
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 11 September 2015
Sec. Food Microbiology
Volume 6 - 2015 | https://doi.org/10.3389/fmicb.2015.00882
 Sophie Roussel*
Sophie Roussel* Benjamin Felix
Benjamin Felix Noémie Vingadassalon
Noémie Vingadassalon Joël Grout
Joël Grout Jacques-Antoine Hennekinne
Jacques-Antoine Hennekinne Laurent Guillier
Laurent Guillier Anne Brisabois
Anne Brisabois Fréderic Auvray
Fréderic Auvray
Staphylococcal food poisoning outbreaks (SFPOs) are frequently reported in France. However, most of them remain unconfirmed, highlighting a need for a better characterization of isolated strains. Here we analyzed the genetic diversity of 112 Staphylococcus aureus strains isolated from 76 distinct SFPOs that occurred in France over the last 30 years. We used a recently developed multiple-locus variable-number tandem-repeat analysis (MLVA) protocol and compared this method with pulsed field gel electrophoresis (PFGE), spa-typing and carriage of genes (se genes) coding for 11 staphylococcal enterotoxins (i.e., SEA, SEB, SEC, SED, SEE, SEG, SEH, SEI, SEJ, SEP, SER). The strains known to have an epidemiological association with one another had identical MLVA types, PFGE profiles, spa-types or se gene carriage. MLVA, PFGE and spa-typing divided 103 epidemiologically unrelated strains into 84, 80, and 50 types respectively demonstrating the high genetic diversity of S. aureus strains involved in SFPOs. Each MLVA type shared by more than one strain corresponded to a single spa-type except for one MLVA type represented by four strains that showed two different-but closely related-spa-types. The 87 enterotoxigenic strains were distributed across 68 distinct MLVA types that correlated all with se gene carriage except for four MLVA types. The most frequent se gene detected was sea, followed by seg and sei and the most frequently associated se genes were sea-seh and sea-sed-sej-ser. The discriminatory ability of MLVA was similar to that of PFGE and higher than that of spa-typing. This MLVA protocol was found to be compatible with high throughput analysis, and was also faster and less labor-intensive than PFGE. MLVA holds promise as a suitable method for investigating SFPOs and tracking the source of contamination in food processing facilities in real time.
Staphylococcal food poisoning is one of the most common food-borne diseases worldwide (Yu et al., 2007; Kadariya et al., 2014). It results from the ingestion of SEs preformed in food and produced by enterotoxigenic strains of CPS (Argudin et al., 2010). SEs are frequent causes of food-borne outbreaks in Europe (EFSA-ECDC, 2013). Two types of SFPOs can be differentiated. Outbreaks for which the evidence implicating a particular food vehicle is strong, based on the assessment of all available data, are referred to as “strong-evidence SFPO,” whereas outbreaks for which no particular food vehicle is suspected or where the evidence implicating a particular food vehicle is weak are referred to as “weak-evidence SFPO” (EFSA, 2011).
Among the seven described species belonging to the CPS group, Staphylococcus aureus ssp. aureus is the main causative agent of SFPOs. To date, 21 SEs have been described: SEA to SElV all possess superantigenic activity whereas only a subset of SEs (i.e., SEA to SEI, SER, SES, and SET) are emetic (Ono et al., 2008). Out of the 21 SEs, 11 (i.e., SEA, SEB, SEC, SED, SEE, SEG, SEH, SEI, SEJ, SEP, SER) are suspected to cause SFPOs (Hennekinne et al., 2011).
Few data is available on the genetic diversity of the strains isolated from SFPOs. Among the molecular methods available, pulsed field gel electrophoresis (PFGE) and Staphylococcus protein A gene (spa) typing have been extremely helpful in short-term investigations and identification of SFPOs (Chiou et al., 2000; Shimizu et al., 2000; Wei and Chiou, 2002; Strommenger et al., 2006; Dyer et al., 2007; Hallin et al., 2007; Kerouanton et al., 2007; Kellermann et al., 2008; Ostyn et al., 2010; Wattinger et al., 2012; Chiang et al., 2014). Although PFGE is highly discriminatory, it remains a time-consuming and labor intensive method. It also requires highly skilled operators and there are no standardized reagents. Moreover, profile interpretation requires several subjective decisions, increasing the variability of the profiles and leading to possible uncertainty about profiles relatedness (Cookson et al., 1996; Murchan et al., 2003).The advantages of spa-typing are its excellent inter-laboratory reproducibility, the portability of the data and its flexible analysis throughput (Koreen et al., 2004; Aires-de-Sousa et al., 2006; Cookson et al., 2007). However, this method is less discriminatory than PFGE for the characterization of food isolates (Babouee et al., 2011).
Identification of SE-encoding genes (se genes) in isolated strains represents a complementary approach for investigating SFPOs. All known se genes are located on mobile genetic elements, including the νSaβ genomic island which contains the enterotoxin gene cluster known as egc (carrying seg and sei), S. aureus pathogenicity islands (SaPIs; carrying seb and sec), prophages (carrying sea, see, and sep), and plasmids (carrying sed, sej, and ser; Argudin et al., 2012). Many PCR assays have been developed to detect se genes in S. aureus strains isolated from contaminated foods (Martin et al., 2004; Morandi et al., 2007; Kadariya et al., 2014). Screening for se genes in the strains involved in SFPOs is useful in two ways. First, the identified se gene may correspond to the type of SE detected in food, thus confirming the result obtained by an immuno-enzymatic method (Ostyn et al., 2010). Second, the se gene identified may correspond to a type of SE known to be emetic, but for which no detection method is available, suggesting the involvement of the corresponding toxin in the outbreak (Kerouanton et al., 2007).
The ANSES Laboratory for Food Safety is the French NRL and the EURL for CPS, including S. aureus and their toxins. One of the EURL activities is to develop and evaluate new molecular methods for bacterial typing and to transfer them to the European NRL network. Simultaneously to the screening for enterotoxins in suspected food, staphylococcal isolates are characterized using (i) spa-typing (ii) PFGE and (iii) a multiplex PCR assay for the detection of se genes coding for 11 SEs. Given the limitations described above, there is still a need for an alternative typing method that would be as discriminatory as PFGE and as portable as spa-typing, at a low cost.
Multiple-locus variable-number tandem-repeat analysis (MLVA) is based on PCR amplification and size analysis of DNA regions containing variable numbers of tandem repeats (VNTRs). MLVA assays offer fast typing of various bacteria, with high resolution (Lindstedt et al., 2013). An assay based on eight VNTR loci was applied on a panel of (i) 1781 S. aureus strains isolated from animal and patients (Schouls et al., 2009) and (ii) 78 strains related to SFPOs, in China, between 2010 and 2012 (Lv et al., 2014). Another MLVA assay targeting 14 loci was used in a survey of 309 strains including clinical methicillin-resistant S. aureus (MRSA) isolates and nasal carriage staphylococcal isolates (Pourcel et al., 2009). Finally, Sobral et al. (2012) proposed a third MLVA protocol based on the detection of 16 VNTR loci, including eight from Schouls et al. (2009) and eight from Pourcel et al. (2009). This protocol was implemented for the characterization of a panel (i) of 251 strains isolated primarily from humans and animals and also, to a lesser extent, from food and food poisoning samples (Sobral et al., 2012) and (ii) of 152 strains isolated from cases of bovine, ovine and caprine mastitits in France (Bergonier et al., 2014).
The aim of this study was to analyze the genetic diversity of a panel of S. aureus strains associated with SFPOs that occurred in France over the past 30 years. More specifically, we assessed the diversity of strains implicated in each outbreak and compared strains obtained from distinct outbreaks. MLVA data generated using the recent protocol of Sobral et al. (2012) were compared with those obtained by PFGE, spa-typing, and se gene detection. In light of our results, we discuss the usefulness of MLVA for routine typing of S. aureus, in terms of discriminatory power, and for investigating SFPOs.
The French NRL has established a large collection of strains isolated from the main food production sectors throughout various French regions, over the past 30 years. This collection also includes clinical strains mainly obtained during collaborative research projects. All the strains have been typed by spa-typing and PFGE and characterized with regard to their se genes. The NRL molecular typing database (BioNumerics software, V 7.1, Applied Maths, Sint-Martens-Latem, Belgium) centralizes the epidemiological information, genotype and phenotype data for all the strains.
A panel of 112 strains isolated from 76 distinct SFPOs that occurred in France from 1981 to 2009 was selected for this study (Table 1). Out of these 112 strains, 13 strains were considered as epidemiologically related because they originated from four distinct “strong evidence” SFPOs (no “3,” “8,” “20,” “102”; Table 2). The epidemiological data regarding these four SFPOs were collected by the local health authorities using interviews or questionnaires. At the same time, tracing back of incriminated food was performed by the local services of the French Ministry in charge of agriculture and food. Three SFPOs (“3,” “8,” “20”) included food and clinical strains isolated within one region. The fourth SFPO (“102”) included only food strains isolated from three different regions in France. For this latter SFPO, a soft cheese made from unpasteurised cow’s milk was identified as the common and single source (Ostyn et al., 2010).
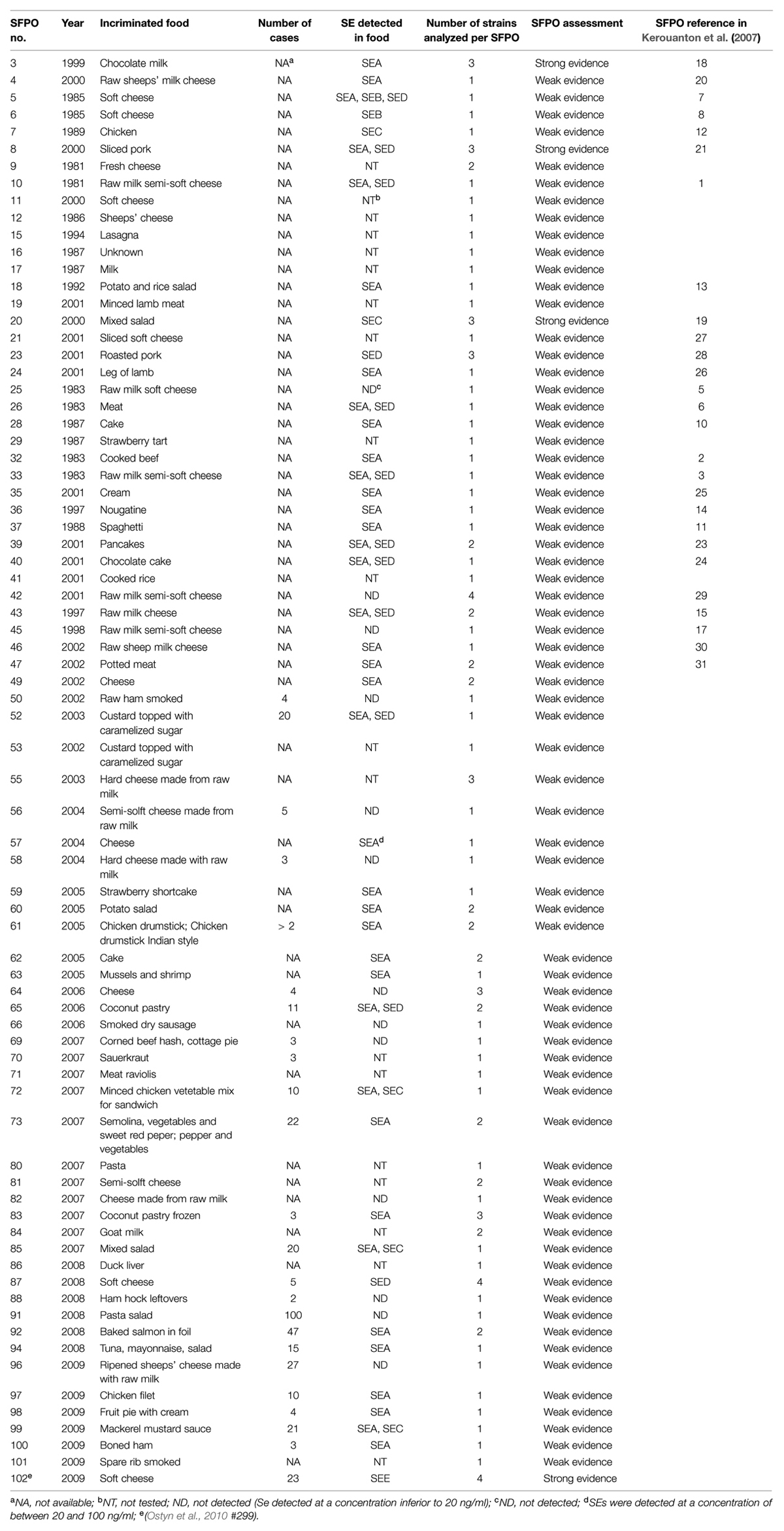
TABLE 1. Description of the 76 SFPOs that occurred in France from 1981 to 2009.
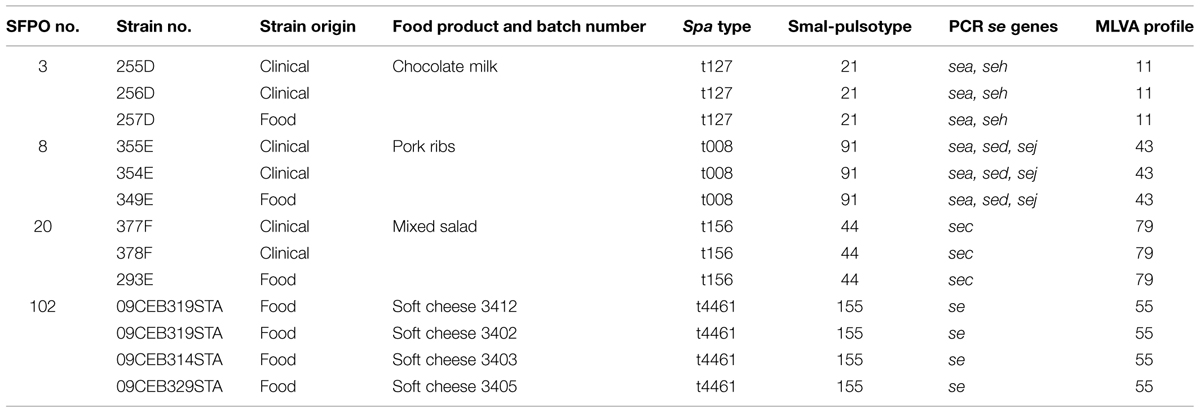
TABLE 2. Typing data on the 13 strains related to four distinct “strong evidence” SFPOs.
The 99 remaining strains were isolated from 72 different “weak evidence” SFPOs that occurred in different locations in France between 1981 and 2009 (Table 1). Out of these 72 “weak evidence” SFPOs, 53 involved only one strain, and the remaining 19 involved between two and four strains isolated from different food samples (Table 3).
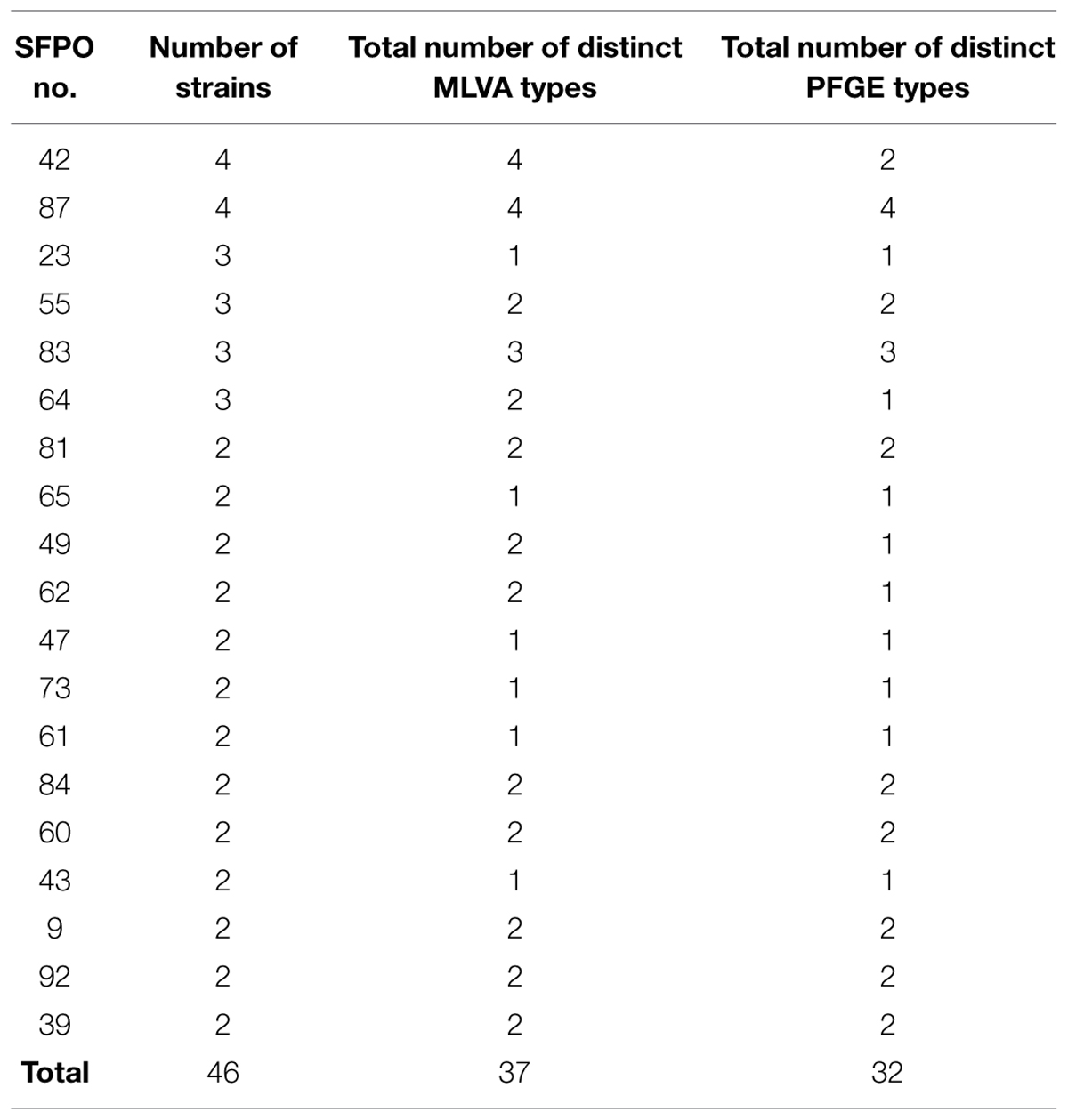
TABLE 3. Multiple-locus variable-number tandem-repeat analysis (MLVA) and pulsed field gel electrophoresis (PFGE) typing data for the 46 strains originating from the 19 “weak evidence” SFPOs including several strains.
To compare the discriminatory power of spa-typing, MLVA and PFGE, 43 additional strains were included in the panel. These 43 strains were selected according to their PFGE profiles to represent the genetic diversity observed in the molecular database. Then, we selected a total panel of 146 epidemiogically unrelated isolates comprising one strain of each of the four “strong evidence” SFPO (i.e., four isolates), 99 strains related to each of the “weak evidence” SFPOs (99 isolates), four strains related to four outbreaks that occurred in Belgium, one strain related to one outbreak in Japan (Omoe et al., 2003; Ono et al., 2008), 31 strains isolated from research projects and seven strains isolated from monitoring programs. This panel included food strains (n = 122 with 112 isolated from SFPOs) and strains isolated from either human cases (n = 19) or animals (n = 5).
All strains were stored in cryobeads at -80°C. They were cultured overnight at 37°C in brain heart infusion (BHI), isolated on a non-selective medium (Milk Plate Count Agar) and incubated at 37°C for 24 h, prior to extraction of total DNA. DNA extraction was performed using the InstaGene kit (Bio-Rad, Marnes-la-Coquette, France) according to the manufacturer’s recommendations. DNA concentrations were adjusted approximately to 100 ng/μl using a Nanodrop1000 spectrophotometer (spectrophotometer, Thermo scientific, Wilmington, DE, USA).
Two multiplex PCR systems were used to detect the genes of 11 types of enterotoxins, i.e., sea, seb, sec, sed, see, seg, seh, sei, sej, sep, and ser. The primers used to amplify the sea, seb, sec, and sed genes were designed by Mehrotra et al. (2000), those for seg, seh, sei, sej, and sep by Bania et al. (2006), those for see by Sharma et al. (2000), and those for ser by Chiang et al. (2008). They were synthesized by Eurofins (MWG Operon, France).
For the multiplex reaction targeting sea, seb, sec, sed, see, and ser genes, the 25 μl reaction mixture contained 1 U Fast Start Taq DNA polymerase (Roche, Diagnostics, Meylan, France), 2.5 mM MgCl2, 0.2 mM dNTPs, 1X PCR buffer, 0.2 μM of each primer for sea, seb, sec, ser, 0.8 μM of each primer for sed, 0.6 μM of each primer for see and 2 μl of DNA. PCR was performed on a Veriti® PCR cycler (Applied Biosystems, Courtaboeuf, France). The thermal cycle included an initial denaturation at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 56°C for 40 s, extension at 72°C for 1 min 30, and a final extension at 72°C for 7 min. The conditions for the multiplex targeting seg, seh, sei, sej, and sep genes were as described above, except that the annealing step was performed at 53°C for 40 s and the reaction mixture included 0.8 μM of each primer for seg, sei, sej, sep and 0.4 μM of each primer for seh. DNA of each isolate was tested by polymerase chain reaction (PCR) targeting the ribosomal RNA 23S gene region specific for S. aureus (Kerouanton et al., 2007).
Five reference S. aureus strains (i.e., FRIS6, 374F, FRI137, HMPL280, FRI326) were used as positive controls. The PCR products were separated by electrophoresis in a 2% agarose gel and visualized using the Gel Doc EQ apparatus (Bio-Rad).
DNA samples were diluted in molecular grade water to obtain solutions at 10 ng/μl. They were used as DNA templates for PCR amplification according to the protocol described by Sobral et al. (2012) with minor modifications to the amplification program: DNA template concentration was set at 10 ng/μl (instead of 5 ng/μl in Sobral’s protocole), touchdown PCR and long range PCR were both set at 18 thermal cycles (instead of 15). Samples were loaded onto an ABI3500® capillary sequencer using a 50 cm capillary filled with performance-optimized polymer 7 (Applied Biosystems) at 60°C for 6200 s with a running voltage of 12 kV, and an injection time and voltage of 10 s and 1.6 kV, respectively.
For each multiplex reaction, 2 μl of purified PCR product was combined with 7.75 μl HiDi formamide and 0.25 μl GS1200LIZ (Applied Biosystems). Samples were loaded onto an ABI3500® capillary sequencer using a 50 cm capillary filled with performance-optimized polymer 7 (Applied Biosystems) at 60°C for 6200 s with a running voltage of 12 kV, and an injection time and voltage of 10 s and 1.6 kV, respectively.
Each run included a negative (water) control to ensure the absence of contamination and a positive control to verify the PCR reaction.
From the panel of 112 strains, 12 strains related to “weak evidence” SFPOs (no 431G, 360F, 338E, 419G, 372F, 402F, 353E, 301E, 384F, 339E, 363F, 399F) that had previously been tested by MLVA by Sobral et al. (2012) were used here as positive controls.
Amplification products were electrophoresed twice in independent runs. At least two independent PCRs were performed from a given DNA extract of the reference strains.
The products of both multiplex PCR amplifications were resolved by capillary electrophoresis, and the alleles from each of the 16 targeted loci were automatically identified. Each VNTR locus was identified according to specific fluorescent dyes and automatically assigned to a DNA fragment size by the GeneMapper software (Applied Biosystems). This size was then converted into an allele designation according to the number of repeats found on the fragment, in associated with a quality index. The typing data file was imported into the NRL molecular database.
Minimum spanning trees were constructed using a categorical coefficient and unweighted pair group method with arithmetic mean (UPGMA) clustering. Allele designations and nomenclature were used according to Sobral et al. (2012). Partial repeats were rounded down to the closest half decimal (e.g., 1.0, 1.5, 2.0).
Any new alleles of unexpected size were sequenced. The loci and flanking regions were amplified in both directions with high-fidelity HotStart Taq Polymerase (Roche Diagnostics). Amplification products were sequenced by Eurofins (MWG Operon, France). The sequence analysis was performed into the NRL molecular typing database.
The initial step of PFGE as defined by the EURL for CPS protocol was performed according to the protocol described by Chung et al. (2000) with minor modifications. Briefly, strains were cultured in liquid BHI (instead of liquid TSB medium). Cell density was determined from a 2 ml suspension (instead of 0.5 ml) at 600 nm (instead of 620 nm). The agarose plugs were prepared in TE buffer (instead of PIV) and the PIV buffer contained 2 mM Tris-HCL and 1 M NaCl (instead of 10 mM Tris, 1 M NaCl). Plug shape was cubic (instead of circular) and EC buffer was incubated for 3 h (instead of 5 h).
The protocol for the subsequent steps of PFGE was based on the recommendations of the Harmony typing group (Murchan et al., 2003). The reference standard, S. aureus NCTC 8325 SmaI profile, was loaded in every fifth or sixth lane. The total running time was 20 h, the first-block switch time was 5–15 s for 8.5 h, and the second-block switch time was 15–60 s for 11.5 h. The voltage applied for the run was 6 V/cm. The CHEF DRIII system (Bio-Rad) was used, with an included angle of 120° and a linear ramp factor.
Gels were stained for 30 min in a 400 ml ultra- pure sterile water solution containing ethidium bromide at 10 mg/ml and banding profiles were visualized under UV light, using the Gel Doc Eq system and Quantity One software (Bio-Rad). DNA profiles were analyzed in the NRL molecular typing database. PFGE pulsotypes were considered as different if there was at least one band different between them (Barrett et al., 2006). Each PFGE profile was arbitrarily assigned to a pulsotype number.
Spa-typing was performed as previously described by Shopsin et al. (1999) and Aires-de-Sousa et al. (2006).
A strain already known for its spa-type (S. aureus Mu50, spa-type t002, Kuroda et al., 2001) was used as a positive control and a reaction without DNA was included within each run as a negative control. The PCR products were migrated on a 2% agarose gel and visualized using the Gel Doc EQ apparatus (Bio-Rad). They were sequenced by Eurofins (Esberg, Germany), on both DNA strands. The sequences were analyzed using BioNumerics software which provides a fully automated workflow, from import of raw sequencer trace files to assignment of repeat codes and spa types using the plug-in spa-typing which connects to SeqNet/Ridom Spa Server1. Each new base composition of the polymorphic repeat found in a strain was assigned a unique repeat code. The succession of repeats in a given strain determines the strain’s spa type. New spa-types were submitted to the SeqNet server2.
The epidemiological concordance of PFGE, spa-typing and MLVA was assessed by testing their capacity to recognize the homogeneity of 13 strains related to four distinct “strong evidence” SFPOs (Table 2) in the same epidemiological groups.
The ability of the methods to discriminate S. aureus strains (i.e., unrelated strains) was assessed by calculating Simpson’s index of diversity (ID; Hunter and Gaston, 1988) with confidence interval calculated according to (Carriço et al., 2006). The ID was calculated from PFGE, spa-typing and MLVA results obtained from the panel of 146 epidemiologically unrelated isolates.
This strain panel was reduced of four strains not typable by spa-typing, and used for congruence test between spa-typing, MLVA and PFGE. The congruence assessments were performed using the adjusted Rand’s coefficient (Carriço et al., 2006, #1093). Adjusted rand coefficient consider (i) the probability that a pair of isolates which is assigned to the same type by one typing method is also typed as identical by the other method, (ii) the probability that a pair of isolates which is assigned to two types by one typing method is also typed as different by the other method and corrects the typing concordance for chance agreement, avoiding the overestimation of congruence between typing methods.
For a finer comparison the adjusted Wallace (AW) coefficients (Severiano et al., 2011, #1095) were also performed. Statistics analysis were performed in BioNumerics software, V 7.1, (Applied Maths) using a script developed by Ana Severiano and João André Carriço available online at http://darwin.phyloviz.net/ComparingPartitions/. The AW coefficient indicates the probability that pairs of isolates which are assigned to the same type by one typing method are also typed as identical by the other and corrects the typing concordance for chance agreement. The AW coefficient is directional, i.e., given a standard method. It considers the probability of two strains having the same type of standard method also sharing the same type of the compared method.
All the strains were identified as S. aureus by the 23S rDNA PCR assay specific for this bacterial species. The ID diversity index was assessed on a panel of 146 epidemiologically unrelated strains, which included 103 strains associated with various French SFPOs. MLVA and PFGE separated the 146 strains into 125 and 118 distinct groups, respectively. Spa-typing separated 142 out of the 146 strains into 71 groups only (Table 4). The four remaining strains could not be typed using this technique. High ID values were observed for MLVA (0.997) and PFGE (0.995) indicating that almost each strain can be distinguished from all other members of the strain panel by these two typing methods. PFGE and MLVA were found to be more discriminatory than spa-typing whose ID value was 0.970 (Table 4).

TABLE 4. Multiple-locus variable-number tandem-repeat analysis, PFGE and spa-typing results from a panel of 146 not epidemiologically related strains.
Four PFGE groups and five MLVA groups were shared mainly into two different spa-types. Eight MLVA groups were shared mainly into two PFGE types. Eleven PFGE groups were shared mainly into two MLVA types (data not shown).
Quantitative determination of concordance of the three methods was calculated (Table 5). Congruence between the three methods was found to be low with adjusted Rand coefficient values ranging from 0.131 to 0.269 (Table 5). The AWMLVA→spa-typing and AWPFGE→spa-typing were respectively higher than AWspa-typing→MLVA and AWspa-typing→PFGE (Table 5) indicating that partitions defined by spa-typing could have been predicted from the results of MLVA or PFGE.
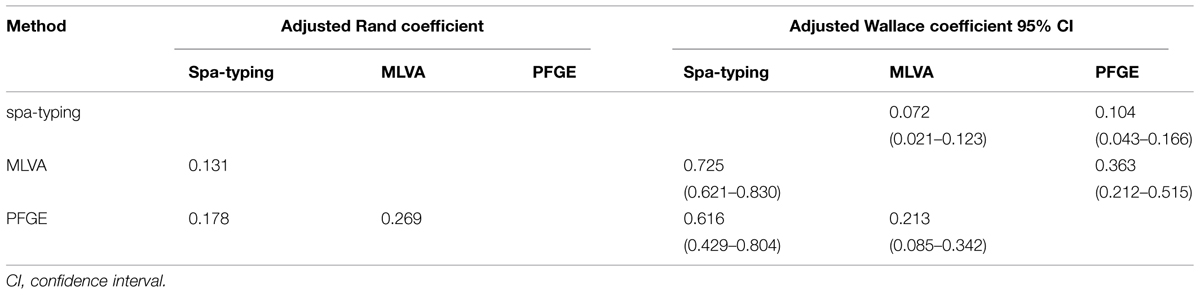
TABLE 5. Congruence between the typing methods using adjusted Rand and adjusted Wallace coefficients.
Typing of 13 S. aureus strains from four “strong evidence” SFPOs showed that the se gene profiles and the MLVA-, PFGE-, and spa- types were indistinguishable within each set of epidemiologically related strains (Table 2). This demonstrated the good epidemiological concordance of the four methods.
A panel of 103 epidemiologically unrelated strains from 76 French SFPOs was selected, containing one strain of each of the four “strong evidence” SFPOs described above (Table 2) and 99 strains from 72 “weak evidence” SFPOs (Table 1). For 12 strains previously tested elsewhere (i.e., 431G, 360F, 338E, 419G, 372F, 402F, 353E, 301E, 384F, 339E, 363F, 399F), the MLVA profiles obtained here were similar to those obtained by Sobral et al. (2012).
Multiple-locus variable-number tandem-repeat analysis separated the 103 strains into 84 different types. The most prevalent MLVA types were “20” and “1,” with each of these two types containing four strains isolated from four different SFPOs that occurred over the periods 1983–2007 and 2000–2008, respectively. The incriminated food was different for each of these SFPOs.
Pulsed field gel electrophoresis separated the 103 strains into 80 pulsotypes. The most prevalent PFGE type was “18” and contained five strains isolated from four distinct SFPOs that occurred between 1999 and 2008.
Out of the 76 different “weak evidence” SFPOs, 19 included several strains (i.e., between two and four strains; Table 3). For 15 of these SFPOs, strains that displayed distinct MLVA profiles also displayed distinct PFGE pulsotypes, and strains with similar MLVA profiles also showed similar PFGE profiles. MLVA data were therefore in agreement with those of PFGE. For three of the 19 SFPOs (i.e., “62,” “49,” “64”), the strains had different MLVA profiles but showed indistinguishable PFGE pulsotypes (Table 3). However, for the SFPOs “49” and “64,” the MLVA profiles obtained were very similar, differing by only 1.5 repeat. For the remaining SFPO (i.e., “42”), the four strains displayed four different MLVA profiles but showed only two distinct PFGE pulsotypes (Table 3).
Spa-typing separated 102 out of the 103 strains into 50 different types; one strain could not be typed. The most frequent spa-types observed were t127 (n = 14) and t008 (n = 10; Figure 1). Five new spa-types were observed for the first time here (i.e., t7667, t7668, t7669, t7671, and t7674). Except for one MLVA type (“20”), all the MLVA types that included several strains corresponded to the same spa-type. The type”20” included one and three strains with two different spa-types, t656 and t008, respectively (Figure 1). These two spa-types were close: they had the same repeat number and only differed at two single nucleotide positions in the second spa repeat.
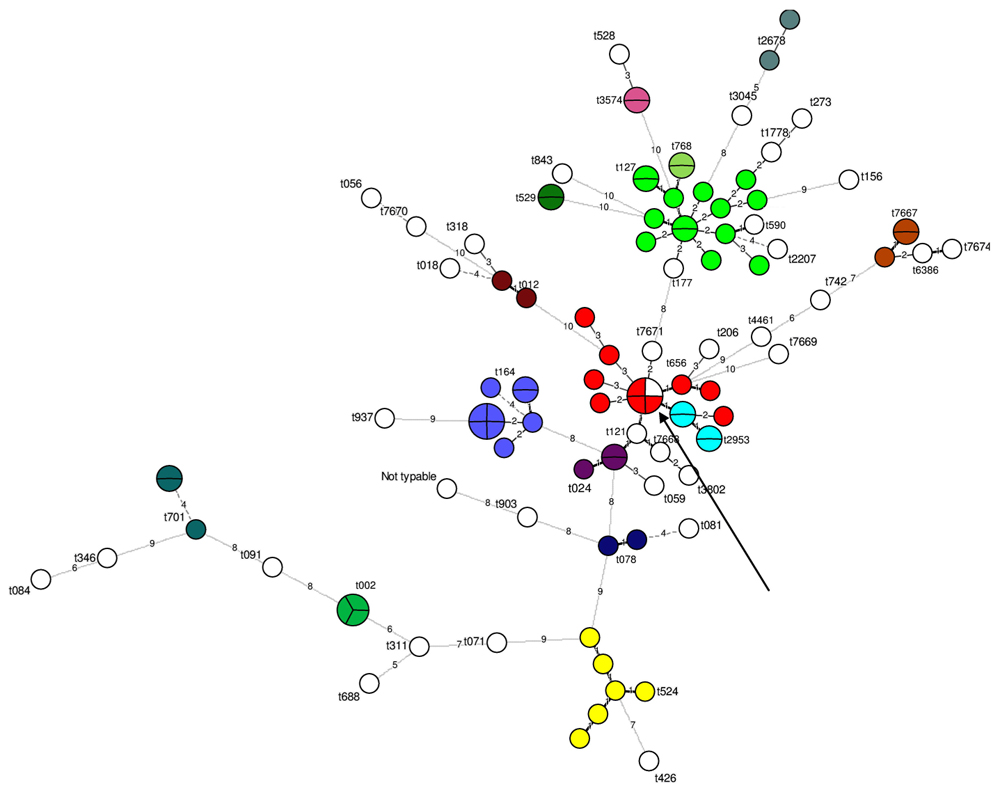
FIGURE 1. Minimum spanning tree of spa-types according to the multiple-locus variable-number tandem-repeat analysis (MLVA) types determined on the 103 strains tested. 102 strains were successfully typed using spa-typing. Each circle represents a particular MLVA type. The size of each circle is proportional to the number of isolates within the MLVA type. The distance between the circles represents the genetic divergence. The divergence is given in number of mutations and is indicated on the branch. Each color represents a different spa-type (n > 1 isolate). All unique (n = 1 isolate) spa-type are shown in white. The arrow shows the single MLVA type including two different spa-types.
Out of the 103 strains tested, 16 strains did not carry any se gene and 87 strains carried at least one se gene. These 87 strains were divided into 20 distinct se gene profiles. The most frequently occurring gene was sea (n = 58), followed by seg (n = 23) and sei (n = 23), by seh (n = 19) and then by sed, sej, ser, sec, sep, seb, and see. Several se genes could be present, and the most frequent associations of se genes detected were sea-seh (n = 15), sea-sed-sej-ser (n = 13) and seg-sei (n = 11). Moreover, 65 strains carried genes corresponding to ‘new’ enterotoxins, i.e., seg, seh, sei, sej, sep, and ser. Eleven strains carried the seg and sei genes and one strain the seg, sei, sej, sep, and ser genes.
The 16 strains for which no se gene were detected were divided into 16 unique MLVA types. The 87 se gene-carrying strains were separated into 68 distinct MLVA-types: 64 MLVA-types correlated with se gene carriage (Figure 2). The four remaining MLVA-types included strains with different se genes as indicated by circles including various colors in the Figure 2. The 21 strains that carried seh either alone or in combination with sea were divided into 19 very close MLVA types that clustered within the same MLVA subgroup (Figure 2). 14 out of the 21 seh-carrying strains that clustered within the same MLVA sub-group possessed the spa type t127 (Figures 1 and 2).
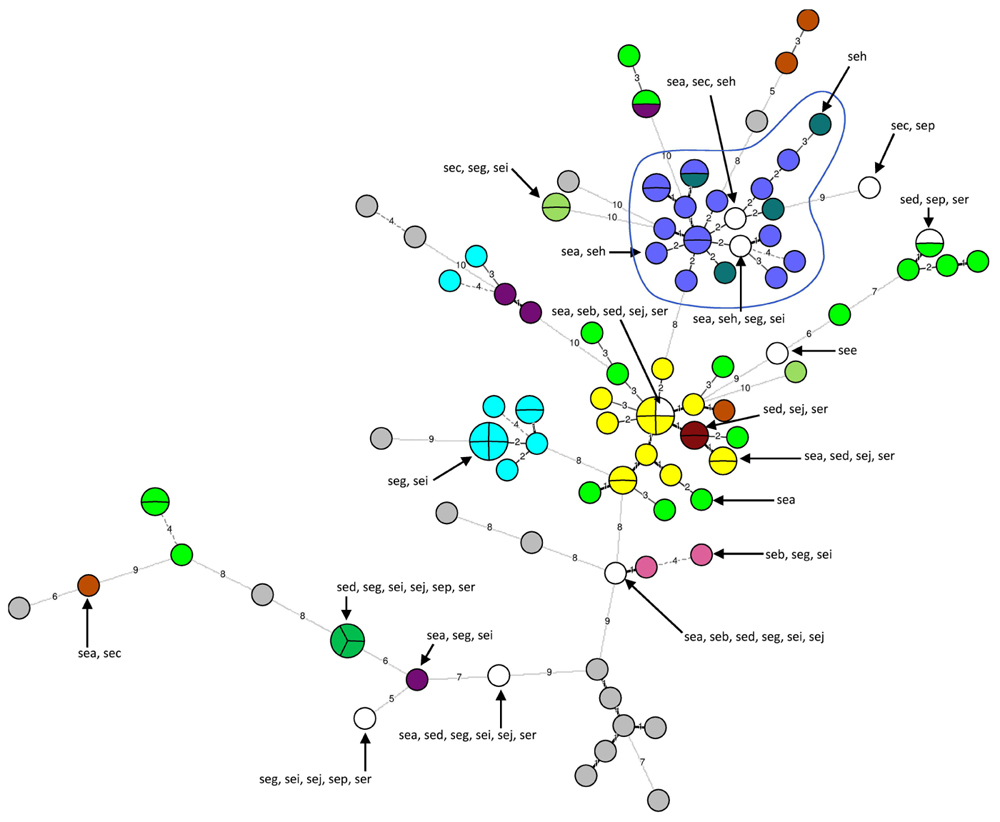
FIGURE 2. Minimum spanning tree of 103 Staphylococcus aureus se gene-carrying strains according to their MLVA types. Each circle represents a particular MLVA type. The size of each circle is proportional to the number of isolates within this MLVA type. The distance between the circles represents the genetic divergence. The divergence is given in number of mutations and is indicated on the branch. Each color represents a particular se gene combination (n > 1 isolate), with unique se combination (n = 1 isolate) shown in white and no se gene shown in gray. Four MLVA-types had strains with different se genes combinations (circles with several colors). The MLVA subgroup that includes seh-carrying strains is highlighted in blue and circled. A subgroup groups strains with MLVA types that differ by a maximum of five mutations.
The aim of this study was to investigate the genetic diversity of 112 S. aureus strains involved in SFPOs in France between 1981 and 2009. The MLVA protocol developed by Sobral et al. (2012) was compared here for the first time with the EURL PFGE protocol. The analysis of a panel of 146 S. aureus strains isolated from different sources (clinical cases, food, animal cases, or SFPOs) showed that MLVA and PFGE have equal discriminatory power.
For the strains related to the strong evidence SFPOs, MLVA, and PFGE typing data demonstrated the good epidemiological concordance of each of the two methods. Moreover, for the strains which were involved in the same “weak-evidence” SFPO MLVA could discriminate strains which had similar PFGE types.
With the MLVA protocol used here, results could be obtained within 48 h, i.e., faster than PFGE analysis which is usually completed in 3 days from receipt of pure culture. Moreover, the method is easy to perform, is readily automatable, and allows high sample throughput. The use of commercially available reagents can help foster standardization. MLVA data are also suitable for electronic transmission between laboratories and are not prone to subjective interpretation. Moreover, this MLVA method benefits from the higher resolution of using a capillary electrophoresis methodology over standard gel-based techniques. The method can therefore be easily implemented by NRLs equipped with a capillary electrophoresis system. One inconvenience could be the use of an expensive system. Nevertheless, this MLVA protocol is also suitable for agarose-gel electrophoresis. Indeed, for each of the 16 VNTRs, repeat size was sufficiently high for accurate band sizing on agarose gels. High-throughput typing on agarose gels can also be facilitated by the use of an automated flow-through gel electrophoresis system (Reskova et al., 2014).
The major drawback of this MLVA assay is the lack of free internet-accessible databases for comparative purposes. To date, the databases used worldwide3 (MLVA.net) centralize profiles obtained with the protocols of Pourcel et al. (2009) and Schouls et al. (2009) using subset of loci. However, the development of an easily accessible database centralizing profiles obtained from the 16 VNTRs with this protocol is in progress and should be available in the coming years.
In this study, 65 out of 87 enterotoxigenic strains carried more than one gene coding for enterotoxins, illustrating the importance of searching for se genes in the strains involved in SFPOs. Our results confirmed the clear predominance of the sea gene (67%) among the SFPO strains and its frequent association with other genes such as seh or sed-sej-ser. The prevalence of the sea gene in the strains linked to SPFOs has been already observed in Spain (Dyer et al., 2007; Sobral et al., 2007; Kellermann et al., 2008), Italy (Morandi et al., 2007), and in Japan (Sato’o et al., 2014). The association of sea with seh has also been observed for strains linked to SFPOs in France (Kerouanton et al., 2007) and recently in Japan (Sato’o et al., 2014).
The three typing method, MLVA, PFGE and spa-typing, show ID greater than 0.90. This value is considered as a cut-off in order to interpret the typing results with confidence (Hunter and Gaston, 1988, #1092; Matallah et al., 2013, #1096). PFGE and MLVA bring more information than spa-typing.
Whatever the typing method used here for the analysis of 112 French SFPOs-isolates, i.e., MLVA, PFGE, spa-typing or se genes detection, a large number of different molecular types was found, highlighting the high genetic diversity of the tested isolates. In this study, associations between MLVA types, se gene combinations and spa-types were identified. All the 21 seh-carrying strains clustered within the same MLVA sub-group, and 14 of these showed spa type t127. These results demonstrated that the strains carrying the seh gene have a genetically related background, as previously suggested by Ruzickova et al. (2008) for 28 enterotoxin H-positive S. aureus strains isolated from food samples in the Czech Republic.
Previously, a high concordance between the MLVA protocol and the MLST method was demonstrated and a distribution signature of clonal complexes typical of human isolates was found for 13 SFPO strains (Sobral et al., 2012). Further comparison of the MLVA data obtained here from 112 SFPO strains with those from a representative panel of human strains could be useful to confirm the hypothesis of a human origin for SFPO strains.
Another investigation could be useful to explore the genetic structure of all the populations of food S. aureus strains associated to SFPO isolated in France over the past 30° years.
By combining various molecular typing methods, we highlighted the high genetic diversity of S. aureus strains involved in SFPO in France over the past 30 years. In addition, the MLVA protocol developed by Sobral et al. (2012) was found highly discriminatory, therefore representing a very interesting alternative to PFGE for establishing epidemiological links during SFPOs investigations. This MLVA method could be used also to better understand the population biology of S. aureus. Before transferring this MLVA protocol through the European NRL network, its reproducibility will need to be assessed in proficiency testing trials, in order to compare and interpret MLVA data and harmonize assignment of MLVA types.
SR participated in the design and coordination of the study, the data interpretation and the draft of all the manuscript. BF participated to the design of the study, the data interpretation under BioNumerics software and the strain typing by PFGE, spa-typing and se genes. NV was in charge of the strain typing by MLVA and MLVA data analysis. JG carried out all the PCR tests at EURL. J-AH took part to the draft of the manuscript. LG carried all the congruence analysis and took part to the draft of the manuscript. AB and FA participated in the design and coordination of the study, and the draft of all the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was conducted as part of the activities of the EURL and the French NRL for CPS. It was supported by a grant from the Directorate-General for Heath and Consumers (DG SANCO) of the European Commission and from ANSES. We thank Dr. Bertrand Lombard (ANSES) for his helpful advice on writing this manuscript.
CPS, coagulase-positive staphylococci; EURL, European Union Reference Laboratory; NRL, National Reference Laboratory; SEs, staphylococcal enterotoxins; SFPO, staphylococcal food poisoning outbreaks.
Aires-de-Sousa, M., Boye, K., De Lencastre, H., Deplano, A., Enright, M. C., Etienne, J., et al. (2006). High interlaboratory reproducibility of DNA sequence-based typing of bacteria in a multicenter study. J. Clin. Microbiol. 44, 619–621. doi: 10.1128/JCM.44.2.619-621.2006
Argudin, M. A., Mendoza, M. C., Gonzalez-Hevia, M. A., Bances, M., Guerra, B., and Rodicio, M. R. (2012). Genotypes, exotoxin gene content, and antimicrobial resistance of Staphylococcus aureus strains recovered from foods and food handlers. Appl. Environ. Microbiol. 78, 2930–2935. doi: 10.1128/AEM.07487-11
Argudin, M. A., Mendoza, M. C., and Rodicio, M. R. (2010). Food poisoning and Staphylococcus aureus enterotoxins. Toxins (Basel) 2, 1751–1773. doi: 10.3390/toxins2071751
Babouee, B., Frei, R., Schultheiss, E., Widmer, A. F., and Goldenberger, D. (2011). Comparison of the DiversiLab repetitive element PCR system with spa typing and pulsed-field gel electrophoresis for clonal characterization of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 49, 1549–1555. doi: 10.1128/JCM.02254-10
Barrett, T. J., Gerner-Smidt, P., and Swaminathan, B. (2006). Interpretation of pulsed-field gel electrophoresis patterns in foodborne disease investigations and surveillance. Foodborne Pathog. Dis. 3, 20–31. doi: 10.1089/fpd.2006.3.20
Bania, J., Dabrowska, A., Korzekwa, K., Zarczynska, A., Bystron, J., Chrzanowska, J., et al. (2006). The profiles of enterotoxin genes in Staphylococcus aureus from nasal carriers. Lett. Appl. Microbiol. 42, 315–320. doi: 10.1111/j.1472-765X.2006.01862.x
Bergonier, D., Sobral, D., Fessler, A. T., Jacquet, E., Gilbert, F. B., Schwarz, S., et al. (2014). Staphylococcus aureus from 152 cases of bovine, ovine and caprine mastitis investigated by Multiple-locus variable number of tandem repeat analysis (MLVA). Vet. Res. 45, 97. doi: 10.1186/s13567-014-0097-4
Carriço, J. A., Silva-Costa, C., Melo-Cristino, J., Pinto, F. R., De Lencastre, H., Almeida, J. S., et al. (2006). Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant streptococcus pyogenes. J. Clin. Microbiol. 44, 2524–2532. doi: 10.1128/JCM.02536-05
Chiang, Y. C., Lai, C. H., Lin, C. W., Chang, C. Y., and Tsen, H. Y. (2014). Improvement of strain discrimination by combination of superantigen profiles, PFGE, and RAPD for Staphylococcus aureus isolates from clinical samples and food-poisoning cases. Foodborne Pathog. Dis. 11, 468–477. doi: 10.1089/fpd.2013.1708
Chiang, Y. C., Liao, W. W., Fan, C. M., Pai, W. Y., Chiou, C. S., and Tsen, H. Y. (2008). PCR detection of Staphylococcal enterotoxins (SEs) N, O, P, Q, R, U, and survey of SE types in Staphylococcus aureus isolates from food-poisoning cases in Taiwan. Int. J. Food Microbiol. 121, 66–73. doi: 10.1016/j.ijfoodmicro.2007.10.005
Chiou, C. S., Wei, H. L., and Yang, L. C. (2000). Comparison of pulsed-field gel electrophoresis and coagulase gene restriction profile analysis techniques in the molecular typing of Staphylococcus aureus. J. Clin. Microbiol. 38, 2186–2190.
Chung, M., De Lencastre, H., Matthews, P., Tomasz, A., Adamsson, I., Aires De Sousa, M., et al. (2000). Molecular typing of methicillin-resistant Staphylococcus aureus by pulsed-field gel electrophoresis: comparison of results obtained in a multilaboratory effort using identical protocols and MRSA strains. Microb. Drug Resist. 6, 189–198. doi: 10.1089/mdr.2000.6.189
Cookson, B. D., Aparicio, P., Deplano, A., Struelens, M., Goering, R., and Marples, R. (1996). Inter-centre comparison of pulsed-field gel electrophoresis for the typing of methicillin-resistant Staphylococcus aureus. J. Med. Microbiol. 44, 179–184. doi: 10.1099/00222615-44-3-179
Cookson, B. D., Robinson, D. A., Monk, A. B., Murchan, S., Deplano, A., De Ryck, R., et al. (2007). Evaluation of molecular typing methods in characterizing a European collection of epidemic methicillin-resistant Staphylococcus aureus strains: the HARMONY collection. J. Clin. Microbiol. 45, 1830–1837. doi: 10.1128/JCM.02402-06
Dyer, M. D., Murali, T. M., and Sobral, B. W. (2007). Computational prediction of host-pathogen protein-protein interactions. Bioinformatics 23, i159–i166. doi: 10.1093/bioinformatics/btm208
EFSA. (2011). Updated technical specifications for harmonised reporting of foodborneoutbreaks through the European Union reporting system in accordance with Directive 2003/99/EC1. EFSA J. 9, 2101.
EFSA-ECDC. (2013). The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2011. EFSA J. 11, 250. doi: 10.2903/j.efsa.2013.3129
Hallin, M., Deplano, A., Denis, O., De Mendonca, R., De Ryck, R., and Struelens, M. J. (2007). Validation of pulsed-field gel electrophoresis and spa typing for long-term, nationwide epidemiological surveillance studies of Staphylococcus aureus infections. J. Clin. Microbiol. 45, 127–133. doi: 10.1128/JCM.01866-06
Hennekinne, J. A., De Buyser, M. L., and Dragacci, S. (2011). Staphylococcus aureus and its food poisoning toxins: characterization and outbreak investigation. FEMS Microbiol Rev. 36, 815–836. doi: 10.1111/j.1574-6976.2011.00311.x
Hunter, P. R., and Gaston, M. A. (1988). Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J. Clin. Microbiol. 26, 2465–2466.
Kadariya, J., Smith, T. C., and Thapaliya, D. (2014). Staphylococcus aureus and staphylococcal food-borne disease: an ongoing challenge in public health. Biomed. Res. Int. 2014, 827965. doi: 10.1155/2014/827965
Kellermann, M. G., Sobral, L. M., Da Silva, S. D., Zecchin, K. G., Graner, E., Lopes, M. A., et al. (2008). Mutual paracrine effects of oral squamous cell carcinoma cells and normal oral fibroblasts: induction of fibroblast to myofibroblast transdifferentiation and modulation of tumor cell proliferation. Oral Oncol. 44, 509–517. doi: 10.1016/j.oraloncology.2007.07.001
Kerouanton, A., Hennekinne, J. A., Letertre, C., Petit, L., Chesneau, O., Brisabois, A., et al. (2007). Characterization of Staphylococcus aureus strains associated with food poisoning outbreaks in France. Int. J. Food Microbiol. 115, 369–375. doi: 10.1016/j.ijfoodmicro.2006.10.050
Koreen, L., Ramaswamy, S. V., Graviss, E. A., Naidich, S., Musser, J. M., and Kreiswirth, B. N. (2004). spa typing method for discriminating among Staphylococcus aureus isolates: implications for use of a single marker to detect genetic micro- and macrovariation. J. Clin. Microbiol. 42, 792–799. doi: 10.1128/JCM.42.2.792-799.2004
Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., et al. (2001). Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240. doi: 10.1016/S0140-6736(00)04403-2
Lindstedt, B. A., Torpdahl, M., Vergnaud, G., Le Hello, S., Weill, F. X., Tietze, E., et al. (2013). Use of multilocus variable-number tandem repeat analysis (MLVA) in eight European countries, 2012. Eurosurveillance 18, 20385.
Lv, G., Xu, B., Wei, P., Song, J., Zhang, H., Zhao, C., et al. (2014). Molecular characterization of foodborne-associated Staphylococcus aureus strains isolated in Shijiazhuang, China, from 2010 to 2012. Diagn. Microbiol. Infect. Dis. 78, 462–468. doi: 10.1016/j.diagmicrobio.2013.12.006
Martin, M. C., Fueyo, J. M., Gonzalez-Hevia, M. A., and Mendoza, M. C. (2004). Genetic procedures for identification of enterotoxigenic strains of Staphylococcus aureus from three food poisoning outbreaks. Int. J. Food Microbiol. 94, 279–286. doi: 10.1016/j.ijfoodmicro.2004.01.011
Matallah, M., Bakhrouf, A., Habeeb, M. A., Turlej-Rogacka, A., Iversen, A., Pourcel, C., et al. (2013). Four genotyping schemes for phylogenetic analysis of Pseudomonas aeruginosa: comparison of their congruence with multi-locus sequence typing. PLoS ONE 8:e82069. doi: 10.1371/journal.pone.0082069
Mehrotra, M., Wang, G., and Johnson, W. M. (2000). Multiplex PCR for detection of genes for Staphylococcus aureus enterotoxins, exfoliative toxins, toxic shock syndrome toxin 1, and methicillin resistance. J. Clin. Microbiol. 38,1032–1035.
Morandi, S., Brasca, M., Lodi, R., Cremonesi, P., and Castiglioni, B. (2007). Detection of classical enterotoxins and identification of enterotoxin genes in Staphylococcus aureus from milk and dairy products. Vet. Microbiol. 124, 66–72. doi: 10.1016/j.vetmic.2007.03.014
Murchan, S., Kaufmann, M. E., Deplano, A., De Ryck, R., Struelens, M., Zinn, C. E., et al. (2003). Harmonization of pulsed-field gel electrophoresis protocols for epidemiological typing of strains of methicillin-resistant Staphylococcus aureus: a single approach developed by consensus in 10 European laboratories and its application for tracing the spread of related strains. J. Clin. Microbiol. 41, 1574–1585.
Omoe, K., Hu, D. L., Takahashi-Omoe, H., Nakane, A., and Shinagawa, K. (2003). Identification and characterization of a new staphylococcal enterotoxin-related putative toxin encoded by two kinds of plasmids. Infect. Immun. 71, 6088–6094. doi: 10.1128/IAI.71.10.6088-6094.2003
Ono, H. K., Omoe, K., Imanishi, K., Iwakabe, Y., Hu, D. L., Kato, H., et al. (2008). Identification and characterization of two novel staphylococcal enterotoxins, types S and T. Infect. Immun. 76, 4999–5005. doi: 10.1128/IAI.00045-08
Ostyn, A., De Buyser, M. L., Guillier, F., Groult, J., Felix, B., Salah, S., et al. (2010). First evidence of a food poisoning outbreak due to staphylococcal enterotoxin type E, France, 2009. Eurosurveillance 15, 19528.
Pourcel, C., Hormigos, K., Onteniente, L., Sakwinska, O., Deurenberg, R. H., and Vergnaud, G. (2009). Improved multiple-locus variable-number tandem-repeat assay for Staphylococcus aureus genotyping, providing a highly informative technique together with strong phylogenetic value. J. Clin. Microbiol. 47, 3121–3128. doi: 10.1128/JCM.00267-09
Reskova, Z., Korenova, J., and Kuchta, T. (2014). Effective application of multiple locus variable number of tandem repeats analysis to tracing Staphylococcus aureus in food-processing environment. Lett. Appl. Microbiol. 58, 376–383. doi: 10.1111/lam.12200
Ruzickova, V., Karpiskova, R., Pantucek, R., Pospisilova, M., Cernikova, P., and Doskar, J. (2008). Genotype analysis of enterotoxin H-positive Staphylococcus aureus strains isolated from food samples in the Czech Republic. Int. J. Food Microbiol. 121, 60–65. doi: 10.1016/j.ijfoodmicro.2007.10.006
Sato’o, Y., Omoe, K., Naito, I., Ono, H. K., Nakane, A., Sugai, M., et al. (2014). Molecular epidemiology and identification of a Staphylococcus aureus clone causing food poisoning outbreaks in Japan. J. Clin. Microbiol. 52, 2637–2640. doi: 10.1128/JCM.00661-14
Schouls, L. M., Spalburg, E. C., Van Luit, M., Huijsdens, X. W., Pluister, G. N., Van Santen-Verheuvel, M. G., et al. (2009). Multiple-locus variable number tandem repeat analysis of Staphylococcus aureus: comparison with pulsed-field gel electrophoresis and spa-typing. PLoS ONE 4:e5082. doi: 10.1371/journal.pone.0005082
Severiano, A., Pinto, F. R., Ramirez, M., and Carriço, J. A. (2011). Adjusted wallace coefficient as a measure of congruence between typing methods. J. Clin. Microbiol. 49, 3997–4000. doi: 10.1128/JCM.00624-11
Sharma, N. K., Rees, C. E., and Dodd, C. E. (2000). Development of a single-reaction multiplex PCR toxin typing assay for Staphylococcus aureus strains. Appl. Environ. Microbiol. 66, 1347–1353. doi: 10.1128/AEM.66.4.1347-1353.2000
Shimizu, A., Fujita, M., Igarashi, H., Takagi, M., Nagase, N., Sasaki, A., et al. (2000). Characterization of Staphylococcus aureus coagulase type VII isolates from staphylococcal food poisoning outbreaks (1980-1995) in Tokyo, Japan, by pulsed-field gel electrophoresis. J. Clin. Microbiol. 38, 3746–3749.
Shopsin, B., Gomez, M., Montgomery, S. O., Smith, D. H., Waddington, M., Dodge, D. E., et al. (1999). Evaluation of protein a gene polymorphic region DNA sequencing for typing of Staphylococcus aureus strains. J. Clin. Microbiol. 37, 3556–3563.
Sobral, A. P., De Oliveira Lima, D. N., Cazal, C., Santiago, T., Das Gracas Granja Mattos, M., Melo, B., et al. (2007). Myxoid liposarcoma of the lip: correlation of histological and cytological features and review of the literature. J. Oral Maxillofac. Surg. 65, 1660–1664. doi: 10.1016/j.joms.2006.06.264
Sobral, D., Schwarz, S., Bergonier, D., Brisabois, A., Fessler, A. T., Gilbert, F. B., et al. (2012). High throughput multiple locus variable number of tandem repeat analysis (MLVA) of Staphylococcus aureus from human, animal and food sources. PLoS ONE 7:e33967. doi: 10.1371/journal.pone.0033967
Strommenger, B., Kettlitz, C., Weniger, T., Harmsen, D., Friedrich, A. W., and Witte, W. (2006). Assignment of Staphylococcus isolates to groups by spa typing, SmaI macrorestriction analysis, and multilocus sequence typing. J. Clin. Microbiol. 44, 2533–2540.
Wattinger, L., Stephan, R., Layer, F., and Johler, S. (2012). Comparison of Staphylococcus aureus isolates associated with food intoxication with isolates from human nasal carriers and human infections. Eur. J. Clin. Microbiol. Infect. Dis. 31, 455–464. doi: 10.1007/s10096-011-1330-y
Wei, H. L., and Chiou, C. S. (2002). Molecular subtyping of Staphylococcus aureus from an outbreak associated with a food handler. Epidemiol. Infect. 128, 15–20. doi: 10.1017/S0950268801006355
Keywords: food poisoning outbreaks, Staphylococcus aureus, MLVA, PFGE, spa-typing, enterotoxin genes, genetic diversity
Citation: Roussel S, Felix B, Vingadassalon N, Grout J, Hennekinne J-A, Guillier L, Brisabois A and Auvray F (2015) Staphylococcus aureus strains associated with food poisoning outbreaks in France: comparison of different molecular typing methods, including MLVA. Front. Microbiol. 6:882. doi: 10.3389/fmicb.2015.00882
Received: 10 June 2015; Accepted: 11 August 2015;
Published: 11 September 2015.
Edited by:
Javier Carballo, University of Vigo, SpainReviewed by:
Kiiyukia Matthews Ciira, Jomo Kenyatta University of Agriculture and Technology, KenyaCopyright © 2015 Roussel, Felix, Vingadassalon, Grout, Hennekinne, Guillier, Brisabois and Auvray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Roussel, ANSES, Laboratoire de sécurité des aliments, 14 Rue Pierre et Marie Curie, 94701 Maisons-Alfort-Cedex, France,c29waGllLnJvdXNzZWxAYW5zZXMuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.