C, and 30 s at 72°C with a quantification measurement at the end of each elongation. A final extension step of 3 min at 72°C was performed prior to a melting curve analysis. This consisted of a denaturation step for 10 s at 95°C followed by an annealing step at 65°C for 1 min prior to a gradual temperature rise to 95°C at a rate of 0.11°C s−1 during which the fluorescence was continuously measured. The number of bacterial 16S rRNA genes and narG genes was determined by comparing the amplification result (Cp) to that of a 10-fold dilution series (101–107 copies μL−1) of Escherichia coli (ATCC 31608) 16S rRNA genes and Pseudomonas aeruginosa (ATCC 15692) narG, respectively, in plasmid. A qPCR assay for the amoA genes was not conducted due to low specificity of the used primers to the amoA gene (see Section Ammonia Oxidizers).
PCR
PCR reactions for denaturing gradient gel electrophoresis (DGGE) analysis of the bacterial 16S rRNA genes, narG and amoA gene profiles in the original groundwater and after the incubation period were performed in 50 μL reactions. The reactions contained 1 u KAPA Hifi—polymerase in 1 × KAPA HF-buffer, 0.6 mM dNTP, 0.4 μM of each relevant primer and 1 μL DNA extract. The PCR program consisted of a 5 min initial denaturation step at 98°C followed by 40 cycles of denaturation at 98°C for 20 s, annealing at 55°C (16S rRNA genes) or 57°C (amoA and narG) for 15 s and elongation at 72°C for 30 s, and a final extension at 72°C for 5 min. For the bacterial 16S rRNA genes, primers U968fGC /U1401r (Nübel et al., 1996) were used; the narG and amoA DGGE PCR products were obtained with primers narG2179F/narG2488R-GC (Pastorelli et al., 2013) and amoA1F/amoA2RGC (Nicolaisen and Ramsing, 2002), respectively.
DGGE
Profiles of bacterial communities having 16S rRNA gene, narG and amoA genes were resolved vs. different treatments using DGGE. The DGGE was performed on a Bio-Rad DCode TM Universal Mutation Detection System (Bio-Rad, USA). For the 16S rRNA gene fragments, a denaturing gradient of 35–65% was used and the electrophoresis was run for 20 h at 85 V at 60°C as described in Nübel et al. (1996). The denaturants used was urea (42 g/100 mL in 100% denaturing solution) and formamide (40% formamide in 100% denaturing solution). The denaturing gradient used for the amoA-gene fragments was 30–70% and the electrophoresis was run for run 6 h with 200 V at 60°C according to Nicolaisen and Ramsing (2002). The narG-gene fragments were subjected to DGGE using a denaturing gradient of 40–70% for 17 h with 70 V at 60°C according to Pastorelli et al. (2013). All gels were made of 8% polyacrylamide.
DGGE bands were detected using SYRB-green I nucleic acid stain (Fisher Scientific, UK) according to the manufacturers protocol and imaged under UV light using a GEL DOC XR+2000 transilluminator (Bio Rad, USA). Prominent DGGE fragments were extracted from the gels using sterile plastic Pasteur pipettes. DNA from the gel plugs was extracted into 20 μL of molecular grade water overnight at 4°C and the DGGE band was reamplified with the relevant primer pair without the GC-clamp, as described above using 2 μL extracted DNA as template. The reamplified DGGE bands were shipped to Macrogen Inc, South Korea on ice packs, for sequencing.
Phylogenetic analyses were performed on the sequences of the DGGE bands with GENEIOUS PRO (Biomatters Ltd, New Zealand). All sequences were The alignments were performed with the MThe alignments were performed with the Mimported into GENEIOUS PRO, and aligned to reference sequences and most closely matching sequences in the NCBI nucleotides database (http://blast.ncbi.nlm.nih.gov/) determined with the blastn tool in GENEIOUS PRO. The alignments were performed with the Mafft tool in GENEIOUS PRO with default settings and the alignments were edited manually. Phylogenetic analyses were performed on the alignments of the 16S rRNA gene sequences using PhyML (Guidon and Gascuel, 2003) with the Jukes-Cantor69 substitution model (Jukes and Cantor, 1969). The narG and amoA gene sequences were translated in to amino acid sequences using translation table 11 prior to alignment with MAFFT and a phylogenetic analysis with PhyML using the WAG substitution model (Whelan and Goldman, 2001). Bootstrap support for nodes were calculated based on 1000 random repeats for all phylogenetic analyses.
Isopycnic Centrifugation and Gradient Fractionation
In order to separate the 15N-containing DNA fraction of the microbial community that had consumed the N substrates from the rest of the community DNA density gradient centrifugation was performed using 5 ml Quick-seal polyallomer tubes (BeckmanCoulter, Brea, CA, USA) in a Beckman ultracentrifuge with a VTI 65.2 vertical tube rotor. Each tube contained a fixed volume of 35 μL extracted DNA suspended in 4.9 mL of CsCl in gradient buffer (5 mL 1 M Tris-HCl, 0.375 g KCl, 0.15 mL EDTA) with a density of 1.725. The DNA was centrifuged for 62 h at 44,100 g at 22°C.
After centrifugation the gradients were apportioned, starting at tube bottoms, into 22 equal fractions of approximately 200 μL. The centrifugation tube was first pierced with hypodermic needles at the top and bottom and the fractions were pushed out through the bottom needle by pushing sterile water mixed with dye at a rate of 200 μL min−1 in to the top of the centrifuge tube using a syringe pump (New Era Pump Systems, Inc., Farmingdale, NY, USA). Gradient formation in the centrifugation was determined by including a control tube without DNA in each run and fractioning the gradient as for the DNA samples. The nd value of each of the control fractions was determined using a refractometer (DR301-95, A. Krüss Optitronic, Germany). In addition, the nd value of the first and last fraction of the DNA sample tubes was also measured in order to ensure that a gradient has been formed during the centrifugation.
Due to the potential for low separation efficiency when there is low concentration of N in the DNA and the high density of GC-rich DNA, fractions with densities 1.725–1.733 were recentrifuged as before, but with the addition of 8 μL (10 mg mL−1) bis-Benzimide in each sample to improve buoyancy of GC-rich DNA as described in Buckley et al. (2007). This was performed according to gradient formation in controls during the first centrifugation. After centrifugation, gradients were apportioned as before directly onto MultiScreenf® Filter Plates (Merck Millipore Ltd., Cork, Ireland) and purified from CsCl salt using a 96-well Vacuum Manifold (Merck Millipore Ltd., Cork, Ireland) with 5 volumes (200 μL) sterile water.
Fractions containing heavy and light DNA were determined by qPCR as described above for the bacterial 16S rRNA gene. DNA from fractions having density of 1.72 and 1.735 were determined to contain DNA fractions of interest and subjected to high throughput (HTP) amplicon sequencing.
High Throughput Amplicon Sequencing and Sequence Analysis
Libraries for 454 HTP amplicon sequencing were prepared by PCR of DNA from the original groundwater used in the experiment, the supplemented incubations and incubation control, and also the heavy-fraction DNA of the Na15NO3,15NH4Cl, Na15NO3+CH4, and15NH4Cl+CH4 treatments. For HTP sequencing, amplicon libraries from the three replicates of each treatment were combined due to the low amount of DNA in the heavy fraction DNA extracts.
Bacterial 16S rRNA fragments covering the V1–V3 variable regions were amplified with primers 8F (Edwards et al., 1998) and P2 (Muyzer et al., 1993) equipped with adapter and MID sequences (tags) at their 5' end in a single round PCR as described in Bomberg et al. (2015). PCRs were performed with KAPA HiFi polymerase (Kapa Biosystems, Inc., Boston, MA, USA) in 1 × HF buffer. Each 50 μL reaction contained 0.5 mM dNTP and 1 μM primer mix. PCR conditions consisted of an initial denaturation step of 30 s at 98°C, followed by 35 cycles of 10 s at 98°C, 15 s at 55°C, and 15 s at 72°C, and a final extension step at 72°C for 5 min. Sequencing of the PCR products was performed at BeckmanCoulters Genomics using the FLX 454 Titanium (454 Life Sciences, Branford, CT, USA).
Sequence reads were analyzed using Mothur (v 1.33.1, Schloss et al., 2009) where the flow-grams were denoised using the default parameters of the sff.multiple workflow in Mothur, trimmed to remove adapter, barcode, and primer sequences, and to exclude sequences that did not meet the quality criteria (i.e., no barcode and primer mismatches, no ambiguous nucleotides, maximum eight-nucleotide-long homopolymer stretches and defined minimum length of 200 bp). The bacterial 16S rRNA sequences were aligned to the Silva v_119 reference alignment (Pruesse et al., 2007) and the alignments were screened to include sequences with defined start positions and minimum end position. The aligned sequences were preclustered prior to chimera detection with the Chimera.slayer command in Mothur and possible chimeric sequences were removed. A distance matrix was calculated for the chimera-filtered sequences using a cut-off of 0.05 without penalty for end gaps. The sequences were clustered into Operational Taxonomic Units (OTUs) according to the distance matrix using the nearest-neighbor method. The representative sequences of the OTUs sharing 97% sequence similarity within each OTU were classified using the Silva v_119 reference database (Pruesse et al., 2007). Alpha diversity analyses were performed on data normalized to 448 sequence reads per sample and rarefaction analyses were performed on the total number of sequence reads per sample.
Accession Numbers
The sequences of the DGGE bands will be deposited in the European Nucleotide Archives (ENA; https://www.ebi.ac.uk/ena) under accession numbers LN866782-LN866813 for 16S rRNA genes, LN866814-LN866821 for amoA genes and LN866822-LN866841 for narG genes. The HTP sequence reads were submitted to ENA under accession numbers ERS739681-ERS739690.
Results
Groundwater Chemistry
At the time of sampling the groundwater contained 0.178 mg L−1 ammonium-N, 0.229 mg L−1 ammonium, <0.040 mg L−1 nitrate, and <0.150 mg L−1 nitrite at pH 8.06. The pH, temperature (T), reduction potential (Eh) and electric conductivity (Ec) of the groundwater was 8.09, 10.3°C, 58 mV vs. Standard Hydrogen Electrode (SHE) and 2.26 mS cm−1, respectively. Other physicochemical parameters are presented in Table 1.
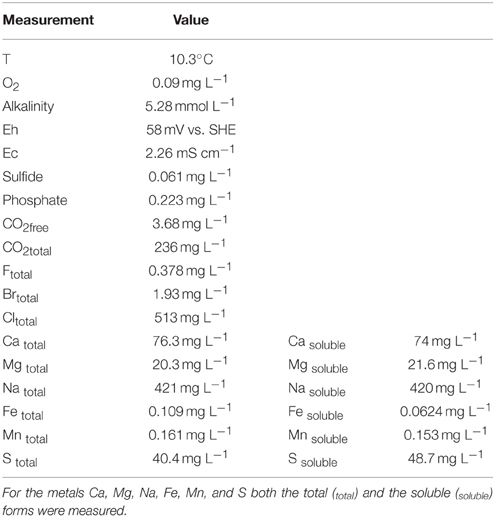
Table 1. The physicochemical composition of the groundwater at the time of sampling.
Consumption of Ammonium and Nitrate
The original concentrations of ammonium and nitrate were low (Figures 1A,C). During 4 weeks of incubation at 12°C the amount of ammonium decreased almost 20% from the original concentration at the time of sampling (Figure 1B). The concentration of nitrate, on the other hand increased 22.5% over the same time period (Figure 1D). After addition of the N substrates, the concentration of ammonia decreased 34.5% in the microcosms that had received ammonium and methane (NH4+CH4) and 51.5% those only receiving ammonium (NH4). However, the ammonium concentration ceased to decrease after 2 weeks of incubation (Figure 1). In the nitrate-supplemented microcosms, the relative concentration of nitrate increased in all treatments after the first addition of nitrate (Figure 1D). However, after 4 weeks of incubation 21.2% of the added nitrate had been consumed in microcosms supplemented with nitrate (NO3) only, while the nitrate concentration in the nitrate+methane (NO3+CH4) remained high.
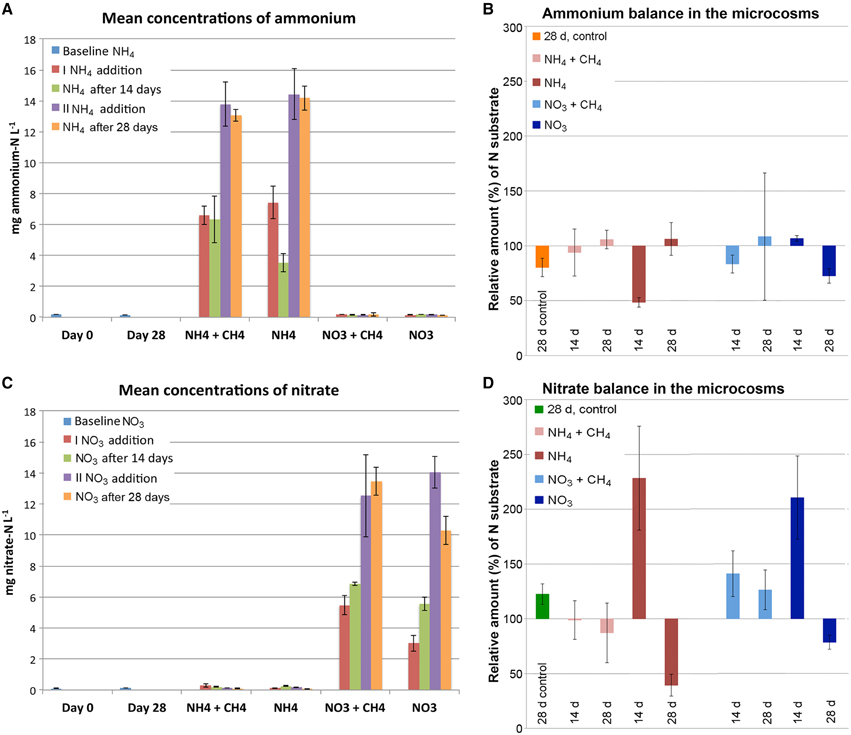
Figure 1. The average concentration (A,C) and relative balance (B,D) of ammonium and nitrate in the microcosms. In (A,C) the average concentration of ammonia or nitrate (mg L−1) is measured from the original groundwater and the 28d controls (blue), directly after the first addition of N-substrate (red), 14d after the first addition of N-substrate (green), directly after the second addition of N-substrate (purple), and at the end of the experiment (orange). In (B,D) the relative change in the concentration of ammonium and nitrate is illustrated for incubations with NH4+CH4 (pink), NH4 (red), NO3+CH4 (light blue) and NO3 (dark blue) after 14 and 28d of incubation. The change in ammonium concentration over 28 days in (B) is shown in orange and the change in nitrate concentration in (D) in green. The relative amount of N substrates in (B,D) above 100% are considered produced and below 100% consumed compared to the amount of N substrates measured directly after addition of substrates.
Over the time of incubation the concentration of nitrate increased with approximately 25% in the unamended control microcosms, while the concentration of ammonium decreased by the same magnitude (Figure 1). The concentration of ammonium in the nitrate-supplemented microcosms increased slightly toward the end of the incubation period in the NO3-supplemented microcosms, and during the first 14 days in the NO3+CH4-supplemented microcosms, but was not extensively produced from the added ammonium (Figure 1B). In the NH4+CH4-supplemented microcosms, on the other hand, the NO3 concentration increased noticeably with approximately 230% during the first 14 days of incubation where after the NO3 was consumed to about 40% of the original NO3 concentration by the end of the incubation period, indicating that NO3 was initially produced from the added NH4 (Figure 1D).
Quantitative Analysis of Microbial Community
There were 7.8 × 104 copies mL−1 of the bacterial 16S rRNA gene in the original groundwater. The addition of NH4+CH4 and NO3 had the greatest effect on the bacterial community and increased the concentration of bacterial 16S rRNA gene copies 12-fold over the 28d incubation period to 9.5 × 105 mL−1 and 9.7 × 105 mL−1, respectively (Figure 2A). In the other treatments the number of bacteria also increased, including the control (unamended) microcosms, by three to five fold.
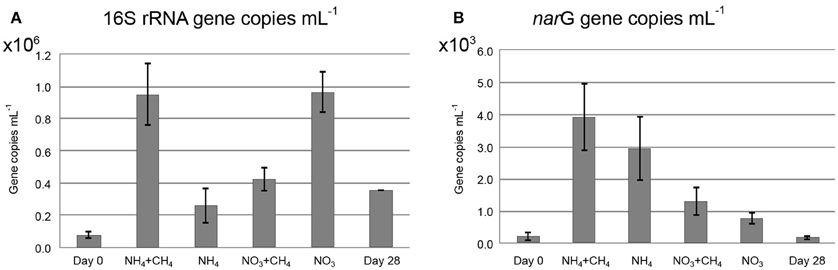
Figure 2. The concentrations of (A) bacterial 16S rRNA gene copies, (B) narG gene copies mL−1 in the original groundwater (Day 0), the incubations with substrates (NH4+CH4, NH4, NO3+CH3, NO3) and without substrates (Day 28).
There were 2.2 × 102 narG gene copies mL−1 corresponding to the population of NRB in the original groundwater (Figure 2B). The NH4+CH4 treatment had the greatest effect on the NRB increasing the concentration of narG genes to 3.9 × 103 copies mL−1 over the course of incubation. In nitrate-treated microcosms the increase in narG genes was 2.7 × 103 copies mL−1. Without substrate additions, the concentration of narG genes did not increase during the incubation time.
Bacterial Community Composition
The number of sequence reads obtained from 454 pyro sequencing ranged from 448 to 5772 sequence reads per sample from the original groundwater and the different microcosms (Table 2). The bacterial community in the original groundwater was diverse (H' = 3.07) according to the 454 amplicon sequence profiles, which showed 125 OTUs belonging to 61 different bacterial genera (Figure 3, Table 2, Table S1). The richness estimates (chao and ace) indicated that only between 21 and 41% of the OTU richness in the original groundwater was captured. Over 28d the microbial OTU diversity increased (H' = 3.51) to altogether 146 OTUs, but the number of identified genera decreased to 43 and according to the richness estimates only 19.2–35.5% of the total OTU richness was obtained. In the substrate amended microcosms the diversity was significantly lower, H' = 0.04–0.70, than that of the original groundwater and the 28d incubation controls. The number of observed OTUs ranged from 2 to 19 and according to the richness estimates between 60.8 and 100% of the total diversity of the bacterial communities was captured (Table 2). The low number of OTUs obtained as well as the low diversity and richness indices indicates that the addition of N substrates enriched a specific population of bacteria involved in N cycling during the incubation.
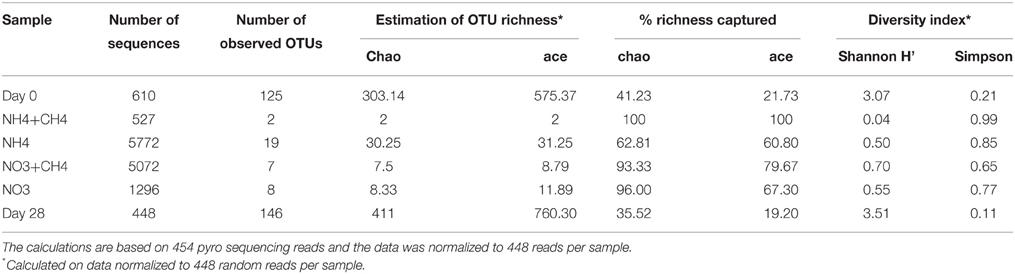
Table 2. Alpha diversity estimates of the original groundwater (Day 0) and after 28 days of incubation in the control microcosms (Day 28) and the microcosms supplemented with NH4+CH4, NH4, NO3+CH4, NO3.
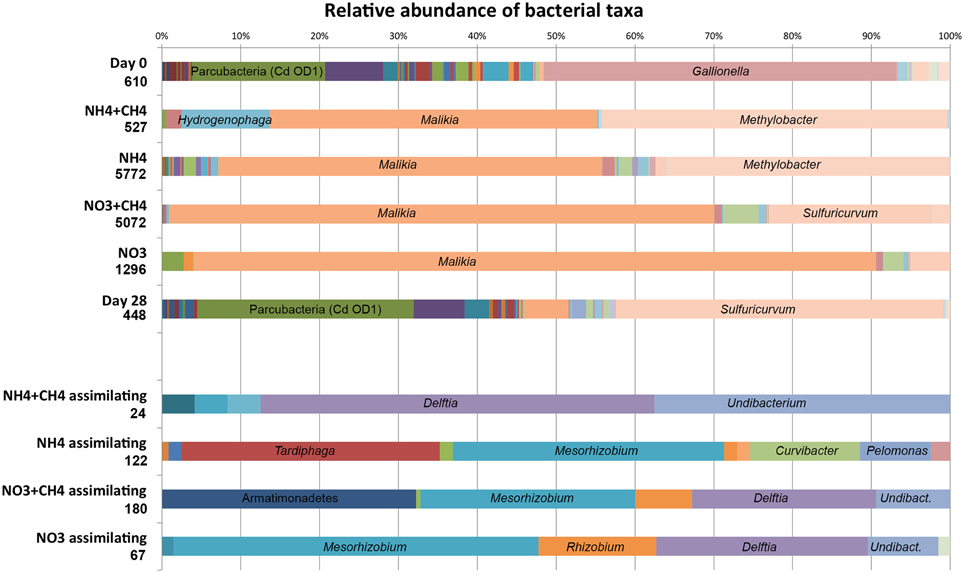
Figure 3. The relative abundance of bacterial genera detected by high throughput amplicon sequencing from the original groundwater (Day 0), N- and C-supplemented microcosms and the non-supplemented control (Day 28). The lower part of the figure shows the relative abundance of nitrogen assimilating bacteria detected from the 15N fractions. The number in by each category indicates the number of sequence reads. The names of the most prominent genera are indicated in the bar graph. More detailed information of the relative abundance of different bacterial taxa detected in the study can be found in Table S1.
Based on pyro sequencing data 12 bacterial genera were present at over 1% relative abundance in the original groundwater. The most abundant bacteria belonged to an unassigned group of Parcubacteria (Rinke et al., 2012) [formerly Candidate division (Cd) OD1], the β-proteobacterial genus Gallionella and the Microgenomates (Rinke et al., 2012; formerly Cd OP11, Figure 3, Table S1). Interestingly, in the 28d incubation controls the relative abundance of Sulfuricurvum increased from 0 to 41.5%, the Parcubacteria group from 16.9 to 27.5% and the Comamonadaceae genus Malikia from 0 to 5.6%. In the NH4 and NH4+CH4 incubations Malikia contributed with 48.7% and 41.6% of the sequence reads, respectively, being the most abundant bacterial group enriched in these microcosms. Methylobacter was the second most abundant bacterial genus contributing with 35.9 and 43.8% of the sequence reads, respectively. In addition, Hydrogenophaga had become enriched (11.2% of the sequence reads) in the NH4+CH4 incubations but was present at below 1% in the NH4 microcosms. The NO3- and NO3+CH4 amendments enriched the Malikia most and 86.5% and 69.2% of the sequence reads belonged to this genus, respectively. In addition, 5.0% and 20.5% of the sequence reads belonged to Sulfuricurvum and 4.4% and 2.6% belonged to Sulfuricella, respectively. Methylobacter contributed with 2.4% of the sequence reads in the NO3+CH4—amended microcosms, but were not found in the microcosms with only NO3. Instead, the NO3—amended microcosms contained Acetobacterium sequences, 2.8%, which were additionally only detected in the original groundwater samples.
The DGGE profiles showing the most abundant taxa in the bacterial communities were similar to the pyro sequencing profiles. The DGGE profiles of the bacterial taxa enriched during incubation were also similar between the different treatments, but specific phylotypes were more clearly detected in comparison to the ones seen in the original groundwater (Figure 4). The genera detected by DGGE affiliated with the β-proteobacterial Hydrogenophaga/Malikia cluster and ε-proteobacterial Sulfuricurvum (Figures 5, 6). The most common bacteria detected by DGGE in all substrate-amended microcosms belonged to ε-proteobacterial Sulfuricurvum and β-proteobacterial Hydrogenophaga/Malikia (Figures 5, 6), although the 454-pyrosequencing detected Sulfuricurvum enriched only in NO3+CH4—amended microcosms and in the 28d controls, and at low abundance in the NH4- and NO3-amended microcosms. DGGE bands belonging to γ-proteobacterial Methylobacter were found in the original groundwater and in the microcosms amended with NH4 and NH4+CH4 in the DGGE analysis (Figures 5, 6). In addition, δ-proteobacterial Desulfocapsa were detected by DGGE in all microcosms although they were not detected by 454 pyro sequencing.
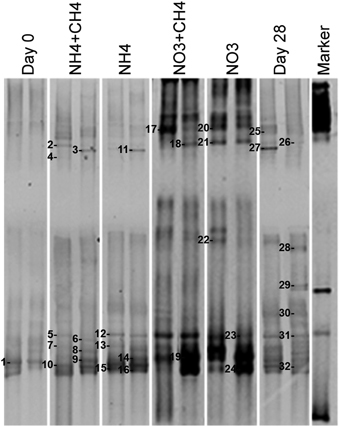
Figure 4. Bacterial community profiles from PCR-DGGE of DNA extracted from original groundwater (Day 0) and supplemented vs. control microcosms (Day 28). The supplemented treatments are shown as described in Figure 2.
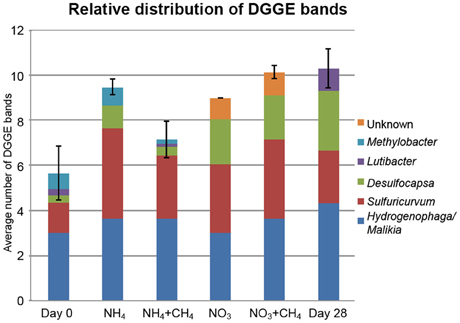
Figure 5. Average number of DGGE bands per replicate treatment (X axis) and the distribution of bacterial taxa identified from the DGGE profiles of the different treatments (Y axis). The number of replicates was 3 (Day 0 and 28) or 6 per treatment (substrate supplemented microcosms). The error bars show standard error of mean (SEM). The supplemented treatments are shown as described in Figure 2.
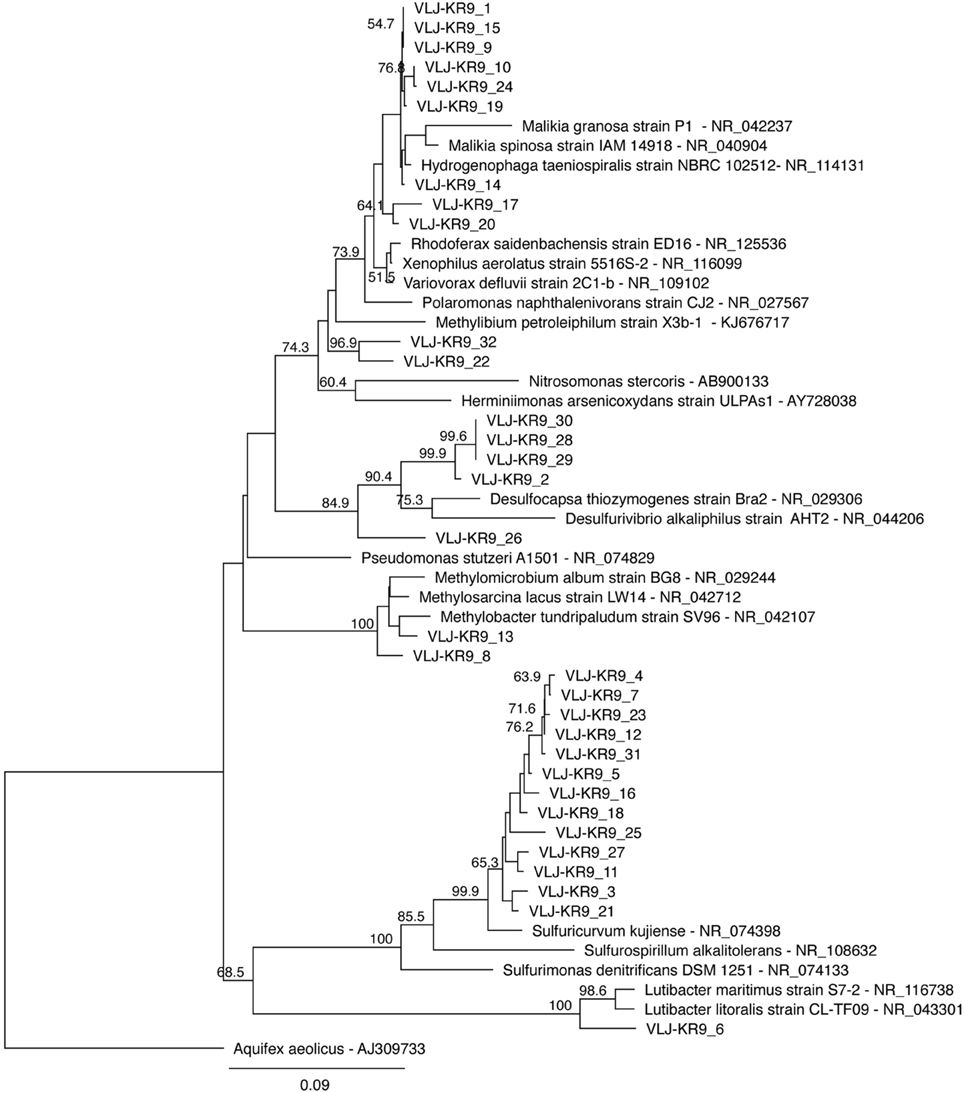
Figure 6. A maximum likelihood tree displaying the sequences obtained from the 16S rRNA gene DGGE bands. The sequences of this study are designated with the identifier VLJ-KR9 and the number at the end of the identifier describes the isolated DGGE band, corresponding to the bands in Figure 4. Bootstrap support for nodes was calculated with 1000 random repeats; nodes with more than 50% support are indicated.
Ammonia Oxidizers
The amoA genes of ammonia oxidizing bacteria were detected by DGGE but of a total of 46 sequenced DGGE bands 8 bands provided a correct amoA sequence (Figure 7). Of these sequences, 7 were obtained from NO3-amended microcosms and one from unamended (control) microcosms. All detected amoA gene fragments were similar to β-proteobacterial Nitrosomonas-type amoA genes (Figure 8).
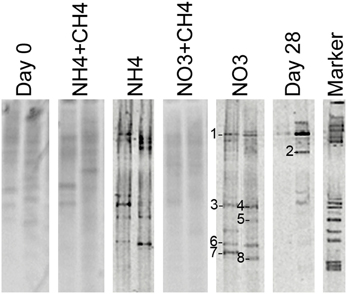
Figure 7. Community profiles of PCR amplicons (amoA-targeted) from DNA of original groundwater (Day 0) and supplemented vs. control microcosms (Day 28). DGGE bands identified by sequencing are indicated by a number to the left of the band. The figure presents two replicate lanes out of 3 (Day 0 and Day 28) or 6 (NH4+CH4, NH4, NO3+CH4, NO3) replicate runs.
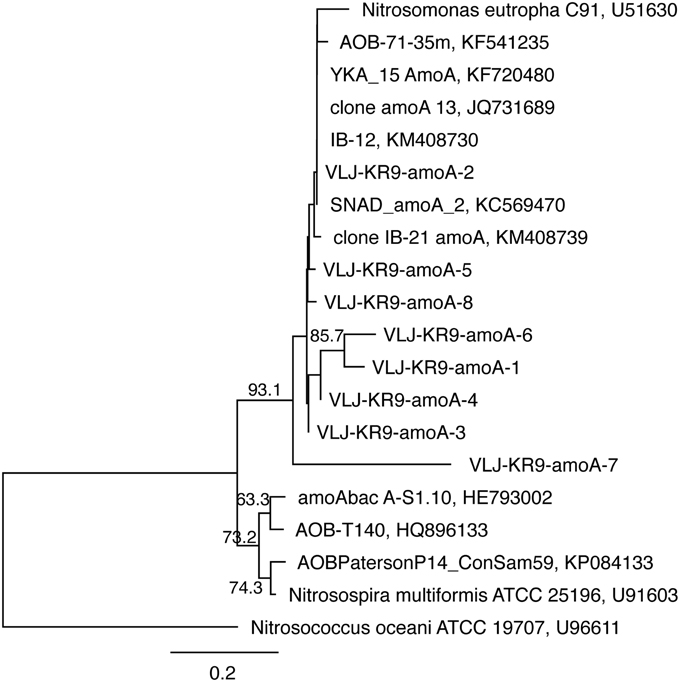
Figure 8. The phylogenetic distribution of the amoA fragments detected in this study presented as a maximum likelihood tree. The gene sequences were translated into amino acid sequences prior to analysis. Bootstrap support for nodes was calculated with 1000 random repeats; nodes with more than 50% support are indicated. Sequences detected in this study are designated with the identifier VLJ-KR9-amoA; the number at the end of the identifier corresponds to the DGGE band indicated in Figure 7.
Nitrate Reducers
narG genes of NRB were detected by DGGE (Figure 9). Altogether 20 bands were successfully sequenced and identified to be narG gene fragments. narG gene sequences belonging to four different taxonomical groups were found (Figure 10). These sequences were similar to narG genes of Pseudomonas (bands 1, 2, 9, 10, 13, 14, 15, 17, 18), Methylibium (bands 3, 6, 8, 11, 19), Alicycliphilus (band 16), Polaromonas (bands 4, 5, 20), Herminiimonas (band 7), and to an uncultured group of bacteria (band 12). Pseudomonas and Methylibium narG genes were frequently detected in all substrate-amended microcosms, while Polaromonas narG was found in microcosms amended with NH4+CH4 or NH4 and in the untreated controls. Alicycliphilus narG was detected only in microcosms supplemented with NO3 and only in one band. narG band 12 was obtained from NH4 supplemented microcosms only, but the position of the DGGE band was similar to many others in the other treatments, which were not successfully sequenced; this indicated that the particular narG type was common in the microcosms.
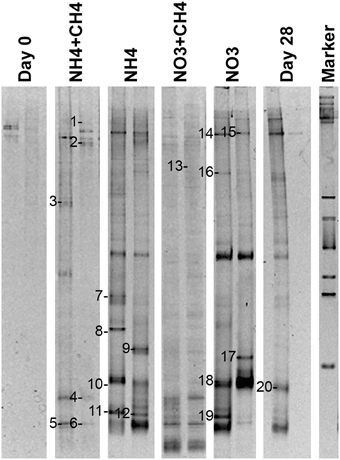
Figure 9. Community profiles of PCR amplicons (narG-targeted) from DNA of original groundwater (Day 0) and supplemented vs. control microcosms (Day 28). DGGE bands identified by sequencing are indicated by a number to the left of the band. The figure presents two replicate lanes out of 3 (Day 0 and Day 28) or 6 (NH4+CH4, NH4, NO3+CH4, NO3) replicate runs.
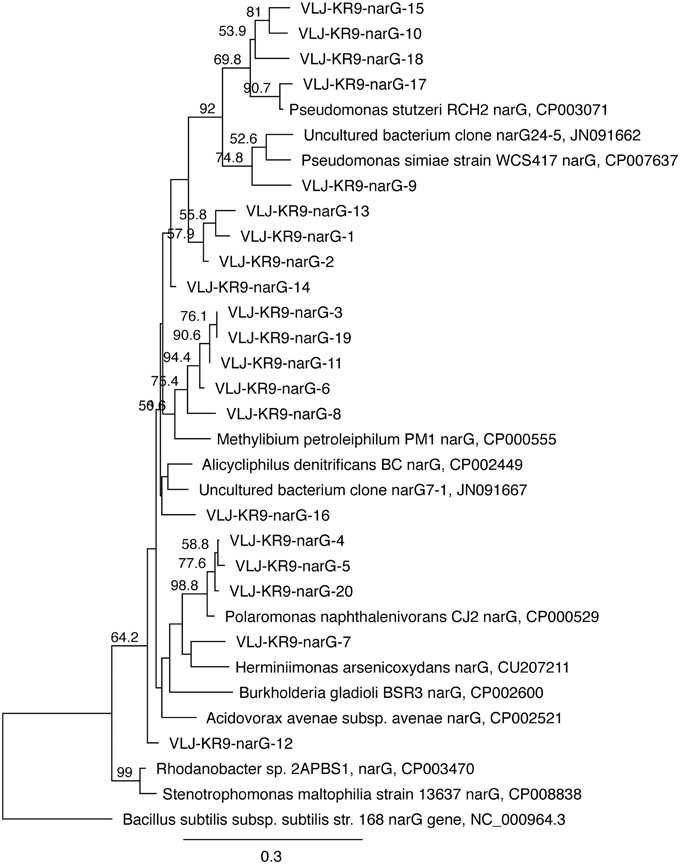
Figure 10. The phylogenetic distribution of the narG fragments detected in this study presented as a maximum likelihood tree. The gene sequences were translated into amino acid sequences prior to analysis. Bootstrap support for nodes was calculated with 1000 random repeats; nodes with more than 50% support are indicated. Sequences detected in this study are designated with the identifier VLJ-KR9-narG; the number at the end of the identifier corresponds to the DGGE band indicated in Figure 9.
Nitrogen Assimilating Bacteria
The bacterial communities corresponding to different treatments changed over time due to the different substrates added to the microcosms. After double isopycnic centrifugation was applied in order to separate first the heavy DNA fraction from the light DNA fraction and then the 15N-enriched heavy DNA from the GC-rich heavy DNA, the amount of DNA left was not adequate to obtain DGGE bands of detectable density. However, 454 amplicon sequencing provided a low number of sequence reads from the 15N-enriched DNA, after pooling the three replicate microcosms within each treatment. Several genera belonging to α-proteobacterial Rhizobiales (Rhizobium, Mesorhizobium, Tardiphaga) and β-proteobacterial Burkholderiales (Delftia, Undibacterium, Curvibacter, Pelomonas) were detected in the 15N-enriched heavy DNA fraction from all amended microcosms (Figure 3; Table S1). In addition, sparse Nitrospirales 16S rRNA gene reads were found in the 15NH4-amended samples, along actinobacterial Propionibacterium from the 15NH4+CH4-amended microcosms (Table S1). In the 15NO3-amended samples, a few reads resembling cyanobacterial sequences were detected, and in the 15NO3+CH4-amended microcosms, an unassigned group of Armatimonadetes constituted the single most abundant 16S rRNA gene type detected in this treatment (Figure 3; Table S1). The corresponding fractions of the 14N substrate-amended microcosms were also tested by PCR, but no PCR fragments were obtained.
Discussion
Nitrogenous compounds, such as nitrate and ammonia may have great effect on the microbial activities in the deep geological repository for low and intermediate level radioactive waste that has been constructed in to the crystalline bedrock of Olkiluoto, Finland. Enhanced microbial activity may cause microbially induced corrosion of the metallic waste, as shown by Rajala et al. (2015b) and affect the long-term safety of the repository. In oligotrophic crystalline bedrock environments methane may be an important component of gases dissolved in the groundwater and may serve as carbon source for the microbial communities and thus increase microbial activity. Nitrate-reducing microbial communities have been found in Olkiluoto deep groundwater by Most Probable Number analysis to depths greater than 300 m (Pedersen, 2008). Olkiluoto groundwater has in general large amounts of dissolved N2 and the amount of CH4 increases with depth from 1 to 1000 μL L−1 groundwater to a depth of 300 m, but increases 100-fold after this depth from where it may diffuse in to more shallow groundwater (e.g., Pedersen, 2008). Different N compounds may be introduced to the repository environment from explosives that have been incompletely detonated during construction of the repository.
In this study, we showed that added nitrate and ammonium had a great effect on the bacterial community in brackish groundwater in Olkiluoto at the depth of ca. 100 m. The bacterial community increased during the 28d incubation as a result amendment with N substrates and methane with the highest increase in the NH4+CH4 and NO3 treatments. The microbial community consumed NH4 during the first 2 weeks of incubation, both with and without CH4, but the consumption decreased toward the end of the incubation period. NO3 consumption was detected at the end of the incubation period, but not together with CH4. This indicates that the NH4- and NO3-consuming populations were not in general affected by CH4. However, the CH4-consuming community despite their intrinsic differences may affect the N-cycling community. The N substrates also decreased the number of detected OTUs, the estimated OTU richness and diversity indices of the microbial communities in the microcosms, while in the unamended 28d control microcosms these factors increased (Table 2). It is thus possible that introduction of N substrates in the repository area may have significant effect on the corrosion rate of the metallic radioactive waste by explicitly increasing specific types of microorganisms.
ε-proteobacteria belonging to the Sulfuricurvum sp. were found to be a main constituent of the bacterial communities in all microcosms according to the DGGE (Figure 5) and the pyro sequencing assay showed that their relative abundance clearly increased in the NO3- and NO3+CH4-amended microcosms and in the 28d controls (Figure 3; Table S1). These bacteria are common inhabitants of groundwater environments (Kodama and Watanabe, 2004; Bomberg et al., 2015) and it has recently been shown that Sulfuricurvum denitrificans use nitrate as electron acceptor. However, some species lack the common cytoplasmic membrane bound Nar nitrate reductase (Handley et al., 2014). In addition, Sievert et al. (2008) speculated that due to the lack of genes for nitrite ammonification (DNRA), S. denitrificans would have to incorporate ammonia from the environment. However, our results did not show any ε-proteobacterial enrichment in the 15N DNA fraction in any of the treatments, indicating that they did not assimilate N from any N-substrates used here. Nevertheless, it should be noted that we obtained very few sequences (24–180 reads per sample) from the 15N-DNA fraction over all, and if ε-proteobacteria incorporated only low amounts of labeled N they may well have gone undetected in this assay. It is also possible that Sulfuricurvum sp. does not use nitrate of ammonium as N source, but only as energy source.
Bacterial taxa affiliating with the Hydrogenophaga/Malikia cluster (Figure 6) was another group that appeared to benefit from N-substrate supplementations, according to the DGGE analyses. In the 454 pyro sequencing Hydrogenophaga/Malikia bacteria were not detected from the original groundwater, but were the dominating bacteria found in all N-substrate amended microcosms (49.6–86.5% of the sequence reads) and their relative abundance in the 28d microcosms also increased (Table S1). Hydrogenophaga and Malikia species reduce nitrate to nitrite (Spring et al., 2005; Yoon et al., 2008). In addition, Hydrogenophaga are able to perform the DNRA process thus producing ammonia from nitrate (Yoon et al., 2008). However, as with Sulfuricurvum sp., Hydrogenophaga/Malikia sequences were undetectable in the 15N-DNA fraction and may thus use other N sources that nitrate or ammonia.
Methylobacter sp. were found in all microcosms except the NO3—amended ones according to the pyro sequencing profiles and in all microcosms that received NH4+CH4, NH4, and NO3+CH4, i.e., in response to NH4 and CH4 according to DGGE (Table S1, Figure 5). Their relative abundance in the NH4+CH4– and NH4–amended microcosms was 43.8 and 35.9% (Figure 3), respectively, and below 2.4% in the other treatments. In general, ammonia has been considered to inhibit methanotrophs. However, methanotrophic bacteria may oxidize ammonium to hydroxylamine using methane monooxygenase (Hanson and Hanson, 1996; Campbell et al., 2011) and in accordance with our results, Zhang et al. (2014) showed that the abundance of Methylobacter increased in landfill cover soil as a result of increased NH4-N fertilization. Interestingly, Nyerges and Stein (2012) showed that ammonium had a distinct competitive effect on the oxidation of methane by methanotrophs.
The abundance of NRB increased in the microcosms during the incubations (Figure 2B). However, the ratio of narG gene to bacterial 16S rRNA gene concentration was generally low. In addition to the membrane bound NAR enzyme many bacteria have a cytoplasmic NAP enzyme. Genes for this enzyme were not targeted in this study, i.e., we could detect only a part of the nitrate-reducing community. The detected narG genes of NRB belonged to γ-proteobacterial Pseudomonas and β-proteobacterial Methylibium, Alicycliphilus, Polaromonas, Herminiimonas, and uncultured β-proteobacteria. Methylibium 16S rRNA gene sequences were not found in any treatment with either of the community profiling methods, although other Burkholderiales clades were detected. In contrast, Pseudomonas 16S rRNA genes or those of closely related groups were not found at all although Pseudomonas has been shown to be a major bacterial group at greater depth in Olkiluoto (Bomberg et al., 2015). Polaromonas narG genes were detected in the NH4+CH4—amended and 28d control microcosms and similar DGGE bands were also faintly seen in most other samples. This is in accordance with the 454 pyro sequencing, which detected Polaromonas 16S rRNA gene sequences from all samples with the exception of the NH4+CH4—amended microcosms. It is possible that the Polaromonas escaped detection because the Hydrogenophaga/Malikia sequences were so dominating in this sample and the overall number of sequences obtained from this sample was low. Herminiimonas narG were found only in the NH4—amended microcosms, but their 16S rRNA gene sequences were detected in the NO3+CH4—amended microcosms. This is not surprising, because even in the sample where Herminiimonas was detected they were present at only 0.04% relative abundance (Table S1) indicating that they belong to the minority groups at this depth in Olkiluoto. The only N assimilating bacterial group for which a functional gene was also detected was the Alicycliphilus. Alicycliphilus narG was detected in the NO3—amended microcosms, but Alicycliphilus 16S rRNA genes were detected in the NH4+CH4 15N SIP fraction. It is possible that this group was present in the other treatments as well, but failed detection due to the overall low number of sequence reads obtained from the 15N-DNA fractions.
The narG gene specific primers we used here appear to have higher affinity to β- and γ-proteobacterial narG genes than to those of the α- and ε-proteobacteria, which may lead to the α- and ε-proteobacteria being under-represented in this study (Nicolaisen and Ramsing, 2002). In addition, different primers were used for the narG qPCR and narG DGGE analyses, which may affect the detection efficiency to different narG genes. In the narG-qPCR assay the size of the produced amplicon was optimized for qPCR and was only 90 bp long. In order to increase sequence length a primer pair producing a 309 bp long amplicon was chosen for the narG DGGE. However, the 90 bp long amplicon produced in the qPCR with primers narG-1960m2f/narG-2050m2F (López-Gutiérrez et al., 2004) have been shown to detect similar narG sequences in Fennoscandian bedrock groundwater (Purkamo et al., 2015; Rajala et al., 2015a) as those detected with primers narG-2179F/narG2488R (Pastorelli et al., 2013) in this study.
amoA gene sequences were only obtained from the microcosms supplemented with NO3 and from the unamended 28d controls and the rest of the sequences were unspecific. All amoA genes were similar to those of Nitrosomonas bacteria. Interestingly, Nitrosomonas 16S rRNA genes were not detected by DGGE or 454 pyro sequencing in any of the samples. However, other closely related Gallionellales groups were detected in all other treatments except the NH4+CH4—amended microcosms (Table S1), and the amoA genes between these groups may be similar to that of the Nitrosomonas. The number of AOB in deep crystalline bedrock groundwater overall is expected to be low, which was shown previously by Purkamo et al. (2015). The fact that amoA genes were only successfully detected in the NO3-amended and 28d control microcosms is curious. It is, however, possible that the DGGE assay for amoA genes was not suitable for our sample matrix. We experienced some issues with the specificity of the primers used and in addition the DGGE bands obtained may have consisted of a mixture of amplicons. In the microcosms from which amoA gene fragments were successfully identified the number of interfering amplicons was probably lower and thus allowing us to detect clean sequences of the gene. Nevertheless, the AOB detected in this study in the NO3-amended and 28d control microcosms may have benefited from the slowly released lower concentration ammonia in the NO3—amended microcosms originating from the conversion of nitrate to ammonia through the DNRA process. In addition, it may also be possible that instead of typical ammonia oxidizers methanotrophic bacteria able to oxidize ammonium using their methane monooxygenase (Hanson and Hanson, 1996; Campbell et al., 2011) increased, and this group would not be detected by the amoA-gene targeted assays.
In the N-assimilating community, bacteria belonging to Rhizobiales genera were the most abundantly detected and were found in the 15N DNA fraction in all of the treatments (Figure 3, Table S1). Rhizobiales represented 72 and 61% of the N-assimilating bacterial community in the in the NH4- and NO3-amended microcosms. In the microcosms that received CH4 in addition to the N-substrates, their relative abundance in the N-assimilating community was lower, 4 and 35% in the NH4+CH4and NO3+CH4 treatments, respectively. In addition to Rhizobiales types, Burkholderiales were detected in the 15N DNA fraction of all treatments, but was especially abundant in the NH4+CH4 incubations. Interestingly, in the original groundwater approximately 8% of the 16S rRNA gene sequence reads obtained by 454 sequencing belonged to Rhizobiales, while by the end of the incubation period Rhizobiales-related sequence reads decreased to <0.5%.
The pyro sequencing and DGGE assays used in this study generally produced similar profiles of the dominating bacterial taxa in the sample although the pyro sequencing provided much more detail as well as abundance information. While DGGE is a visual method for fast detection of microbial community profiles and is excellent for comparison of microbial community development between a low number of samples, it has some restrictions. It could be concluded from e.g., the present work and from other groundwater studies (e.g., Bomberg et al., 2014) that the method generally only presents the main groups of the community. In addition, many microbial groups dominating in a specific environment have been shown to go undetected by the DGGE method due to their incompatibility with the method of detection, low amounts of target template or high level of intra-species microdiversity, which makes optimization of the DGGE conditions very demanding (e.g., Costa et al., 2006; Alonso-Sáez et al., 2007; Sánchez et al., 2009). This was the case also here, where the DGGE failed to detect e.g., the Parcubacteria present at high relative abundance in both the original groundwater and the 28d incubation control (Table S1). However, the pyro sequencing approach failed to detect Sulfuricurvum in the original groundwater and the NH4+CH4-supplemented microcosms, although they were found in all samples according to the DGGE analysis.
The primers chosen for a study may also greatly affect the results. In this study we showed that microbial profile results depend on the detection method and primers used for characterizing microbial communities. For example the primers U968fGC/U1401r (Nübel et al., 1996) used for DGGE detected Desulfocapsa in the 28d incubation controls, but the primers 8F (Edwards et al., 1998) and P2 (Muyzer et al., 1993) used for 454-pyrosequencing did not.
Conclusions
The bacterial community of groundwater from 100 m depth in the VLJ-cave situated in the crystalline bedrock of the Fennoscandian Shield was affected by ammonia and nitrate supplementation. The bacterial population increased when N compounds were available, enriching specific bacterial groups. Known NRB belonging to ε-proteobacterial Sulfuricurvum and β-proteobacterial Hydrogenophaga/Malikia clades were greatly enriched when groundwater microcosms were supplemented with ammonium or nitrate with or without methane. Methylobacter was enriched in microcosms that had received ammonium with and without the addition of methane, indicating that the Methylobacter may also oxidize ammonia. In addition, it was demonstrated that bacterial clades belonging to Rhizobiales and Burkholderiales from nitrogen-poor deep crystalline bedrock groundwater were able to assimilate N from added nitrate and ammonium substrates. Here, the study of the N cycle in crystalline bedrock groundwater was restricted to specific enzymes of specific bacterial groups. In future investigations the N cycling genes of different taxonomic groups as well as different enzymes, such as the soluble nitrate reductase NAP, should be included. It would also be of value to investigate the N2 fixation and anaerobic ammonia oxidation (ANAMMOX) and archaeal ammonia oxidation in order to obtain a complete picture of the N cycle in deep crystalline bedrock environments. Nevertheless, we show here that despite the low abundance of N cycling bacteria in the pristine groundwater of Olkiluoto, N assimilating, ammonia oxidizing and nitrate reducing bacteria increase when the concentration of ammonia and nitrate increase, which may influence the long-term safety of the underground repository for low and intermediate level radioactive waste.
Author Contributions
HK, PR, LC, and MB conceived, designed and performed the experiments, analyzed the data and wrote the paper.
Funding
The work was performed on funding provided by the Finnish Research Program on Nuclear Waste Management 2011–2014 (project MICCU), and the Academy of Finland (project 261220).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The assistance of the staff of TVO Olkiluoto Nuclear Power Plant during sampling is gratefully acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01079
References
Alonso-Sáez, L., Balagué, V., Sà, E. L., Sánchez, O., González, J. M., Pinhassi, J., et al. (2007). Seasonality in bacterial diversity in north-west Mediterranean coastal waters: assessment through clone libraries, fingerprinting and FISH. FEMS Microbiol. Ecol. 60, 98–112. doi: 10.1111/j.1574-6941.2006.00276.x
Beller, H. R., Madrid, V., Hudson, G. B., McNab, W. W., and Carlsen, T. (2004). Biogeochemistry and natural attenuation of nitrate in groundwater at an explosives test facility. Appl. Geochem. 19, 1483–1494. doi: 10.1016/j.apgeochem.2003.12.010
Bomberg, M., Nyyssönen, M., Nousiainen, A., Hultman, J., Paulin, L., Auvinen, P., et al. (2014). Evaluation of molecular techniques in characterization of deep terrestrial biosphere. Open J. Ecol. 4, 468–487. doi: 10.4236/oje.2014.48040
Bomberg, M., Nyyssönen, M., Pitkänen, P., Lehtinen, A., and Itävaara, M. (2015). Active microbial communities inhabit sulphate-methane interphase in deep bedrock fracture fluids in Olkiluoto, Finland. BMRI 2015:979530. doi: 10.1155/2015/979530
Bothe, H., Ferguson, S. J., and Newton, W. E. (2007). Biology of Nitrogen Cycle. Amsterdam: Elsevier Science.
Buckley, D. H., Huangyutitham, V., Hsu, S. F., and Nelson, T. A. (2007). Stable isotope probing with 15N achieved by disentangling the effects of genome G+C content and isotope enrichment on DNA density. Appl. Environ. Microbiol. 73, 3189–3195. doi: 10.1128/AEM.02609-06
Campbell, B. J., Engel, A. S., Porter, M. L., and Takai, K. (2006). The versatile epsilon-proteobacteria: key players in sulphidic habitats. Nat. Rev. Microbiol. 4, 458–468. doi: 10.1038/nrmicro1414
Campbell, M. A., Nyerges, G., Kozlowski, J. A., Poret-Peterson, A. T., Stein, L. Y., and Klotz, G. M. (2011). Model of the molecular basis for hydroxylamine oxidation and nitrous oxide production in methanotrophic bacteria. FEMS Microbiol. Lett. 322, 82–89. doi: 10.1111/j.1574-6968.2011.02340.x
Costa, R., Götz, M., Mrotzek, N., Lottmann, J., Berg, G., and Smalla, K. (2006). Effects of site and plant species on rhizosphere structure revealed by molecular analysis of microbial guilds. FEMS Microbiol. Ecol. 56, 236–249. doi: 10.1111/j.1574-6941.2005.00026.x
D'Hondt, S., Rutherford, S., and Spivack, A. (2002). Metabolic activity of subsurface life in deep-sea sediments. Science 295, 2067–2070. doi: 10.1126/science.1064878
Edwards, U., Rogall, T., Blöcker, H., Emde, M., and Böttger, E. C. (1998). Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 17, 7843–53. doi: 10.1093/nar/17.19.7843
Guidon, S., and Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Handley, K. M., Bartels, D., O'Loughlin, E. J., Williams, K. H., Trimble, W. L., Skinner, K., et al. (2014). The complete genome sequence for putative H2-and S-oxidizer Candidatus Sulfuricurvum sp., assembled de novo from an aquifer-derived metagenome. Environ. Microbiol. 16, 3443–3462. doi: 10.1111/1462-2920.12453
Iker, B. C., Kambesis, P., Oehrle, S. A., Groves, C., and Barton, H. A. (2010). Microbial atrazine breakdown in a karst groundwater system and its effect on ecosystem energetics. J. Environ. Qual. 19, 509–518. doi: 10.2134/jeq2009.0048
Jukes, T. H., and Cantor, C. R. (1969). “Evolution of protein molecules,” in Mammalian protein metabolism, ed H. N. Munro (New York, NY: Academic Press), 21–123.
Kodama, Y., and Watanabe, K. (2004). Sulfuricurvum kujiense gen. nov., sp. nov., a facultatively anaerobic, chemolithoautotrophic, sulfur-oxidizing bacterium isolated from an underground crude-oil storage cavity. ISME J. 54, 2297–2300. doi: 10.1099/ijs.0.63243-0
López-Gutiérrez, J., Henry, S., Hallet, S., Martin-Laurent, F., Catroux, G., and Philippot, L. (2004). Quantification of a novel group of nitrate-reducing bacteria in the environment by real-time PCR. J. Microbiol. Methods 57, 399–407. doi: 10.1016/j.mimet.2004.02.009
Luque-Almagro, V. M., Gates, A. J., Moreno-Vivián, C., Ferguson, S. J., Richardson, D. J., and Roldan, M. D. (2011). Bacterial nitrate assimilation: gene distribution and regulation. Biochem. Soc. Trans. 39, 1838–1843. doi: 10.1042/BST20110688
Muyzer, G., de Waal, E. C., and Uitterlinden, A. G. (1993). Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59, 695–700.
Nicolaisen, M., and Ramsing, N. (2002). Denaturing gradient gel electrophoresis (DGGE) approaches to study the diversity of ammonia-oxiding bacteria. J. Microbiol. Methods 50, 189–203. doi: 10.1016/S0167-7012(02)00026-X
Nübel, U., Engelen, B., Felske, A., Snaidr, J., Wieshuber, A., Amann, R. I., et al. (1996). Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J. Bacteriol. 78, 5636–5643.
Nyerges, G., and Stein, L. Y. (2012). Ammonia cometabolism and product inhibition vary considerably among species of methanotrophic bacteria. FEMS Microbiol. Ecol. Lett. 297, 131–136. doi: 10.1111/j.1574-6968.2009.01674.x
Nyyssönen, M., Hultman, J., Ahonen, L., Kukkonen, I., Paulin, L., Laine, P., et al. (2014). Taxonomically and functionally diverse microbial communities in deep crystalline rocks of the Fennoscandian Shield. ISME J. 8, 126–138. doi: 10.1038/ismej.2013.125
Pastorelli, R., Piccolo, R., Simoncini, S., and Landi, S. (2013). New Primers for Denaturing gradient gel electrophoresis analysis of nitrate-reduction bacterial community in soil. Pedoshere 23, 340–349. doi: 10.1016/S1002-0160(13)60025-9
Pedersen, K. (2008). Numbers, biomass and cultivable diversity of microbial populations relate to depth and borehole-specific conditions in groundwater from depths of 4–450 min Olkiluoto, Finland. ISME J. 2, 760–775. doi: 10.1038/ismej.2008.43
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Purkamo, L., Bomberg, M., Nyyssönen, M., Kukkonen, I., Ahonen, L., and Itävaara, M. (2015). Heterotrophic communities supplied by ancient organic carbon predominate in deep fennoscandian bedrock fluids. Microb. Ecol. 69, 319–332. doi: 10.1007/s00248-014-0490-6
Rajala, P., Bomberg, M., Kietäväinen, R., Kukkonen, I., Ahonen, L., Nyyssönen, M., et al. (2015a). Deep subsurface microbes rapidly reactivate in the presence of C-1 Compounds. Microorganisms 3, 17–33. doi: 10.3390/microorganisms3010017
Rajala, P., Carpén, L., Vepsäläinen, M., Raulio, M., Sohlberg, E., and Bomberg, M. (2015b). Microbially induced corrosion of carbon steel in deep groundwater environment. Front. Microbiol. 6:647. doi: 10.3389/fmicb.2015.00647
Ray, K., Painter, R., and Byl, T. (2010). Ammonia Oxidation by Bacteria Collected from a Karst-Bedrock Well. USGS Scientific Investigations Report 2008–5023, U.S. Geological Survey.
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J.-F., et al. (2012). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
Sánchez, O., Gasol, J. M., Balagué, V., Massana, R., Mas, J., and Pedrós-Alió, C. (2009). Influence of primer mismatch and microdiversity on DGGE results: a case study with SAR11. Aquat. Microb. Ecol. 54, 211–216. doi: 10.3354/ame01267
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing MOTHUR: open-source, platform-independent, community-supported software for describing and comparing microbial communities Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Sievert, S. M., Scott, K. M., Klotz, M. G., Chain, P. S., Hauser, L. J., Hemp, J., et al. (2008). Genome of the epsilonproteobacterial chemolithoautotroph Sulfurimonas denitrificans. Appl. Environ. Microbiol. 74, 1145–1156. doi: 10.1128/AEM.01844-07
Spring, S., Wagner, M., Schumann, P., and Kämpfer, P. (2005). Malikia granosa gen. nov., sp. nov., a novel polyhydroxylalkanoate- and polyphosphate-accumulating bacterium isolated from activated sludge, and reclassification of Pseudomonas spinosa as Malikia spinosa comb. nov. Int. J. Syst. Evol. Microbiol. 55, 621–629. doi: 10.1099/ijs.0.63356-0
Wang, F., Zhou, H., Meng, J., Peng, X., Jiang, L., Sun, P., et al. (2009). GeoChip-based analysis of metabolic diversity of microbial communities at the Juan de Fuca Ridge hydrothermal vent. Proc. Natl. Ac. Sci. U.S.A. 106, 4840–4845. doi: 10.1073/pnas.0810418106
Weisburg, W. G., Barns, S. M., Pelletier, D. A., and Lane, D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703.
Whelan, S., and Goldman, N. (2001). A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699. doi: 10.1093/oxfordjournals.molbev.a003851
Yoon, K.-S., Tsukada, N., Sakai, Y., Ishii, M., Igarashi, Y., and Nishihara, H. (2008). Isolation and characterization of a new facultatively autotrophic hydrogen-oxidizing Betaproteobacterium, Hydrogenophaga sp. AH-24. FEMS Microbiol. Lett. 278, 94–100. doi: 10.1111/j.1574-6968.2007.00983.x
Keywords: nitrate reducer, ammonia oxidizer, stable isotope probing, nitrogen cycle, terrestrial deep biosphere, groundwater, crystalline bedrock, nitrogen assimilation
Citation: Kutvonen H, Rajala P, Carpén L and Bomberg M (2015) Nitrate and ammonia as nitrogen sources for deep subsurface microorganisms. Front. Microbiol. 6:1079. doi: 10.3389/fmicb.2015.01079
Received: 03 August 2015; Accepted: 21 September 2015;
Published: 15 October 2015.
Edited by:
Lisa Y. Stein, University of Alberta, CanadaReviewed by:
Jennifer F. Biddle, University of Delaware, USARichard S. Winder, Natural Resources Canada, Canada
Copyright © 2015 Kutvonen, Rajala, Carpén and Bomberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Malin Bomberg, VTT Technical Research Centre of Finland, PO Box 1000, Tietotie 2, FI-02044 VTT Espoo, Finland,bWFsaW4uYm9tYmVyZ0B2dHQuZmk=
†Present Address: Heini Kutvonen, United Medix Laboratories Ltd., Espoo, Finland