 Md. Habibur Rahaman
Md. Habibur Rahaman Tarequl Islam
Tarequl Islam Rita R. Colwell
Rita R. Colwell Munirul Alam
Munirul Alam- 1Department of Biology and Chemistry, North South University, Dhaka, Bangladesh
- 2Enteric and Food Microbiology Lab, Center for Communicable Diseases, International Center for Diarrheal Disease Research, Dhaka, Bangladesh
- 3Maryland Pathogen Research Institute, University of Maryland, College Park, MD, USA
- 4Center for Bioinformatics and Computational Biology, University of Maryland, College Park, MD, USA
Vibrio cholerae, the etiological agent of cholera, has been a scourge for centuries. Cholera remains a serious health threat for developing countries and has been responsible for millions of deaths globally over the past 200 years. Identification of V. cholerae has been accomplished using a variety of methods, ranging from phenotypic strategies to DNA based molecular typing and currently whole genomic approaches. This array of methods has been adopted in epidemiological investigations, either singly or in the aggregate, and more recently for evolutionary analyses of V. cholerae. Because the new technologies have been developed at an ever increasing pace, this review of the range of fingerprinting strategies, their relative advantages and limitations, and cholera case studies was undertaken. The task was challenging, considering the vast amount of the information available. To assist the study, key references representative of several areas of research are provided with the intent to provide readers with a comprehensive view of recent advances in the molecular epidemiology of V. cholerae. Suggestions for ways to obviate many of the current limitations of typing techniques are also provided. In summary, a comparative report has been prepared that includes the range from traditional typing to whole genomic strategies.
Introduction
Cholera is a severe and watery form of diarrhea caused by the pathogen, Vibrio cholerae. Clinical manifestations range from voluminous stool, hypo-volemic shock, to acidosis (Kaper et al., 1995). Severe fluid loss can lead to death within a day of onset (Sack et al., 2004). To date, seven pandemic outbreaks of cholera have been classified by year of commencement, namely 1817, 1829, 1852, 1861, 1881, 1899, and 1961 (Stine and Morris, 2014). The current seventh pandemic is believed to have originated in Indonesia (Barua, 1972) and continued in other continents, typical of pandemics. The current cholera pandemic is reported to be the most extensive, in terms of duration and geography (Faruque and Mekalanos, 2012). Earlier pandemics were speculated to have originated in the Ganges Delta region of the Indian subcontinent (Singh and Mohapatra, 2008). In addition to recorded pandemics, many localized outbreaks have also affected regions with severe outcomes, as occurred with re-emergence of cholera in Latin America in 1991 and explosive outbreaks of cholera in Orissa 1999, Dhaka 2006, Zimbabwe 2008, Haiti 2010, and Kenya 2010 (Singh and Mohapatra, 2008; Stine and Morris, 2014). All of these events highlight the epidemiological importance of this global disease. Ranges of typing and whole genomic approaches have been adopted for effective tracking of the evolution of V. cholerae, an overview of which is provided here, showing the overall impact on the changing paradigm of V. cholerae epidemiology and evolution.
Cholera Epidemiology
Cholera, a curse for centuries, is speculated to have originated before the time of Hippocrates and Buddha (Barua, 1992). However, it was not until 1854, when the classical epidemiological study of John Snow mapped the waterborne transmission of cholera in London (Snow, 1855), even before the notion of microbial causation of cholera. Subsequently, Filippo Pacini identified V. cholerae as the causative agent of cholera in 1855 (Bentivoglio and Pacini, 1995), a finding that was overshadowed when Robert Koch was able to grow the “Vibrio comma” in culture in 1884 (Koch, 1884; Figure 1). All of these findings overruled many of the misguided notions about cholera, as it was once thought that cholera was spread by “miasma” or “fog of rivers” (Sack et al., 2004).
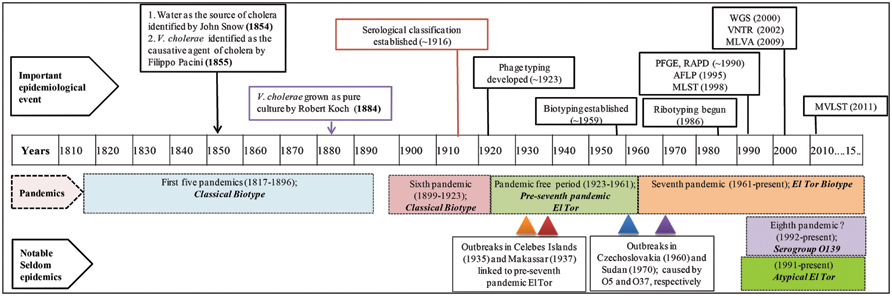
FIGURE 1. Timeline of cholera epidemiology since 1817. The upper panel shows important scientific advances that have changed the landscape of cholera epidemiology. Pandemics and their putative causative strains are shown in shaded boxes with dashed boarders. The lowermost panels show infrequent outbreaks and their causative strains, with triangles indicating time of outbreaks. This timeline has been adapted from Banerjee et al. (2014). PFGE, pulsed-field gel electrophoresis; RAPD, random amplification of polymorphic DNA; AFLP, amplified fragment length polymorphism; MLST, multi-locus sequence typing; WGS, whole genome sequencing; VNTRs, variable number of tandem repeats; MLVA, multi-locus variable tandem repeat; and MVLST, multi-virulence locus sequencing typing.
During the first six cholera pandemics, V. cholerae classical biotype was isolated globally between 1817 and 1923 (Blake, 1994; Dziejman et al., 2002). Based on historical records, the period between 1923 and 1961 was pandemic free, although a major epidemic outbreak was recorded in the Celebes Islands (currently Indonesia) in 1935 caused by a new biotype, ‘El Tor’ (V. cholerae MAK757). The ‘El Tor’ biotype was also isolated in Makassar, Indonesia (strain M66-2) during a cholera outbreak in 1937. Both outbreaks were not pandemics and de Moor described them as ‘Paracholera’ and van Loghem as ‘Enteritis choleriformis El Tor,’ but the MAK757 and M66-2 isolates were subsequently labeled as pre-seventh pandemic El Tor (Barua, 1992; Banerjee et al., 2014; Figure 1).
The seventh cholera pandemic is reported to have been caused by the El Tor biotype that gradually replaced classical strains. This was the only pandemic that had originated outside of India, namely in Sulawesi, Indonesia, in 1961 and was isolated from cholera patients in territories of Asia by 1966. Until 1971, cholera outbreaks were few, while in Africa and Europe, an upsurge was recorded and until then, cholera had not been reported in those countries for more than 100 years (Reeves and Lan, 1998). After a lull, El Tor again caused a massive outbreak in 1991 in Peru, the first cholera epidemic in Latin America since 1895 (Seas et al., 2000). In 1992, a new variant of the seventh pandemic strain appeared in Madras and spread rapidly in Asia, raising concerns about the beginning of an eighth pandemic of cholera (Siddique et al., 1996; Figure 1). This new pathogenic serotype is referred to as V. cholerae O139 Bengal, the O1 antigen of the prototypic seventh pandemic strain having been replaced by the O139 antigen, hence a new serogroup (Albert, 1994). Later, the emergence of new variants of V. cholerae O1 harboring traits of both classical and El Tor biotypes was recorded (Nair et al., 2002, 2006; Ansaruzzaman et al., 2004), collectively referred to as ‘atypical El Tor’ (Safa et al., 2010). The ongoing seventh pandemic is the longest of the cholera pandemics and is believed to continue because the causative agent, El Tor and its derivatives are presumed better adapted for global dissemination compared to their classical predecessors (Faruque et al., 1998; Safa et al., 2010). Although V. cholerae strains belonging to O1 and O139 serogroups have been responsible for both major epidemics and endemic cholera, other serogroups are referred to as V. cholerae non-O1/non-O139. They have also caused cholera, but rarely epidemics, an example of which occurred in the 1960 and 1970s, when V. cholerae O5 and O37 serogroups caused explosive outbreaks of cholera in Czechoslovakia and Sudan, respectively (Aldová et al., 1968; Kamal, 1971; Yamamoto et al., 1995; Figure 1).
Although seven cholera pandemics have been recorded since 1817, isolates from the first five pandemics are not available because V. cholerae was first isolated and identified only at the time of the fifth cholera pandemic (Koch, 1884). Evolution of diagnostic and fingerprinting strategies since then have enabled study of sixth and seventh pandemic strains in detail (Reeves and Lan, 1998). In this review, we have grouped fingerprinting methods into three categories, phenotypic/traditional, pre-genomic, and genomic, showing estimated time of development and the influence of each on cholera epidemiology (Figure 1).
Phenotypic Fingerprinting
Phenotype based traditional typing methods, namely serotype, biotype, antibiogram, and phagetyping have been used for many years, although eventually challenged because of their relatively low discriminatory power (Bhowmick et al., 2011). Among phenotypic strategies, serological classification of cholera vibrios was established during the sixth cholera pandemic (Greig, 1916). Later, the most widely used typing scheme was developed by Sakazaki and Shimada (1977) who used anti-sera of heat killed microbes. The scheme identified 138 V. cholerae O serogroups, to which the O139 serogroup was added (Bhattacharya et al., 1993). In the USA, a similar scheme using anti-sera of live organisms was also used for serological classification of V. cholerae (Smith, 1979). All of these serological classifications are based on differences in the sugar composition of the heat-stable surface somatic ‘O’ antigen of V. cholerae (Gardner and Venkatraman, 1935). Moreover, based on antigenic factors, V. cholerae O1 has been differentiated into three serotypes, referred to as Ogawa, Inaba, and Hikojima (Banerjee et al., 2014; Figure 2).
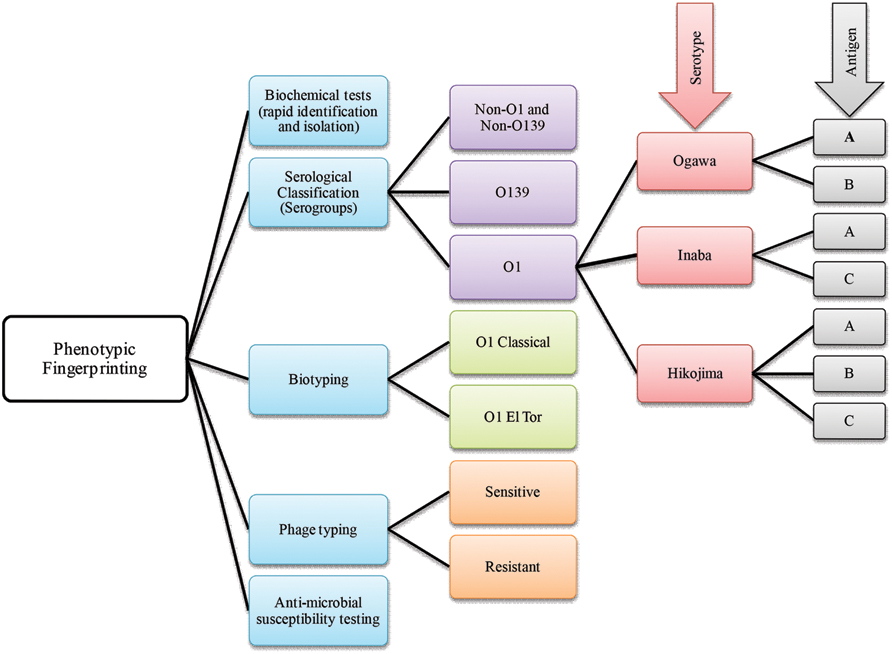
FIGURE 2. A brief scheme that includes phenotype based fingerprinting strategies employed in the classification of Vibrio cholerae.
Historically, two major biotypes of V. cholerae, Classical and El Tor, are recognized. El Tor biotypes are generally hemolytic, whereas classical biotypes are usually non-hemolytic (Pollitzer et al., 1959). Since exceptions to every rule may be found in biology sooner or later, it is not surprising that most El Tor biotype strains are non-hemolytic except isolates obtained in the early 1960s (Barrett and Blake, 1981). Therefore, hemolysis biotyping has limited utility because of inconsistency. The hemeagglutinating property of vibrios was explored by Doorenbos in 1932 (Felsenfeld, 1966), and later applied as a biotyping tool, using chicken red blood cells which agglutinate with El Tor but not with classical V. cholerae (Finkelstein and Mukerjee, 1963). In addition, the Voges–Proskauer test, resistance to polymyxin B (Han and Khie, 1963), and modified CAMP test (Lesmana et al., 1994) have been used for determining V. cholerae biotypes.
Vibrio cholerae belonging to the same serotype or biotype can be further differentiated by phage typing. One of the earlier studies on cholera phages was carried out by Nobechi in 1923 and has been summarized (Pollitzer et al., 1959). Subsequently, phage typing was done using Mukerjee’s cholera phages (Basu and Mukerjee, 1968). The FK phages of Takeya et al. (1981) later were adopted to differentiate classical and El Tor biotypes. In both cases, phage susceptibility was used as a method for strain differentiation. Mukerjee’s phage typing scheme was efficient when used to study the initial spread of V. cholerae O1 El Tor (Faruque, 2014) also, phage typing has proven useful when phenotypic fingerprinting was inconclusive. Over the course of time, phage typing schemes proved inadequate for typing the large number of emerging V. cholerae strains. The fact that the number of well characterized Vibrio phages was limited and consensus for typing schemes was not uniform, posed major challenges to the use of phage sensitivity and resistance as a major typing tool (Kaper et al., 1995). Interestingly, new phage typing schemes for V. cholerae O1 El Tor (Chattopadhyay et al., 1993) and O139 strains (Chakrabarti et al., 2000) have been developed that appear to overcome some of the limitations.
Antimicrobial susceptibility has been used to characterize Vibrio spp. and their susceptibility to various antibiotics is measured by using a battery of antibiotics at various concentrations. The resistances are scored to generate a fingerprint, i.e., an antibiotic resistance profile, which is subjected to cluster analysis, and comparison to a reference database for microbial tracking (Scott et al., 2002). Changing antibiotic resistance patterns has been described as a hallmark of cholera epidemiology and is associated with the substantial mobility of genetic elements harboring antibiotic resistance genes in V. cholerae (Faruque et al., 1998). In addition to the standard disk diffusion method recommended by the Clinical Laboratory and Standard Institute (CLSI), a rapid Vitek susceptibility system has been developed to determine antibiotic resistance profiles of V. cholerae O1, O139, and Non-O1 (Sciortino et al., 1996).
Although phenotypic fingerprinting strategies have been used routinely for identification and tracking of V. cholerae since the beginning of the seventh pandemic, these tools have major disadvantages, including unstable phenotypes, lower sensitivity, and limited specificity.
Pre-genomic Era of Fingerprinting
Relatedness and differences among bacterial isolates derived from molecular signatures at the DNA level have been used for molecular fingerprinting (Sabat et al., 2013). The applicability of a given fingerprinting method depends on its discriminatory power, that is, its ability to distinguish epidemiologically unrelated isolates and determine those closely related. In addition, speed, reproducibility, portability among laboratories, cost and ease of interpretation is major considerations (Struelens, 1996; van Belkum et al., 2007). Molecular methods have proven effective for determining genetic changes that result in displacement of an existing serogroup with a newly emerging serogroup.
Stine and Morris (2014) proposed acqisition of a novel genetic element(s) leads to an increase in bacterial fitness which, in turn, causes a selective sweep (a local or global outbreak). The seventh cholera pandemic was suggested to comprise four such selective sweeps. The first selective sweep is considered to be associated with emergence of V. cholerae O1 El Tor in 1961, whereby the ancestor of V. cholerae O1 El Tor strains acquired Vibrio seventh pandemic Islands (VSPs) I and II (Dziejman et al., 2002). The second selective sweep was considered to have begun with acquisation in 1981 of the novel sxt element. The sxt element harbors several antibiotic resistance genes that provide selective advantages to the host and is present in almost all post-1990 V. cholerae O1 clinical isolates (Waldor et al., 1996). The third selective sweep is considered to have been triggered by replacement of V. cholerae O1 serogroup encoding genes with those for O139, resulting in an outbreak of cholera across the Indian subcontinent (Bik et al., 1995; Comstock et al., 1996; Mooi and Bik, 1997). The fourth, or most recent selective sweep, is proposed to have been initiated by replacement of the El Tor ctx allele with the classical ctx allele in an El Tor background (Raychoudhuri et al., 2009), presumed to be associated with the acute form of cholera (Nair et al., 2002). Molecular fingerprinting has been used to elucidate all of these marker events in cholera epidemiology leading to a growing interest in its clinical applications. In fact, several DNA-based strategies have been used for molecular typing of V. cholerae, exemplified by pulsed-field gel electrophoresis (PFGE), ribotyping, random amplification of polymorphic DNA (RAPD), AFLP, enterobacterial repetitive intergenic consensus sequence-PCR (ERIC-PCR), VNTRs, multi-locus sequence typing (MLST), and multi-locus variable tandem repeat analysis (MLVA; Bhowmick et al., 2011; Figure 3). To underscore the impact of these methods on cholera epidemiology, publications that have cited molecular fingerprinting strategies are summarized as shown in Figure 4.
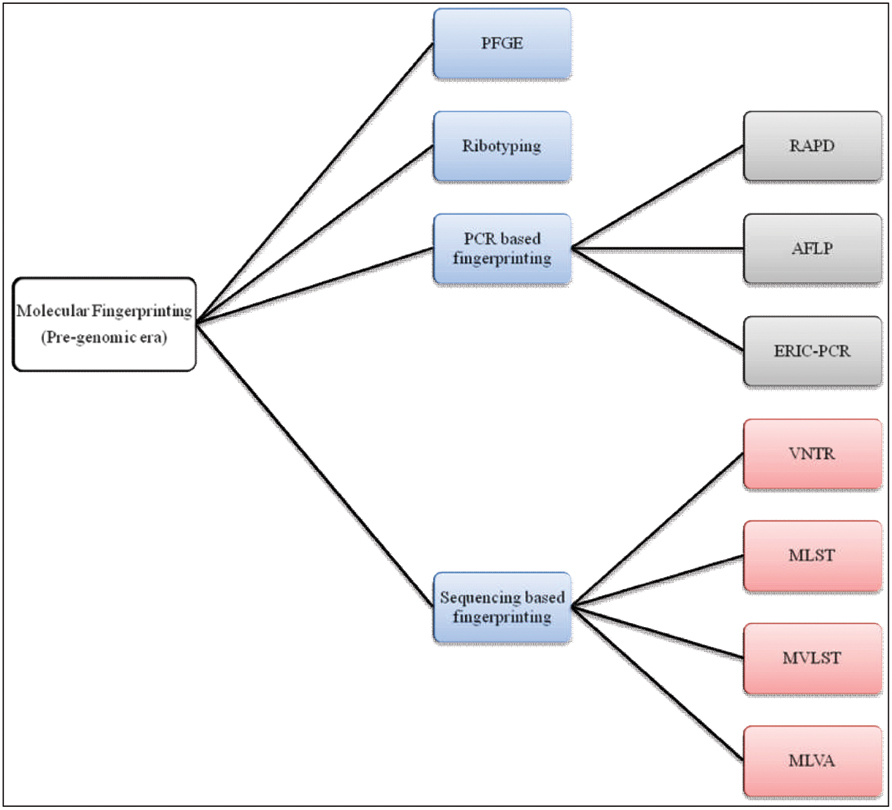
FIGURE 3. A chart of molecular fingerprinting strategies used in epidemiological investigations of V. cholerae.
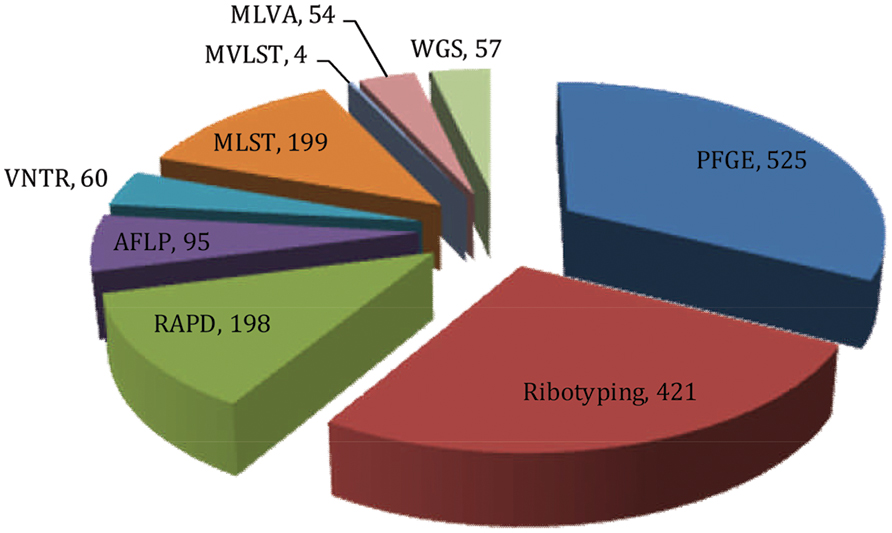
FIGURE 4. Pie chart showing the number of publications retrieved from the literature database (HighWire and PubMed) that cited corresponding fingerprinting tools employed in the molecular epidemiology of V. cholerae (Data retrieved up to July, 2015).
Pulsed-Field Gel Electrophoresis
Among the various molecular fingerprinting methods, PFGE has proven most effective in epidemiological investigation of V. cholerae and is considered the ‘gold standard’ of typing (Tenover et al., 1997; Sabat et al., 2013). The method was used in epidemiological investigations of various bacterial species during the 1990s (Arbeit et al., 1990; Prévost et al., 1991; Gordillo et al., 1993; Tenover et al., 1995). Using the searching keyword ‘PFGE AND V. cholerae,’ 525 publications have been retrieved from the HighWire literature database1 in which PubMed2 was also included. The very large number of publications indicates the importance and influence of this method for molecular typing of V. cholerae.
Pulsed-field gel electrophoresis requires highly purified genomic DNA which is chopped with restriction endonuclease and the restriction fragments separated by applying alternating electric fields, i.e., a ‘pulsed-field,’ to obtain better resolution of separated fragments. Larger DNA fragments, with sizes ranging from 30 kb to 1 Mb, can be resolved using this typing approach (Goering, 2010). PFGE has been used frequently because of its high epidemiological concordance and excellent discriminatory power to distinguish among closely related isolates. Excellent typability, intra-laboratory reproducibility, and less expensive bench-top deployment provide added value (Sabat et al., 2013). However, the huge amount of PFGE-typing data generated over the last decade has challenged the scientific community to seek a common platform to improve inter-laboratory comparability. A robust, reproducible, and standardized PFGE protocol was sought (Cooper et al., 2006) and the networks, PulseNet (Swaminathan et al., 2001), and Harmony (Murchan et al., 2003), were formed.
An earlier study highlighted the usefulness of PFGE, notably in revealing clonality among isolates obtained from two well-defined cholera outbreaks in Malaysia. Restriction endonuclease analysis (REA) patterns of V. cholerae O1 El Tor strains from the two outbreaks were found to be similar but not identical. They varied widely from strains isolated from sporadic cases of cholera (Mahalingam et al., 1994). Cameron et al. (1994) undertook a comparative study that included PFGE, multi-locus enzyme electrophoresis (MEE) and ribotyping, using 180 isolates of V. cholerae O1. They concluded PFGE profiling was more discriminating for the V. cholerae O1 serogroup, compared to the other subtyping assays and recommended it as an effective tool for epidemiological surveillance (Cameron et al., 1994). PFGE based molecular typing provided new insight into the 1991 cholera epidemic in Mexico in a study carried out by Alam et al. (2012), who suggested there was a regional signature in the evolution of classical biotypes in the Americas, independent from the geographically ecosystem of Asia. Recently, PFGE analysis of Mexican isolates collected between 1998 and 2008 revealed a progression of CTX+ El Tor harboring truncated CTX prophage, the predominant cause of endemic cholera in Mexico despite altered El Tor (carrying classical CTX allele) being the predominant cholera causing agent worldwide. This was reported to be a key historical event in the global epidemiology of cholera and PFGE was concluded to be an effective fingerprinting tool (Alam et al., 2014).
Although, PFGE has been widely used in molecular epidemiology studies of V. cholerae, some of its drawbacks have to be taken into consideration. The method is technically demanding, labor-intensive, relatively slow, and has limited data portability, compared to sequence-based methods. In addition, inter-laboratory comparability of complex PFGE-patterns is technically challenging. Single mutations at cutting sites can result in altered restriction profiles and PFGE cannot distinguish among nearly identical band sizes differing by less than 5% (Sabat et al., 2013). Nonetheless, PFGE is used in combination with other typing strategies that compensate for many of its limitations. Teh et al. (2010) demonstrated that PFGE in combination with multi-locus VNTR (MLVA) can provide more information on epidemiological relatedness and differences among isolates from different sources or geographical regions.
Ribotyping
In a search using the keywords ‘Ribotyping AND V. cholerae,’ 421 publications were retrieved from the literature database, indicating the significance of this method in defining the biogeography and molecular epidemiology of V. cholerae. Since the emergence of ribotyping in Grimont and Grimont (1986), many studies showed that the technique alone or in combination with other methods had been effective in studies of the molecular evolution of V. cholerae (Lan and Reeves, 1998; Pourshafie et al., 2000, 2002).
In general, four major consecutive steps are usually employed in ribotyping: (a) restriction digestion of the bacterial chromosome; (b) gel electrophoresis of the resulting fragments; (c) transfer of the fragments to a membrane and; (d) hybridization of the fragments with labeled probes complementary to the 16S and 23S rRNAs (Grimont and Grimont, 1986). However, wider interest led to adaptation of this procedure. To some extent, the name ‘ribotyping’ has been a misnomer. Based on in silico genomics, Bouchet et al. (2008) suggested that ribotypes are derived from restriction fragment length polymorphisms (RFLPs) of neutrally evolving housekeeping genes that flank rRNA genes, rather than directly from rRNA gene sequences that serve solely as conserved and linked tags. Using intergenic polymorphisms, i.e., the spaces between 16S and 23S rRNA genes, has also been adopted mainly in Europe for microbial fingerprinting, e.g., Clostridium difficile as described elsewhere (Sabat et al., 2013).
Popovic et al. (1993) proposed a standardized scheme for ribotyping used in epidemiological investigations of V. cholerae. Under their scheme, data collected during a 60 years follow up study were analyzed. A total of 214 V. cholerae O1 strains were isolated from 35 countries and 14 states of USA and grouped into 27 BglI ribotypes. Interestingly, it was found that strains causing the fifth, sixth, and ongoing seventh pandemic could be grouped into different ribotypes. Among the classical biotypes, seven different but similar ribotypic patterns (1a–1g) were observed, whereas, six different ribotypes with three subtypes were found among seventh pandemic strains. It was suggested that wide circulation of different clones favored persistence of V. cholerae in the environment but certain clones were restricted to specific regions, i.e., ribotype-5 was predominant in Latin America and ribotype-8 was restricted to central Africa (Popovic et al., 1993; Thompson et al., 2004). Later, Faruque et al. (1997) classified V. cholerae O1 El Tor strains into five ribotypes based on BglI restriction profiles. V. cholerae El Tor strains isolated before emergence of O139 were grouped as ‘ribotype I–IV’ and post-O139 isolates were grouped as ‘ribotype V’ (Faruque et al., 1997), which had not been described by Popovic et al. (1993).
An earlier ribotyping study had shown an association between clinical and environmental isolates of V. cholerae O1 during an 8 years follow-up study in Australia (Desmarchelier et al., 1995). A recent study undertaken in Bangladesh reported V. cholerae toxigenic O1 and O139 environmental strains shared similar ribotype profiles with pandemic strains. Furthermore, the V. cholerae non-O1, non-O139, and TCP- non-toxigenic O1 strains diverged widely from seventh pandemic O1 and O139 strains. Hence, it was suggested that there was heterogeneity among the environmental V. cholerae population in a cholera endemic area like Bangladesh (Faruque et al., 2004). Another earlier case study provided evidence that the presence of 11 ribotypic polymorphic restriction sites in seventh pandemic isolates can be used to distinguish them from sixth and pre-seventh pandemic isolates. However, ribotyping does not necessarily represent bacterial evolution because the pattern obtained may be the result of recombination events (Karaolis et al., 1994).
Polymerase Chain Reaction (PCR) Fingerprinting
Random Amplification of Polymorphic DNA
Random amplification of polymorphic DNA is one of the earlier PCR based fingerprinting strategies which had been used to characterize and trace the phylogeny of diverse bacterial species of epidemiological importance. The technique was first developed in the 1990s by two independent groups (Welsh and McClelland, 1990; Williams et al., 1990). The search keywords ‘RAPD AND V. cholerae’ was used to retrieve 198 publications from literature databases (as described above). These publications amplify the importance of this tool in studying V. cholerae molecular epidemiology. RAPD employs several arbitrary and short oligonucleotide primers (9–10 bases in length) to hybridize chromosomal DNA randomly at a lower annealing temperature. If two RAPD primers can hybridize in proper orientation and proximity (within a few kilo-base span); the fragments will be amplified, corresponding to the distance between the two primers. Resulting amplified fragments are resolved by agarose gel electrophoresis to generate a semi-unique profile of the RAPD reaction which, in theory, is characteristic of the particular bacterial isolates. Relationships between V. cholerae isolates can be determined by comparing their unique RAPD fingerprinting profiles. However, a standardized RAPD protocol has not been used to form a data bank for identification of V. cholerae O1 strains, as was studied in Brazil (Leal et al., 2004). Moreover, RAPD is sensitive to technical variations introduced by laboratory personnel, different DNA samples, and different sources of enzyme and primers. These can generate different banding patterns, hence poor reproducibility (Meunier and Grimont, 1993).
Amplified Fragment Length Polymorphism (AFLP)
Amplified fragment length polymorphism (AFLP) is another molecular fingerprinting approach that utilizes a subset of genomic fragments generated by restriction digestion. The technique was developed in Vos et al. (1995) and since then has been used for molecular typing of diverse bacterial species. From a literature database search using the keywords ‘AFLP AND V. cholerae,’ 95 publications were retrieved, highlighting the impact of this method for V. cholerae molecular epidemiology and evolution. In the AFLP method, genomic DNA is cleaved with two restriction enzymes and the sticky ends of the restriction fragments are ligated with double stranded adaptors. Subsequently, restriction fragments are selectively amplified by PCR, wherein the flanking adaptor sequences are used as the primer binding sites. Initial cycles of PCR are run with stringency to ensure fidelity of the reaction or to avoid non-specific amplification. The amplified fragments are resolved by an automated DNA sequencer and the resulting banding patterns used to decipher genetic relatedness among bacterial isolates (Mortimer and Arnold, 2001). An exemplar study used AFLP based typing to uncover genetic relatedness among environmental and clinical V. cholerae isolates and concluded that pathogenic strains may have arisen from non-toxigenic strains in the aquatic environment (Jiang et al., 2000). Another study reported AFLP can be used to decipher variation among seventh pandemic clones more effectively, compared to ribotyping. Furthermore, a temporal pattern of change was found among clones of V. cholerae in which strains isolated between the 1960 and 1970s clustered distinctly, when compared with isolates collected between the 1980 and 1990s (Lan and Reeves, 2002).
Sequencing based Fingerprinting
Variable Number of Tandem Repeats Analysis
Many pathogenic V. cholerae O1 and O139 isolates demonstrate similar genetic profiles by PFGE (Basu et al., 2000), CTX-genotyping (Basu et al., 2000; Mukhopadhyay et al., 2001), and ribotyping (Faruque et al., 2000). Sequencing based genotypic approaches subsequently have been adopted to obviate the limitations of these methods. Among those strategies, variable number of tandem repeats (VNTRs) or simple sequence repeats (SSRs) provide a source of high genomic polymorphism to distinguish rigorously within and among V. cholerae isolates. Literature database searches, using the keywords ‘VNTR and V. cholerae’ retrieved 60 publications showing a growing interest in VNTR as a fingerprinting tool.
Variable number of tandem repeats is short DNA sequence motifs that are repeated in tandem at a specific locus and represent a distinctive hereditary feature of an individual isolate. The first report of a VNTR locus in V. cholerae was published in 2002, in which the locus was designated VcA, located on chromosome 2, and consisting of a TGCTGT repeat (Vodop’ianov et al., 2002). The usefulness of VcA VNTR was further tested and the conclusion was that the discriminatory ability of VcA VNTR was better than PFGE (Bhowmick et al., 2010). Another study identified 17 VNTR loci that could be used to differentiate V. cholerae isolates not discriminated by PFGE. Two of the loci were reported to be stable during serial passage under specific culture conditions (Danin-Poleg et al., 2007). VNTRs have also been shown to be effective in assessing genetic relatedness among outbreak isolates from geographic co-located Bangladeshi villages and within a short time frame (Stine et al., 2008). The spread of specific genotypes of V. cholerae O1 and O139 was tracked spatiotemporally across another cholera endemic country, namely India, by exploiting VNTR as an effective fingerprinting tool (Ghosh et al., 2008).
Multi-locus Sequence Typing
Multi-locus sequence typing is a sequencing based fingerprinting tool of particular interest since its development in Maiden et al. (1998). It provides sequence-based resolution, is informational, and is technically feasible. Literature database search using the keywords ‘MLST AND V. cholerae’ retrieved 199 publications showing applications of MLST as a fingerprinting tool. V. cholerae shows variation in three housekeeping genes, gyrB, pgm, and recA, presumed to be highly conserved. Two earlier studies had used MLST based molecular typing to analyze evolutionary relationships among V. cholerae clones isolated from different geographic regions (Byun et al., 1999; Farfán et al., 2002). Kotetishvili et al. (2003) used MLST, along with PFGE, to characterize 22 V. cholerae isolates representing epidemic and non-epidemic serogroups and concluded MLST to be more discriminatory than PFGE. Another study used MLST of nine genetic loci and found Mozambique isolates shared the same unique sequence type (ST) as V. cholerae O1 El Tor strain N16961, a seventh pandemic isolate (Lee et al., 2006). Along with utilizing housekeeping genes, a derivative of the MLST scheme was developed that is based entirely on virulence genes and has been coined multi-virulence locus sequencing typing (MVLST). A recent study showed MVLST is more discriminatory than traditional MLST, because MVLST differentiates outbreak strains and toxigenic from non-toxigenic subtypes. In combination, they proved to be more discriminatory and informative in studying local epidemiology of V. cholerae (Teh et al., 2011).
Multi-locus Variable Tandem Repeat Analysis
Multi-locus variable tandem repeat analysis is a sequencing based typing tool that targets five or six loci in the V. cholerae genome. Each locus harbors a tandem repeat of six or seven nucleotides which can be repeated 4–31 times. The length of the tandem repeats represents allele numbers in the corresponding locus, which in turn is used to infer a five-digit genotype, i.e., for the five different loci. Strains derived from the same ancestor are presumed to be part of a clonal complex and, therefore, presumed to differ only at a single locus in a five-digit genotype. In contrast, genotypes that differ at two or more loci are considered clonally unrelated, hence evolved from different ancestors (Stine and Morris, 2014). A literature database search using the keywords ‘MLVA AND V. cholerae’ retrieved 54 publications where MLVA was used as a fingerprinting tool.
One MLVA study showed the presence of five different clonal complexes in a set of Kenyan isolates (Mohamed et al., 2012) which was contrary to the hypothesis of a clonal introduction of outbreak causing strains (Stine and Morris, 2014). A recent study in Bangladesh exploited MLVA to determine clonal relationship between environmental and clinical V. cholerae O1 isolates from outbreaks in two geographically distinct locations. The study shed light on the mechanism of an accelerated mode of cholera transmission, i.e., person-to-person transmission, compared to slow mode transmission, namely the person-to-aquatic environment-to-person pathway (Rashed et al., 2014). Lam et al. (2012) showed that MLVA can be effective in resolution of closely related seventh pandemic clones which had been grouped on the basis of SNP profiles. An earlier study had developed a simple and highly discriminating MLVA strategy for connecting clinical and environmental V. cholerae isolates, using 12 VNTR loci among which six were found to be polymorphic. For those six loci, a higher discriminatory power (Simpsons Diversity Index = 0.99) was calculated for 142 environmental and clinical V. cholerae strains isolated from diverse geographic regions. MLVA was documented to be a potential fingerprinting tool for tracking the source of V. cholerae (Olsen et al., 2009).
Fingerprinting in the Era of Genomics
Although molecular fingerprinting has been used for many years to study the molecular epidemiology of V. cholerae, thoroughness and resolution of these methods are limited. For instance, it was not until the late 1990s that the presence of two chromosomes in Vibrio species had been described by physical mapping (Trucksis et al., 1998; Yamaichi et al., 1999). The genomic era was launched by whole genome sequencing (WGS) of V. cholerae O1 El Tor N16961 by the joint consortium of The Institute for Genomic Research, the University of Maryland, and Harvard Medical School (Heidelberg et al., 2000). Functional annotation of the genome sequences and holistic resolution at the nucleotide level eased the way to high-throughput assays. Microarrays (Dziejman et al., 2002), parallel WGS (Grim et al., 2010; Mutreja et al., 2011), and hybrid de novo assembly of second and third generation sequencing data (Bashir et al., 2012) have since been used to redefine the intricate mechanisms of cholera pathogenesis and corollary outbreaks of the disease. A significant number of reports of whole genome based molecular epidemiology of V. cholerae indicate the field is growing rapidly.
After the initial WGS of V. cholerae by Heidelberg et al. (2000), comparative genomic analysis was conducted using classical, pre-pandemic, and pandemic El Tor, and two non-toxigenic V. cholerae N16961 strains in which a whole genome microarray of a seventh pandemic strain was used as a reference (Dziejman et al., 2002). All strains had shown remarkable similarity in gene content and each strain harbored at least 99% of the genes present in the reference strain. The V. cholerae O139 strains were found to be similar to the V. cholerae O1 El Tor strains and very likely were clonally derived from El Tor after a change in LPS. A seventh pandemic island was found only in the pandemic El Tor and O139 strains, but not in the classical or pre-pandemic El Tor strains. Genes encoded in the seventh pandemic island may function in persistence in the environment, hence a potential mechanism for replacement of classical strains by El Tor strains. These genes may allow seventh pandemic strains to withstand nutrient depletion or other environmental stresses or may be involved in interactions with non-human aquatic hosts (Chun et al., 2009).
Availability of extensive microbial genomic strain sequences led to the coining of a new terminology, the ‘pan-genome,’ comprising the core genome (indispensable genes shared by all strains) and the dispensable genome, i.e., genes not shared by all strains or genes that are unique to an individual strain (Tettelin et al., 2005). Both core and pan-genome genes are taken into consideration to deduce bacterial phylogenies. The core genome is more informative for determining phylogenetic relationships than ribotyping which exploits only variations in housekeeping genes flanking 16S/23S rRNA genes (Robins and Mekalanos, 2014). Chun et al. (2009) did a comparative genomic analysis of 23 V. cholerae strains and identified 2,432 common core genes, or orthologs, and 6,953 total unique genes in their pan-genome.
In the post-genomic era, second-generation (or ‘next-generation’) sequencing has been used extensively over the last decade for bacterial WGS, though their shorter read lengths remain as a major limitation. Next-generation sequencing merely produces draft or incomplete genomes, their higher contig numbers and varying qualities pose major challenges in genome assembly and closure (Land et al., 2014). As a result, majority of the currently available Vibrio genome sequences are reported to be incomplete draft sequences (Chapman et al., 2015). In contrary, the third generation (or ‘single molecule’) sequencing offers longer read lengths and potentially produces a finished or complete genome within a few hours. One of the first ‘single molecule’ sequencing platforms was whole genome optical mapping (Schwartz et al., 1993), in which the complete genomic map is deduced from a number of partial restriction maps, thus obviating the need for a physical map. It has proven effective for capturing and validating genomic complexities to produce a finished or complete genome (Niedringhaus et al., 2011). However, the dearth of standard statistical package and bioinformatic tools to analyze the myriad of sequencing data, sensitivity to stochastic insertion and deletion (indels) mutations, lower throughput and relatively higher expenses are among the major challenges for the routine application of the third generation sequencing, till now (Land et al., 2015).
The finished or complete genome is the ultimate target as it can aid in the deployment of in silico genomics precisely by using robust genome relatedness indices, e.g., average nucleotide identity (ANI; Konstantinidis et al., 2006). ANI values can potentially define the identity or similarity between two bacterial genomes by calculating the proportion of DNA shared by the genomes, and have been proposed as a ‘next-generation gold standard’ for species demarcation in the post-genomic era (Kim et al., 2014). ANI values coupled with comparative genomics have already been in use for identifying the close relatives of V. cholerae from environmental and clinical samples (Haley et al., 2010; Hasan et al., 2010; Kirchberger et al., 2014). Although the finished genome of V. cholerae is still in infancy, it has the potential to complement many of the traditional fingerprinting tools namely MLST and Ribotyping. For example, instead of typing 16S and 23S rRNA operons or other housekeeping genes such as gyrB, pgm, and recA by sequencing, those can be mapped directly from the complete genome reads to decipher variations related to the molecular evolution of V. cholerae (Kwong et al., 2015).
Genomics and Haitian Cholera: A Case Study
Genomic tools potentiated an exploration of the mode of emergence and transmission of V. cholerae in the Haitian cholera epidemic that erupted in 2010 (Chin et al., 2011). This was the first whole genome study and showed strains of V. cholerae isolated during the Haitian cholera epidemic were related to South Asian seventh-pandemic O1 El Tor strains. It was hypothesized that these strains may have been introduced by human activities. Haitian strains were subsequently shown to be distinct from Latin American and East African strains of V. cholerae (Chin et al., 2011). The hypothesis of human transmission of a clonal strain from the South Asian region was supported by other investigators (Ali et al., 2011; Hendriksen et al., 2011; Reimer et al., 2011). Whole genome sequence typing (WGST) of Haitian and Nepalese strains indicated that they were related. Hendriksen et al. (2011) concluded that the 2010 cholera epidemic was initiated by Nepalese peacekeepers. However, based on whole genomic phylogeny and core genome SNPs, Reimer et al. (2011) concluded that the Haitian strain of V. cholerae O1 was genetically related to Indian and Cameroon strains. Chin et al. (2011) further concluded that Haitian cholera isolates could have originated from South Asia, not Nepal.
Hasan et al. (2012) showed both Haitian V. cholerae O1 and V. cholerae non-O1/O139 isolates had been isolated from Haitian cholera patient samples, based on WGS. Furthermore, they found non-O1/O139 strains were the sole pathogen in 21% of clinical specimens that had been analyzed (Hasan et al., 2012). They suggested autochthonous V. cholerae non-O1/O139 may serve as a reservoir for horizontally transmitted genes, as well as being pathogenic as has been reported elsewhere (Haley et al., 2012). In addition, a hydro-climatological study reinforced the concept of environmental factors playing a role in the intensity of the Haitian cholera outbreak (Jutla et al., 2013). The whole genomic study undertaken by Katz et al. (2013) spurred further debate concerning Haitian cholera. They compared 23 Haitian V. cholerae-O1 genomes with 108 available V. cholerae genomes isolated from geographically distinct sources and concluded the Haitian isolates were nearly identical to Nepalese strains, consistent with the previous conclusion of Hendriksen et al. (2011). However, in contradiction to this conclusion, they also showed the Haiti isolates to be clearly distinguishable from isolates circulating elsewhere in the world. Katz et al. (2013) concluded the Haitian epidemic V. cholerae isolates were untransformable, with insertion of horizontally transmitted DNA was not detected. They did not examine V. cholerae non-O1 isolated in Haiti. However, the concept of a single source introduction of Haitian cholera from Nepal (Grad and Waldor, 2013) remains to be proven as the sole cause of all cholera and cholera-like diarrheal disease in Haiti in 2010.
Thus, even though whole genome analyses provided evidence for a major role of V. cholerae O1 in the Haiti epidemic, the issue is not settled. Mutreja et al. (2011) pointed to the cradle of seventh pandemic cholera by studying 136 WGSs of V. cholerae seventh pandemic El Tor isolates collected over the past 40 years, including 18 previously published genomes of V. cholerae El Tor and classical biotypes. They traced three independent overlapping waves of cholera descending from a 1950s ancestor isolated in the Bay of Bengal region and also suggested transcontinental outbreaks of cholera that appear to have been caused by genetically similar V. cholerae. This can be interpreted equally well, as evidence of the ubiquitous nature of this bacterium that has been shown to be autochthonous to the aquatic environment (Colwell, 1996).
Conclusion
Technological advances, including automation of genomic fingerprinting and next generation sequencing have significantly improved cholera epidemiology. High-throughput sequencing has triggered output of an enormous amount of data. Bioinformatic tools are being developed that help to advance the molecular biology revolution. However, no single fingerprinting method is sufficient, as each method has its advantages and limitations. Nevertheless, a combination of methods can provide robust information and ensure resolution of epidemiological controversies.
Phenotypic fingerprinting continues to be useful for initial isolation, identification, and classification of V. cholerae. Molecular fingerprinting, i.e., VNTR and MLVA, can be useful in combination with WGS to achieve more precise discrimination in outbreaks caused by V. cholerae (Hasan et al., 2012) and become complementary to whole genome analyses. Although WGS promises to deliver highest resolution for a genomic-based epidemiology, differential annotation of whole genome data has generated intense debate exemplified by cholera in Haiti in 2010. Inter-laboratory comparative studies of WGS data no doubt will prove useful in reconciling the issues. A common platform for sharing whole genomic data is, therefore, strongly recommended. The newer generation of sequencing technologies has the potential to generate complete genomes in increasing frequency to allow precise genomic typing in silico, which will be complementary to the conventional typing strategies. Although cost of long read third generation sequencing is relatively high now, the cost is continuing to reduce to the point where it will soon be uniformly affordable. For now, it is concluded that combinations of phenotypic and molecular fingerprinting and WGS offer strategies for improved tracking, monitoring, and treating cholera.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
International Center for Diarrheal Disease Research, Bangladesh is thankful to the Governments of Australia, Bangladesh, Canada, Sweden, and the UK for providing core/unrestricted supports.
Footnotes
References
Alam, M., Islam, M. T., Rashed, S. M., Johura, F. T., Bhuiyan, N. A., Delgado, G., et al. (2012). Vibrio cholerae classical biotype strains reveal distinct signatures in Mexico. J. Clin. Microbiol. 50, 2212–2216. doi: 10.1128/JCM.00189-12
Alam, M., Rashed, S. M., Mannan, S. B., Islam, T., Lizarraga-Partida, M. L., Delgado, G., et al. (2014). Occurrence in Mexico, 1998-2008, of Vibrio cholerae CTX+ El Tor carrying an additional truncated CTX prophage. Proc. Natl. Acad. Sci. U.S.A. 111, 9917–9922. doi: 10.1073/pnas.1323408111
Aldová, E., Láznicková, K., Stěpánková, E., and Lietava, J. (1968). Isolation of nonagglutinable vibrios from an enteritis outbreak in Czechoslovakia. J. Infect. Dis. 118, 25–31. doi: 10.1093/infdis/118.1.25
Ali, A., Chen, Y., Johnson, J. A., Redden, E., Mayette, Y., Rashid, M. H., et al. (2011). Recent clonal origin of cholera in Haiti. Emerg. Infect. Dis. 17, 699–701. doi: 10.3201/eid1704.101973
Ansaruzzaman, M., Bhuiyan, N. A., Nair, B. G., Sack, D. A., Lucas, M., Deen, J. L., et al. (2004). Cholera in Mozambique, variant of Vibrio cholerae. Emerg. Infect. Dis. 10, 2057–2059. doi: 10.3201/eid1011.040682
Arbeit, R. D., Arthur, M., Dunn, R., Kim, C., Selander, R. K., and Goldstein, R. (1990). Resolution of recent evolutionary divergence among Escherichia coli from related lineages: the application of pulsed field electrophoresis to molecular epidemiology. J. Infect. Dis. 161, 230–235. doi: 10.1093/infdis/161.2.230
Banerjee, R., Das, B., Balakrish Nair, G., and Basak, S. (2014). Dynamics in genome evolution of Vibrio cholerae. Infect. Genet. Evol. 23, 32–41. doi: 10.1016/j.meegid.2014.01.006
Barrett, T. J., and Blake, P. A. (1981). Epidemiological usefulness of changes in hemolytic activity of Vibrio cholerae biotype El Tor during the seventh pandemic. J. Clin. Microbiol. 13, 126–129.
Barua, D. (1972). The global epidemiology of cholera in recent years. Proc. R. Soc. Med. 65, 423–428.
Barua, D. (1992). “History of cholera,” in Cholera, eds D. Barua and W. B. III Greenough (New York, NY: Plenum Publishing), 1–35.
Bashir, A., Klammer, A. A., Robins, W. P., Chin, C. S., Webster, D., Paxinos, E., et al. (2012). A hybrid approach for the automated finishing of bacterial genomes. Nat. Biotechnol. 30, 701–707. doi: 10.1038/nbt.2288
Basu, A., Garg, P., Datta, S., Chakraborty, S., Bhattacharya, T., Khan, A., et al. (2000). Vibrio cholerae O139 in Calcutta, 1992-1998: incidence, antibiograms, and genotypes. Emerg. Infect. Dis. 6, 139–147. doi: 10.3201/eid0602.000206
Basu, S., and Mukerjee, S. (1968). Bacteriophage typing of Vibrio eltor. Experientia 24, 299–300. doi: 10.1007/BF0215283
Bentivoglio, M., and Pacini, P. (1995). Filippo Pacini: a determined observer. Brain Res. Bull. 38, 161–165. doi: 10.1016/0361-9230(95)00083-Q
Bhattacharya, M. K., Bhattacharya, S. K., Garg, S., Saha, P. K., Dutta, D., Nair, G. B., et al. (1993). Outbreak of Vibrio cholerae non-O1 in India and Bangladesh. Lancet 341, 1346–1347. doi: 10.1016/0140-6736(93)90855-B
Bhowmick, T. S., Das, M., and Sarkar, B. L. (2010). Evaluation of VcA VNTR as a strain-typing and phylogeny study method of Vibrio cholerae strains. Epidemiol. Infect. 138, 1637–1649. doi: 10.1017/S0950268810000427
Bhowmick, T. S., Das, M., and Sarkar, B. L. (2011). Genotypic characterization of Vibrio cholerae isolates using several DNA fingerprint techniques. Future Microbiol. 6, 29–42. doi: 10.2217/fmb.10.159
Bik, E. M., Bunschoten, A. E., Gouw, R. D., and Mooi, F. R. (1995). Genesis of the novel epidemic Vibrio cholerae O139 strain: evidence for horizontal transfer of genes involved in polysaccharide synthesis. EMBO J. 14, 209–216.
Blake, P. A. (1994). “Historical perspectives on pandemic cholera,” in Vibrio cholerae and Cholera: Molecular to Global Perspectives, eds K. Wachsmuth, P. A. Blake, and Ø. Olsvik (Washington, DC: American Society for Microbiology), 293–295.
Bouchet, V., Huot, H., and Goldstein, R. (2008). Molecular genetic basis of ribotyping. Clin. Microbiol. Rev. 21, 262–273. doi: 10.1128/CMR.00026-07
Byun, R., Elbourne, L. D., Lan, R., and Reeves, P. R. (1999). Evolutionary relationships of pathogenic clones of Vibrio cholerae by sequence analysis of four housekeeping genes. Infect. Immun. 67, 1116–1124.
Cameron, D. N., Khambaty, F. M., Wachsmuth, I. K., Tauxe, R. V., and Barrett, T. J. (1994). Molecular characterization of Vibrio cholerae O1 strains by pulsed-field gel electrophoresis. J. Clin. Microbiol. 32, 1685–1690.
Chakrabarti, A. K., Ghosh, A. N., Nair, G. B., Niyogi, S. K., Bhattacharya, S. K., and Sarkar, B. L. (2000). Development and evaluation of a phage typing scheme for Vibrio cholerae O139. J. Clin. Microbiol. 38, 44–49.
Chapman, C., Henry, M., Bishop-Lilly, K. A., Awosika, J., Briska, A., Ptashkin, R. N., et al. (2015). Scanning the landscape of genome architecture of non-O1 and non-O139 Vibrio cholerae by whole genome mapping reveals extensive population genetic diversity. PLoS ONE 10:e0120311. doi: 10.1371/journal.pone.0120311
Chattopadhyay, D. J., Sarkar, B. L., Ansari, M. Q., Chakrabarti, B. K., Roy, M. K., Ghosh, A. N., et al. (1993). New phage typing scheme for Vibrio cholerae O1 biotype El Tor strains. J. Clin. Microbiol. 31, 1579–1585.
Chin, C. S., Sorenson, J., Harris, J. B., Robins, W. P., Charles, R. C., Jean-Charles, R. R., et al. (2011). The origin of the Haitian cholera outbreak strain. N. Engl. J. Med. 364, 33–42. doi: 10.1056/NEJMoa1012928
Chun, J., Grim, C. J., Hasan, N. A., Lee, J. H., Choi, S. Y., Haley, B. J., et al. (2009). Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 106, 15442–15447. doi: 10.1073/pnas.0907787106
Colwell, R. R. (1996). Global climate and infectious disease: the cholera paradigm. Science 274, 2025–2031. doi: 10.1126/science.274.5295.2025
Comstock, L. E., Johnson, J. A., Michalski, J. M., Morris, J. G. Jr., and Kaper, J. B. (1996). Cloning and sequence of a region encoding a surface polysaccharide of Vibrio cholerae O139 and characterization of the insertion site in the chromosome of Vibrio cholerae O1. Mol. Microbiol. 19, 815–826. doi: 10.1046/j.1365-2958.1996.407928.x
Cooper, K. L., Luey, C. K., Bird, M., Terajima, J., Nair, G. B., Kam, K. M., et al. (2006). Development and validation of a PulseNet standardized pulsed-field gel electrophoresis protocol for subtyping of Vibrio cholerae. Foodborne Pathog. Dis. 3, 51–58. doi: 10.1089/fpd.2006.3.51
Danin-Poleg, Y., Cohen, L. A., Gancz, H., Broza, Y. Y., Goldshmidt, H., Malul, E., et al. (2007). Vibrio cholerae strain typing and phylogeny study based on simple sequence repeats. J. Clin. Microbiol. 45, 736–746. doi: 10.1128/JCM.01895-06
Desmarchelier, P. M., Wong, F. Y., and Mallard, K. (1995). An epidemiological study of Vibrio cholerae O1 in the Australian environment based on rRNA gene polymorphisms. Epidemiol. Infect. 115, 435–446. doi: 10.1017/S0950268800058593
Dziejman, M., Balon, E., Boyd, D., Fraser, C. M., Heidelberg, J. F., and Mekalanos, J. J. (2002). Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc. Natl. Acad. Sci. U.S.A. 99, 1556–1561. doi: 10.1073/pnas.042667999
Farfán, M., Miñana-Galbis, D., Fusté, M. C., and Lorén, J. G. (2002). Allelic diversity and population structure in Vibrio cholerae O139 Bengal based on nucleotide sequence analysis. J. Bacteriol. 184, 1304–1313. doi: 10.1128/JB.184.5.1304-1313.2002
Faruque, S. M. (2014). Role of phages in the epidemiology of cholera. Curr. Top. Microbiol. Immunol. 379, 165–180. doi: 10.1007/82_2013_358
Faruque, S. M., Ahmed, K. M., Abdul Alim, A. R., Qadri, F., Siddique, A. K., and Albert, M. J. (1997). Emergence of a new clone of toxigenic Vibrio cholerae O1 biotype El Tor displacing V. cholerae O139 Bengal in Bangladesh. J. Clin. Microbiol. 35, 624–630.
Faruque, S. M., Albert, M. J., and Mekalanos, J. J. (1998). Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 62, 1301–1314.
Faruque, S. M., Chowdhury, N., Kamruzzaman, M., Dziejman, M., Rahman, M. H., Sack, D. A., et al. (2004). Genetic diversity and virulence potential of environmental Vibrio cholerae population in a cholera-endemic area. Proc. Natl. Acad. Sci. U.S.A. 101, 2123–2128. doi: 10.1073/pnas.0308485100
Faruque, S. M., and Mekalanos, J. J. (2012). Phage-bacterial interactions in the evolution of toxigenic Vibrio cholerae. Virulence 3, 556–565. doi: 10.4161/viru.22351
Faruque, S. M., Saha, M. N., Asadulghani Bag, P. K., Bhadra, R. K., Bhattacharya, S. K., et al. (2000). Genomic diversity among Vibrio cholerae O139 strains isolated in Bangladesh and India between 1992 and 1998. FEMS Microbiol. Lett. 184, 279–284. doi: 10.1111/j.1574-6968.2000.tb09027.x
Felsenfeld, O. (1966). A review of recent trends in cholera research and control. With an annex on the isolation and identification of cholera vibrios. Bull. World Health Organ. 34, 161–195.
Finkelstein, R. A., and Mukerjee, S. (1963). Hemagglutination a rapid method for differentiating Vibrio cholerae and El Tor Vibrios. Exp. Biol. Med. 112, 355–359. doi: 10.3181/00379727-112-28043
Gardner, A. D., and Venkatraman, K. V. (1935). The antigens of the cholera group of vibrios. J. Hyg. 35, 262–282. doi: 10.1017/S0022172400032265
Ghosh, R., Nair, G. B., Tang, L., Morris, J. G., Sharma, N. C., Ballal, M., et al. (2008). Epidemiological study of Vibrio cholerae using variable number of tandem repeats. FEMS Microbiol. Lett. 288, 196–201. doi: 10.1111/j.1574-6968.2008.01352.x
Goering, R. V. (2010). Pulsed field gel electrophoresis: a review of application and interpretation in the molecular epidemiology of infectious disease. Infect. Genet. Evol. 10, 866–875. doi: 10.1016/j.meegid.2010.07.023
Gordillo, M. E., Singh, K. V., Baker, C. J., and Murray, B. E. (1993). Typing of group B streptococci: comparison of pulsed-field gel electrophoresis and conventional electrophoresis. J. Clin. Microbiol. 31, 1430–1434.
Grad, Y. H., and Waldor, M. K. (2013). Deciphering the origins and tracking the evolution of cholera epidemics with whole-genome-based molecular epidemiology. Mbio 4, e00670-13. doi: 10.1128/mBio.00670-13
Greig, E. D. W. (1916). The serological investigation and classification of cholera-like vibrios isolated from water in Calcutta. Indian J. Med. Res. 3, 628–637.
Grim, C. J., Hasan, N. A., Taviani, E., Haley, B., Chun, J., Brettin, T. S., et al. (2010). Genome sequence of hybrid Vibrio cholerae O1 MJ-1236, B-33, and CIRS101 and comparative genomics with V. cholerae. J. Bacteriol. 192, 3524–3533. doi: 10.1128/JB.00040-10
Grimont, F., and Grimont, P. A. (1986). Ribosomal ribonucleic acid gene restriction patterns as potential taxonomic tools. Ann. Inst. Pasteur Microbiol. 137B, 165–175. doi: 10.1016/S0769-2609(86)80105-3
Haley, B. J., Chen, A., Grim, C. J., Clark, P., Diaz, C. M., Taviani, E., et al. (2012). Vibrio cholerae in an historically cholera-free country. Environ. Microbiol. Rep. 4, 381–389. doi: 10.1111/j.1758-2229.2012.00332.x
Haley, B. J., Grim, C. J., Hasan, N. A., Choi, S. Y., Chun, J., Brettin, T. S., et al. (2010). Comparative genomic analysis reveals evidence of two novel Vibrio species closely related to V. cholerae. BMC Microbiol. 10:154. doi: 10.1186/1471-2180-10-154
Han, G. K., and Khie, T. S. (1963). A new method for the differentiation of Vibrio comma and Vibrio El Tor. Am. J. Hyg. 77, 184–186.
Hasan, N. A., Choi, S. Y., Eppinger, M., Clark, P. W., Chen, A., Alam, M., et al. (2012). Genomic diversity of 2010 Haitian cholera outbreak strains. Proc. Natl. Acad. Sci. U.S.A. 109, E2010–E2017. doi: 10.1073/pnas.1207359109
Hasan, N. A., Grim, C. J., Haley, B. J., Chun, J., Alam, M., Taviani, E., et al. (2010). Comparative genomics of clinical and environmental Vibrio mimicus. Proc. Natl. Acad. Sci. U.S.A. 107, 21134–21139. doi: 10.1073/pnas.1013825107
Heidelberg, J. F., Eisen, J. A., Nelson, W. C., Clayton, R. A., Gwinn, M. L., Dodson, R. J., et al. (2000). DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406, 477–484. doi: 10.1038/35020000
Hendriksen, R. S., Price, L. B., Schupp, J. M., Gillece, J. D., Kaas, R. S., Engelthaler, D. M., et al. (2011). Population genetics of Vibrio cholerae from Nepal in 2010: evidence on the origin of the Haitian outbreak. Mbio 2, e00157-11. doi: 10.1128/mBio.00157-11
Jiang, S. C., Matte, M., Matte, G., Huq, A., and Colwell, R. R. (2000). Genetic diversity of clinical and environmental isolates of Vibrio cholerae determined by amplified fragment length polymorphism fingerprinting. Appl. Environ. Microbiol. 66, 148–153. doi: 10.1128/AEM.66.1.140-147.2000
Jutla, A., Whitcombe, E., Hasan, N., Haley, B., Akanda, A., Huq, A., et al. (2013). Environmental factors influencing epidemic cholera. American J. Trop. Med. Hyg. 89, 597–607. doi: 10.4269/ajtmh.12-0721
Kamal, A. M. (1971). Outbreak of gastroenteritis by nonagglutinable (NAG) vibrios in the Republic of Sudan. J. Egypt. Publ. Health Assoc. 46, 125–173.
Karaolis, D. K., Lan, R., and Reeves, P. R. (1994). Molecular evolution of the seventh-pandemic clone of Vibrio cholerae and its relationship to other pandemic and epidemic V. cholerae isolates. J. Bacteriol. 176, 6199–6206.
Katz, L. S., Petkau, A., Beaulaurier, J., Tyler, S., Antonova, E. S., Turnsek, M. A., et al. (2013). Evolutionary dynamics of Vibrio cholerae O1 following a single-source introduction to Haiti. mBio 4:e00398-13. doi: 10.1128/mBio.00398-13
Kim, M., Oh, H. S., Park, S. C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64(Pt 2), 346–351. doi: 10.1099/ijs.0.059774-0
Kirchberger, P. C., Turnsek, M., Hunt, D. E., Haley, B. J., Colwell, R. R., Polz, M. F., et al. (2014). Vibrio metoecus sp. nov., a close relative of Vibrio cholerae isolated from coastal brackish ponds and clinical specimens. Int. J. Syst. Evol. Microbiol. 64(Pt 9), 3208–3214. doi: 10.1099/ijs.0.060145-0
Koch, R. (1884). An address on cholera and its Bacillus. Br. Med. J. 2, 403–407. doi: 10.1136/bmj.2.1235.403
Konstantinidis, K. T., Ramette, A., and Tiedje, J. M. (2006). The bacterial species definition in the genomic era. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 361, 1929–1940. doi: 10.1098/rstb.2006.1920
Kotetishvili, M., Stine, O. C., Chen, Y., Kreger, A., Sulakvelidze, A., Sozhamannan, S., et al. (2003). Multilocus sequence typing has better discriminatory ability for typing Vibrio cholerae than does pulsed-field gel electrophoresis and provides a measure of phylogenetic relatedness. J. Clin. Microbiol. 41, 2191–2196. doi: 10.1128/JCM.41.5.2191-2196.2003
Kwong, J. C., McCallum, N., Sintchenko, V., and Howden, B. P. (2015). Whole genome sequencing in clinical and public health microbiology. Pathology 47, 199–210. doi: 10.1097/PAT.0000000000000235
Lam, C., Octavia, S., Reeves, P. R., and Lan, R. (2012). Multi-locus variable number tandem repeat analysis of 7th pandemic Vibrio cholerae. BMC Microbiol. 12:82. doi: 10.1186/1471-2180-12-82
Lan, R., and Reeves, P. R. (1998). Recombination between rRNA operons created most of the ribotype variation observed in the seventh pandemic clone of Vibrio cholerae. Microbiology (Read. Engl.) 144(Pt 5), 1213–1221. doi: 10.1099/00221287-144-5-1213
Lan, R., and Reeves, P. R. (2002). Pandemic spread of cholera: genetic diversity and relationships within the seventh pandemic clone of Vibrio cholerae determined by amplified fragment length polymorphism. J. Clin. Microbiol. 40, 172–181. doi: 10.1128/JCM.40.1.172-181.2002
Land, M., Hauser, L., Jun, S. R., Nookaew, I., Leuze, M. R., Ahn, T. H., et al. (2015). Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genomics 15, 141–161. doi: 10.1007/s10142-015-0433-4
Land, M. L., Hyatt, D., Jun, S. R., Kora, G. H., Hauser, L. J., Lukjancenko, O., et al. (2014). Quality scores for 32,000 genomes. Stand. Genomic Sci. 9, 20. doi: 10.1186/1944-3277-9-20
Leal, N. C., Sobreira, M., Leal-Balbino, T. C., de Almeida A. M., de Silva M. J., Mello, D. M., et al. (2004). Evaluation of a RAPD-based typing scheme in a molecular epidemiology study of Vibrio cholerae O1, Brazil. J. Appl. Microbiol. 96, 447–454. doi: 10.1111/j.1365-2672.2004.02090.x
Lee, J. H., Han, K. H., Choi, S. Y., Lucas, M. E., Mondlane, C., Ansaruzzaman, M., et al. (2006). Multilocus sequence typing (MLST) analysis of Vibrio cholerae O1 El Tor isolates from Mozambique that harbour the classical CTX prophage. J. Med. Microbiol. 55(Pt 2), 165–170. doi: 10.1099/jmm.0.46287-0
Lesmana, M., Subekti, D., Tjaniadi, P., and Pazzaglia, G. (1994). Modified CAMP test for biogrouping Vibrio cholerae O1 strains and distinguishing them from strains of V. cholerae non-O1. J. Clin. Microbiol. 32, 235–237.
Mahalingam, S., Cheong, Y. M., Kan, S., Yassin, R. M., Vadivelu, J., and Pang, T. (1994). Molecular epidemiologic analysis of Vibrio cholerae O1 isolates by pulsed-field gel electrophoresis. J. Clin. Microbiol. 32, 2975–2979.
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Meunier, J. R., and Grimont, P. A. (1993). Factors affecting reproducibility of random amplified polymorphic DNA fingerprinting. Res. Microbiol. 144, 373–379. doi: 10.1016/0923-2508(93)90194-7
Mohamed, A. A., Oundo, J., Kariuki, S. M., Boga, H. I., Sharif, S. K., Akhwale, W., et al. (2012). Molecular epidemiology of geographically dispersed Vibrio cholerae, Kenya, January 2009-May 2010. Emerg. Infect. Dis. 18, 925–931. doi: 10.3201/eid1806.111774
Mooi, F. R., and Bik, E. M. (1997). The evolution of epidemic Vibrio cholerae strains. Trends Microbiol. 5, 161–165. doi: 10.1016/S0966-842X(96)10086-X
Mortimer, P., and Arnold, C. (2001). FAFLP: last word in microbial genotyping? J. Med. Microbiol. 50, 393–395. doi: 10.1099/0022-1317-50-5-393
Mukhopadhyay, A. K., Chakraborty, S., Takeda, Y., Nair, G. B., and Berg, D. E. (2001). Characterization of VPI pathogenicity island and CTXphi prophage in environmental strains of Vibrio cholerae. J. Bacteriol. 183, 4737–4746. doi: 10.1128/JB.183.16.4737-4746.2001
Murchan, S., Kaufmann, M. E., Deplano, A., de Ryck, R., Struelens, M., Zinn, C. E., et al. (2003). Harmonization of pulsed-field gel electrophoresis protocols for epidemiological typing of strains of methicillin-resistant Staphylococcus aureus: a single approach developed by consensus in 10 European laboratories and its application for tracing the spre. J. Clin. Microbiol. 41, 1574–1585. doi: 10.1128/JCM.41.4.1574-1585.2003
Mutreja, A., Kim, D. W., Thomson, N. R., Connor, T. R., Lee, J. H., Kariuki, S., et al. (2011). Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477, 462–465. doi: 10.1038/nature10392
Nair, G. B., Faruque, S. M., Bhuiyan, N. A., Kamruzzaman, M., Siddique, A. K., and Sack, D. A. (2002). New variants of Vibrio cholerae O1 biotype El Tor with attributes of the classical biotype from hospitalized patients with acute diarrhea in Bangladesh. J. Clin. Microbiol. 40, 3296–3299. doi: 10.1128/JCM.40.9.3296-3299.2002
Nair, G. B., Qadri, F., Holmgren, J., Svennerholm, A. M., Safa, A., Bhuiyan, N. A., et al. (2006). Cholera due to altered El Tor strains of Vibrio cholerae O1 in Bangladesh. J. Clin. Microbiol. 44, 4211–4213. doi: 10.1128/JCM.01304-06
Niedringhaus, T. P., Milanova, D., Kerby, M. B., Snyder, M. P., and Barron, A. E. (2011). Landscape of next-generation sequencing technologies. Anal. Chem. 83, 4327–4341. doi: 10.1021/ac2010857
Olsen, J. S., Aarskaug, T., Skogan, G., Fykse, E. M., Ellingsen, A. B., and Blatny, J. M. (2009). Evaluation of a highly discriminating multiplex multi-locus variable-number of tandem-repeats (MLVA) analysis for Vibrio cholerae. J. Microbiol. Methods 78, 271–285. doi: 10.1016/j.mimet.2009.06.011
Pollitzer, R., Swaroop, S., and Burrows, W. (1959). Cholera. Monogr. Ser. World Health Organ. 58, 1001–1019.
Popovic, T., Bopp, C., Olsvik, O., and Wachsmuth, K. (1993). Epidemiologic application of a standardized ribotype scheme for Vibrio cholerae O1. J. Clin. Microbiol. 31, 2474–2482.
Pourshafie, M., Grimont, F., Kohestani, S., and Grimont, P. A. (2002). A molecular and phenotypic study of Vibrio cholerae in Iran. J. Med. Microbiol. 51, 392–398. doi: 10.1099/0022-1317-51-5-392
Pourshafie, M. R., Grimont, F., Saifi, M., and Grimont, P. A. (2000). Molecular epidemiological study of Vibrio cholerae isolates from infected patients in Teheran, Iran. J. Med. Microbiol. 49, 1085–1090. doi: 10.1099/0022-1317-49-12-1085
Prévost, G., Pottecher, B., Dahlet, M., Bientz, M., Mantz, J. M., and Piémont, Y. (1991). Pulsed field gel electrophoresis as a new epidemiological tool for monitoring methicillin-resistant Staphylococcus aureus in an intensive care unit. J. Hosp. Infect. 17, 255–269. doi: 10.1016/0195-6701(91)90270-I
Rashed, S. M., Azman, A. S., Alam, M., Li, S., Sack, D. A., Morris, J. G. Jr., et al. (2014). Genetic variation of Vibrio cholerae during outbreaks, Bangladesh, 2010-2011. Emerg. Infect. Dis. 20, 54–60. doi: 10.3201/eid2001.130796
Raychoudhuri, A., Patra, T., Ghosh, K., Ramamurthy, T., Nandy, R. K., Takeda, Y., et al. (2009). Classical ctxB in Vibrio cholerae O1, Kolkata, India. Emerg. Infect. Dis. 15, 131–132. doi: 10.3201/eid1501.080543
Reeves, P. R., and Lan, R. (1998). Cholera in the 1990s. Br. Med. Bull. 54, 611–623. doi: 10.1093/oxfordjournals.bmb.a011714
Reimer, A. R., Van Domselaar, G., Stroika, S., Walker, M., Kent, H., Tarr, C., et al. (2011). Comparative genomics of Vibrio cholerae from Haiti, Asia, and Africa. Emerg. Infect. Dis. 17, 2113–2121. doi: 10.3201/eid1711.110794
Robins, W. P., and Mekalanos, J. J. (2014). Genomic science in understanding cholera outbreaks and evolution of Vibrio cholerae as a human pathogen. Curr. Top. Microbiol. Immunol. 379, 211–229. doi: 10.1007/82_2014_366
Sabat, A. J., Budimir, A., Nashev, D., Sá-Leão, R., van Dijl, J. M., Laurent, F., et al. (2013). Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill. 18, 20380.
Sack, D. A., Sack, R. B., Nair, G. B., and Siddique, A. K. (2004). Cholera. Lancet 363, 223–233. doi: 10.1016/S0140-6736(03)15328-7
Safa, A., Nair, G. B., and Kong, R. Y. C. (2010). Evolution of new variants of Vibrio cholerae O1. Trends Microbiol. 18, 46–54. doi: 10.1016/j.tim.2009.10.003
Sakazaki, R., and Shimada, T. (1977). Serovars of Vibrio cholerae identified during 1970-1975. Jpn. J. Med. Sci. Biol. 30, 279–282. doi: 10.7883/yoken1952.30.279
Schwartz, D. C., Li, X., Hernandez, L. I., Ramnarain, S. P., Huff, E. J., and Wang, Y. K. (1993). Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science 262, 110–114. doi: 10.1126/science.8211116
Sciortino, C. V., Johnson, J. A., and Hamad, A. (1996). Vitek system antimicrobial susceptibility testing of O1, O139, and non-O1 Vibrio cholerae. J. Clin. Microbiol. 34, 897–900.
Scott, T. M., Rose, J. B., Jenkins, T. M., Farrah, S. R., and Lukasik, J. (2002). Microbial source tracking: current methodology and future directions. Appl. Environ. Microbiol. 68, 5796–5803. doi: 10.1128/AEM.68.12.5796-5803.2002
Seas, C., Miranda, J., Gil, A. I., Leon-Barua, R., Patz, J., Huq, A., et al. (2000). New insights on the emergence of cholera in Latin America during 1991: the Peruvian experience. Am. J. Trop. Med. Hyg. 62, 513–517.
Siddique, A. K., Akram, K., Zaman, K., Mutsuddy, P., Eusof, A., and Sack, R. B. (1996). Vibrio cholerae O139: how great is the threat of a pandemic? Trop. Med. Int. Health 1, 393–398. doi: 10.1046/j.1365-3156.1996.d01-54.x
Singh, D. V., and Mohapatra, H. (2008). Application of DNA-based methods in typing Vibrio cholerae strains. Future Microbiol. 3, 87–96. doi: 10.2217/17460913.3.1.87
Snow, J. (1855). On the Mode of Communication of Cholera (Google eBook), John Churchill. Available at: http://books.google.com/books?hl=en&lr=&id=-N0_AAAAcAAJ&pgis=1 [Accessed April 30, 2014].
Stine, O. C., Alam, M., Tang, L., Nair, G. B., Siddique, A. K., Faruque, S. M., et al. (2008). Seasonal cholera from multiple small outbreaks, rural Bangladesh. Emerg. Infect. Dis. 14, 831–833. doi: 10.3201/eid1405.071116
Stine, O. C., and Morris, J. G. (2014). Circulation and transmission of clones of Vibrio cholerae during cholera outbreaks. Curr. Top. Microbiol. Immunol. 379, 181–193. doi: 10.1007/82_2013_360
Struelens, M. J. (1996). Consensus guidelines for appropriate use and evaluation of microbial epidemiologic typing systems. Clin. Microbiol. Infect. 2, 2–11. doi: 10.1111/j.1469-0691.1996.tb00193.x
Swaminathan, B., Barrett, T. J., Hunter, S. B., Tauxe, R. V., and CDC PulseNet Task Force (2001). PulseNet: the molecular subtyping network for foodborne bacterial disease surveillance, United States. Emerg. Infect. Dis. 7, 382–389. doi: 10.3201/eid0703.017303
Takeya, K., Otohuji, T., and Tokiwa, H. (1981). FK phage for differentiating the classical and El T or groups of Vibrio cholerae. J. Clin. Microbiol. 14, 222–224.
Teh, C. S. J., Chua, K. H., and Thong, K. L. (2010). Multiple-locus variable-number tandem repeat analysis of Vibrio cholerae in comparison with pulsed field gel electrophoresis and virulotyping. J. Biomed. Biotechnol. 2010, 817190. doi: 10.1155/2010/817190
Teh, C. S. J., Chua, K. H., and Thong, K. L. (2011). Genetic variation analysis of Vibrio cholerae using multilocus sequencing typing and multi-virulence locus sequencing typing. Infect. Genet. Evol. 11, 1121–1128. doi: 10.1016/j.meegid.2011.04.005
Tenover, F. C., Arbeit, R. D., and Goering, R. V. (1997). How to select and interpret molecular strain typing methods for epidemiological studies of bacterial infections: a review for healthcare epidemiologists. molecular typing working group of the society for healthcare epidemiology of America. Infect. Control Hosp. Epidemiol. 18, 426–439. doi: 10.2307/30141252
Tenover, F. C., Arbeit, R. D., Goering, R. V., Mickelsen, P. A., Murray, B. E., Persing, D. H., et al. (1995). Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J. Clin. Microbiol. 33, 2233–2239.
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Thompson, F. L., Iida, T., and Swings, J. (2004). Biodiversity of vibrios. Microbiol. Mol. Biol. Rev. 68, 403–431. doi: 10.1128/MMBR.68.3.403-431.2004
Trucksis, M., Michalski, J., Deng, Y. K., and Kaper, J. B. (1998). The Vibrio cholerae genome contains two unique circular chromosomes. Proc. Natl. Acad. Sci. U.S.A. 95, 14464–14469. doi: 10.1073/pnas.95.24.14464
van Belkum, A., Tassios, P. T., Dijkshoorn, L., Haeggman, S., Cookson, B., Fry, N. K., et al. (2007). Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Clin. Microbiol. Infect. 13(Suppl. 3), 1–46.
Vodop’ianov, S. O., Oleinikov, I. P., Goncharov, E. K., Duvanova, O. V., Mishan’kin, M. B., Suchkov, I., et al. (2002). Variable tandem repeat VcA of Vibrio cholerae. Mol. Biol. 36, 1074–1079.
Vos, P., Hogers, R., Bleeker, M., Reijans, M., van de Lee, T., Hornes, M., et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 23, 4407–4414. doi: 10.1093/nar/23.21.4407
Waldor, M. K., Tschäpe, H., and Mekalanos, J. J. (1996). A new type of conjugative transposon encodes resistance to sulfamethoxazole, trimethoprim, and streptomycin in Vibrio cholerae O139. J. Bacteriol. 178, 4157–4165.
Welsh, J., and McClelland, M. (1990). Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res. 18, 7213–7218. doi: 10.1093/nar/18.24.7213
Williams, J. G., Kubelik, A. R., Livak, K. J., Rafalski, J. A., and Tingey, S. V. (1990). DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 18, 6531–6535. doi: 10.1093/nar/18.22.6531
Yamaichi, Y., Iida, T., Park, K. S., Yamamoto, K., and Honda, T. (1999). Physical and genetic map of the genome of Vibrio parahaemolyticus: presence of two chromosomes in Vibrio species. Mol. Microbiol. 31, 1513–1521. doi: 10.1046/j.1365-2958.1999.01296.x
Keywords: cholera, V. cholerae, molecular fingerprinting, PFGE, MLST, MLVA, whole genome sequencing
Citation: Rahaman MH, Islam T, Colwell RR and Alam M (2015) Molecular tools in understanding the evolution of Vibrio cholerae. Front. Microbiol. 6:1040. doi: 10.3389/fmicb.2015.01040
Received: 23 July 2015; Accepted: 13 September 2015;
Published: 06 October 2015.
Edited by:
Frank T. Robb, University of Maryland, USACopyright © 2015 Rahaman, Islam, Colwell and Alam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Munirul Alam, Enteric and Food Microbiology Lab, Center for Communicable Diseases, International Center for Diarrheal Disease Research, 68 Shahid Tajuddin Ahmed Sarani, Mohakhali, Dhaka, Bangladesh, munirul@icddrb.org