 Catharina H. M. Lüdeke
Catharina H. M. Lüdeke Narjol Gonzalez-Escalona
Narjol Gonzalez-Escalona Markus Fischer2
Markus Fischer2 Jessica L. Jones
Jessica L. Jones
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 10 June 2015
Sec. Food Microbiology
Volume 6 - 2015 | https://doi.org/10.3389/fmicb.2015.00564
This article is part of the Research Topic Ecology, virulence and detection of pathogenic and pandemic Vibrio parahaemolyticus View all 13 articles
Vibrio parahaemolyticus is a leading cause of seafood-borne infections in the US. This organism has a high genetic diversity that complicates identification of strain relatedness and epidemiological investigations. However, sequence-based analysis methods are promising tools for these identifications. In this study, Multi-Locus Sequence Typing (MLST) and Multiple-Locus Variable-Number Tandem-Repeat Analysis (MLVA) was performed on 58 V. parahaemolyticus isolates (28 of oyster and 30 of clinical origin), to identify differences in phylogeny. The results obtained by both methods were compared to Pulsed-Field Gel Electrophoresis (PFGE) patterns determined in a previous study. Forty-one unique sequence types (STs) were identified by MLST among the 58 isolates. Almost half of the isolates (22) belonged to a new ST and added to the MLST database. A ST could not be generated for 5 (8.6%) isolates, primarily due to an untypable recA locus. Analysis with eBURST did not identify any clonal complex among the strains analyzed and revealed 37 singeltons with 4 of them forming 2 groups (1 of them SLV, and the other a DLV). An established MLVA assay, targeting 12 total genes through three separate 4-plex PCRs, was successfully adapted to high resolution melt (HRM) analysis with faster and easier experimental setup; resulting in 58 unique melt curve patterns. HRM-MLVA was capable of differentiating isolates within the same PFGE cluster and having the same ST. Conclusively, combining the three methods PFGE, MLST, and HRM-MLVA, for the phylogenetic analysis of V. parahaemolyticus resulted in a high resolution subtyping scheme for V. parahaemolyticus. This scheme will be useful as a phylogenetic research tool and as an improved method for outbreak investigations for V. parahaemolyticus.
Vibrio parahaemolyticus can cause acute gastroenteritis associated with consumption of raw or undercooked seafood (Nishibuchi and DePaola, 2005). Presently, this bacterium represents the most common cause for seafood-associated infections in the United States (Iwamoto et al., 2010). V. parahaemolyticus has shown a high rate of recombination and mutation which leads to a high genetic diversity (Gonzalez-Escalona et al., 2008). V. parahaemolyticus isolates are frequently characterized for their virulence gene profile, serotype, ribotype, and/or Pulsed-Field Gel Electrophoresis (PFGE) pattern for research studies and epidemiological investigations (Broberg et al., 2011; Jones et al., 2012; Paranjpye et al., 2012; Banerjee et al., 2014; Xu et al., 2015). However, there are several phylogenetic and evolutionary methodologies for differentiation of V. parahaemolyticus, such as Multi-Locus Sequence Typing (MLST) and Multiple-Locus Variable-Number Tandem-Repeat Analysis (MLVA), which may provide greater discrimination.
For this study, we selected two sequence-based typing methods, MLST and MLVA. MLST is based on sequence diversity of loci which are generally well-conserved, or under purifying selection, as is the case with housekeeping genes. MLST is a frequently used typing method for many organisms and the development of a public database (PubMLST) has simplified sequence analysis and identification of evolutionary relationships within bacterial species (Maiden et al., 1998; Perez-Losada et al., 2013). MLST was selected to examine the current strain collection based on the previous success of the technique for characterizing diverse environmental and clinical V. parahaemolyticus isolates (Gonzalez-Escalona et al., 2008) and to add a diverse set of isolates to the MLST database. MLVA has also been used to distinguish isolates with little genetic variation. MLVA uses PCR for amplification of size polymorphisms in several Variable-Number Tandem-Repeat (VNTR) loci (Lindstedt, 2005). The VNTRs are highly polymorphic and are well suited for differentiation of bacterial isolates (Lindstedt, 2005; van Belkum, 2007). MLVA was selected for use in this study as it is able to differentiate between indistinguishable PFGE patterns for V. parahaemolyticus (Hayat et al., 1993; Harth-Chu et al., 2009) or identical MLST sequence types (STs) in other organisms (Maiden et al., 1998, 2013). Recently, MLVA was used for epidemiological analysis for discrimination of clinical and environmental V. parahaemolyticus isolates with indistinguishable Direct Genome Restriction Enzyme Analysis (DGREA) patterns (Harth-Chu et al., 2009; Garcia et al., 2012). Generally, the PCR products from MLVA are separated by agarose gel or capillary electrophoresis (CE) (Lindstedt et al., 2013). However, differentiation of amplification products using high resolution melt (HRM) analysis has been described for MLVA assays in other organisms (Fortini et al., 2007).
The objective of this study was to evaluate the combined use of MLST and the HRM-MLVA sequence-based methods for discrimination of environmental and clinical V. parahaemolyticus isolates previously characterized by other fingerprinting methods, such as PFGE (Ludeke et al., 2014). MLST was selected to identify phylogenetic relationships while MLVA was applied with the hypothesis that it will further discriminate isolates with identical STs. In order to achieve this objective, an HRM-MLVA protocol for rapid and simple characterization of V. parahaemolyticus isolates was developed. This is the first study, to the authors' knowledge, to use the combined approach of the subtyping methods MLST, HRM-MLVA, and PFGE for differentiation of V. parahaemolyticus isolates.
For the MLST and MLVA analysis, 58 V. parahaemolyticus isolates were selected; among those were 28 environmental (oyster) and 30 clinical isolates. Isolates were selected to represent multiple collection states and serotypes (Table 1). Each isolate was inoculated into Luria Bertani broth with 1% NaCl and incubated with shaking overnight at 35°C. Afterwards, 1 mL of the overnight culture was transferred to a 1.5 mL microcentrifuge tube, heated to 100°C for 10 min, and placed in ice for 5 min. The samples were stored at −20°C until used as a PCR template.
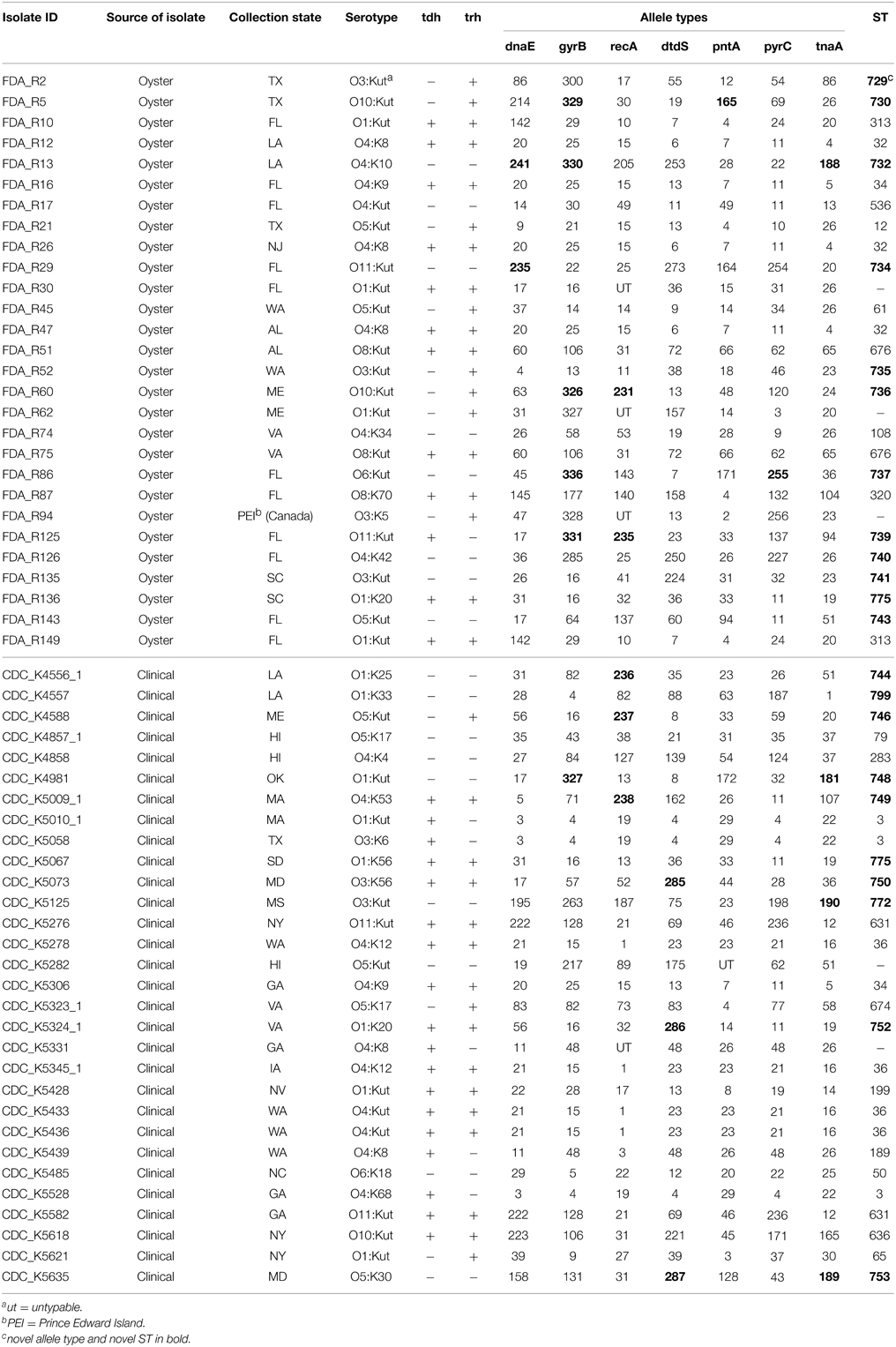
Table 1. Isolates used in this study and their sequence types (ST).
MLST was performed as described in the protocol for Vibrio parahaemolyticus (Gonzalez-Escalona et al., 2008). The PCR products were purified using a PCR purification kit (Qiagen, Valencia, CA) with a total elution volume of 25 μL. The purified samples were sequenced on an ABI 3730 × l sequencer at McLab (South San Francisco, CA). Sequences were analyzed with BioEdit software 7.1.9 (Abbott, Carlsbad, CA). The allelic and sequence type (ST) identification was determined using the MLST database (http://pubmlst.org/vparahaemolyticus). In the cases where whole genome sequence data was available, sequences were de novo assembled using CLC Genomics Workbench software 7.0.3 (CLCbio, Germantown, MD) and consensus assemblies submitted to the MLST database for allelic and ST identification.
For identification of clonal complexes, eBURST version 3 was used (http://eburst.mlst.net/). As reported previously, two different STs were considered single-locus variant (SLV) when they differed by a single locus; a double-locus variant (DLV) has two different loci (Gonzalez-Escalona et al., 2008). To be part of a clonal complex, isolates needed to share at least six out of seven alleles.
The minimum evolution tree of the concatenated sequences of the seven loci was built based on the method of Kimura-2-parameter in Mega 6 (Tamura et al., 2013). The ratio between the number of synonymous and non-synonymous substitutions, showing the type of selection at each locus, was calculated using the method of Nei and Gojobori in Mega 6. The hypotheses of neutrality (dS = dN), purifying selection (dS/dN > 1), and positive selection (dS/dN < 1) were tested.
Three multiplex real-time-PCR assays using HRM curve analysis were performed with the LightCycler® 480 High Resolution Melting Master Kit (Roche, Indianapolis, IN). The primer sequences utilized in this study are listed in Table 2 and have been previously described (Harth-Chu et al., 2009). These primers were utilized as unmodified oligonucleotides since HRM was used as the detection method rather than CE. Each reaction mixture for the multiplex PCR A and B (Multi A and B) had a final volume of 20 μL and consisted of: 1X master mix solution (Roche), 2 mM MgCl2 (Roche), 0.2 μM each primer (Integrated DNA Technologies; IDT, Coralville, IA), and 5 μL boiled template. For multiplex PCR C (Multi C), reaction concentrations were the same as for Multi A and B, except for primer pair VP2-07; 0.4 μM of each primer was used. The temperature program was as described previously for Multi A and B (Harth-Chu et al., 2009): Initial denaturation at 95°C for 15 min, followed by 20 cycles of a touchdown PCR consisting of denaturation at 94°C for 30 s, annealing starting at 63°C and decreasing 0.2°C per cycle for 1.5 min, and elongation at 72°C for 1 min. A final 10 cycles of denaturation at 94°C for 30 s, annealing at 59°C for 1.5 min, and elongation at 72°C for 1 min was used. The cycling program of Multi C included the same initial denaturation as for Multi A and B; cycling consisted of 30 cycles of denaturation at 94°C for 30 s, annealing at 61°C for 1.5 min, and elongation at 72°C for 1 min. A final annealing step at 60°C for 30 min completed both cycling programs. The melt curve analysis was performed using the Roche High Resolution Melting Kit protocol. The temperature program for the HRM analysis started at 95°C for 1 min with a ramp of 4.4°C per s, followed by 40°C for 1 min with a ramp of 2.2°C per s, 60°C for 1 s with a ramp of 4.4°C per s and a continuous step at 95°C. For the PCR amplification and HRM analysis a LightCycler® 480 (Roche) was used.
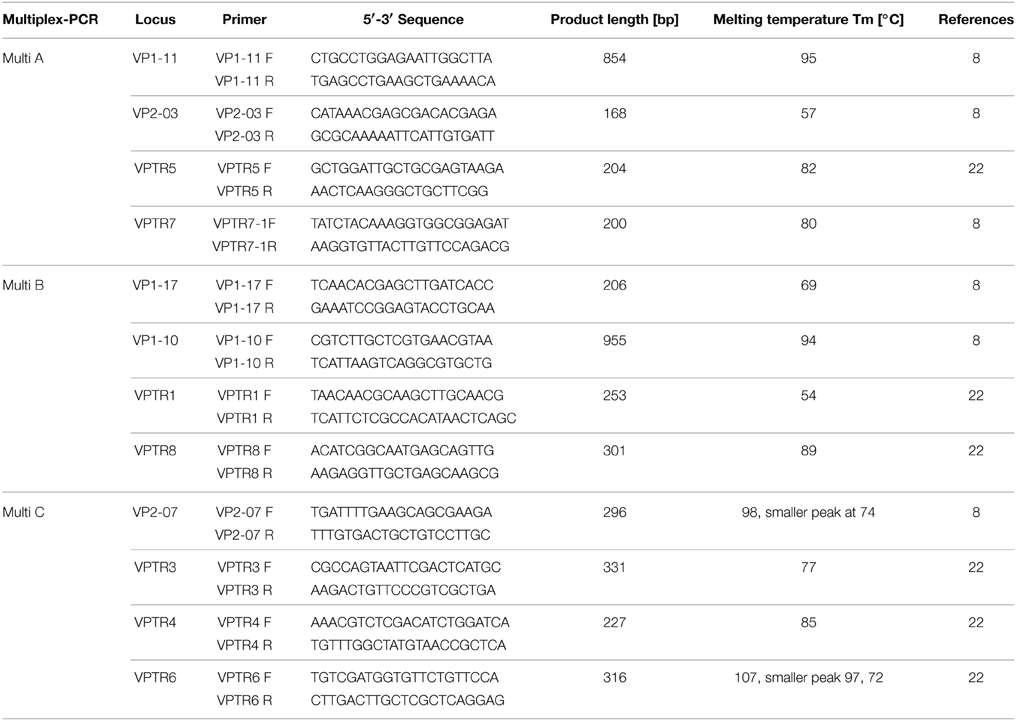
Table 2. Primer sequences including their amplification product length and melting temperature for MLVA multiplex PCR.
Due to overlapping peaks in Multi A and C, the presence of the target loci VNTR5 and VNTR7, VNTR3, and VP2-07 were confirmed using simplex real-time-PCR for each gene with the same PCR reaction conditions and cycling parameters as described for the multiplex, but with omission of the three other primer sets. The melt curves were analyzed with BioNumerics software 6.6 with a customized script (Applied Maths, Austin, TX). This script compares the melting curves of each multiplex PCR, as well as a combination of all curves from the three multiplex PCRs. The combined dendrogram of all three multiplex PCR was built based on the Pearson correlation of average trend curves in BioNumerics as well. This dendrogram was also converted to a rendered rooted tree.
From the 58 V. parahaemolyticus isolates analyzed, MLST resulted in 41 different STs (Table 1). Four (6.9%) and one (1.7%) of the isolates were untypeable for recA and pntA, respectively. For those strains, no ST could be assigned. Twelve (42.9%) of the oyster and ten (40.0%) of the clinical isolates were a novel ST. The most frequently identified STs were ST36 (13.3%) and ST3 (10.0%) in clinical isolates and ST32 (10.7%), ST313 (7.1%), and ST676 (7.1%) in oyster isolates.
All loci showed ratios of synonymous and non-synonymous substitutions (dN/dS) below 1 and therefore under purifying selection, as expected for housekeeping genes. eBURST analysis divided the 53 isolates for which a ST could be identified into 37 singletons and two groups: one SLV and one DLV (data not shown). No clonal complexes could be identified; demonstrating that none of the STs identified in this study share more than six alleles and, therefore, belong to different V. parahaemolyticus lineages.
A minimum evolution tree was constructed using the concatenated sequences of each allele (Figure 1). The isolates grouped into two main clusters, or lineages (I and II), with each lineage containing ST of clinical and oyster isolates. Isolates with the same ST generally had the same serotype; ST631 isolates possessed serotype O11:Kut, ST676 were serotype O8:Kut, ST36 were serotype O4:K12 or O4:Kut, and ST313 were serotype O1:Kut. However, the three ST3 isolates had all different serotypes.
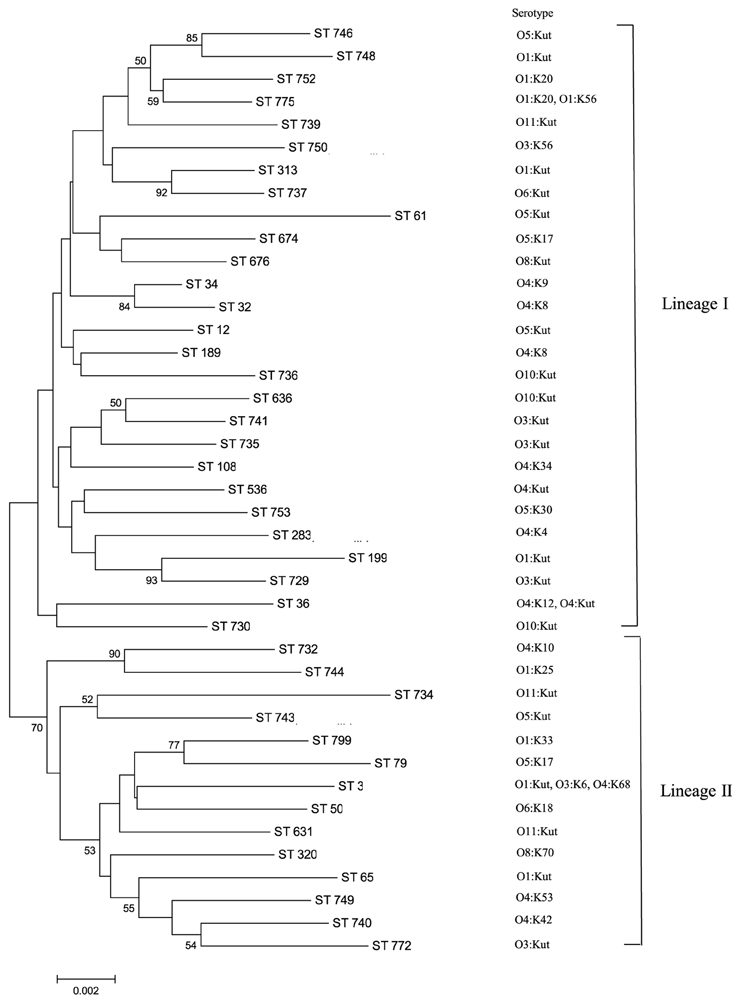
Figure 1. MLST minimum evolution tree of the 58 V. parahaemolyticus isolates. The tree was built with Mega six software using concatenated sequences. The scale represents the evolutionary distance and the branches show bootstrap values above 50%.
The 58 V. parahaemolyticus isolates from this study were further analyzed by MLVA with HRM analysis. Three multiplex PCRs covering twelve different loci were used. Each multiplex PCR generated reproducible melting curve profiles of select isolates (data not shown). Table 3 shows the percentage of isolates from which each target sequence in the MLVA scheme was amplified. From the Multi A, VP2-03, VPTR7, or VP1-11 was not amplified in 10% and 14.3%, 46.6% and 39.3%, or 3.3% and 3.6% of clinical and oyster isolates, respectively; VPTR5 was amplified from all isolates. In Multi B, VP1-10 or VPTR8 was not present in 96.7% and 17.9%, 23.3% and 17.9% of clinical and oyster isolates, respectively; VPTR1 and VP1-17 was amplified in all isolates with the exception of 6.7% of clinical isolates not amplifying VPTR1. From Multi C, VPRT6 was not amplified in 83.3% and 85.7% of clinical and oyster isolates, respectively. All isolates amplified VPTR4, VPTR3, and VP2-07, with the exception of 3.6% of oyster isolates for VPTR4.
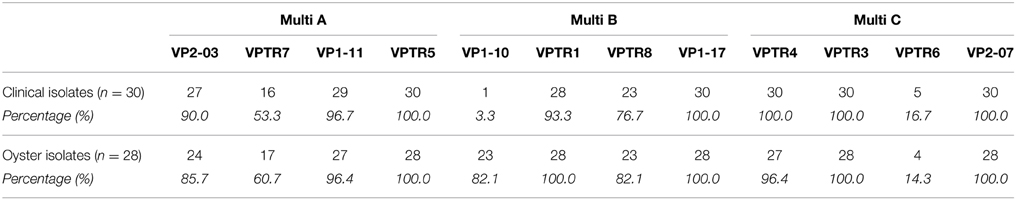
Table 3. Presence of individual MLVA genes in clinical and oyster isolates.
Using the trend curves of each individual multiplex PCR, dendrograms were constructed (data not shown). Each multiplex PCR generally clustered the isolates based on their serotypes and ST. The individual multiplex PCR dendrograms demonstrated the ability of MLVA to differentiate between the same ST (data not shown).
Based on the hypothesis MLVA can differentiate isolates with the same ST and PFGE pattern, these isolates' MLVA patterns were compared to the MLST data, as well as previously published PFGE results (Ludeke et al., 2014). To compare these methods, dendrograms were built of the combined melting curves from the three MLVA multiplex PCRs and correlated to the PFGE cluster and ST of each isolate. MLVA allowed further differentiation of isolates with identical STs and PFGE clusters (Figure 2). Specifically, the isolates with ST3 and ST36 share the same PFGE cluster, but were distinguishable by MLVA melting curve profiles (Figure 3). The dendrogram with only ST3 and ST36 isolates showed ST-specific clusters, but separation within those clusters based on the combined melting curves of MLVA.

Figure 2. Combined dendrogram of MLVA melting curves of the three multiplex PCRs built with BioNumerics software version 6.6. using Pearson correlation and the unweighted pair group method using arithmetic averages (UPGMA). Isolates originated from oysters starting with “FDA,” isolates from clinical origin labeled with “CDC.” The PFGE pattern designations are as previously reported (Ludeke et al., 2014).

Figure 3. Dendrogram of MLVA melting curves of the three multiplex PCRs for the isolates carrying ST3 and ST36 built with BioNumerics software version 6.6. using Pearson correlation and the unweighted pair group method using arithmetic averages (UPGMA). The PFGE cluster designations are as previously reported (Ludeke et al., 2014).
This study analyzed 58 V. parahaemolyticus isolates by MLST and a newly-developed HRM-MLVA assay to investigate the relatedness of the isolates. The seven gene MLST protocol reported in a previous study was employed in this study (Gonzalez-Escalona et al., 2008). A different MLST method using ten housekeeping genes was described (Yan et al., 2011). Both methods report the same level of discrimination; however, the seven gene protocol was selected due to the availability of a public repository for the data (http://pubmlst.org/vparahaemolyticus), which allows comparisons to be made with isolates analyzed by other researchers. With 22 novel STs, this study substantially contributed to the diversity in the MLST database. Four of our isolates were untypeable for recA. In a recently published study, a V. parahaemolyticus strain contained a recA gene that was fragmented by a 30 kb DNA insertion (Gonzalez-Escalona et al., 2015). It is possible a similar insertion exists in the recA gene of some of the strains in the current study, but further analysis is needed to confirm.
ST3, ST32, and ST36 were the STs that occurred most often in our isolates, as well as in the public database. In our study, the fourth most frequent ST observed was ST676, which is one of the novel STs reported here. Two of these STs (ST3 and ST36) have been reported as part of clonal complexes CC3 and CC36, respectively (Gonzalez-Escalona et al., 2008), and correlated with outbreaks in multiple countries, including the US and Chile (Fuenzalida et al., 2006; Martinez-Urtaza et al., 2013). ST3 was identified as the ancestral ST of CC3 with ST27, ST42, and ST51 as SLVs. None of other STs from CC3 were identified in this study. A previous study using MLST on a set of clinical and environmental isolates from the Pacific Northwest Region of the United States showed that some environmental isolates were of ST3, suggesting a higher potential for virulence than other environmental isolates (Turner et al., 2013). In this study, three clinical isolates were ST3 and no direct relationship to environmental or other clinical clades were observed.
We developed an HRM-MLVA method, based on an existing MLVA method that uses CE, for subtyping of V. parahaemolyticus and to differentiate between similar PFGE patterns or STs. The CE method provides the actual number of tandem repeats while HRM does not. However, the HRM analysis still recognizes allelic variants and is able to distinguish between otherwise indistinguishable strains. For example, the ST3, ST32, and ST36 strains in this study also shared common PFGE profiles, but each isolate produced a unique HRM curve combination. This resolving power of HRM-MLVA is similar to previous reports of CE-MLVA, where Chilean isolates which shared a DGREA pattern and were ST3 could be differentiated by CE-MLVA (Gonzalez-Escalona et al., 2008; Harth-Chu et al., 2009). These data demonstrate that the HRM-MLVA method developed in this study provides similar discrimination as previously reported for the CE-MLVA method and is suitable examination of V. parahaemolyticus isolates. Additionally, the use of HRM-MLVA on the LightCycler®480 saves the step of electrophoretic detection, thus minimizing the potential for cross contamination by PCR amplicons during that additional handling step.
The loci amplified in this study by MLVA are coding proteins such as a putative hemolysin (VPTR4) and putative collagenase (VPTR3) (Kimura et al., 2008). Most of these genes, could be amplified from the current strain selection with the exception of VPTR7 (Multi A), VP1-10 (Multi B), and VPTR6 (Multi C). Nearly all clinical isolates failed to amplify the VP1-10 locus and approximately half failed to amplify the locus VPTR7. A failure to amplify VPTR7 from some shellfish isolates has been reported previously (Harth-Chu et al., 2009). Fewer than 20% of isolates amplified VPTR6 in this study. Additionally, a previous study found VPTR6 to be one of the few loci with high genetic diversity (Harth-Chu et al., 2009). Together, these data suggest that these two loci might not be suitable targets for future MLVA studies, especially for environmental isolate screening. Nonetheless, the HRM-MLVA method successfully discriminated between otherwise indistinguishable V. parahaemolyticus isolates.
Previous studies have employed multiple methods for characterization and subtyping of V. parahaemolyticus isolates from various sources. For example, DePaola et al. used a combination of serotyping and ribotyping to identify types more highly associated with clinical isolates (DePaola et al., 2003). Turner et al. utilized REP-PCR, as fingerprint-based subtyping method, followed by MLST to identify region-specific clades of V. parahaemolyticus (Turner et al., 2013). Banerjee et al. employed PFGE, MLST, serotyping, and ribotyping to examine clinical V. parahaemolyticus isolates and provided combinatorial analysis to determine relatedness (Banerjee et al., 2014). However, none of these previous studies utilized the combination of two highly discriminatory, sequence-based methods as does the current study.
This combined method approach described here using PFGE, MLST, and MLVA has not been previously reported for discrimination of V. parahaemolyticus isolates, but is similar to approaches used for other organisms: for example, the Centers for Disease Control and Prevention has used a combination of PFGE followed by MLVA for epidemiological investigations of STEC O157 to discriminate between closely related isolates (Hyytia-Trees et al., 2006). Also, a combination of MLST and MLVA has been used as an epidemiological tool for distinguishing between clones of Listeria monocytogenes (Chenal-Francisque et al., 2013).
This study used a combined method approach to increase the discrimination of V. parahaemolyticus isolates. MLST was able to determine the identity and the phylogenetic relatedness of the isolates in this collection. As hypothesized, the developed HRM-MLVA method further refined the relationship of isolates by being able to distinguish between isolates with indistinguishable PFGE groupings or STs. Our data demonstrates that a combination of PFGE, MLST, and HRM-MLVA would be the most suitable approach for outbreak and evolutionary investigations of V. parahaemolyticus, due to the high resolution provided. In instances where further discrimination is needed, and if available, next generation sequence data could be used to determine relatedness or to generate subtyping results in silico.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This project was supported by an appointment of CL to the Research Fellowship Program for the Center for Food Safety and Applied Nutrition administered by the Oak Ridge Associated Universities through a contract with the FDA.
Banerjee, S. K., Kearney, A. K., Nadon, C. A., Peterson, C. L., Tyler, K., Bakouche, L., et al. (2014). Phenotypic and genotypic characterization of Canadian clinical isolates of Vibrio parahaemolyticus collected from 2000 to 2009. J. Clin. Microbiol. 52, 1081–1088. doi: 10.1128/JCM.03047-13
Broberg, C. A., Calder, T. J., and Orth, K. (2011). Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect. 13, 992–1001. doi: 10.1016/j.micinf.2011.06.013
Chenal-Francisque, V., Diancourt, L., Cantinelli, T., Passet, V., Tran-Hykes, C., Bracq-Dieye, H., et al. (2013). Optimized Multilocus variable-number tandem-repeat analysis assay and its complementarity with pulsed-field gel electrophoresis and multilocus sequence typing for Listeria monocytogenes clone identification and surveillance. J. Clin. Microbiol. 51, 1868–1880. doi: 10.1128/JCM.00606-13
DePaola, A., Ulaszek, J., Kaysner, C. A., Tenge, B. J., Nordstrom, J. L., Wells, J., et al. (2003). Molecular, serological, and virulence characteristics of Vibrio parahaemolyticus isolated from environmental, food, and clinical sources in North America and Asia. Appl. Environ. Microbiol. 69, 3999–4005. doi: 10.1128/AEM.69.7.3999-4005.2003
Fortini, D., Ciammaruconi, A., De, S. R., Fasanella, A., Battisti, A., D'Amelio, R., et al. (2007). Optimization of high-resolution melting analysis for low-cost and rapid screening of allelic variants of Bacillus anthracis by multiple-locus variable-number tandem repeat analysis. Clin. Chem. 53, 1377–1380. doi: 10.1373/clinchem.2007.085993
Fuenzalida, L., Hernandez, C., Toro, J., Rioseco, M. L., Romero, J., and Espejo, R. T. (2006). Vibrio parahaemolyticus in shellfish and clinical samples during two large epidemics of diarrhoea in southern Chile. Environ. Microbiol. 8, 675–683. doi: 10.1111/j.1462-2920.2005.00946.x
Garcia, K., Gavilan, R. G., Hofle, M. G., Martinez-Urtaza, J., and Espejo, R. T. (2012). Microevolution of pandemic Vibrio parahaemolyticus assessed by the number of repeat units in short sequence tandem repeat regions. PLoS ONE 7:e30823. doi: 10.1371/journal.pone.0030823
Gonzalez-Escalona, N., Gavilan, R. G., Brown, E. W., and Martinez-Urtaza, J. (2015). Transoceanic spreading of pathogenic strains of Vibrio parahaemolyticus with distinctive genetic signatures in the recA gene. PLoS ONE 10:e0117485. doi: 10.1371/journal.pone.0117485
Gonzalez-Escalona, N., Martinez-Urtaza, J., Romero, J., Espejo, R. T., Jaykus, L. A., and DePaola, A. (2008). Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 190, 2831–2840. doi: 10.1128/JB.01808-07
Harth-Chu, E., Espejo, R. T., Christen, R., Guzman, C. A., and Hofle, M. G. (2009). Multiple-locus variable-number tandem-repeat analysis for clonal identification of Vibrio parahaemolyticus isolates by using capillary electrophoresis. Appl. Environ. Microbiol. 75, 4079–4088. doi: 10.1128/AEM.02729-08
Hayat, U., Reddy, G. P., Bush, C. A., Johnson, J. A., Wright, A. C., Morris, J. G., et al. (1993). Capsular types of Vibrio vulnificus: an analysis of strains from clinical and environmental sources. J. Infect. Dis. 168, 758–762. doi: 10.1093/infdis/168.3.758
Hyytia-Trees, E., Smole, S. C., Fields, P. A., Swaminathan, B., and Ribot, E. M. (2006). Second generation subtyping: a proposed PulseNet protocol for multiple-locus variable-number tandem repeat analysis of Shiga toxin-producing Escherichia coli O157 (STEC O157). Foodborne. Pathog. Dis. 3, 118–131. doi: 10.1089/fpd.2006.3.118
Iwamoto, M., Ayers, T., Mahon, B. E., and Swerdlow, D. L. (2010). Epidemiology of seafood-associated infections in the United States. Clin. Microbiol. Rev. 23, 399–411. doi: 10.1128/CMR.00059-09
Jones, J. L., Ludeke, C. H., Bowers, J. C., Garrett, N., Fischer, M., Parsons, M. B., et al. (2012). Biochemical, serological, and virulence characterization of clinical and oyster Vibrio parahaemolyticus isolates. J. Clin. Microbiol. 50, 2343–2352. doi: 10.1128/JCM.00196-12
Kimura, B., Sekine, Y., Takahashi, H., Tanaka, Y., Obata, H., Kai, A., et al. (2008). Multiple-locus variable-number of tandem-repeats analysis distinguishes Vibrio parahaemolyticus pandemic O3:K6 strains. J. Microbiol. Methods 72, 313–320. doi: 10.1016/j.mimet.2007.12.014
Lindstedt, B. A. (2005). Multiple-locus variable number tandem repeats analysis for genetic fingerprinting of pathogenic bacteria. Electrophoresis 26, 2567–2582. doi: 10.1002/elps.200500096
Lindstedt, B. A., Torpdahl, M., Vergnaud, G., Le, H. S., Weill, F. X., Tietze, E., et al. (2013). Use of multilocus variable-number tandem repeat analysis (MLVA) in eight European countries, 2012. Euro. Surveill. 18:20385.
Ludeke, C. H., Fischer, M., LaFon, P., Cooper, K., and Jones, J. L. (2014). Suitability of the molecular subtyping methods intergenic spacer region, direct genome restriction analysis, and pulsed-field gel electrophoresis for clinical and environmental Vibrio parahaemolyticus isolates. Foodborne. Pathog. Dis. 11, 520–528. doi: 10.1089/fpd.2013.1728
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Maiden, M. C., Jansen van Rensburg, M. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley, K. A., et al. (2013). MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736. doi: 10.1038/nrmicro3093
Martinez-Urtaza, J., Baker-Austin, C., Jones, J. L., Newton, A. E., Gonzalez-Aviles, G. D., and DePaola, A. (2013). Spread of Pacific Northwest Vibrio parahaemolyticus strain. N. Engl. J. Med. 369, 1573–1574. doi: 10.1056/NEJMc1305535
Nishibuchi, M., and DePaola, A. (2005). “Vibrio spp.,” in Foodborne Pathogens: Microbiology and Molecular Biology, eds P. M. Fratamico, A. K. Bhunia, and J. L. Smith (Norfolk: Caister Academic Press), 251–271.
Paranjpye, R., Hamel, O. S., Stojanovski, A., and Liermann, M. (2012). Genetic diversity of clinical and environmental Vibrio parahaemolyticus strains from the Pacific Northwest. Appl. Environ. Microbiol. 78, 8631–8638. doi: 10.1128/AEM.01531-12
Perez-Losada, M., Cabezas, P., Castro-Nallar, E., and Crandall, K. A. (2013). Pathogen typing in the genomics era: MLST and the future of molecular epidemiology. Infect. Genet. Evol. 16, 38–53. doi: 10.1016/j.meegid.2013.01.009
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Turner, J. W., Paranjpye, R. N., Landis, E. D., Biryukov, S. V., Gonzalez-Escalona, N., Nilsson, W. B., et al. (2013). Population structure of clinical and environmental Vibrio parahaemolyticus from the Pacific Northwest coast of the United States. PLoS ONE 8:e55726. doi: 10.1371/journal.pone.0055726
van Belkum, A. (2007). Tracing isolates of bacterial species by multilocus variable number of tandem repeat analysis (MLVA). FEMS Immunol. Med. Microbiol. 49, 22–27. doi: 10.1111/j.1574-695X.2006.00173.x
Xu, F., Ilyas, S., Hall, J. A., Jones, S. H., Cooper, V. S., and Whistler, C. A. (2015). Genetic characterization of clinical and environmental Vibrio parahaemolyticus from the Northeast USA reveals emerging resident and non-indigenous pathogen lineages. Front. Microbiol. 6:272. doi: 10.3389/fmicb.2015.00272
Keywords: Vibrio parahaemolyticus, phylogenetics, MLVA, MLST, HRM
Citation: Lüdeke CHM, Gonzalez-Escalona N, Fischer M and Jones JL (2015) Examination of clinical and environmental Vibrio parahaemolyticus isolates by multi-locus sequence typing (MLST) and multiple-locus variable-number tandem-repeat analysis (MLVA). Front. Microbiol. 6:564. doi: 10.3389/fmicb.2015.00564
Received: 27 March 2015; Accepted: 22 May 2015;
Published: 10 June 2015.
Edited by:
Indrani Karunasagar, Karnataka Veterinary Animal and Fisheries Sciences University, IndiaCopyright © 2015 Lüdeke, Gonzalez-Escalona, Fischer and Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica L. Jones, Gulf Coast Seafood Laboratory, Division of Seafood Science and Technology, Food and Drug Administration, 1 Iberville Dr, Dauphin Island, AL 36528, USA,amVzc2ljYS5qb25lc0BmZGEuaGhzLmdvdg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.