 José E. Belizário
José E. Belizário Mauro Napolitano
Mauro Napolitano
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 06 October 2015
Sec. Evolutionary and Genomic Microbiology
Volume 6 - 2015 | https://doi.org/10.3389/fmicb.2015.01050
This article is part of the Research TopicFrom Genes to Species: Novel Insights from MetagenomicsView all 20 articles
The human body is the residence of a large number of commensal (non-pathogenic) and pathogenic microbial species that have co-evolved with the human genome, adaptive immune system, and diet. With recent advances in DNA-based technologies, we initiated the exploration of bacterial gene functions and their role in human health. The main goal of the human microbiome project is to characterize the abundance, diversity and functionality of the genes present in all microorganisms that permanently live in different sites of the human body. The gut microbiota expresses over 3.3 million bacterial genes, while the human genome expresses only 20 thousand genes. Microbe gene-products exert pivotal functions via the regulation of food digestion and immune system development. Studies are confirming that manipulation of non-pathogenic bacterial strains in the host can stimulate the recovery of the immune response to pathogenic bacteria causing diseases. Different approaches, including the use of nutraceutics (prebiotics and probiotics) as well as phages engineered with CRISPR/Cas systems and quorum sensing systems have been developed as new therapies for controlling dysbiosis (alterations in microbial community) and common diseases (e.g., diabetes and obesity). The designing and production of pharmaceuticals based on our own body’s microbiome is an emerging field and is rapidly growing to be fully explored in the near future. This review provides an outlook on recent findings on the human microbiomes, their impact on health and diseases, and on the development of targeted therapies.
The evolution of Homo sapiens is linked to a mutualistic partnership with the human gut microbiota. The human genome is part of a collective genome of complex commensal, symbiotic, and pathogenic microbial community that colonizes the human body. Our microbiome includes not only bacteria, but also viruses, protozoans, and fungi (Backhed et al., 2012). Bacteria are a vast group of living organisms considered a domain of life in themselves (Woese et al., 1990). They are classified using DNA-based tests, morphologically and biochemically based on cell wall type, cell shape, oxygen requirements, endospore production, motility, and energy requirements. Hans Christian Gram (1850–1938), a Danish scientist, discovered that the presence of high levels of peptidoglycan (50–90%) produced a dark violet color, while low levels (<10%) resulted in reddish/pinkish colors, which are the respective staining of Gram-positive and Gram-negative bacteria. The Gram-negative cell wall is also characterized by the presence of lipopolysaccharides (LPSs). Based on their capacity to produce energy in presence or absence of oxygen, bacteria can also be classified as aerobic, anaerobic or facultative anaerobic. In addition to the generation of ATP via aerobic or anaerobic respiration, bacteria can also produce energy via fermentation. Facultative anaerobic bacteria are able to generate ATP with or without oxygen, while obligated anaerobic bacteria do not tolerate it and only survive in anaerobiosis. Lactobacillus, Staphylococcus, and Escherichia coli are examples of facultative anaerobic bacteria. Bacteroides, on the other hand, are obligated anaerobic species. In inflamed tissues, the enterocytes produce reactive oxygen species (ROS) and kill anaerobic bacteria increasing the abundance of aerobic and facultative species.
Bacteria are classified phylogenetically based on the analysis of nucleotide sequences of small subunit ribosomal RNA operons, mainly variable regions of the bacterial specific ribosomal RNA, 16S rRNA (Woese, 1987; Woese et al., 1990; Marchesi et al., 1998). Currently, the Bacteria domain is divided into many phyla; however, the majority of microbes forming the human microbiota can be assigned to four major phyla: Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria (Zoetendal et al., 2008; Arumugam et al., 2011; Segata et al., 2012). Firmicutes and Bacteroidetes represent more than 90% of the relative abundance of the gut microbiome (Arumugam et al., 2011; Segata et al., 2012). Firmicutes are a diverse phylum composed mainly of the Bacilli and Clostridia classes. They are Gram-positive, anaerobic (Clostridia) and obligate or facultative aerobes (Bacilli) characterized by a low GC content. Bacteria of Clostridium species produce endospores in order to survive to adverse (aerobic) conditions (Paredes-Sabja et al., 2014). The phylum Bacteroidetes is composed of Gram-negative, non-spore forming anaerobic bacteria that tolerate the presence of oxygen but cannot use it for growth. Actinobacteria (e.g., Bifidobacterium) are Gram-positive, multiple branching rods, non-motile, non-spore-forming, and anaerobic bacteria. Proteobacteria (e.g., Escherichia, Klebsiella, Enterobacter) are aerobic or facultative anaerobic, Gram-negative, non-spore-forming rod bacteria, which inhabit the intestinal tract of all vertebrates.
Recent survey studies on the variation of human microbiomes concluded that European individuals could be classified in up to three enterotypes based on 16S rRNA gene data and functional metagenome (whole genome shotgun) data (Arumugam et al., 2011; Koren et al., 2013). An enterotype refers to the relative abundance of specific bacterial taxa within the gut microbiomes of humans. The functional metagenome of each enterotype revealed differences in the proportions of genes involved in carbohydrate versus protein metabolism, which is consistent with diets of different populations (Arumugam et al., 2011; Koren et al., 2013). People differ by species composition, distribution, diversity, and numbers of bacteria (Yatsunenko et al., 2012). The dietary habits are the critical contributing factor. Diversity (microbiome variation and complexity) increases from birth and reaches its highest point in early adulthood, thereafter declining with old age. However, larger longitudinal studies that include more populations, such as South Americans, Indians and Africans need to be done to identify the actual structure and biological impact of the distinct human microbiomes. These studies may also reveal how evolution of life-styles modulated ancestral and modern human microbiomes. Here we will present and discuss recent advances of microbiome studies and the strategies for the development of innovative pharmaceuticals based on emerging population and individual microbiota genomic information.
The recent development of next generation sequencing (NGS) technologies such as 454, Solexa/Illumina, Ion Torrent and Ion Proton sequencers and the parallel expansion of powerful bioinformatics programs made possible the genomic analysis of over 1,000 prokaryotic and 100 eukaryotic organisms, including over 1,200 complete human genomes (Flintoft, 2012; Belizario, 2013). Metagenomics is a biotechnological approach to study genomic sequences of uncultivated microbes directly from their natural sources (Wooley et al., 2010). This allows the simultaneous analysis of microbial diversity connecting it to specific functions in different environments, such as soil, marine environments, and human body habitats (Ley et al., 2008; Robinson et al., 2010; Culligan et al., 2014). Using these novel methods, scientists have provided evidence for the existence of more than one thousand microorganism species living in our body (Arumugam et al., 2011; Segata et al., 2012) and an estimation of 107 to 109 different species of bacteria living on earth (Curtis et al., 2002). More important, the metagenomics approach has the potential to uncover entirely novel genes, gene families, and their encoded proteins, which might be of biotechnological and pharmaceutical relevance.
Currently several international projects aimed at the characterization of the human microbiota are being carried out. The Human Microbiome Project (HMP) is a research initiative of the National Institute of Health (NIH) in the United States, which aims to characterize the microbial communities found in several different sites of the human body (Turnbaugh et al., 2007; Backhed et al., 2012; Human Microbiome Project, 2012a,b). MetaHIT (Metagenomics of the Human Intestinal Tract) is a project financed by the European Commission and is under management of a consortium of 13 European partners from academia and the industry. The International Human Microbiome Consortium (IHMC) is composed of European, Canadian, Chinese, and US scientific institutions1.
A simple molecular approach to explore the microbial diversity is based on the analysis of variable regions of 16S rRNA gene using “universal” primers which are complementary to highly conserved sequences among the homologous 16S rRNA genes (Marchesi et al., 1998; Culligan et al., 2014). These genes contain nine hypervariable regions (V1–V9) whose sequence diversity is appropriated for characterizing bacterial community compositions in complex samples (Guo et al., 2013; Jiang et al., 2014; Montassier et al., 2014). DNA sequences obtained with this approach can be mapped onto a reference set of known bacterial genomes. For this purpose useful bioinformatics tools and databases are available. For example, the SILVA database2 is a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data that helps determine an optimal alignment for the different sequence regions (Pruesse et al., 2007). First released in 1995, The Ribosomal Database Project (RDP) is another database that provides high quality alignments of archaeal and bacterial 16S rRNA sequences as well as fungal 28S rRNA sequences (Maidak et al., 1996). The microbial profiling and phylogenetic clustering of microbiomes of the American and European population have been already deposited and are free for consultation3,4. The HMP projects and other independent projects have been generating an enormous amount of metagenomic data and the assemblies of microbiome data is being undertaken by the Genomes OnLine Database5. The data management system for cataloging and continuous monitoring of worldwide sequencing projects contains data from over 4000 metagenome sequencing projects, in which more than 1500 are aimed at the characterization of host associated metagenomes (Human Microbiome Jumpstart Reference Strains et al., 2010; Fodor et al., 2012; Reddy et al., 2015).
The first release of the HMP database included microbiome data of nasal passages, the oral cavity, skin, gastrointestinal tract, and urogenital tract (Human Microbiome Project, 2012a,b). Figure 1 schematically summarizes the data of these studies. The results of over 690 human microbiomes have shown that the majority of bacteria of the gut microbiome belongs to four phyla: Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria (Human Microbiome Project, 2012a,b). Only a fraction of microbes identified so far have been successfully cultured, and thousands are yet to be fully sequenced for a deeper taxonomic resolution (strains and subspecies) and functional analysis at the genomic level (Qin et al., 2010; Robinson et al., 2010; Abubucker et al., 2012; Flintoft, 2012; Zhou et al., 2013).
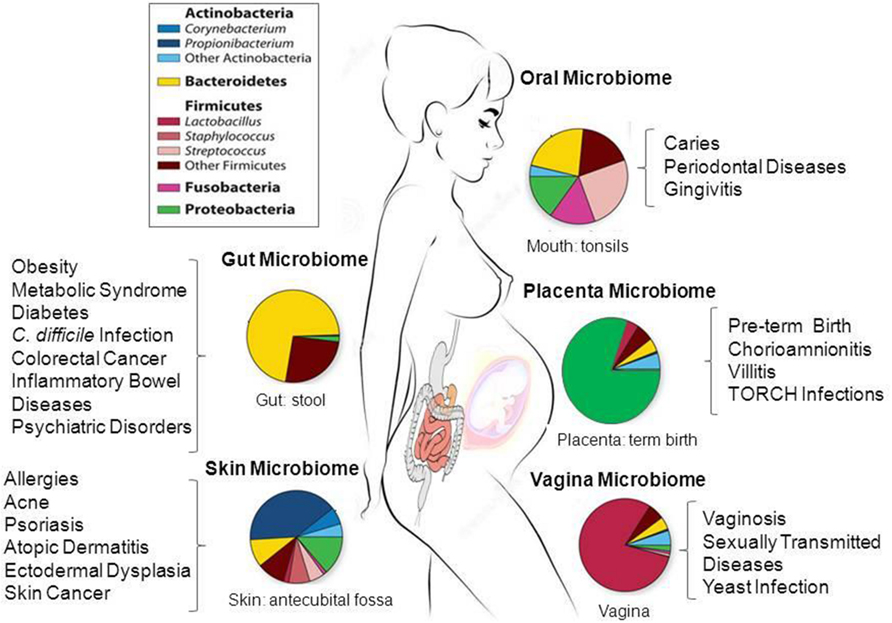
FIGURE 1. Taxonomic distribution, prevalence and abundance of microbial taxa that inhabit healthy human body sites as defined in the human microbiome projects (HMP). The colored rectangles denote phylum/class and genera. Clinical studies of the microbiome will help to elucidate the link between microbes and the promotion of a large number of diseases and pathological conditions as shown in the figure. The images were adapted from NIH HMP (http://www.hmpdacc.org/) and National Human Genome Research Institute (https://www.genome.gov/). TORCH, Toxoplasmosis, Oher infections (coxsackievirus, HIV, syphilis, etc), Rubella, Cytomegalovirus, Herpes simplex.
The metagenome wide association studies in development in many countries are promising in predicting new diagnostic and prognostic tools for numerous human disorders. The results of these studies will dramatically increase our knowledge of diseases linked to microbial composition (Qin et al., 2010; Clemente et al., 2012; Flintoft, 2012; Gevers et al., 2012). In order to better understand the host-gene-microbial interactions and the role of non-pathogenic and pathogenic strains in large populations, we need to compare microbiome profiles across multiple body sites and microbiome datasets under environmentally controlled normal and disease conditions. In the following sections, we will provide a synthesis of the recent studies on the human microbiomes identified in some major body sites.
The HMPs have shown that the human gut harbors one of the most complex and abundant ecosystems colonized by more than 100 trillion microorganisms (Human Microbiome Project, 2012a,b). In adults, the majority of the bacteria found in the gut belong to two bacterial phyla, the gram-negative Bacteroidetes and the gram-positive, Firmicutes; and the others represented at subdominant levels are the Actinobacteria, Fusobacteria, and Verrucomicrobia phyla, but this varies dramatically among individuals (Eckburg et al., 2005; Zoetendal et al., 2008; Arumugam et al., 2011; Backhed et al., 2012; Segata et al., 2012). For instance, the most abundant genera from the Bacteroidetes phylum are Bacteroides and Prevotella species, which represent 80% of all Bacteroidetes in fecal samples. Nonetheless, many of the taxa numerically underrepresented and less-abundant bacterial species exert fundamental functions at a particular location in the gut. To better define these different microbial colonization and microbiota structure in different cohorts arose the concept of ‘enterotype clusters’ that allow the classification of each individual based on the relative abundance of specific bacterial taxa in fecal samples, and their microbial metabolic and functional pathways (Arumugam et al., 2011; Backhed et al., 2012; Koren et al., 2013). The results of metagenomic sequencing of fecal samples from European, American, and Japanese subjects confirmed the three robust clusters dominated by Bacteroides (enterotype 1), Prevotella (enterotype 2), and Ruminococcus (enterotype 3), each one characterized by specific taxonomic composition and relative abundance of metabolic pathways. For example, enterotype 1 was enriched in biosynthesis of biotin, riboflavin pantothenate and ascorbate; enterotype 2 in biosynthesis of thiamine and folate. Enterotype 3 showed high abundance of genes involved in haem biosynthesis pathway. Although in one of these studies (Arumugam et al., 2011) it was confirmed that a set of 12 genes correlated with age and a set of three functional modules with the body mass index (BMI), further studies will be required to determine if specific microbiome and/or enterotype is associated with gender, BMI, health status, diet, and age of individuals (Arumugam et al., 2011; Backhed et al., 2012; Koren et al., 2013).
Although stable over long periods, the composition and functions of the gut microbiome may be influenced by a number of factors including genetics, mode of delivery, age, diet, geographic location, and medical treatments (Clemente et al., 2012; Brown et al., 2013). The intestinal microbiota is acquired in the postnatal periods of time, consisting of a wide variety of bacteria that plays different functions in the human host, including nutrient absorption, protection against pathogens, and modulation of the immune system (Brown et al., 2013). The gut is an anaerobic environment in which indigenous species have co-evolved with the host. The aerobic pathogenic species cannot invade and colonize it; however, anaerobic and facultative pathogenic species can invade it causing diseases. High diversity defines healthy human gut microbiomes, whereas reduction in diversity may be associated with dysbiosis (Manichanh et al., 2006; Backhed et al., 2012). Dysbiosis refers to an imbalance in the microbiome structure that results from an abnormal ratio of commensal and pathogenic bacterial species. Many studies have suggested a possible direct relationship between dysbiosis and inflammatory and metabolic diseases such as is inflammatory bowel diseases (IBD) including colitis and Crohn’s disease (CD), obesity and cancer (Clemente et al., 2012; Sartor and Mazmanian, 2012; Brown et al., 2013). However, investigation of such a complex ecosystem is difficult and it is still not easy to define how shifts in microbial composition and member abundance can lead to diseases. Induction of some IBD has been linked to a reduction of Firmicutes and Bacteroidetes and an expansion of Proteobacteria. For example, Faecalibacterium prausnitzii, a prominent member of Clostridium group IV (Firmicutes), protective and anti-inflammatory commensal bacterium, is frequently reduced in CD patients (Sokol et al., 2008; Sartor and Mazmanian, 2012). Despite these advances, it should be noted that microbiota composition varies between different locations in the gastrointestinal tract (Eckburg et al., 2005; Zoetendal et al., 2008; Arumugam et al., 2011; Cucchiara et al., 2012; Segata et al., 2012; Lepage et al., 2013). Most studies in the literature have explored only fecal microbiota. Fecal samples contain between 1,000 and 1,150 bacterial species, and up to 55% are uncultivable and thus uncharacterized (Zoetendal et al., 2008; Qin et al., 2010; Segata et al., 2012; Zhou et al., 2014). Our knowledge is especially limited when it comes to the other parts of the GI tract, a potential source of uncharacterized microbial species, which is largely due to sampling constraints.
The dysregulation of the intestinal immune system can also trigger microbial dysbiosis (Clemente et al., 2012; Sartor and Mazmanian, 2012; Brown et al., 2013). Many different inflammatory diseases are characterized by mutations or loss of some innate response genes in lymphoid tissues, Paneth cells, smaller Peyer’s patches and mesenteric lymph nodes (Clemente et al., 2012; Frantz et al., 2012; Sartor and Mazmanian, 2012). The growth of microbiota communities is under control of distinct subfamilies of host genes encoding antimicrobial peptides (AMPs). AMPs are the most ancient component of the innate host response against bacterial infections (Guani-Guerra et al., 2010; Ostaff et al., 2013). When bacteria colonize a given human habitat, the expression of AMPs, including α and β defensins and cathelicidins, is upregulated in order to limit the spreading of bacteria. The equilibrium between the immune system and immunoregulatory functions of bacteria appears to be a delicate balance in which the loss of a specific species can lead to an overreaction or suppression of the innate immune system (Round and Mazmanian, 2009; Clemente et al., 2012; Sartor and Mazmanian, 2012; Brown et al., 2013). Intestinal epithelial cells (IECs) form a physical and immunological barrier that separate luminal bacteria from underlying immune cells in the intestinal mucosa. IECs and hematopoietic cells express a variety of receptors called pattern recognition receptors (PRRs) that mediate the interactions between the immune system and the commensal microbiota (Frantz et al., 2012). Toll-like receptors (TLRs) and nuclear oligomerization domain-like receptors (NLRs) are examples of PRR that recognize unique microbial molecules named microbe-associated molecular patterns (MAMPs) including lipopolysaccharides (LPS), lipid A, peptidoglycans, flagella, and microbial RNA/DNA. These receptors activate inflammasomes and thereby the production of cytokines TNF-α and IL-1β (Brown et al., 2013; Sangiuliano et al., 2014). Myeloid differentiation factor MyD88 is an adaptor protein that is essential for TLRs signaling and host-microbial interactions and tissue homeostasis (Sangiuliano et al., 2014). Mice lacking MyD88 in IECS (IEC-Myd88-/- mice) display intestinal barrier disruption, deficiency in the production of pro-inflammatory cytokines and AMPs and overgrowth of several enteric bacterial pathogens (Frantz et al., 2012). It will be important to understand when and how dysbiosis and genetic defects in mucosa-IECs and innate regulatory mechanisms can lead to development of infectious or inflammatory diseases.
Microorganisms synthesize a wide range of low-molecular weight signaling molecules (metabolites), many of which are similar to metabolites produced by human cells (Wikoff et al., 2009). The maintenance of a stable, fermentative gut microbiota requires diets rich in whole plant foods particularly high in dietary fibers and polyphenols (Zoetendal et al., 2008). Under anaerobic conditions, species belonging to the Bacteroides genus, and to the Clostridiaceae and Lactobacillaceae families, produce short-chain fatty acids (SCFAs). Acetate (with two carbons), propionate (with three carbons), and butyrate (with four carbons) are SCFA used by the epithelial cells of the colon (colonocytes) and act as a major player in maintenance of gut homeostasis (Meijer et al., 2010). SCFAs induce the secretion of glucagon-like peptide (GLP-1) and peptide YY (PYY), which increase nutrient absorption from the intestinal lumen. This is a key process in controlling mucosal proliferation, differentiation and maintenance of mucosal integrity (Round and Mazmanian, 2009). Individuals colonized by bacteria of the genera Faecalibacterium, Bifidobacterium, Lactobacillus, Coprococcus, and Methanobrevibacter have significantly less of a tendency to develop obesity-related diseases like type-2-diabetes and ischemic cardiovascular disorders (Ley et al., 2006; Le Chatelier et al., 2013). These species are characterized by high production of lactate, propionate and butyrate as well as higher hydrogen production rates, which are known to inhibit biofilm formation and activity of pathogens, including Staphylococcus aureus, in the gut (Le Chatelier et al., 2013). Genetic and diet-induced mouse models of obesity have shown that the Bacteroidetes/Firmicutes ratio is decreased in obese animals compared to non-obese animals, which is consistent with what has been observed in human obese subjects (Ley et al., 2005, 2006; Le Chatelier et al., 2013; Verdam et al., 2013). However, controversies exist regarding the human data on gut microbiota composition in relation to obesity (Turnbaugh et al., 2009; De Filippo et al., 2010; Verdam et al., 2013). The intestinal microbiota changes in obese mice may increase the intestinal permeability and inflammation locally and in adipose tissues (Cani and Delzenne, 2011; Kootte et al., 2012). As discussed in recent studies, the microbial-derived LPS released through circulation may promote low-grade inflammatory process and metabolic disturbances related to obesity, such as insulin resistance and type-2 diabetes (Cani and Delzenne, 2011; Kootte et al., 2012). Despite our currently incomplete understanding of the mechanisms, there have been high expectations that targeted changes in microbiota by the rational use of prebiotics and probiotics might abolish metabolic alterations associated with obesity (Cani and Delzenne, 2011; Kootte et al., 2012).
The first study based on pyrosequencing of barcoded 16S rRNA genes of vaginal microbiota performed on samples from North-American women revealed the inherent differences within and between women in different ethnic groups (Ravel et al., 2011). The vaginal microbial composition from three vaginal sites (mid-vagina, cervix, and introitus) has been compared to the buccal mucosa and the perianal region in recent studies (Fettweis et al., 2012; Romero et al., 2014; Vaginal Microbiome consortium6). These studies have shown that the vagina possesses over 200 phylotypes and that the most predominant belong to the phyla Firmicutes, Bacteroidetes, Actinobacteria, and Fusobacteria (Ravel et al., 2011; Romero et al., 2014). The vagina has low pH due to secretion of lactic acid and hydrogen peroxide by Lactobacillus sp. If Lactobacillus decreases under the effects of antibiotics, Gardnerella vaginalis and Peptostreptococcus anaerobius, Prevotella sp., Mobiluncus sp. Sneathia, Atopobium vaginae, Ureaplasma, Mycoplasma, and numerous fastidious or uncultivated anaerobes can cause bacterial vaginosis (BV). BV is an ecological disorder of the vaginal microbiota that affects millions of women annually, and is associated with numerous adverse health outcomes including preterm birth and acquisition of sexually transmitted infections, e.g., HIV, Neisseria gonorrhoeae, Chlamydia trachomatis, and HSV-2 (Kenyon et al., 2013). Lactobacillus morphotypes predominate in normal grade 1. BVs grade 3 and higher are characterized by a reduced number of lactobacilli and increased diversity, especially high concentration of Gram-negative bacteria and coccobacillus (e.g., G. vaginalis and G. mobiluncus) and Peptostreptococcus (Delaney and Onderdonk, 2001). The results of microbiome studies of the vagina are showing different patterns and imbalances in bacterial communities associated with BVs, as well as those associated with non-infectious pathological states that predict increased risk for infertility, spontaneous abortion, and preterm birth.
Advances in microbiological diagnostic techniques have shown the complex interaction between the oral microbiota and the host (Segata et al., 2012; Jiang et al., 2014; Perez-Chaparro et al., 2014). Bacteria, fungi, archaea, viruses, and protozoa are part of the oral microbiome. The HMP investigated bacterial communities in nine intraoral sites: buccal mucosa, hard palate, keratinized gingiva, palatine tonsils, saliva, sub-and supra gingival plaque, throat, and tongue dorsum (Human Microbiome Project, 2012a,b). Over 300 genera, belonging to more than 20 bacterial phyla were identified (Zhou et al., 2013; Jiang et al., 2014). However, only a limited number of species find proper conditions to colonize the root canal system (Zhou et al., 2013; Perez-Chaparro et al., 2014). The microbiota of periodontitis or caries is usually complex consisting of Gram-negative anaerobic bacteria such as Porphyromonas gingivalis, Treponema denticola, Prevotella intermedia, Tannerella forsythia, and Agregatibacter actinomycetemcomitans (Mason et al., 2013; Jiang et al., 2014). Most early data on the endodontic microbiota were obtained by culture-based method and it is likely that not-yet-cultivable and unknown species of bacteria play a role in oral microbial shift toward a disease (Mason et al., 2013; Zaura et al., 2014). As expected, deep DNA sequencing data revealed a larger number of taxa involved in endodontic infections. Species of phyla Bacteroidetes, Firmicutes, Proteobacteria, Spirochaetes, Synergistetes, and Candidatus Saccharibacteria were more frequently found. All these studies on bacterial diversity in endodontic infections revealed high inter-subject variability, indicating the need for further studies using homogenous diagnosis criteria in a significant number of healthy subjects (Mason et al., 2013; Jiang et al., 2014; Perez-Chaparro et al., 2014).
The skin is the human body’s largest organ, colonized by over 100 microbial phylotypes, most of which are harmless or even beneficial to their host (Rosenthal et al., 2011; Ladizinski et al., 2014; Zhou et al., 2014). Phylotypes, microbial abundance and diversity differ in relation to skin color, race, and geographic location (Grice et al., 2009; Rosenthal et al., 2011). Colonization is influenced by the ecology and the epidermis layers of the skin surface. Therefore it is highly variable depending on topographical location, endogenous host factors and exogenous environmental factors. The Actinobacteria phylum is the most abundant on the skin. Gram-positive Staphylococcus epidermidis and Propionibacterium acnes are predominant on human epithelia and in sebaceous follicles, respectively. Propionibacterium acnes colonizes healthy pores and is responsible for the production of SCFAs and thiopeptides, which inhibit the growth of Staphylococcus aureus and Streptococcus pyogenes. However, depending on the host’s immune system, the overgrowth and clogging of pores allow subsequent colonization of S. epidermidis and Staphylococcus aureus. Atopic dermatitis is one chronic inflammatory condition of the skin that occurs in many children and adults (Grice et al., 2009). Staphylococcus sp. Corynebacterium sp. and the fungi Candida sp, and Malassezia sp. are also frequently associated with a number of skin diseases, including atopic dermatitis and abnormal flaking and itching of the scalp (Grice et al., 2009).
The skin microbiota is under autonomous control of the local cutaneous immune system, thus it is independent of the systemic immune response which is modulated by the gut microbiota (Naik et al., 2012). The major innate mechanism of antimicrobial defense on the skin consists of AMPs, for example defensins, cathelicidin LL-37 and dermcidin (Guani-Guerra et al., 2010). These peptides are emerging as important tools in the control of skin pathogenic bacteria as well as bacteria involved in diseases of the lung and gastrointestinal tract. Many AMPs bind to the phospholipid membrane surfaces, forming ion-channels and pores causing leakage and cell death (Guani-Guerra et al., 2010; Ostaff et al., 2013). However, their specific immunomodulatory roles in innate immune defense against bacterial and viral infection remain poorly understood (Ostaff et al., 2013; Wang, 2014). An enhanced understanding of the skin microbiome is necessary to gain insight into AMPs and innate response in human skin disorders. The cutaneous inflammatory disorders such as atopic dermatitis, psoriasis, eczema, and primary immunodeficiency syndromes have been associated with dysbiosis in the cutaneous microbiota. The skin commensals promote effector T cell response, via their capacity to control the NF-κB signaling and the production of cytokines TNF-α and IL-1β (Hooper et al., 2012). The binding of the skin microbiota components to TLRs or NLRs allows a sustainable homeostasis toward innate and adaptive immunity within a complex epithelial barrier throughout distinct topographical skin sites.
Historically, the fetus and intrauterine environment were considered sterile. However, the first profile of microbes in healthy term pregnancies identified a unique microbiome niche in normal placenta, composed of non-pathogenic commensal microbiota from the Firmicutes, Tenericutes, Proteobacteria, Bacteroidetes, and Fusobacteria phyla (Aagaard et al., 2014). This study describes the microbial communities of 320 placental specimens and, despite the expected differences between individuals, the taxonomic classification of the placental microbiome bears most similarity to the non-pregnant oral microbiome, in particular to those associated with tongue, tonsils, and gingival plaques. One predominant species was Fusobacterium nucleatum, a Gram-negative oral anaerobe. E. coli was also found in placenta; however, it is not present in the oral microbiome (Aagaard et al., 2014). The authors suggested a possible hematological spread of oral microbiome during early vascularization and placentation. The pathways related with the metabolism of cofactors and vitamins were the most abundant among placental functional gene profiles, which is different from the metabolic pathways found in other body sites (Aagaard et al., 2014).
The balance of the different microbe species in and on the human body changes throughout life and particularly in different stages of pregnancy (Qin et al., 2010; Human Microbiome Project, 2012a,b). It is well known that preterm delivery (<37 weeks) causes substantial neonatal mortality and morbidity (DiGiulio et al., 2008). Placentas from normal deliveries and preterm deliveries contained different populations of microbial species (Groer et al., 2014). The gram-negative bacillus Durkholderia was associated with preterm delivery and the gram-positive, rod-shaped, facultative anaerobic bacteria Paenibacillus with term delivery (Aagaard et al., 2014). Consistent with other studies, an enrichment in Streptococci, Acinetobacter and Klebsiella was also demonstrated in women with history of antenatal infection (Aagaard et al., 2014).
The presence of different microbes in amniotic fluid, umbilical cord blood, meconium (first stool), placental and fetal membranes suggested the existence of various routes and mechanisms by which bacteria from different microbiota translocate to placenta and babies (DiGiulio et al., 2008). Studies in mice have demonstrated the placental transmission from mother’s oral microbiota (Fardini et al., 2010). Many of these organisms are transmitted to babies during nursing. Babies born vaginally have more diverse gut microbial communities similar to their mother’s vaginal microbiota, while microbiomes of babies delivered by Cesarean section are similar to skin microbiota (Dominguez-Bello et al., 2010). The lack of exposure to maternal vaginal microbiome might explain why cesarean section babies are at greater risk of developing type 1 diabetes, celiac disease, asthma, and obesity (DiGiulio et al., 2008; Dominguez-Bello et al., 2010). Breastfed babies’ microbiome is enriched with Lactobacillus and Bifidobacterium species whereas microbiome of babies fed with formula/solid food are enriched with Enterococci, Enterobacteria, Bacteroides, Clostridia, and Streptococci (Guaraldi and Salvatori, 2012; Palmer et al., 2012; Thompson et al., 2015). The transition from breast milk to solid foods is associated with acquisition of a more adulthood-like microbiome; however, infectious diseases, antibiotic use and the characteristics of the diet can interfere with babies’ microbiota composition (Thompson et al., 2015). Together, these findings emphasize the need for further studies on placental microbiome for elucidating more mechanisms to be explored in the prevention and treatment of babies from preterm birth and other diseases.
Metagenomics has proven to be a powerful tool in determining the diversity and abundance of microbes in the human body. The microbiome databases have been explored as sources of interesting targets to drug development (Cani and Delzenne, 2011; Collison et al., 2012; Haiser and Turnbaugh, 2012; Carr et al., 2013; Wallace and Redinbo, 2013). Therapeutic interventions in the microbiome can be directed against molecular entities, such as essential and antibiotic resistance genes to quorum sensing systems components used to control microbial networking behaviors, including the chemical communication and production of virulence factors (Collison et al., 2012). In the next sections, we will present and discuss strategies to discover novel antimicrobial targets as well as dietary interventions and microbial modification genetic tools to eliminate pathogenic microorganisms and to control dysbiosis.
Searching of essential genes for bacterial growth and viability is the first step for identifying potential drug targets (Wallace and Redinbo, 2013). Computational analyses can provide candidate targets in microbial community of pharmacological significance for controlling bacterial species involved in chronic diseases, metabolic, and cardiovascular diseases as well as drug metabolism (Collison et al., 2012). The metagenomic databases are critical for constructing gene and protein networks and an initial framework for drug target screening (Collison et al., 2012; Carr et al., 2013; Manor and Borenstein, 2015). Several bioinformatics approaches have been used to identify microbial gene essentiality and putative new classes and functions for unique microbial genes in the metagenomic databases. HUMAnN is a program for metagenomic functional reconstruction to directly associate community functions with habitat and host phenotype. This program has been used to compare functional diversity and organismal ecology in the human microbiome (Abubucker et al., 2012). About 20% of all genes in a strain are essential and this has gained interest in drug discovery research (Christen et al., 2011). In vitro transposition and genetic transformation of the wild-type bacteria using a transposon library is a reliable experimental approach to uncover gene essentiality (van Opijnen et al., 2009). ESSENTIALS is another software for rapid analysis of high throughput transposon insertion sequencing data and discovery of essential genes (Zomer et al., 2012).
The majority of unique targets found in microbes’ genomes are genes responsible for the metabolism of carbohydrates, amino acids, xenobiotics, methanogenesis, and the biosynthesis of vitamins and isoprenoids. These genes are either non-homologous or orthologous to those encompassed in human genome. Vitamin biosynthetic pathways constitute a major source of potential drug targets. Most bacteria synthesize thiamine de novo, whereas humans depend on dietary uptake. Folic acid (vitamin B9) is an indispensable cofactor, which plays a key role in the methylation cycle and in DNA biosynthesis. Enzymes of the folate biosynthesis pathway, for example, dihydrofolate reductase, have been an attractive pharmaceutical targets for inhibiting folate synthesis. Sulfanilamide and trimethoprim are examples of effective antimicrobials used in a broad range of infectious diseases. Niacin (vitamin B3) participates in the biosynthesis of nicotinamide adenine dinucleotide (NAD+), a coenzyme essential in electron transport reactions in cell metabolism processes. Bacterial NAD+ kinases have been explored as targets for inhibiting bacterial growth. Methionine is not synthesized de novo in humans, and is supplied by diet. In contrast, most bacteria need to synthesize methionine to survive. S-adenosylmethionine synthetase, a key enzyme in methionine biosynthesis, is one drug target whose great potential has been explored against various pathogens. New drugs, for example platensimycin and platencin, that inhibit the microbial fatty acid synthesis (FAS) pathway by targeting key FAS enzymes have been successfully developed (Parsons et al., 2014). A recent survey identified 127 orthologous groups conserved in both human and human commensal gut microflora that are not suitable targets for drug development. However among these, the 20 aminoacyl-tRNA synthetases (aaRSs), which encode essential enzymes for protein synthesis, can be used since bacterial and eukaryotic AaRS have different specificity for tRNAs (Ochsner et al., 2007; Mobegi et al., 2014). These are only few examples of attractive targets for drug development; however, metagenomic data will open new frontiers for discovery of essential genes.
The structure of the microbial community is maintained by specific microbial communication, cell signaling through cell-to-cell contact, metabolic interactions, and quorum sensing (Wright, 2010). Species within a bacterial community are either susceptible or resistant to epithelial innate AMPs and/or chemical antibiotics (Seo et al., 2010; Wozniak and Waldor, 2010; Sommer and Dantas, 2011). Bacterial genomes acquired resistance and metabolic genes from mobile genetic elements (MGE), including conjugative transposons, also called integrative conjugative elements (ICE), which are horizontally transferred by bacteriophages and plasmids (Wozniak and Waldor, 2010). Antibiotic resistance genes encoded in microbial genomes include multidrug efflux transporters, tetracycline resistance genes, vancomycin resistance genes, and beta-lactamases. In addition, a number of microbial genes and products, including bacteriocins, lysins, holins, restriction/modification endonuclease systems, and other virulence factors contribute to resistance to antibiotics (Dawid et al., 2007; Seo et al., 2010; Wozniak and Waldor, 2010; Smillie et al., 2011). Targeted (PCR-based) and functional metagenomic approaches have been used to track the presence of resistance genes or their families in different ecosystems (Mullany, 2014). A method to specifically trap plasmids containing antibiotic resistance genes called transposon-aided capture (TRACA) has been developed (Jones and Marchesi, 2007; Mullany, 2014). In this method, the plasmids are tagged with transposons that contain a selectable marker and a replication origin, which facilitate acquisition of plasmids from the human gut metagenomic DNA extracts, and subsequent maintenance and selection in an E. coli host.
Most of the antibiotics used to fight bacterial infections today are derived from soil microbes. Penicillin, the first true antibiotic, came from the soil fungus Penicillium (Kardos and Demain, 2011). To investigate the role of soil microbiota as a reservoir of genes encoding antibiotic resistance in the metagenomic data set, the ORFs found on contigs and on unassembled reads were compared with 3,000 known antibiotic resistance genes (Forsberg et al., 2014). It was concluded that most of the identified soil bacteria resistance genes were not typically close to known human pathogen resistance genes, suggesting little sharing between soil and gut bacterial species. A study on the microbiome of uncontacted Amerindians, members of a Yanomami isolated village living in the Amazon region has revealed the highest diversity of bacteria and genetic functions in fecal, oral, and skin bacterial microbiome ever reported compared with the US group (Clemente et al., 2015). Despite their isolation and no known exposure to commercial antibiotics, they carry functional antibiotic resistance genes with over >95% amino acid identity to those that confer resistance to semisynthetic and synthetic antibiotic monobactam and ceftazidime (Clemente et al., 2015). This finding provided important insights into how westernization impacts on the heritability of the microbiome among populations (Yatsunenko et al., 2012). There is evidence suggesting that exposure to microbes from animal gut microbiomes and within our indoor spaces (house, office, schools, cars, etc.) may become new sources for antibiotics and antibiotic resistance genes to human populations (Wright, 2010; Sommer and Dantas, 2011; Forslund et al., 2013). These discoveries emphasize the importance of continued functional investigations on antibiotic resistance reservoirs in metagenomic data from isolated ancestral and modern populations with a given disease.
The term “Quorum Sensing” (QS) indicates systems used by bacteria to communicate with each other in order to synchronize their gene expression activities and behave in unison as a group (Miller and Bassler, 2001; Waters and Bassler, 2005; Hense and Schuster, 2015). This mechanism controls the synthesis of secreted products, disease-causing virulence factors, and many metabolites, including bacterial antibiotics that target competing bacteria, and substances that suppress the immune system (Miller and Bassler, 2001; Waters and Bassler, 2005). Thus, an alternative to killing or inhibiting growth of pathogenic bacteria is targeting these key regulatory systems (Finch et al., 1998; Defoirdt et al., 2010). Metagenomic studies have identified the genetic and phenotypic diversity of quorum-sensing systems that co-evolved with pathogenic species (Joelsson et al., 2006; Kimura, 2014). QS system was first described in marine bacteria Vibrio harveyi and V. fischeri, which use LuxI and LuxR proteins to control the expression of the luciferase enzyme for emitting luminesce upon reaching a critical mass or “quorum” (Nealson and Hastings, 1979). These bacteria secrete in the extracellular environment a small molecule, an acylated homoserine lactone (AHL), called autoinducer 1 (AI-1), to communicate with members of the same species (intraspecific communication; Miller and Bassler, 2001; Waters and Bassler, 2005; Ng and Bassler, 2009). After its discovery in marine bacteria, QS systems have been identified in more than 70 different bacterial species, including Streptococcus pneumoniae, Bacillus subtilis, and Staphylococcus aureus (Miller and Bassler, 2001; Waters and Bassler, 2005; Ng and Bassler, 2009). The QS systems control not only bioluminescence, but also other cooperative processes such as sporulation, conjugation, nutrient acquisition, biofilm formation, bio-corrosion, and antibiotics and toxins (Waters and Bassler, 2005; Kimura, 2014; Hense and Schuster, 2015). Remarkably, bacteria not only can communicate with members of the same species, but they are also able to sense the presence of different species in a community (interspecific communication). This interspecific communication is performed using a second type of autoinducer (AI-2). Thus, while each bacterial species has its own AI-1 to talk intraspecifically, AI-2 is common to all Gram-negative and Gram-positive bacteria. In fact AI-2 is not a single molecule but rather it refers to a group of molecules belonging to the family of interconverting furanones derived from 4,5-dihydroxy-2,3-pentanedione (DPD), whose biosynthesis is under the control of the enzyme LuxS (Xavier and Bassler, 2003). Development of novel compounds able to disrupt QS mechanisms has been carried out in recent years. For example QS quenching enzymes like lactonases and acylases are able to degrade acylated homoserine lactone (Dong and Zhang, 2005). A series of compounds, named halogenated furanones produced by many microbial species, mostly belonging to the Proteobacteria, can interfere with AHL and AI-2 QS pathways in Gram-negative and Gram-positive bacteria (Manefield et al., 2002; Rasko et al., 2008; Kayumov et al., 2014). Identification of the chemical signals, receptors, target genes, and mechanisms of signal transduction involved in quorum sensing are essential to our understanding how bacterial cell-cell communication may be used in preventing colonization by pathogenic bacteria. More data from metagenomic and metabolomics studies will help to decode the bacterial cross-talk and microbiome-immune system interplay, and particularly, distinctive regulatory mechanisms.
Antibiotics have been used to treat infectious diseases over the past century. However, it is clear that antibiotic treatment can render individuals more susceptible to infections (Dethlefsen et al., 2008; Forslund et al., 2013). High doses and frequent use of antibiotics can disrupt and destabilize the normal bowel microbiota, predisposing patients to develop Clostridium difficile infections. Up to 35% of these patients develop a chronic recurrent pattern of disease. Fecal bacteriotherapy is the transplantation of liquid suspension of stool from a donor (usually a family member) and has been used successfully in severe cases of recurrent C. difficile relapse (Gough et al., 2011; Rupnik, 2015). However, many problems exist with this therapy since it can increase the risks of transmitting other pathogens (Brandt and Reddy, 2011).
Fecal transplantation studies in mice showed that transferring the microbiota from lean and fat mice to germ-free mice induces greater weight gain in those receiving the microbiota from fat donors (Ley et al., 2006). The discovery of the link between lean-associated microbiome has opened new possibility of using transplanted microbiota to treat metabolic disorders in humans.
Probiotics are defined as live microorganisms that ultimately improve the balance of the intestinal flora, thus fostering healthy gut functions through a healthy gut microbiome (reviewed in Gareau et al., 2010; Whelan and Quigley, 2013). There are several in vitro assays to validate the actual in vivo efficacy of probiotic microorganisms, which include specific biological criteria, such as resistance to low gastric pH and capacity to reach the intestines alive to exert beneficial effects on the human body (Papadimitriou et al., 2015). Probiotic microorganisms are mainly lactic acid-producing bacteria of Lactobacillus and Bifidobacterium genera. Other microorganisms, such as the yeast Saccharomyces boulardii and the bacteria E. coli Nissle 1917, Streptococcus thermophilus, F. parausnitzii and Bacillus polyfermenticus have also been investigated. The beneficial therapeutic effects and mechanisms of action of Lactobacilli and bifidobacteria in patients with gastrointestinal diseases have long been demonstrated (Ng et al., 2009). These probiotics can prevent or ameliorate clinical symptoms of irritable bowel syndrome, inflammatory and necrotizing enterocolitis and acute diarrhea (Ng et al., 2009; Gareau et al., 2010; Whelan and Quigley, 2013). It was found that they could regulate the balance of intestinal microbiota by physically blocking the adhesion of pathogenic species onto epithelial cells. This is directly mediated by means of increases in the production of a mucosal barrier by goblet epithelial cells (Etzold et al., 2014). In addition, they can regulate epithelial permeability by enhancing the formation of tight-junctions between cells (Ng et al., 2009). Their immune-modulatory effects are associated with a decrease in the production of pro-inflammatory cytokines, as well as the microbial peptides bacteriocins (Ng et al., 2009; Whelan and Quigley, 2013).
The use of probiotics is not limited to gastrointestinal disorders. Studies evaluating their application in dermatology, urology and dentistry have been increasing (Vuotto et al., 2014). Bifidobacterium bifidum has been used in the prevention and treatment of infantile eczema. Intra-vaginal administration of Lactobacillus rhamnosus GR-1 and L. fermentum RC-14 were shown to have a positive effect on the prevention of recurrent BV and candidiasis (Anukam et al., 2006; Vuotto et al., 2014). Consumption of probiotics can be effective in the prevention of dental caries and periodontal diseases (Pandey et al., 2015). The continuous consumption of Yakult’s L. casei strain Shirota (LcS), one of the most popular probiotics, in adequate amounts, may reduce the risk of cancers by modulating immune function (Ishikawa et al., 2005). Finally, the treatment of obese mice with Bifidobacterium infantis was shown to reduce the production of pro-inflammatory cytokines and white adipose tissue weight (Cani et al., 2007). The effect of the endogenous host microbiota on obesity and beneficial role of probiotics including L. rhamnosus and gasseri and Bifidobacterium lactis in the treatment of adiposity and obesity has been reviewed elsewhere (Mekkes et al., 2014). This is a new area under intense investigation.
Prebiotics are functional food ingredients that can change the composition and/or the activity of the colonic flora (Roberfroid, 2000; Roberfroid, 2007; Brownawell et al., 2012). The dietary supplementation with prebiotics can promote the growth of beneficial bacteria such as lactobacilli and bifidobacteria strains (Roberfroid, 2000, 2007). Poorly digestible carbohydrates (fibers), such as resistant starch, non-starch polysaccharides (e.g., celluloses, hemicelluloses, pectins, and gums), oligosaccharides and polyphenols are resistant to gastric acidity, gastrointestinal absorption, and non-digestible by hydrolysis by mammalian enzymes. Colonic bacteria through carbohydrate hydrolyzing enzymes and fermentation produce hydrogen, methane, carbon dioxide, and SCFA, which can affect host energy levels and gut hormone regulation (Slavin, 2013). The most commonly used prebiotics are fructo-oligosaccharides (FOS) and trans-galacto-oligosaccharides (TOS), for example inulin (Roberfroid, 2000). However, not all dietary carbohydrates are prebiotics (Roberfroid, 2007). Mixtures of probiotic and prebiotic ingredients have been used to selectively stimulate growth or activity of health-promoting bacteria. In conclusion, it appears that the therapeutic use of pro- and prebiotics will find more applications in the near future when large-scale clinical trials and metagenomic surveys will determine which microbes are active, which are damaged, and which may respond to a given prebiotic, probiotic or synbiotic (synergic association of probiotic and prebiotic) at the genomic level (Nagata et al., 2011).
Phage therapy consists of using bacterial viruses bacteriophages, (also known as phages) as antimicrobial agents (Sulakvelidze et al., 2001; Abedon, 2014). Bacteriophages attach to specific receptors present in the host membrane and then inject their genetic material into the bacterium. Viral proteins are then synthetized using the host’s translational machinery. Phage infection can result in lysis, lysogeny or resistance. Lytic bacteriophages induce host cell death and breakdown in order to spread the infection whereas lysogenic (or temperate) phages insert their genome into the host DNA. Resistance may be acquired during replicative cycles by gene transposition or recombination.
Phage therapy can potentially have beneficial impact on human microbiomes and host health (Koskella and Meaden, 2013). The host specificity greatly limits the types of bacteria that will enter into contact with a particular phage, therefore avoiding the elimination of non-pathogenic species (Koskella and Meaden, 2013). However in order to choose a specific phage to use as a therapeutic agent, it is necessary to know the pathogen causing a given disease. When this is not the case, the use of a cocktail of different species of phages would broaden the range of action but could also have a possible negative effect on the microbial communities (Chan et al., 2013). The synergistic use of phages and low dose of antibiotics, a strategy named Phage-Antibiotic Synergy (PAS), could be useful in certain clinical situations (Comeau et al., 2007).
Bacteria have evolved various mechanisms of defense against phage infections, which act at different levels. In fact they can prevent phage attachment by mutation/loss of membrane receptors or block phage DNA entry with the aid of specific membrane proteins. Furthermore, Bacteria and Archaea developed an intrinsic innate immunity mechanism, which allows them to remember phage infection by capturing short DNA sequences from phage genetic material. These viral sequences are integrated as spacer sequences into their own chromosome, specifically into an array of repeated sequences called Clustered Regularly Interspaced Short Palindromic Repeats or CRISPR, with the help of the proteins encoded by Cas (CRISPR-associated) family of genes (Garneau et al., 2010; van der Oost et al., 2014).
CRISPR loci consist of short (~24–48 nucleotides) repeats separated by similarly sized, unique spacers found in genomes of Archaea (~90%), and Bacteria (~40%) (Garneau et al., 2010; van der Oost et al., 2014). Cas genes encode a large and heterogeneous family of proteins with functional domains typical of nucleases, helicases, polymerases, and polynucleotide-binding proteins. Upon invasion, the host organism samples and integrates in its genome short fragments of the foreign DNA, called protospacers, thus creating immunity against that particular infective agent. The protospacer is flanked by the repeated regions, and transcribed with them into a CRISPR-RNA (crRNA), which guides specific nucleases to a target DNA containing regions complementary to the protospacer. Upon recognition, nucleases cleave invasive DNA preventing it to replicate and blocking infection. Three different types and eleven subtypes of CRIPSR/Cas system can be classified based on their Cas protein repertoire and mechanisms of action (Plagens et al., 2015). A detailed list of type I, II, and III CRISPR-Cas systems is also available at the CRISPRdb website7. Type I systems are characterized by the molecular machinery named a Cascade complex (CRISPR-associated complex for antiviral defense) which displays nickase and exonuclease activities. Type III systems are characterized by the presence of Cas10 (the signature protein) and associated proteins. The systems are subclassifed as type III-A (CSM) and type III-B (CMR), depending on their specificity for DNA or RNA targets. In addition, types I and III share a variable number of repeat associated mysterious protein (RAMP) subunits (Rouillon et al., 2013; Plagens et al., 2015).
Type II is the simplest CRISPR-Cas system that is characterized by the presence of dsDNA endonuclease Cas9 and the transactivating CRISPR-RNA (tracrRNA). The tracrRNA anneals with the invariable regions of mature crRNA creating RNA heterodimers which, in turn, forms a nucleoprotein complex with Cas9, guiding it to the target DNA. Cas9 recognizes and binds to a specific 5′-NGG-3′ motif, called protospacer adjacent motif (PAM). Then the complex searches for a sequence complementary to the spacer portion of crRNA. Cas9 contains two nuclease domains, namely RuvC and HNH, and produces a double strand break in the target. Subsequently, cleaved DNA becomes a substrate of the bacterial DNA repair mechanisms, either non-homologous end joining (NHEJ) or homologous recombination (HR). NHEJ is an imperfect repair system and may cause insertion or deletion (indels) of base pairs, as well as single nucleotide polymorphisms (SNPs). However, high-fidelity HR repair may occur if a sequence complementary to the cleaved fragment is provided. The relative simplicity of the mechanism of action and the peculiarities of Cas9 make the CRISPR/Cas9 system an ideal tool for a vast assortment of procedures, particularly for genomic editing (reviewed in Ma et al., 2014; Selle and Barrangou, 2015; Xiao-Jie et al., 2015). A considerable amount of work in this field has been already done in different organisms, especially eukaryotes, using engineered versions of CRISPR/Cas9. On the other hand, despite its enormous potential, manipulation of bacterial genomes by CRISPR/Cas9 has so far been scarcely executed (Selle and Barrangou, 2015). CRISPR/Cas9 can be used to selectively deplete a given bacterial community of a particular harmful strain or species (Vercoe et al., 2013; Gomaa et al., 2014; Yosef et al., 2015). It has been shown that there is an inverse correlation between the presence of CRISPR loci and acquired antibiotic resistance in Enterococcus fecalis (Palmer and Gilmore, 2010), indicating that the use of antibiotics may increase the ability of bacteria to acquire drug resistance-encoding plasmids. CRISPR/Cas9 system can be used to introduce specific mutations into essential, antibiotic resistance, and virulence genes. It has been already shown that by providing in trans a DNA (linear or plasmid) homologous to the target sequence, it is possible to introduce very specific mutations to the desired target (Marraffini and Sontheimer, 2008; Jiang et al., 2013; Yosef et al., 2015). Also CRISPR/Cas9 has the potential to directly modulate the expression of particular genes. An engineered version of Cas9 lacking the nuclease activity but still retaining its binding capacity (dCas9) has already been created to repress bacterial transcription by binding to promoter regions or within a ORF, thus blocking transcriptional initiation and elongation, respectively. dCas9 can also be fused to regulatory domains in order to switch on/off the expression of specific genes (Bikard et al., 2013; Qi et al., 2013). In the near future the engineering of commensal bacteria with improved properties using a CRISPR/Cas system may constitute an effective vaccination tool in public health for prevention of diseases. However, despite great advances, still much work needs to be done in order to improve target specificity and delivering efficiency.
A more complete perspective on how phage therapy and CRISPR/Cas9 systems can be employed to combat pathogenic species within our bodies, especially antibiotic-resistant bacterial pathogens needs the expansion of in vitro, ex vivo, and in silico approaches (Fritz et al., 2013). Several publicly available methods for hit-specific retrieval of protospacers in the reference microbiomes have already been developed (Bi et al., 2012). Over 123,003 protospacers have been predicted based on 690 phage genomes (Zhang et al., 2013). The functional exploration of pathogen-specific bacteriophages and gene therapy depends on development of relevant animal models including transgenic and bacteria-free animals (Fritz et al., 2013). Finally, we will need to confirm the results in the proof-of-concept in well-designed clinical trials. In Figure 2, we graphically summarize some of the pharmacological approaches discussed in this article.
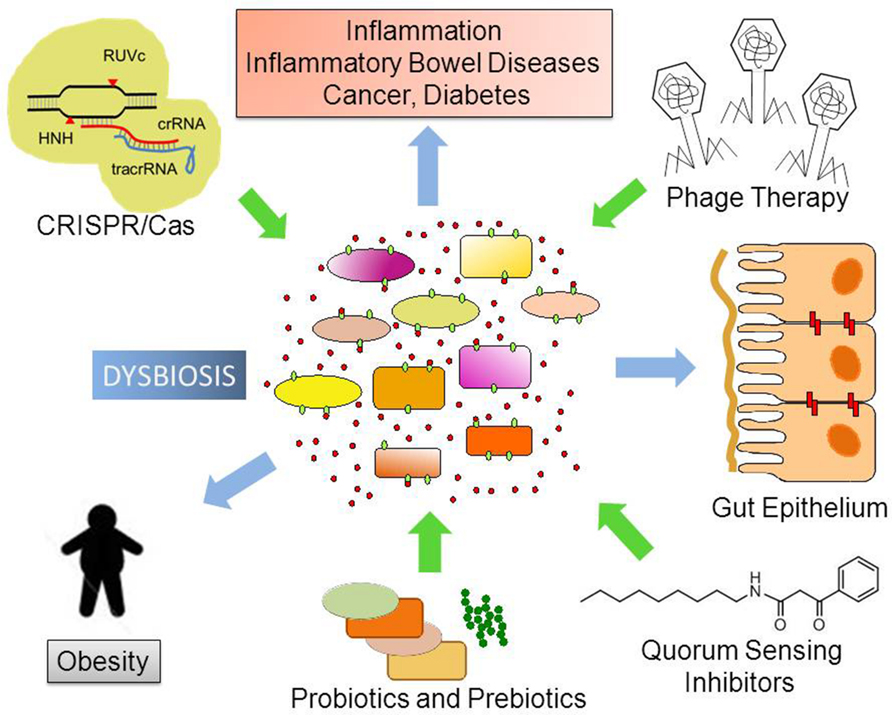
FIGURE 2. Possible future therapeutic approaches to control dysbiosis. Disturbances in the ecological community of commensal, symbiotic, and pathogenic microorganisms may favor dysbiosis. This leads to increased bacterial translocation and/or release of microorganism-associated molecular patterns (MAMPs), which activate Toll-like receptors (TLRs) in several cell types. Local and body-wide immune system activities promote inflammation, which ultimately leads to chronic diseases. Novel therapeutic approaches like phage therapy, disruption of Quorum Sensing and the use of the biotechnological tool CRISPR/Cas9 to edit microbial genomes have the potential to target specific bacterial taxa thus helping to re-establish homeostasis and microbiome balance.
To assess the interaction of the human body with pharmaceuticals, we need to understand the complex relationship between ecology, physiology, and pharmacology (Rahman et al., 2007; Flintoft, 2012; Haiser and Turnbaugh, 2012). From pharmacogenomic studies it is clear that sequence variations in drug target proteins, drug-metabolizing enzymes, and drug transporters can alter drug efficacy, produce side effects, causing variable drug responses in individual patients (Wilson and Nicholson, 2009). Microorganisms participate in a very wide range of biotransformations, including hydrolysis, and processing of glutathione conjugates of xenobiotics excreted in the bile (Johnson et al., 2012). Hence, the determination of the genetic variability of human microbiomes has potential to predict the efficacy, bioavailability and individual response variability in drug therapy.
Finally, further studies are needed to elucidate whether the vast number of functional microbiota gene-products exerts unknown off-target effects and how they can negatively or positively affect drug responses. These are the major research challenges for exploring the potential of metagenomics to better understand microbial ecology and to translate the molecular and genomic data into pharmacomicrobiomics (Saad et al., 2012). According to this new ecological paradigm, competency in knowledge, skills, and attitudes as well as integrated environmental conscience and social responsibility are essential for professionals who will in the future create and develop a new generation of green and sustainable pharmaceutical products, as shown in Figure 3.
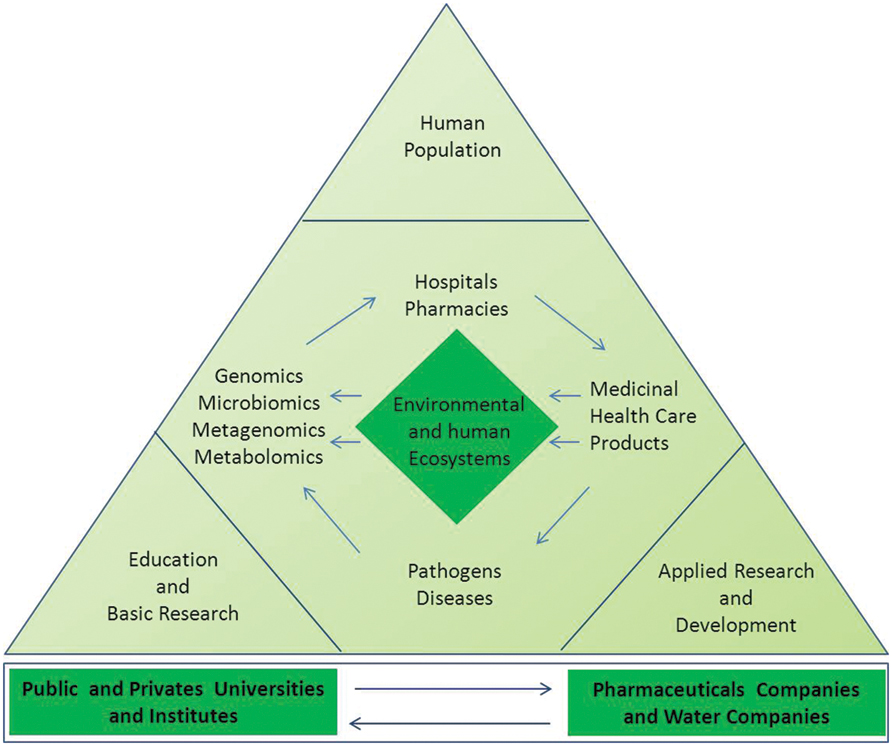
FIGURE 3. Actions and molecular approaches aiming to protect the environmental and human microbial ecosystems. The measurements of ecological, phylometagenomic, and microbial metabolic variations in the microbiomes require a specialized and complex set of knowledge. Collaboration between universities, research entities, non-governmental organizations (NGO), and the pharmaceutical industry professionals are key for evaluating both biological and pharmaceutical impacts in the ecosystems and elucidating the mechanism-of-action of new compounds in the host and its microbiomes. The utility of metagenomic functional reconstruction for direct association of community functions with habitat and host phenotype will be critical for proper study designs and production of greener pharmaceutical products for future personalized medicine.
Recent advances in microbiome sequencing projects revealed the high complexity of microbial communities in various human body sites. They have confirmed the critical roles of the human-microbiota ecosystems in health-promoting or disease-causing processes. These studies have highlighted the unexpected and wide-ranging consequences of eliminating certain bacteria living in our body.
While the natural variation of the human microbiota has yet to be fully determined, the annotation and analyses of a large number of human microbiomes have shown that the presence or absence of specific microbial species categorizes human individuals based on enterotypes. It is likely that cultivated and uncultivated microbes will contribute to discovering new fundamental biomarkers for specific human disorders and that they may become better discriminatory tools than human-based ones.
Changes in the stability and dynamic of numerous microbial communities have been associated with several diseases, including type II diabetes, obesity, fatty liver disease, irritable bowel syndrome, and IBDs and even certain cancers. However, further studies need to be done in order to confirm whether low bacterial diversity increases the chances to develop such diseases and metabolic perturbations.
The use of antibiotics compromises genome defense and increases the ability to acquire antibiotic resistance. Prebiotics, probiotics, synbiotics, phage therapy, quorum sensing systems, and CRISPR/Cas systems have been proposed as tools to control and modulate microbial communities. Engineering of pathogen-specific bacteriophages and production of pharmaceuticals based on our own body’s microbiome will be possible and fully explored in the near future. The use of novel pharmaceuticals and nutraceuticals to modulate microbial colonization and development of a healthy gut microbial community in early childhood will support healthy adult human body functions and prevent the occurrence of several diseases.
The authors conducted the literature review process, grading, and categorizing criteria, and quality of selected articles. The authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank colleagues of Institute of Biomedical Sciences of the University of São Paulo for insights and productive discussions. This work was supported by grants from Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP, proc. 2015/1177-8, 2015/18647-6) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Aagaard, K., Ma, J., Antony, K. M., Ganu, R., Petrosino, J., and Versalovic, J. (2014). The placenta harbors a unique microbiome. Sci. Transl. Med. 6:237ra265. doi: 10.1126/scitranslmed.3008599
Abedon, S. T. (2014). Phage therapy: eco-physiological pharmacology. Scientifica (Cairo) 2014:581639. doi: 10.1155/2014/581639
Abubucker, S., Segata, N., Goll, J., Schubert, A. M., Izard, J., Cantarel, B. L., et al. (2012). Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 8:e1002358. doi: 10.1371/journal.pcbi.1002358
Anukam, K. C., Osazuwa, E., Osemene, G. I., Ehigiagbe, F., Bruce, A. W., and Reid, G. (2006). Clinical study comparing probiotic Lactobacillus GR-1 and RC-14 with metronidazole vaginal gel to treat symptomatic bacterial vaginosis. Microbes Infect. 8, 2772–2776. doi: 10.1016/j.micinf.2006.08.008
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Backhed, F., Fraser, C. M., Ringel, Y., Sanders, M. E., Sartor, R. B., Sherman, P. M., et al. (2012). Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe 12, 611–622. doi: 10.1016/j.chom.2012.10.012
Belizario, J. E. (2013). The humankind genome: from genetic diversity to the origin of human diseases. Genome 56, 705–716. doi: 10.1139/gen-2013-0125
Bi, D., Xu, Z., Harrison, E. M., Tai, C., Wei, Y., He, X., et al. (2012). ICEberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Res. 40, D621–D626. doi: 10.1093/nar/gkr846
Bikard, D., Jiang, W., Samai, P., Hochschild, A., Zhang, F., and Marraffini, L. A. (2013). Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41, 7429–7437. doi: 10.1093/nar/gkt520
Brandt, L. J., and Reddy, S. S. (2011). Fecal microbiota transplantation for recurrent Clostridium difficile infection. J. Clin. Gastroenterol. 45(Suppl.), S159–S167. doi: 10.1097/MCG.0b013e318222e603
Brown, C. T., Sharon, I., Thomas, B. C., Castelle, C. J., Morowitz, M. J., and Banfield, J. F. (2013). Genome resolved analysis of a premature infant gut microbial community reveals a Varibaculum cambriense genome and a shift towards fermentation-based metabolism during the third week of life. Microbiome 1:30. doi: 10.1186/2049-2618-1-30
Brownawell, A. M., Caers, W., Gibson, G. R., Kendall, C. W., Lewis, K. D., Ringel, Y., et al. (2012). Prebiotics and the health benefits of fiber: current regulatory status, future research, and goals. J. Nutr. 142, 962–974. doi: 10.3945/jn.112.158147
Cani, P. D., and Delzenne, N. M. (2011). The gut microbiome as therapeutic target. Pharmacol. Ther. 130, 202–212. doi: 10.1016/j.pharmthera.2011.01.012
Cani, P. D., Neyrinck, A. M., Fava, F., Knauf, C., Burcelin, R. G., Tuohy, K. M., et al. (2007). Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50, 2374–2383. doi: 10.1007/s00125-007-0791-0
Carr, R., Shen-Orr, S. S., and Borenstein, E. (2013). Reconstructing the genomic content of microbiome taxa through shotgun metagenomic deconvolution. PLoS Comput. Biol. 9:e1003292. doi: 10.1371/journal.pcbi.1003292
Chan, B. K., Abedon, S. T., and Loc-Carrillo, C. (2013). Phage cocktails and the future of phage therapy. Future Microbiol 8, 769–783. doi: 10.2217/fmb.13.47
Christen, B., Abeliuk, E., Collier, J. M., Kalogeraki, V. S., Passarelli, B., Coller, J. A., et al. (2011). The essential genome of a bacterium. Mol. Syst. Biol. 7:528. doi: 10.1038/msb.2011.58
Clemente, J. C., Pehrsson, E. C., Blaser, M. J., Sandhu, K., Gao, Z., Wang, B., et al. (2015). The microbiome of uncontacted Amerindians. Sci. Adv. 1:e1500183. doi: 10.1126/sciadv.1500183
Clemente, J. C., Ursell, L. K., Parfrey, L. W., and Knight, R. (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270. doi: 10.1016/j.cell.2012.01.035
Collison, M., Hirt, R. P., Wipat, A., Nakjang, S., Sanseau, P., and Brown, J. R. (2012). Data mining the human gut microbiota for therapeutic targets. Brief. Bioinform. 13, 751–768. doi: 10.1093/bib/bbs002
Comeau, A. M., Tetart, F., Trojet, S. N., Prere, M. F., and Krisch, H. M. (2007). Phage-antibiotic synergy (PAS): beta-lactam and quinolone antibiotics stimulate virulent phage growth. PLoS ONE 2:e799. doi: 10.1371/journal.pone.0000799
Cucchiara, S., Stronati, L., and Aloi, M. (2012). Interactions between intestinal microbiota and innate immune system in pediatric inflammatory bowel disease. J. Clin. Gastroenterol. 46(Suppl.), S64–S66. doi: 10.1097/MCG.0b013e31826a857f
Culligan, E. P., Sleator, R. D., Marchesi, J. R., and Hill, C. (2014). Metagenomics and novel gene discovery: promise and potential for novel therapeutics. Virulence 5, 399–412. doi: 10.4161/viru.27208
Curtis, T. P., Sloan, W. T., and Scannell, J. W. (2002). Estimating prokaryotic diversity and its limits. Proc. Natl. Acad. Sci. U.S.A. 99, 10494–10499. doi: 10.1073/pnas.142680199
Dawid, S., Roche, A. M., and Weiser, J. N. (2007). The blp bacteriocins of Streptococcus pneumoniae mediate intraspecies competition both in vitro and in vivo. Infect. Immun. 75, 443–451. doi: 10.1128/IAI.01775-05
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696. doi: 10.1073/pnas.1005963107
Defoirdt, T., Boon, N., and Bossier, P. (2010). Can bacteria evolve resistance to quorum sensing disruption? PLoS Pathog. 6:e1000989. doi: 10.1371/journal.ppat.1000989
Delaney, M. L., and Onderdonk, A. B. (2001). Nugent score related to vaginal culture in pregnant women. Obstet. Gynecol. 98, 79–84. doi: 10.1016/S0029-7844(01)01402-8
Dethlefsen, L., Huse, S., Sogin, M. L., and Relman, D. A. (2008). The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6:e280. doi: 10.1371/journal.pbio.0060280
DiGiulio, D. B., Romero, R., Amogan, H. P., Kusanovic, J. P., Bik, E. M., Gotsch, F., et al. (2008). Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture-based investigation. PLoS ONE 3:e3056. doi: 10.1371/journal.pone.0003056
Dominguez-Bello, M. G., Costello, E. K., Contreras, M., Magris, M., Hidalgo, G., Fierer, N., et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U.S.A. 107, 11971–11975. doi: 10.1073/pnas.1002601107
Dong, Y. H., and Zhang, L. H. (2005). Quorum sensing and quorum-quenching enzymes. J. Microbiol. 43, 101–109.
Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., et al. (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638. doi: 10.1126/science.1110591
Etzold, S., Kober, O. I., Mackenzie, D. A., Tailford, L. E., Gunning, A. P., Walshaw, J., et al. (2014). Structural basis for adaptation of lactobacilli to gastrointestinal mucus. Environ. Microbiol. 16, 888–903. doi: 10.1111/1462-2920.12377
Fardini, Y., Chung, P., Dumm, R., Joshi, N., and Han, Y. W. (2010). Transmission of diverse oral bacteria to murine placenta: evidence for the oral microbiome as a potential source of intrauterine infection. Infect. Immun. 78, 1789–1796. doi: 10.1128/IAI.01395-09
Fettweis, J. M., Serrano, M. G., Sheth, N. U., Mayer, C. M., Glascock, A. L., Brooks, J. P., et al. (2012). Species-level classification of the vaginal microbiome. BMC Genomics 13(Suppl. 8):S17. doi: 10.1186/1471-2164-13-S8-S17
Finch, R. G., Pritchard, D. I., Bycroft, B. W., Williams, P., and Stewart, G. S. (1998). Quorum sensing: a novel target for anti-infective therapy. J. Antimicrob. Chemother. 42, 569–571. doi: 10.1093/jac/42.5.569
Flintoft, L. (2012). Disease genomics: associations go metagenome-wide. Nat. Rev. Genet. 13, 756–757. doi: 10.1038/nrg3347
Fodor, A. A., DeSantis, T. Z., Wylie, K. M., Badger, J. H., Ye, Y., Hepburn, T., et al. (2012). The “most wanted” taxa from the human microbiome for whole genome sequencing. PLoS ONE 7:e41294. doi: 10.1371/journal.pone.0041294
Forsberg, K. J., Patel, S., Gibson, M. K., Lauber, C. L., Knight, R., Fierer, N., et al. (2014). Bacterial phylogeny structures soil resistomes across habitats. Nature 509, 612–616. doi: 10.1038/nature13377
Forslund, K., Sunagawa, S., Kultima, J. R., Mende, D. R., Arumugam, M., Typas, A., et al. (2013). Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 23, 1163–1169. doi: 10.1101/gr.155465.113
Frantz, A. L., Rogier, E. W., Weber, C. R., Shen, L., Cohen, D. A., Fenton, L. A., et al. (2012). Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol. 5, 501–512. doi: 10.1038/mi.2012.23
Fritz, J. V., Desai, M. S., Shah, P., Schneider, J. G., and Wilmes, P. (2013). From meta-omics to causality: experimental models for human microbiome research. Microbiome 1:14. doi: 10.1186/2049-2618-1-14
Gareau, M. G., Sherman, P. M., and Walker, W. A. (2010). Probiotics and the gut microbiota in intestinal health and disease. Nat. Rev. Gastroenterol. Hepatol. 7, 503–514. doi: 10.1038/nrgastro.2010.117
Garneau, J. E., Dupuis, M. E., Villion, M., Romero, D. A., Barrangou, R., Boyaval, P., et al. (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. doi: 10.1038/nature09523
Gevers, D., Pop, M., Schloss, P. D., and Huttenhower, C. (2012). Bioinformatics for the human microbiome project. PLoS Comput. Biol. 8:e1002779. doi: 10.1371/journal.pcbi.1002779
Gomaa, A. A., Klumpe, H. E., Luo, M. L., Selle, K., Barrangou, R., and Beisel, C. L. (2014). Programmable removal of bacterial strains by use of genome-targeting CRISPR-Cas systems. MBio 5:e928-13. doi: 10.1128/mBio.00928-13
Gough, E., Shaikh, H., and Manges, A. R. (2011). Systematic review of intestinal microbiota transplantation (fecal bacteriotherapy) for recurrent Clostridium difficile infection. Clin. Infect. Dis. 53, 994–1002. doi: 10.1093/cid/cir632
Grice, E. A., Kong, H. H., Conlan, S., Deming, C. B., Davis, J., Young, A. C., et al. (2009). Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192. doi: 10.1126/science.1171700
Groer, M. W., Luciano, A. A., Dishaw, L. J., Ashmeade, T. L., Miller, E., and Gilbert, J. A. (2014). Development of the preterm infant gut microbiome: a research priority. Microbiome 2:38. doi: 10.1186/2049-2618-2-38
Guani-Guerra, E., Santos-Mendoza, T., Lugo-Reyes, S. O., and Teran, L. M. (2010). Antimicrobial peptides: general overview and clinical implications in human health and disease. Clin. Immunol. 135, 1–11. doi: 10.1016/j.clim.2009.12.004
Guaraldi, F., and Salvatori, G. (2012). Effect of breast and formula feeding on gut microbiota shaping in newborns. Front. Cell Infect. Microbiol. 2:94. doi: 10.3389/fcimb.2012.00094
Guo, F., Ju, F., Cai, L., and Zhang, T. (2013). Taxonomic precision of different hypervariable regions of 16S rRNA gene and annotation methods for functional bacterial groups in biological wastewater treatment. PLoS ONE 8:e76185. doi: 10.1371/journal.pone.0076185
Haiser, H. J., and Turnbaugh, P. J. (2012). Is it time for a metagenomic basis of therapeutics? Science 336, 1253–1255. doi: 10.1126/science.1224396
Hense, B. A., and Schuster, M. (2015). Core principles of bacterial autoinducer systems. Microbiol. Mol. Biol. Rev. 79, 153–169. doi: 10.1128/MMBR.00024-14
Hooper, L. V., Littman, D. R., and Macpherson, A. J. (2012). Interactions between the microbiota and the immune system. Science 336, 1268–1273. doi: 10.1126/science.1223490
Human Microbiome Jumpstart Reference Strains, C., Nelson, K. E., Weinstock, G. M., Highlander, S. K., Worley, K. C., Creasy, H. H., et al. (2010). A catalog of reference genomes from the human microbiome. Science 328, 994–999. doi: 10.1126/science.1183605
Human Microbiome Project, C. (2012a). A framework for human microbiome research. Nature 486, 215–221. doi: 10.1038/nature11209
Human Microbiome Project, C. (2012b). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Ishikawa, H., Akedo, I., Otani, T., Suzuki, T., Nakamura, T., Takeyama, I., et al. (2005). Randomized trial of dietary fiber and Lactobacillus casei administration for prevention of colorectal tumors. Int. J. Cancer 116, 762–767. doi: 10.1002/ijc.21115
Jiang, W., Bikard, D., Cox, D., Zhang, F., and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239. doi: 10.1038/nbt.2508
Jiang, W., Ling, Z., Lin, X., Chen, Y., Zhang, J., Yu, J., et al. (2014). Pyrosequencing analysis of oral microbiota shifting in various caries states in childhood. Microb. Ecol. 67, 962–969. doi: 10.1007/s00248-014-0372-y
Joelsson, A., Liu, Z., and Zhu, J. (2006). Genetic and phenotypic diversity of quorum-sensing systems in clinical and environmental isolates of Vibrio cholerae. Infect. Immun. 74, 1141–1147. doi: 10.1128/IAI.74.2.1141-1147.2006
Johnson, C. H., Patterson, A. D., Idle, J. R., and Gonzalez, F. J. (2012). Xenobiotic metabolomics: major impact on the metabolome. Annu. Rev. Pharmacol. Toxicol. 52, 37–56. doi: 10.1146/annurev-pharmtox-010611-134748
Jones, B. V., and Marchesi, J. R. (2007). Transposon-aided capture (TRACA) of plasmids resident in the human gut mobile metagenome. Nat. Methods 4, 55–61. doi: 10.1038/nmeth964
Kardos, N., and Demain, A. L. (2011). Penicillin: the medicine with the greatest impact on therapeutic outcomes. Appl. Microbiol. Biotechnol. 92, 677–687. doi: 10.1007/s00253-011-3587-6
Kayumov, A. R., Khakimullina, E. N., Sharafutdinov, I. S., Trizna, E. Y., Latypova, L. Z., Thi Lien, H., et al. (2014). Inhibition of biofilm formation in Bacillus subtilis by new halogenated furanones. J. Antibiot. (Tokyo) 68, 297–301. doi: 10.1038/ja.2014.143
Kenyon, C., Colebunders, R., and Crucitti, T. (2013). The global epidemiology of bacterial vaginosis: a systematic review. Am. J. Obstet. Gynecol. 209, 505–523. doi: 10.1016/j.ajog.2013.05.006
Kimura, N. (2014). Metagenomic approaches to understanding phylogenetic diversity in quorum sensing. Virulence 5, 433–442. doi: 10.4161/viru.27850
Kootte, R. S., Vrieze, A., Holleman, F., Dallinga-Thie, G. M., Zoetendal, E. G., de Vos, W. M., et al. (2012). The therapeutic potential of manipulating gut microbiota in obesity and type 2 diabetes mellitus. Diabetes Obes. Metab. 14, 112–120. doi: 10.1111/j.1463-1326.2011.01483.x
Koren, O., Knights, D., Gonzalez, A., Waldron, L., Segata, N., Knight, R., et al. (2013). A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput. Biol. 9:e1002863. doi: 10.1371/journal.pcbi.1002863
Koskella, B., and Meaden, S. (2013). Understanding bacteriophage specificity in natural microbial communities. Viruses 5, 806–823. doi: 10.3390/v5030806
Ladizinski, B., McLean, R., Lee, K. C., Elpern, D. J., and Eron, L. (2014). The human skin microbiome. Int. J. Dermatol. 53, 1177–1179. doi: 10.1111/ijd.12609
Le Chatelier, E., Nielsen, T., Qin, J., Prifti, E., Hildebrand, F., Falony, G., et al. (2013). Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546. doi: 10.1038/nature12506
Lepage, P., Leclerc, M. C., Joossens, M., Mondot, S., Blottiere, H. M., Raes, J., et al. (2013). A metagenomic insight into our gut’s microbiome. Gut 62, 146–158. doi: 10.1136/gutjnl-2011-301805
Ley, R. E., Backhed, F., Turnbaugh, P., Lozupone, C. A., Knight, R. D., and Gordon, J. I. (2005). Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. U.S.A. 102, 11070–11075. doi: 10.1073/pnas.0504978102
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Ma, Y., Zhang, L., and Huang, X. (2014). Genome modification by CRISPR/Cas9. FEBS J. 281, 5186–5193. doi: 10.1111/febs.13110
Maidak, B. L., Olsen, G. J., Larsen, N., Overbeek, R., McCaughey, M. J., and Woese, C. R. (1996). The ribosomal database project (RDP). Nucleic Acids Res. 24, 82–85. doi: 10.1093/nar/24.1.82
Manefield, M., Rasmussen, T. B., Henzter, M., Andersen, J. B., Steinberg, P., Kjelleberg, S., et al. (2002). Halogenated furanones inhibit quorum sensing through accelerated LuxR turnover. Microbiology 148(Pt 4), 1119–1127. doi: 10.1099/00221287-148-4-1119
Manichanh, C., Rigottier-Gois, L., Bonnaud, E., Gloux, K., Pelletier, E., Frangeul, L., et al. (2006). Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55, 205–211. doi: 10.1136/gut.2005.073817
Manor, O., and Borenstein, E. (2015). MUSiCC: a marker genes based framework for metagenomic normalization and accurate profiling of gene abundances in the microbiome. Genome Biol. 16:53. doi: 10.1186/s13059-015-0610-8
Marchesi, J. R., Sato, T., Weightman, A. J., Martin, T. A., Fry, J. C., Hiom, S. J., et al. (1998). Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl Environ. Microbiol. 64, 795–799.
Marraffini, L. A., and Sontheimer, E. J. (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. doi: 10.1126/science.1165771
Mason, M. R., Nagaraja, H. N., Camerlengo, T., Joshi, V., and Kumar, P. S. (2013). Deep sequencing identifies ethnicity-specific bacterial signatures in the oral microbiome. PLoS ONE 8:e77287. doi: 10.1371/journal.pone.0077287
Meijer, K., de Vos, P., and Priebe, M. G. (2010). Butyrate and other short-chain fatty acids as modulators of immunity: what relevance for health? Curr. Opin. Clin. Nutr. Metab. Care 13, 715–721. doi: 10.1097/MCO.0b013e32833eebe5
Mekkes, M. C., Weenen, T. C., Brummer, R. J., and Claassen, E. (2014). The development of probiotic treatment in obesity: a review. Benef. Microbes 5, 19–28. doi: 10.3920/BM2012.0069
Miller, M. B., and Bassler, B. L. (2001). Quorum sensing in bacteria. Annu. Rev. Microbiol. 55, 165–199. doi: 10.1146/annurev.micro.55.1.165
Mobegi, F. M., van Hijum, S. A., Burghout, P., Bootsma, H. J., de Vries, S. P., van der Gaast-de Jongh, C. E., et al. (2014). From microbial gene essentiality to novel antimicrobial drug targets. BMC Genomics 15:958. doi: 10.1186/1471-2164-15-958
Montassier, E., Batard, E., Massart, S., Gastinne, T., Carton, T., Caillon, J., et al. (2014). 16S rRNA gene pyrosequencing reveals shift in patient faecal microbiota during high-dose chemotherapy as conditioning regimen for bone marrow transplantation. Microb. Ecol. 67, 690–699. doi: 10.1007/s00248-013-0355-4
Mullany, P. (2014). Functional metagenomics for the investigation of antibiotic resistance. Virulence 5, 443–447. doi: 10.4161/viru.28196
Nagata, S., Asahara, T., Ohta, T., Yamada, T., Kondo, S., Bian, L., et al. (2011). Effect of the continuous intake of probiotic-fermented milk containing Lactobacillus casei strain Shirota on fever in a mass outbreak of norovirus gastroenteritis and the faecal microflora in a health service facility for the aged. Br. J. Nutr. 106, 549–556. doi: 10.1017/S000711451100064X
Naik, S., Bouladoux, N., Wilhelm, C., Molloy, M. J., Salcedo, R., Kastenmuller, W., et al. (2012). Compartmentalized control of skin immunity by resident commensals. Science 337, 1115–1119. doi: 10.1126/science.1225152
Nealson, K. H., and Hastings, J. W. (1979). Bacterial bioluminescence: its control and ecological significance. Microbiol. Rev. 43, 496–518.
Ng, S. C., Hart, A. L., Kamm, M. A., Stagg, A. J., and Knight, S. C. (2009). Mechanisms of action of probiotics: recent advances. Inflamm. Bowel Dis. 15, 300–310. doi: 10.1002/ibd.20602
Ng, W. L., and Bassler, B. L. (2009). Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 43, 197–222. doi: 10.1146/annurev-genet-102108-134304
Ochsner, U. A., Sun, X., Jarvis, T., Critchley, I., and Janjic, N. (2007). Aminoacyl-tRNA synthetases: essential and still promising targets for new anti-infective agents. Expert Opin. Investig. Drugs 16, 573–593. doi: 10.1517/13543784.16.5.573
Ostaff, M. J., Stange, E. F., and Wehkamp, J. (2013). Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol. Med. 5, 1465–1483. doi: 10.1002/emmm.201201773
Palmer, D. J., Metcalfe, J., and Prescott, S. L. (2012). Preventing disease in the 21st century: the importance of maternal and early infant diet and nutrition. J. Allergy Clin. Immunol. 130, 733–734. doi: 10.1016/j.jaci.2012.06.038
Palmer, K. L., and Gilmore, M. S. (2010). Multidrug-resistant enterococci lack CRISPR-cas. mBio 1:e00227-10. doi: 10.1128/mBio.00227-10
Pandey, V., Berwal, V., Solanki, N., and Malik, N. S. (2015). Probiotics: healthy bugs and nourishing elements of diet. J. Int. Soc. Prev. Community Dent. 5, 81–87. doi: 10.4103/2231-0762.155726
Papadimitriou, K., Zoumpopoulou, G., Foligne, B., Alexandraki, V., Kazou, M., Pot, B., et al. (2015). Discovering probiotic microorganisms: in vitro, in vivo, genetic and omics approaches. Front. Microbiol. 6:58. doi: 10.3389/fmicb.2015.00058
Paredes-Sabja, D., Shen, A., and Sorg, J. A. (2014). Clostridium difficile spore biology: sporulation, germination, and spore structural proteins. Trends Microbiol. 22, 406–416. doi: 10.1016/j.tim.2014.04.003
Parsons, J. B., Broussard, T. C., Bose, J. L., Rosch, J. W., Jackson, P., Subramanian, C., et al. (2014). Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 111, 10532–10537. doi: 10.1073/pnas.1408797111
Perez-Chaparro, P. J., Goncalves, C., Figueiredo, L. C., Faveri, M., Lobao, E., Tamashiro, N., et al. (2014). Newly identified pathogens associated with periodontitis: a systematic review. J. Dent. Res. 93, 846–858. doi: 10.1177/0022034514542468
Plagens, A., Richter, H., Charpentier, E., and Randau, L. (2015). DNA and RNA interference mechanisms by CRISPR-Cas surveillance complexes. FEMS Microbiol. Rev. 3, 442–463. doi: 10.1093/femsre/fuv019
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. M., Ludwig, W., Peplies, J., et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. doi: 10.1093/nar/gkm864
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., et al. (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. doi: 10.1016/j.cell.2013.02.022
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
Rahman, S. Z., Khan, R. A., Gupta, V., and Uddin, M. (2007). Pharmacoenvironmentology–a component of pharmacovigilance. Environ. Health 6:20. doi: 10.1186/1476-069X-6-20
Rasko, D. A., Moreira, C. G., Li de, R., Reading, N. C., Ritchie, J. M., Waldor, M. K., et al. (2008). Targeting QseC signaling and virulence for antibiotic development. Science 321, 1078–1080. doi: 10.1126/science.1160354
Ravel, J., Gajer, P., Abdo, Z., Schneider, G. M., Koenig, S. S., McCulle, S. L., et al. (2011). Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1), 4680–4687. doi: 10.1073/pnas.1002611107
Reddy, T. B., Thomas, A. D., Stamatis, D., Bertsch, J., Isbandi, M., Jansson, J., et al. (2015). The genomes OnLine Database (GOLD) v.5: a metadata management system based on a four level (meta)genome project classification. Nucleic Acids Res. 43, D1099–D1106. doi: 10.1093/nar/gku950
Roberfroid, M. B. (2000). Prebiotics and probiotics: are they functional foods? Am. J. Clin. Nutr. 71(6 Suppl.), 1682S–1687S.
Robinson, C. J., Bohannan, B. J., and Young, V. B. (2010). From structure to function: the ecology of host-associated microbial communities. Microbiol. Mol. Biol. Rev. 74, 453–476. doi: 10.1128/MMBR.00014-10
Romero, R., Hassan, S. S., Gajer, P., Tarca, A. L., Fadrosh, D. W., Nikita, L., et al. (2014). The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2:4. doi: 10.1186/2049-2618-2-4
Rosenthal, M., Goldberg, D., Aiello, A., Larson, E., and Foxman, B. (2011). Skin microbiota: microbial community structure and its potential association with health and disease. Infect. Genet. Evol. 11, 839–848. doi: 10.1016/j.meegid.2011.03.022
Rouillon, C., Zhou, M., Zhang, J., Politis, A., Beilsten-Edmands, V., Cannone, G., et al. (2013). Structure of the CRISPR interference complex CSM reveals key similarities with cascade. Mol. Cell. 52, 124–134. doi: 10.1016/j.molcel.2013.08.020
Round, J. L., and Mazmanian, S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9, 313–323. doi: 10.1038/nri2515
Rupnik, M. (2015). Toward a true bacteriotherapy for Clostridium difficile infection. N. Engl. J. Med. 372, 1566–1568. doi: 10.1056/NEJMcibr1500270
Saad, R., Rizkallah, M. R., and Aziz, R. K. (2012). Gut Pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes. Gut Pathog. 4:16. doi: 10.1186/1757-4749-4-16
Sangiuliano, B., Perez, N. M., Moreira, D. F., and Belizario, J. E. (2014). Cell death-associated molecular-pattern molecules: inflammatory signaling and control. Mediators Inflamm. 2014:821043. doi: 10.1155/2014/821043
Sartor, R. B., and Mazmanian, S. K. (2012). Intestinal microbes in inflammatory bowel diseases. Am. J. Gastroenterol. Suppl. 1, 15–21. doi: 10.1038/ajgsup.2012.4
Segata, N., Haake, S. K., Mannon, P., Lemon, K. P., Waldron, L., Gevers, D., et al. (2012). Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 13:R42. doi: 10.1186/gb-2012-13-6-r42
Selle, K., and Barrangou, R. (2015). Harnessing CRISPR-Cas systems for bacterial genome editing. Trends Microbiol. 23, 225–232. doi: 10.1016/j.tim.2015.01.008
Seo, H. S., Xiong, Y. Q., Mitchell, J., Seepersaud, R., Bayer, A. S., and Sullam, P. M. (2010). Bacteriophage lysin mediates the binding of Streptococcus mitis to human platelets through interaction with fibrinogen. PLoS Pathog. 6:e1001047. doi: 10.1371/journal.ppat.1001047
Slavin, J. (2013). Fiber and prebiotics: mechanisms and health benefits. Nutrients 5, 1417–1435. doi: 10.3390/nu5041417
Smillie, C. S., Smith, M. B., Friedman, J., Cordero, O. X., David, L. A., and Alm, E. J. (2011). Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480, 241–244. doi: 10.1038/nature10571
Sokol, H., Pigneur, B., Watterlot, L., Lakhdari, O., Bermudez-Humaran, L. G., Gratadoux, J. J., et al. (2008). Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. U.S.A. 105, 16731–16736. doi: 10.1073/pnas.0804812105
Sommer, M. O., and Dantas, G. (2011). Antibiotics and the resistant microbiome. Curr. Opin. Microbiol. 14, 556–563. doi: 10.1016/j.mib.2011.07.005
Sulakvelidze, A., Alavidze, Z., and Morris, J. G. (2001). Bacteriophage therapy. Antimicrob. Agents Chemother. 45, 649–659. doi: 10.1128/AAC.45.3.649-659.2001
Thompson, A. L., Monteagudo-Mera, A., Cadenas, M. B., Lampl, M. L., and Azcarate-Peril, M. A. (2015). Milk- and solid-feeding practices and daycare attendance are associated with differences in bacterial diversity, predominant communities, and metabolic and immune function of the infant gut microbiome. Front. Cell Infect. Microbiol. 5:3. doi: 10.3389/fcimb.2015.00003
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi: 10.1038/nature07540
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiome project. Nature 449, 804–810. doi: 10.1038/nature06244
van der Oost, J., Westra, E. R., Jackson, R. N., and Wiedenheft, B. (2014). Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 12, 479–492. doi: 10.1038/nrmicro3279
van Opijnen, T., Bodi, K. L., and Camilli, A. (2009). Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767–772. doi: 10.1038/nmeth.1377
Vercoe, R. B., Chang, J. T., Dy, R. L., Taylor, C., Gristwood, T., Clulow, J. S., et al. (2013). Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet. 9:e1003454. doi: 10.1371/journal.pgen.1003454
Verdam, F. J., Fuentes, S., de Jonge, C., Zoetendal, E. G., Erbil, R., Greve, J. W., et al. (2013). Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity (Silver Spring) 21, E607–E615. doi: 10.1002/oby.20466
Vuotto, C., Longo, F., and Donelli, G. (2014). Probiotics to counteract biofilm-associated infections: promising and conflicting data. Int. J. Oral Sci. 6, 189–194. doi: 10.1038/ijos.2014.52
Wallace, B. D., and Redinbo, M. R. (2013). The human microbiome is a source of therapeutic drug targets. Curr. Opin. Chem. Biol. 17, 379–384. doi: 10.1016/j.cbpa.2013.04.011
Wang, G. (2014). Human antimicrobial peptides and proteins. Pharmaceuticals (Basel) 7, 545–594. doi: 10.3390/ph7050545
Waters, C. M., and Bassler, B. L. (2005). Quorum sensing: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 21, 319–346. doi: 10.1146/annurev.cellbio.21.012704.131001
Whelan, K., and Quigley, E. M. (2013). Probiotics in the management of irritable bowel syndrome and inflammatory bowel disease. Curr. Opin. Gastroenterol. 29, 184–189. doi: 10.1097/MOG.0b013e32835d7bba
Wikoff, W. R., Anfora, A. T., Liu, J., Schultz, P. G., Lesley, S. A., Peters, E. C., et al. (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. U.S.A. 106, 3698–3703. doi: 10.1073/pnas.0812874106
Wilson, I. D., and Nicholson, J. K. (2009). The role of gut microbiota in drug response. Curr. Pharm. Des. 15, 1519–1523. doi: 10.2174/138161209788168173
Woese, C. R., Kandler, O., and Wheelis, M. L. (1990). Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. U.S.A. 87, 4576–4579. doi: 10.1073/pnas.87.12.4576
Wooley, J. C., Godzik, A., and Friedberg, I. (2010). A primer on metagenomics. PLoS Comput. Biol. 6:e1000667. doi: 10.1371/journal.pcbi.1000667
Wozniak, R. A., and Waldor, M. K. (2010). Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 8, 552–563. doi: 10.1038/nrmicro2382
Wright, G. D. (2010). Antibiotic resistance in the environment: a link to the clinic? Curr. Opin. Microbiol. 13, 589–594. doi: 10.1016/j.mib.2010.08.005
Xavier, K. B., and Bassler, B. L. (2003). LuxS quorum sensing: more than just a numbers game. Curr. Opin. Microbiol. 6, 191–197. doi: 10.1016/S1369-5274(03)00028-6
Xiao-Jie, L., Hui-Ying, X., Zun-Ping, K., Jin-Lian, C., and Li-Juan, J. (2015). CRISPR-Cas9: a new and promising player in gene therapy. J. Med. Genet. 52, 289–296. doi: 10.1136/jmedgenet-2014-102968
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. doi: 10.1038/nature11053
Yosef, I., Manor, M., Kiro, R., and Qimron, U. (2015). Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. U.S.A. 112, 7267–7272. doi: 10.1073/pnas.1500107112
Zaura, E., Nicu, E. A., Krom, B. P., and Keijser, B. J. (2014). Acquiring and maintaining a normal oral microbiome: current perspective. Front. Cell Infect. Microbiol. 4:85. doi: 10.3389/fcimb.2014.00085
Zhang, Q., Rho, M., Tang, H., Doak, T. G., and Ye, Y. (2013). CRISPR-Cas systems target a diverse collection of invasive mobile genetic elements in human microbiomes. Genome Biol. 14, R40. doi: 10.1186/gb-2013-14-4-r40
Zhou, Y., Gao, H., Mihindukulasuriya, K. A., La Rosa, P. S., Wylie, K. M., Vishnivetskaya, T., et al. (2013). Biogeography of the ecosystems of the healthy human body. Genome Biol. 14:R1. doi: 10.1186/gb-2013-14-1-r1
Zhou, Y., Mihindukulasuriya, K. A., Gao, H., La Rosa, P. S., Wylie, K. M., Martin, J. C., et al. (2014). Exploration of bacterial community classes in major human habitats. Genome Biol. 15, R66. doi: 10.1186/gb-2014-15-5-r66
Zoetendal, E. G., Rajilic-Stojanovic, M., and de Vos, W. M. (2008). High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 57, 1605–1615. doi: 10.1136/gut.2007.133603
Keywords: microbiome, metagenomics, phage therapy, CRISPR/Cas system, quorum sensing, pharmacomicrobiomics
Citation: Belizário JE and Napolitano M (2015) Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front. Microbiol. 6:1050. doi: 10.3389/fmicb.2015.01050
Received: 12 May 2015; Accepted: 14 September 2015;
Published: 06 October 2015.
Edited by:
Eric Altermann, AgResearch Ltd, New ZealandReviewed by:
M Andrea Azcarate-Peril, University of North Carolina at Chapel Hill, USACopyright © 2015 Belizário and Napolitano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José E. Belizário, Department of Pharmacology, Institute of Biomedical Sciences, University of São Paulo, Avenida Lineu Prestes, 1524, CEP 05508-900, São Paulo, SP, Brazil, amViZWxpemFAdXNwLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.