
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 12 May 2015
Sec. Plant Pathogen Interactions
Volume 6 - 2015 | https://doi.org/10.3389/fmicb.2015.00440
This article is part of the Research TopicEmerging tools for emerging symbioses—using genomics applications to studying endophytesView all 14 articles
 Raheleh Sheibani-Tezerji1,2
Raheleh Sheibani-Tezerji1,2 Muhammad Naveed1†
Muhammad Naveed1† Marc-André Jehl2
Marc-André Jehl2 Angela Sessitsch1
Angela Sessitsch1 Thomas Rattei2*
Thomas Rattei2* Birgit Mitter1*
Birgit Mitter1*The seed as a habitat for microorganisms is as yet under-explored and has quite distinct characteristics as compared to other vegetative plant tissues. In this study, we investigated three closely related P. ananatis strains (named S6, S7, and S8), which were isolated from maize seeds of healthy plants. Plant inoculation experiments revealed that each of these strains exhibited a different phenotype ranging from weak pathogenic (S7), commensal (S8), to a beneficial, growth-promoting effect (S6) in maize. We performed a comparative genomics analysis in order to find genetic determinants responsible for the differences observed. Recent studies provided exciting insight into the genetic drivers of niche adaption and functional diversification of the genus Pantoea. However, we report here for the first time on the analysis of P. ananatis strains colonizing the same ecological niche but showing distinct interaction strategies with the host plant. Our comparative analysis revealed that genomes of these three strains are highly similar. However, genomic differences in genes encoding protein secretion systems and putative effectors, and transposase/integrases/phage related genes could be observed.
Bacterial endophytes have been defined as “bacteria, which for all or part of their life cycle invade the tissues of living plants and cause unapparent and asymptomatic infections entirely within plant tissues, but cause no symptoms of disease” (Wilson, 1995). Based on this definition, endophytes are clearly distinct from plant pathogens. However, bacteria can exist in plants in quiescence but proliferate and become detrimental to the host under certain conditions such as plant growth perturbations (Kloepper et al., 2013). Moreover, plant-pathogen interactions are often plant species specific and bacteria that are pathogenic to one plant species can exhibit an endophytic lifestyle in other plants (Bashan et al., 1982). On the other hand, it has been shown that plants respond differently to endophytes and plant pathogens (Bordiec et al., 2011). A promising approach in revealing differences in the host interaction strategies of pathogens and plant beneficial bacteria might be the comparison of functionalities and gene content of closely related bacterial strains that show different modes of interaction with host plants. Genome sequencing provides detailed information on the genes present in bacteria and offers a basis for comparative genomics that aids in revealing differences in the host interaction strategies of pathogens and plant growth promoting bacteria.
The seed as a habitat for microorganisms is under-explored, although the first report of bacteria colonizing seeds dates back to the 1970s (Mundt and Hinkle, 1976). Only few studies have been performed on seed endophytes (Compant et al., 2011; Johnston-Monje and Raizada, 2011; Hardoim et al., 2012) and the origin of endophytes is under debate. A few studies suggest that at least some bacterial endophytes are vertically transmitted (Johnston-Monje and Raizada, 2011). The seed has quite distinct characteristics as compared to other vegetative plant tissues and one would expect that it also harbors distinct microbial communities. Based on cultivation-based analysis it has been reported that Gammaproteobacteria represent the most abundant class of maize seed endophytes, comprising mostly Pantoea and Enterobacter (Johnston-Monje and Raizada, 2011). Similarly, Rijavec et al. (2007) identified Pantoea as a major genus among endophytes isolated from maize seeds.
Pantoea ananatis is a bacterial species that was originally discovered in pineapple in the Philippines, in 1928 (Serrano, 1928). Members of this species have been shown to infect many mono- and dicotyledonous plant species, such as onion, rice, melon, sudan grass, tomato, and sorghum (Stall et al., 1969; Wells et al., 1987; Gitaitis and Gay, 1997; Azad et al., 2000; Cother et al., 2004; Cota et al., 2010). In maize P. ananatis is the causing agent of the foliar disease termed maize white spot disease (Paccola-Meirelles et al., 2001). P. ananatis strains display a wide range of ecological versatility, as they are commonly recovered from water, soil, insects, and plants (De Maayer et al., 2014). Depending on their host and ecological niches, P. ananatis strains can show different life styles such as mutualistic, saprophytic and pathogenic life styles (Coutinho and Venter, 2009). De Maayer et al. (2012a) showed that the Large Pantoea Plasmid (LLP-1) plays a crucial role in niche adaption and functional diversification of the genus Pantoea. By analyzing the pan-genome of eight sequenced P. ananatis strains De Maayer et al. (2014) identified a large number of proteins in this species with orthologs restricted to bacteria associated either with plants, animals or insects. The mechanisms of the diverse interactions between P. ananatis and the host are still poorly understood and only little is known on the genetic traits underlying plant pathogenic or beneficial activity. Shyntum et al. (2014) showed that type IV section system could play a role in pathogenicity and niche adaptation. Genome analysis of the plant growth promoting strain P. ananatis B1-9 that has been isolated from the rhizosphere of green onions in Korea indicates that the strain lacks traits related to pathogenicity. Furthermore, it harbors genes that are putatively involved in plant growth stimulation and yield improvement (Kim et al., 2012).
In this work, we studied three endophytic P. ananatis strains (S6, S7, S8) isolated from maize seeds. Although they were isolated from seeds of healthy plants, they showed distinct characteristics in regard to plant growth and health. Strain S6 exhibited clear beneficial effects on maize growth, whereas S8 had hardly any effect and is considered as neutral and S7 caused disease symptoms known from P. ananatis infections. Therefore, this closely related group of strains represents a promising model to unravel genetic determinants in P. ananatis responsible for beneficial and pathogenic effects. Consequently, we functionally characterized the strains by testing for various known plant growth-promoting characteristics as well as for their effect on plant growth, and performed a comparative genome analysis to elucidate genetic features determining the type of plant-microbe interaction.
Seeds of the maize cultivars (Helmi, Morignon, Pelicon, and Peso) were obtained from local farmers in Seibersdorf, Austria. Maize seeds with no cracks or other visible deformations were surface-sterilized with 70% ethanol for 3 min and 5% sodium hypochlorite for 5 min, and followed by repeated washing with sterile distilled water (3 times for 1 min). The efficacy of surface sterilization was checked by plating 3–5 seeds and aliquots of the final rinse onto 10% tryptic soy agar plates, and incubated for 3 days at 28 ± 1°C. The medium was checked daily for bacterial or fungal growth.
Seed-borne bacteria were isolated following the procedure described by Rijavec et al. (2007) with some modifications. For isolation, 50 surface-sterilized seeds of each cultivar were crushed and blended aseptically in 90 mL of half strength nutrient broth (Difco, Detroit, Michigan) for 5 min. The blend was then incubated at room temperature for 4 h on a rotary shaker (VWR International GmbH, Austria) at 100 r min−1. Half strength nutrient broth containing 200 mg/L cycloheximide was inoculated with a series of the incubation mixture (10:1 mL ratio) and further incubated for 4 days on a rotary shaker at room temperature. Aliquots were taken from Erlenmeyer flasks with observed microbial growth and plated onto R2A (Difco, Detroit, Michigan). Plates were incubated at 28°C for 24–48 h. One hundred colonies were picked, and pure cultures were obtained by further streaking on agar plates. Single colonies were picked, inoculated in LB broth and incubated with shaking at 28°C for 24–48 h. Bacterial strains were preserved at −80°C as saturated cultures containing 20% (w/v) glycerol.
For phylogenetic identification of maize seed endophytes we performed partial 16S rDNA (V1 to V3) PCR and sequencing as described by Reiter and Sessitsch (2006). Sequencing was performed by LGC Genomics (Berlin, Germany).
Inoculum of the selected strains (S6, S7, S8) were prepared in 100 mL 10% tryptic soy broth in 250 mL Erlenmeyer flasks and incubated at 28 ± 2°C for 48 h in an orbital shaking incubator (VWR International, GmbH) at 180 r min−1. The optical density of the broth was adjusted to 0.5 measured at λ 600 nm using spectrophotometer (Gene Quant Pro, Gemini BV, The Netherlands) to obtain an uniform population of bacteria [108–109 colony-forming units (CFU) mL−1] in the broth at the time of inoculation.
Seeds were surface-sterilized by dipping them in 70% ethanol for 3 min and then in a 5% sodium hypochlorite solution for 5 min and subsequently thoroughly washing with sterilized distilled water. The efficacy of surface sterilization was checked by plating seeds, and aliquots of the final rinse onto 10% tryptic soy agar. Samples were considered to be successfully sterilized when no colonies were observed on the tryptic soy agar plates after inoculation for 3 days at 28°C. Surface-disinfected seeds of three maize cultivars (DaSilvie, Kaleo, and Mazurka) were immersed in the bacterial suspensions for 1 h. For the uninoculated control, sterilized tryptic soy broth was used for the seed treatment. Fifteen seeds per treatment were planted in plastic trays with sterilized compost (Blumenerde, COMPO SANA®) and trays were arranged using a randomized design with 3 replications resulting in total number of 45 seeds per treatment. The experiment was conducted for 24 days and data of shoot and root length as well as biomass were recorded.
Color and shape of bacterial colonies, growth behavior in different pHs, salt concentrations and C sources as well as aggregate and biofilm formation and motility were tested following the procedures described by Naveed et al. (2014). Biochemical testing of oxidase, catalase, gelatin hydrolysis, and casein hydrolysis activity of the selected strains was performed according to Naveed et al. (2014).
Strains were tested for activities known to be involved in plant growth regulation and/or rhizosphere competence such as ACC-deaminase activity, auxin production, phosphate solubilization (organic/inorganic P) and siderophore production as well as ammonia, hydrogen cyanide and PHB production as described by Naveed et al. (2014).
Bacterial cell wall hydrolyzing activities such as amylase, cellulase, chitinase, lipase, pectinase, phosphatase, protease, and xylanase were screened on diagnostic plates as described by Naveed et al. (2014).
Antibiotic resistance was tested individually on tryptic soy agar plates containing the antibiotics ampicillin, cycloheximide, gentamycin, kanamycin, chloramphenicol, rifampicin, spectinomycin, streptomycin or tetracycline respectively at the following concentrations: 25, 50, 75, 100 μg ml−1. The plates were incubated at 28 ± 2°C for 5 days and resistance was observed in terms of bacterial growth.
The antagonistic activities of bacterial isolates were screened against plant pathogenic fungi (Fusarium caulimons, Fusarium graminarium, Fusarium oxysporum, Fusarium solani, Rhizoctonia solani, Thielaviopsis basicola) and oomycetes Phytophthora infestans, Phytophthora citricola, Phytophthora cominarum). Antagonistic activity of the bacterial isolates against fungi and oomycetes was tested by the dual culture technique on potato dextrose agar (PDA) and yeast malt agar (YMA) media as described by Naveed et al. (2014).
The data of plant growth parameters and colonization were subjected to analyses of variance (ANOVA). The means were compared with Least Significant Difference (LSD) testing (p < 0.05) to detect statistical significance among treatments (Steel et al., 1997). Statistical analyses were conducted using SPSS software version 19 (IBM SPSS Statistics 19, USA).
For DNA isolation, the bacterial strains were grown by loop-inoculating one single colony in 5 mL LB broth. The bacterial cultures were incubated at 28 ± 2°C overnight at 180 rpm in a shaking incubator. The overnight cultures were used to inoculate 50 mL fresh LB broth and again incubated at 28 ± 2°C overnight at 180 rpm in a shaking incubator. Bacterial cells were harvested by centrifugation at 4700 rpm for 10 min at 4°C. DNA was extracted from bacterial cell pellets according to the following protocol: The cell pellet was washed with 5 mL lysis buffer (0.1 M NaCl; 0.05 M EDTA, pH 8.0), resuspended in 4 mL lysis buffer containing lysozyme (20 mg mL-1; Roche Diagnostics, Mannheim, Germany) and incubated at 37°C for 20 min. Then 300 μl of 10% sarkosyl was added and placed on ice for 5 min. DNA was extracted with phenol-chloroform-isoamylalcohol (25:24:1, Fluka, Sigma-Aldrich Co.) and re-extracted with chloroform (1:1, Merck, Darmstadt, Germany) followed by precipitation with 0.1 volume of 3 M sodium acetate (pH 5.2) and 2.5 volume of ice-cold absolute ethanol (Merck, Darmstadt, Germany) at −20°C overnight. DNA pellets were washed with 1 ml of 70% ethanol and dissolved in 100 μL TE buffer (10 mM Tris-HCl, pH 7.5; 1 mM EDTA, pH 8·0). DNA was treated with RNase A (final concentration 0.2 gl −1; Invitrogen, Carlsbad, CA) for 90 min at 37°C. DNA quality was analyzed by electrophoresis (80 V) on 0.8% (w/v) agarose gels stained with ethidium bromide. DNA concentration was measured using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
Genome sequencing of the three strains of P. ananatis (S6, S7, and S8) was done by GATC Biotech AG (Konstanz, Germany) using a Roche/454 GS-FLX system. After sequencing, pairwise analysis of average nucleotide identity (ANI) was performed between the P. ananatis strains with closed genome sequences and the strains S6, S7, and S8 draft genomes individually as described previously (Goris et al., 2007).
The raw reads from sequencing projects have been deposited at the European Nucleotide Archive (ENA, http://www.ebi.ac.uk/ena/data/view/) under the following project accession numbers: P. ananatis S6, PRJEB7511; P. ananatis S7, PRJEB7512, and P. ananatis S8, PRJEB7513. Genome assemblies are available in ENA under accession numbers CVNF01000001 to CVNF01000077 for P. ananatis S6, CVNG01000001 to CVNG01000071 for P. ananatis S7 and CVNH01000001 to CVNH01000061 for P. ananatis S8. The contigs were assembled using AMOScmp comparative assembler (Pop et al., 2004) and the Roche GS de novo assembler package (Newbler v2.6) in the 454 GS-FLXTM system (http://www.454.com/), indipendently. For AMOScmp assembly, four complete genomes of P. ananatis strains (P. ananatis AJ13355, P. ananatis LMG20103, P. ananatis LMG5342 and P. ananatis PA13) were used as potential reference genomes. As the assemblies based on P. ananatis AJ13355 resulted into highest coverage and mapping quality, this genome was used as reference genome for AMOScmp. The result of quality and coverage control of the assembly of each P. ananatis genome sequence was calculated using Qualimap v.1.0 (Garcia-Alcalde et al., 2012). In repetitive regions, such as rRNA operons, the assembly was further evaluated based on the read coverage distribution. Whole genome comparisons between P. ananatis S6, S7, and S8 strains were performed using Mauve v.2.3.1 (Darling et al., 2004). In Mauve, the Progressive Mauve algorithm was used to order the contigs against P. ananatis AJ13355 as reference genome. Genome assemblies are accessible via http://fileshare.csb.univie.ac.at/pantoea/.
Five complete genomes of P. ananatis strains with different life styles and environmental origin were used in the comparative genomics and phylogenetic analysis. P. ananatis PA13 (accession numbers CP003085 and CP003086) is known as a pathogen of rice causing grain and sheath rot (Choi et al., 2012). P. ananatis AJ13355 (accession numbers AP012032 and AP012033) shows saprophytic life style and was isolated from soil (Hara et al., 2012). P. ananatis LMG20103 (accession number CP001875) is a pathogenic strain causing the severe blight and dieback of Eucalyptus (De Maayer et al., 2010). P. ananatis LMG5342 (accession numbers HE617160 and HE617161) is an opportunistic human pathogen reported from clinical isolations (De Maayer et al., 2012b). P. vagans C9-1 (accession numbers CP002206, CP001893, CP001894, and CP001895) is known as a common plant epiphyte (Smits et al., 2010).
We constructed a phylogenetic tree for P. ananatis S6, S7, S8 and the Pantoea genomes mentioned above. P. vagans C9-1 was included as outgroup. Mauve v2.3.1 (Darling et al., 2004) was used to identify specific and shared SNPs between all compared genomes. The alignments of the genomes were checked manually to eliminate possible false positive SNPs in less conserved regions, particularly if they occur in direct neighborhood of insertions and deletions. The obtained SNPs were filtered based on the position of phylogenetic markers of P. ananatis AJ13355 as reference [identified by AMPHORA2; Wu and Scott (2012)] to get the core SNPs of the genome sequences of P. ananatis strains. Afterwards, the phylogenetic tree was computed with Geneious 8.0 (Kearse et al., 2012) using 1000 runs for bootstrapping.
Gene prediction and annotation were obtained from the in-house ConsPred workflow. ConsPred consists of two phases: ab initio as well as homology-based predictions. Ab initio predictions are followed by Genemark.hmm (Lukashin and Borodovsky, 1998), Glimmer (Delcher et al., 2007), Prodigal (Hyatt et al., 2010), Critica (Badger and Olsen, 1999) and additional homology based information derived from a BLAST search against the NCBI non-redundant sequence database (NR) (Sayers et al., 2012). Protein domains were predicted by InterProScan (Zdobnov and Apweiler, 2001). For protein sequences without significant hits in NR, functional annotation of protein-coding genes was obtained by a similarity search against the UniProt/SwissProt database (Uniprot consortium, 2009). Non protein-coding elements such as tRNA and rRNA were predicted using tRNAScan and RNAmmer tools, respectively (Lowe and Eddy, 1997; Lagesen et al., 2007). Non-coding RNA genes (ncRNAs) were identified and annotated by a search against RFAM database (Griffiths-Jones et al., 2005).
To check for the completeness of housekeeping genes in the genomes of strains S6, S7, and S8 we used AMPHORA2 (Wu and Scott, 2012) with 31 bacterial phylogenetic marker genes for inferring phylogenetic information.
To identify the plasmid sequences within the assembled contigs we compared the plasmid sequence of the closest reference genome (P. ananatis AJ13355) to the assembly of P. ananatis S6, S7 and S8 strains using Mauve v2.3.1 (Darling et al., 2004).
To visualize the coverage of the plasmids in the draft genome sequences, the plasmid sequence of P. ananatis AJ13355 were used as reference for comparative circular alignments of the three P. ananatis S6, S7, and S8 strains using the BLAST Ring Image Generator (Stothard and Wishart, 2005; Alikhan et al., 2011).
Paralogous and orthologous clusters were identified using OrthoMCL (Li et al., 2003) using the predicted proteomes of seven P. ananatis strains (P. ananatis AJ13355, P. ananatis LMG20103, P. ananatis LMG5342, P. ananatis PA13 and P. ananatis S6, S7, and S8 strains) which initially required an all-vs.-all blastp (E-value cut-off of 1 × 10−5). Then the mcl clustering algorithm was used to deduce the relationship between genes.
To identify eukaryotic-like protein domains (ELDs) in protein sequences in the genomes of strains S6, S7, and S8, those genomes were included in the individual ELD calculation procedure of the Effective web-portal (Jehl et al., 2011). The approach detects protein domains that are present in eukaryotic organisms and significantly enriched in pathogenic and symbiotic compared to non-pathogenic, non-host-associated bacteria. Using default settings, all eukaryotic-like protein domains with an enrichment score greater or equal to 4 were considered for comparison regarding functional differences in P. ananatis strains of diverse phenotype.
In a previous study, we isolated 90 bacterial strains from seeds of healthy maize plants grown at organic farming fields in Austria. Thirty-seven of these strains shared highest 16S rDNA sequence homology with P. ananatis strains (data not shown). Ten strains were randomly selected and tested for effects on seedling growth of maize grown in sterile hydroponic cultures (for a description see Naveed et al., 2014). Along with strains that did not influence maize seedling growth we found one strain with clear detrimental effect and other strains that promoted maize seedling growth (data not shown). One representative of each group was selected and further tested on maize grown in compost. Strain S6 significantly increased seedling growth in all three maize cultivars tested compared to the control (Figure S1; Table 1). Depending on the plant variety root- and shoot-dry biomass was increased up to 47 and 41%, respectively. Root and shoot length was increased up to 57 and 41%, respectively. Strain S8 showed positive effects on plant growth in cultivar DaSilvie only but did not significantly affect growth of the cultivars Kolea and Mazurka (Table 1). In contrast, strain S7 had a negative effect on seedling growth in all the maize cultivars with the effect being significant in DaSilvie and Kolea and less pronounced in Mazurka (Table 1; Figure S1). Apart from reduced biomass S7 treated plants showed white streaks on leaves (Figure S2).
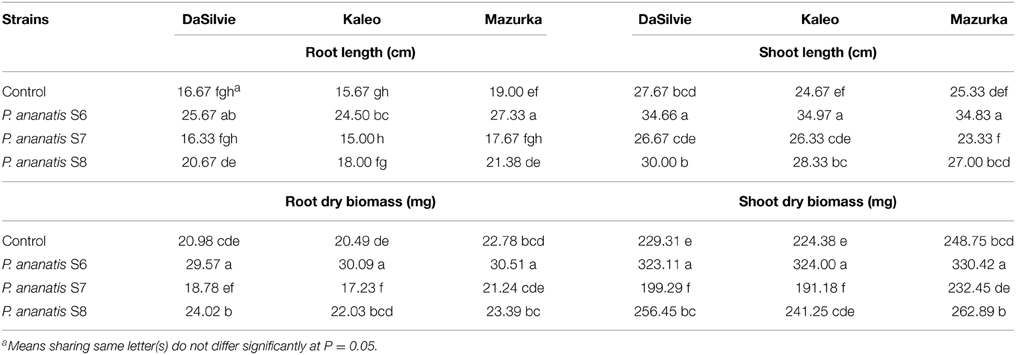
Table 1. Effect of inoculation with seed-associated endophytic bacteria on root/shoot length and biomass of maize seedlings.
A range of activities known to contribute to plant growth promotion, stress tolerance or biocontrol was tested. The results of functional characterization are summarized in Table 2. All strains exhibited ACC-deaminase activity and showed auxin, NH3 and siderophore production (qualitative). All three strains showed P-solubilization and were able to produce AHL and PHB. S6, S7, and S8 behaved similar in tests for motility and chemotaxis as well as the biochemical characters mentioned in Table 2. No strain produced EPS in our assays. Lipase, pectinase, phosphatase and xylanase activity was detected in all strains, whereas none of the strains showed amylase, cellulose, chitinase or protease activity. Strain S6 showed in vitro antagonistic activity against all bacterial pathogens tested but F. solani. Strain S7 inhibited growth of F. oxysporum, T. basicola, and P. citricola in our assays, whereas strain S8 negatively affected growth of F. graminarium, F. oxysporum, R. solani, and P. citricola.
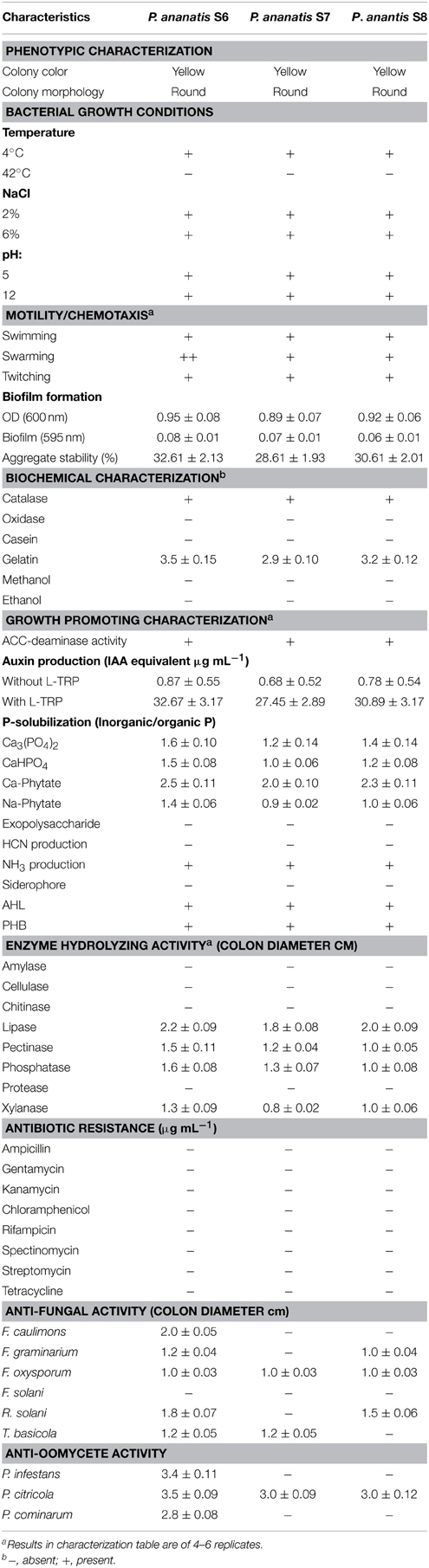
Table 2. Physico-chemical and growth-promoting characteristics of maize seed-borne endophytic bacteria.
Genomic DNA of strains S6, S7, and S8 was sequenced and the generated raw reads represented 230, 76, and 79 million bases respectively (Table 3). The number of sequenced reads varied from 570,490 in strain S6 with an average length of 406 bp to 174,500 and 179,051 in S7 and S8 respectively, with an average length of 441 bp in both strains.
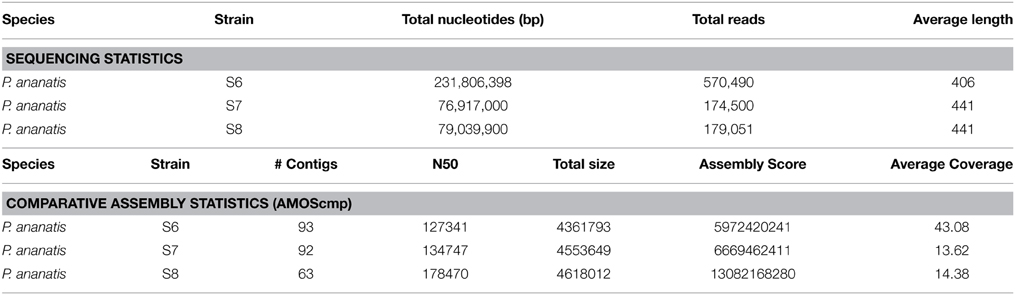
Table 3. Genome characteristics of sequencing and assembly of three strains of P. ananatis S6, S7, and S8.
The pairwise comparision of average nucleotide identity (ANI) of the draft genomes of strains S6, S7, and S8 with the P. ananatis AJ13355 genome showed that the similarity of the analyzed strains and strain AJ13355 exceeds 99% (Supplementary Table 1).
Comparative sequence assembly was performed by AMOScmp program (Pop et al., 2004) as a conservative method that uses the most similar available complete genome sequence as a reference to assemble the 454 reads (Table 3). The P. ananatis S6, S7, and S8 draft genomes consist of 93, 92, and 63 contigs, respectively, and range from 4.3 to 4.6 Mb in length.
De novo assembly resulted in almost the same coverage assembly but less assembly score and N50 value in comparison to AMOScmp assembler.
The comparison of draft genome assembly for P. ananatis S6, S7, and S8 against P. ananatis AJ13355 as reference genome are shown in Figure 1, illustrating a higher degree of genome conservation among the strains S6, S7, S8, as compared to P. ananatis AJ13355 (Figure 1). Phylogenetic analysis revealed a close relationship between strains S6, S7, S8, and the other four genomes of P. ananatis in comparison to P. vagans (Figure 2).
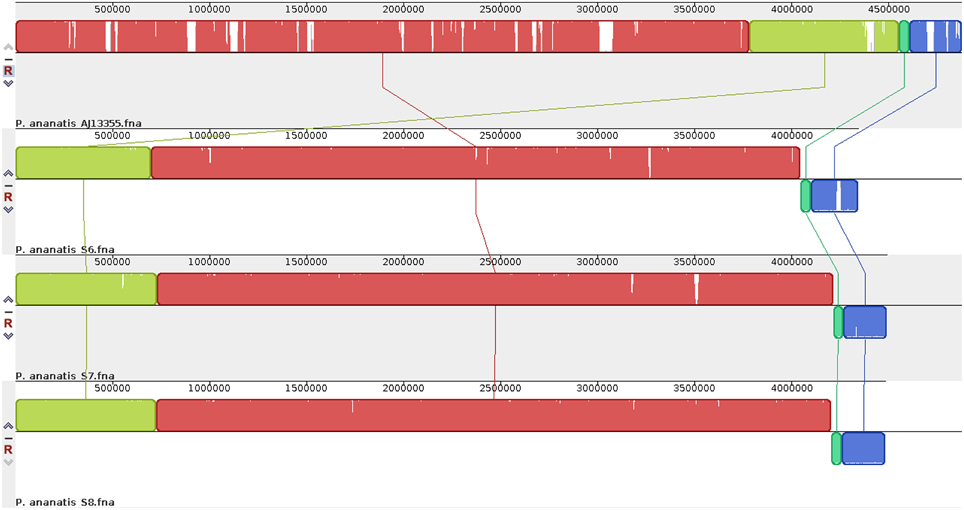
Figure 1. Genome-scale comparison for draft genome sequences of the three P. ananatis strains (S6, S7, and S8) and complete genome sequence of P. ananatis AJ13355. Homologous DNA regions among the strains are marked by the same colored blocks, while gaps correspond to non-homologous regions. The figure was generated using nucleotide sequences of the genomes using Mauve v2.3.1.
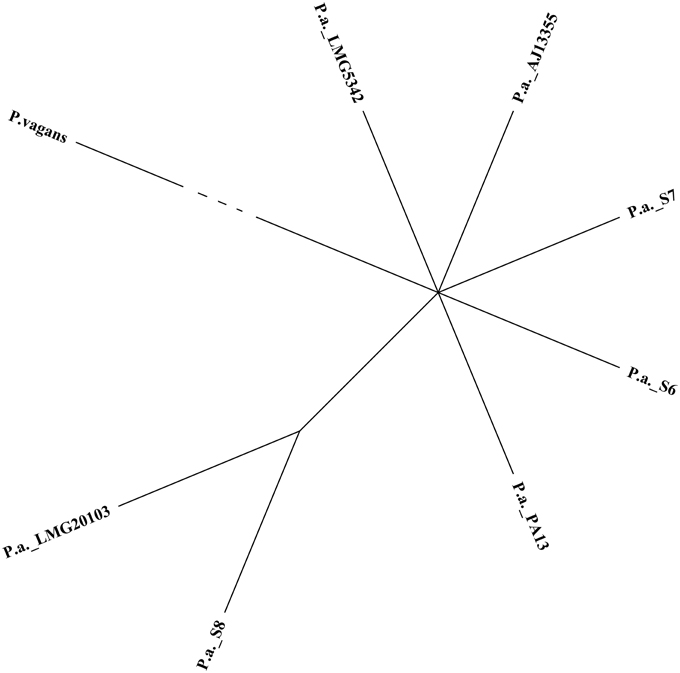
Figure 2. Phylogenetic trees of three strains of P. ananatis S6, S7 and S8 with four closed genomes from Pantoea genus. Pantoea vagans was included as an out group (edge has been shortened).
The genome annotation of P. annanatis S6, S7, and S8 resulted in different numbers of protein-coding genes. The genome of strain S6 consists of 4.375 predicted coding sequences (CDSs), while S7 and S8 contain 4.516 and 4.528 predicted CDSs, respectively (Table 4). Seven 16S rRNA, seven 23S rRNA and eight 5S rRNA genes are encoded in each of the P. annanatis strains. In total all tRNA genes for 33 different anticodons were found in all P. annanatis strains. The results of annotation analysis of three P. ananatis S6, S7, and S8 strains and reannotation of P. ananais strains (P. ananatis AJ13355, P. ananatis LMG20103, P. ananatis LMG5342, and P. ananatis PA13) are summarized in Table 4.
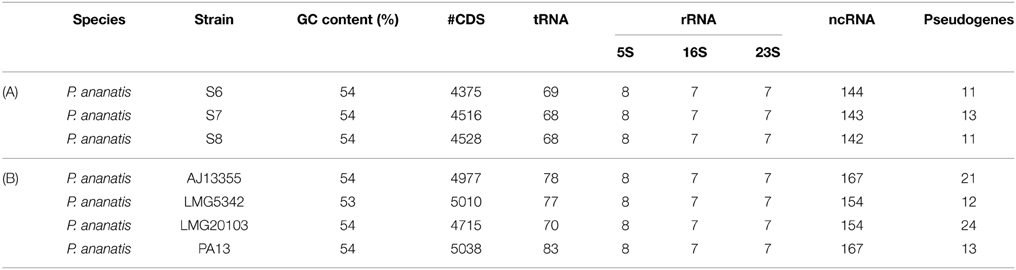
Table 4. Comparison of (A) draft genome annotation of three P. ananatis S6, S7, and S8 strains and (B) re-annotation of four complete genome of P. ananatis strains.
The fact that tRNA genes for all essential amino acids, the 16S rRNA gene and 31 housekeeping genes were found in the draft genomes of strains S6, S7, and S8 indicates that the genomes are close to complete. Moreover, the overall pattern of distribution of housekeeping genes and the gene copy number are identical to other members of the Enterobacteriaceae family.
To verify the sequence quality generated by 454 sequencing technology we identified putative pseudogenes represented by frameshifts in the draft genomes of P. ananatis. The low number of pseudogenes in the genomes of strains S6, S7, and S8 (11, 13, and 11 respectively) indicated that the genome draft has sufficient quality for further comparative genomics analysis (Table 4).
Five, six, and seven contigs in P. ananatis S6, S7, and S8 genome sequences, respectively, were homologous with the plasmid sequence of P. ananatis AJ13355 (Supplementary Table 2). In total, 287, 271, and 276 genes were identified in the plasmid contigs of strains S6, S7, and S8. The core factors identified on the large universal Pantoea plasmid LPP-1 (De Maayer et al., 2012a) such as genes coding for thiamine biosynthesis proteins (thiOSF), pigment biosynthetic protiens (crtEXYIBZ), arbutin/cellobiose/salicin transport and catabolism components (ascBFG), malate:quinone oxidoreductase (mqo), 1,3-diaminopropane production (dat, ddc) and branched-chain amino acid transport protein (azlDC) are present on the plasmid sequences of P. ananatis S6, S7, and S8 (Supplementary Table 1).
Comparative circular blast alignments of the plasmid sequences in Figure 3 shows high homology between plasmid sequences of P. ananatis S6, S7, and S8 as compared to the P. ananatis AJ13355 plasmid sequence (Stothard and Wishart, 2005; Alikhan et al., 2011) (Figure 3).
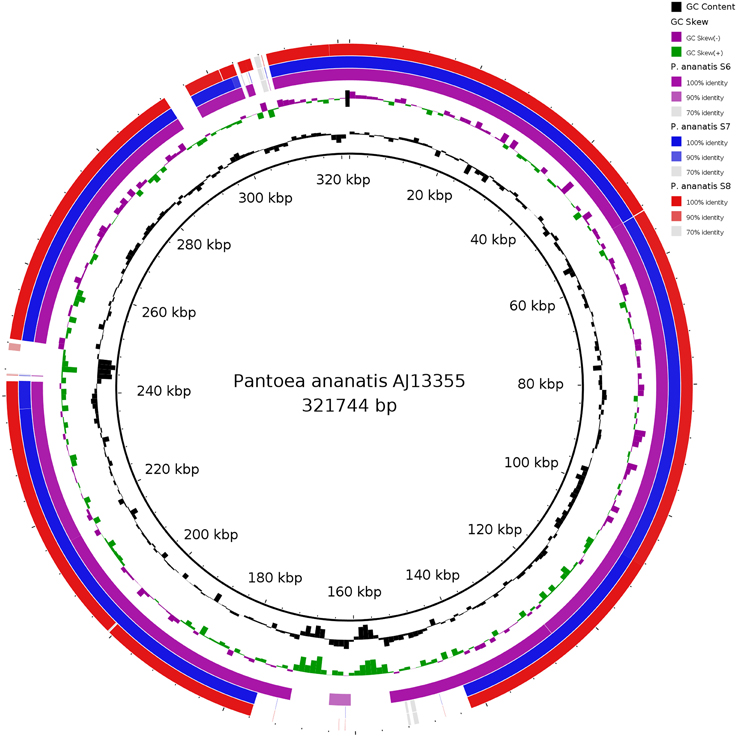
Figure 3. Comparison of the circular genome map of plasmid sequences of three P. ananatis S6, S7, and S8 genome structures with the known P. ananatis AJ13355 plasmid sequence as reference genome using blast ring image generator (BRIG). The inner circle shows the scale (bp). The first and the second rings show the GC content (black) and GC skew (purple/green), respectively, with respect to the reference genome. The 3rd, 4th, and 5th rings show BLAST comparisons of P. ananatis strains S6, S7, and S8 plasmid sequences, respectively.
To identify the core P. ananatis genome, we clustered orthologous groups from genes predicted in the seven P. ananatis genomes of this study (P. ananatis AJ13355, P. ananatis LMG20103, P. ananatis LMG5342, P. ananatis PA13 and strains S6, S7, S8) using OrthoMCL (Li et al., 2003). Of the total 33,159 protein-coding genes in all P. ananatis strains, 31,987 genes clustered into 4959 gene families. Out of these, 27,578 genes representing 3785 gene families, were common to all P. ananatis strains, hereafter referred to as the core P. ananatis proteome (Figure 4). Fifty-three clusters were shared between P. ananatis S6 and S7. P. ananatis S7 and S8 have 207 clusters in common while P. ananatis S6 and S8 shared 79 clusters (Figure 5).
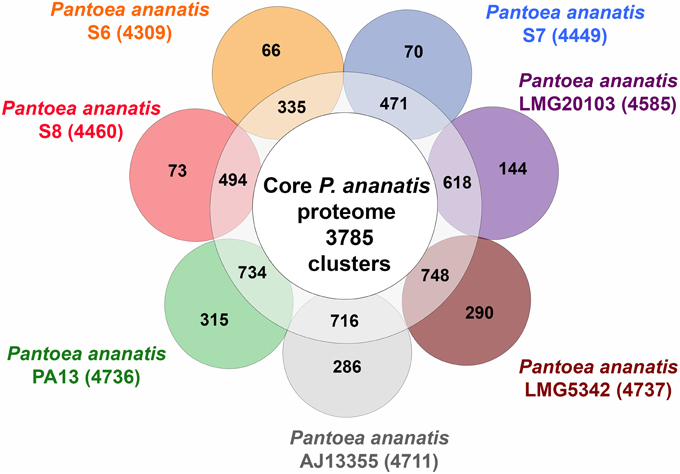
Figure 4. Clusters of orthologous gene families in seven P. ananatis strains identified by OrthoMCL. The inner circle shows the core proteome shared between all strains. The numbers of gene clusters shared between specific strains are shown in the ring. The specific proteins for each strain are indicated in each of the outer circles. The numbers outside the Venn diagram show the total number of genes (in parentheses) for each strain.
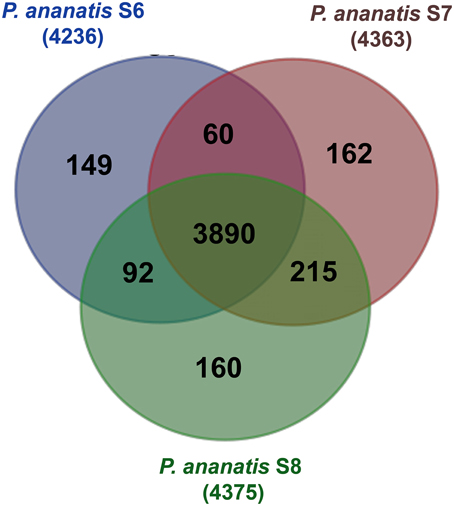
Figure 5. Venn diagram of OrthoMCL cluster distribution across three P. ananatis S6, S7, and S8 strains identified by OrthoMCL. The number of core proteome clusters, gene families shared between the species and the specific proteins for each strain is indicated in each of the components. The numbers outside the Venn diagram show the total number of genes (in parentheses) for each strain.
To understand the functions of shared and specific genes between the P. ananatis strains, we analyzed the functional categories of the respective P. ananatis gene clusters based on the NOG annotations (Jensen et al., 2008).
As expected, the core P. ananatis genes were categorized in functions involved in metabolism, cellular processes and signaling activity, information storage and processing (Supplementary Tables 3, 5). The beneficial P. ananatis S6 specific genes encode proteins with putative functions in metabolism, signal transduction and information storage and processing. Whereas, pathogenic P. ananatis S7 specific genes were specifically involved in cell cycle control, cell division, chromosome partitioning and amino acid transport. The commensal P. ananatis S8 specific genes were responsible for transcription and amino acid transport (Supplementary Tables 4, 6).
Functional annotations of orthologous groups on the predicted proteomes of S6, S7, and S8 and the published P. ananatis genomes revealed functions that were common within all the genomes. This analysis also indicated gene families that cause differences among the strains on the functional level. The distribution of genes in COG functional categories is shown in Figure 6.
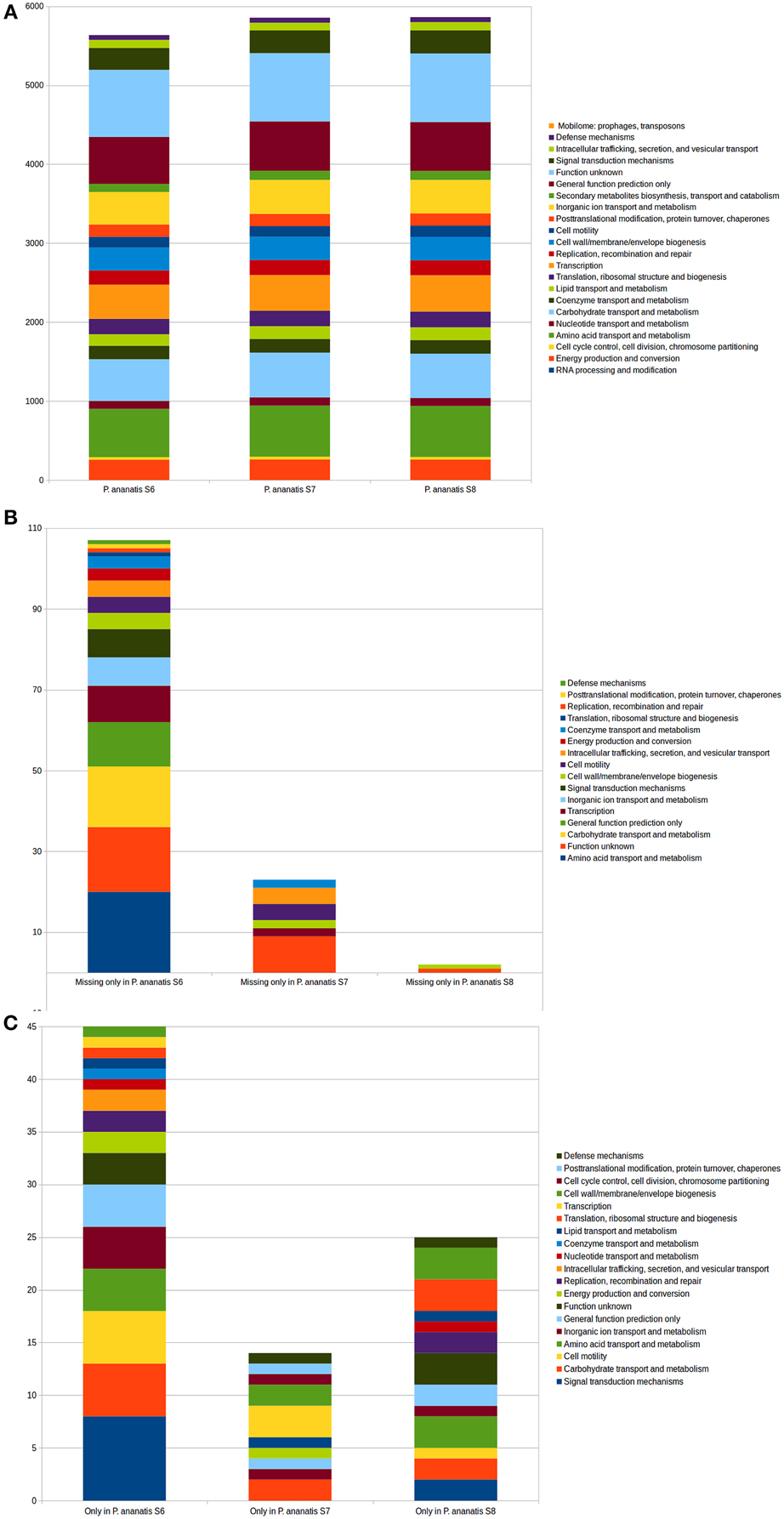
Figure 6. Functional COG categories in the genomes of the three P. ananatis strains S6, S7, and S8. (A) Comparison of the COG categories in the genomes of the three P. ananatis strains S6, S7, and S8. (B) The COG categories that present in two of the P. ananatis strains but are absent in the third strain (S6, S7, or S8). (C) The COG categories existing in only one of the P. ananatis strains (S6, S7, or S8).
Type IV pilus biogenesis proteins such as PilNQCWTZ, type IV pilus secretin PilQ, pili assembly chaperone and prepilin type IV endopeptidase were found in the core proteome of P. ananatis strains (Supplementary Table 5). Interestingly, two genes related to pili assembly chaperon and fimbrial-type adhesion (the uncharacterized fimbrial chaperone YhcA and F17a-A fimbrial protein) were found in all P. ananatis strains but missing from beneficial P. ananatis S6 and two other genes related to the pili (fimbrial chaperone YfcS and chaperone protein PapD) were absent in pathogenic P. ananatis S7 but found in all other P. ananatis strains (Supplementary Table 7; Figure 6B).
Transposases related proteins such as tyrosine recombinase XerD, tyrosine recombinase XerC and site-specific recombinase XerD were identified in the core proteome of all P. ananatis strains. The only difference seen was theYhgA-like transposase that was found only in the commensal P. ananatis S8 (Supplementary Tables 5, 6; Figures 6A,C).
Virulence associated genes on mobile genetic elements showed that phage/bacteriophage related proteins such as bacteriophage P2 (GpU), bacteriophage tail protein Gp41 and phage tail tape measure protein are present in P. ananatis S7 and S8 and all other P. ananatis strains but do not have orthologs in the beneficial P. ananatis strain S6. The bacteriophage T7, Gp4, DNA primase/helicase is presented only in commensal P. ananatis S8 strain. Orthologous for integrases were not found in the beneficial strain S6 but were presented in the other strains (Supplementary Tables 6, 7; Figures 6B,C).
The chemotaxis related proteins such as chemotaxis methyl-accepting receptor (CheR) and chemotaxis proteins (CheVWY) were identified in the core proteome of the P. ananatis strains. The methyl-accepting chemotaxis signaling proteinI TSR is missing in the beneficial P. ananatis S6 strain but this strain contains the methyl-accepting chemotaxis signaling protein (MCP) which has the same activity in transducing the signal to downstream signaling proteins in the cytoplasm (Supplementary Tables 5, 6; Figures 6A,C).
The orthologous groups that are related to flagellar structures in the core P. ananatis proteome consists of flagella basal body P-ring formation proteins FlgAC; flagellar assembly proteins FliH; flagellar basal body rod protein components FlaE, FlgJ; flagellar hook-basal body complex proteins FliELK, FlgCK and flagellar biosynthesis proteins FlhAQRO. Other main flagellar related proteins are FliJ, FlhE flagellar motor protein MotA/MotB and FliNGMSTZ identified in the core proteome of P. ananatis strains (Supplementary Table 5; Figure 6A).
Gene families for T6SS loci were found on the core proteome of all seven P. ananatis strains investigated in this study. These common genes encoding DotU (COG2885), ATPase ClpV1 (COG0542), FHA domain-containing protein (COG3456), IcmF (COG3523), lipoprotein SciN (COG3521), lysozyme-related protein (impF) (COG3518), OmpA/MotB domain (COG3455), T6SS RhsGE-associated Vgr family subset (COG3501), T6SS-associated BMAA0400 (COG3913), T6SS -associated ImpA (COG3515) and Hcp1 (COG3157) (Supplementary Table 5; Figure 6A). Our analysis showed also that the outer component of the T6SS, which consist of two proteins, VgrG (COG3501) and Hcp (COG3157) have also been identified as secreted effectors of the T6SS in some of the P. ananatis strains (Supplementary Table 7; Figure 6B). The effector protein genes hcp1, hcp1_2, and hcp1_3 loci are presented in six P. ananatis strains but absent in the pathogenic strain P. ananatis S7 (Table 5). The HcpC as major exported protein is missing in commensal P. ananatis S8 and pathogenic strains P. ananatis S7 and LMG5342, however it was present in the beneficial S6 strains, P. ananatis AJ13355 and pathogenic strains of P. ananatis PA13 and P. ananatis LMG20103 (Supplementary Table 7; Figure 6B).

Table 5. Hemolysin co-regulated effector proteins (Hcp) presented in the Type VI secretion system identified in orthologous clusters of P. ananatis strains.
We identified eukaryotic-like protein domains (ELDs) in strains S6, S7, and S8 by applying the prediction framework of the Effective web-portal (Jehl et al., 2011). The prediction assigns a eukaryotic-like domain enrichment score (ELD score) to each protein domain, reflecting the maximal enrichment of that domain in any pathogen or symbiont compared to the background frequency of the protein domain in non-pathogenic, non-host-associated bacteria. A high ELD score equals strong enrichment of the protein domain in pathogenic/symbiotic bacteria and suggests an important functional role of the secreted protein in the interaction with the host cell. All ELDs with a significant ELD score greater or equal to 4 were considered to investigate the genomic variance of P. ananatis strains S6, S7, and S8 that cause different phenotypes in the host plant.
In summary, 29 different ELDs were predicted (Table 6). The majority, i.e., 26 ELDs are shared between all three genomes, supporting the assumption of a high average functional similarity of effector proteins. One eukaryotic-like protein domain, the tRNA delta-isopentenylpyrophosphate (IPP) transferase domain (PF01715) was exclusively found in the genome of the beneficial maize seed strain P. ananatis S6. IPP transferases are involved in the modification of tRNAs and convert A(37) to isopentenyl A(37). Another one was unique in the pathogenic strain S7 and contains the C terminal part of a GMP synthase (PF00958). This enzyme belongs to the family of ligases and is involved the biosynthesis of the nucleic acid guanine. A eukaryotic-like domain containing the signature of the collagen-binding domain of bacterial collagenases (PF12904) was found in S7, S8 and all other P. ananatis genomes but was absent in S6.
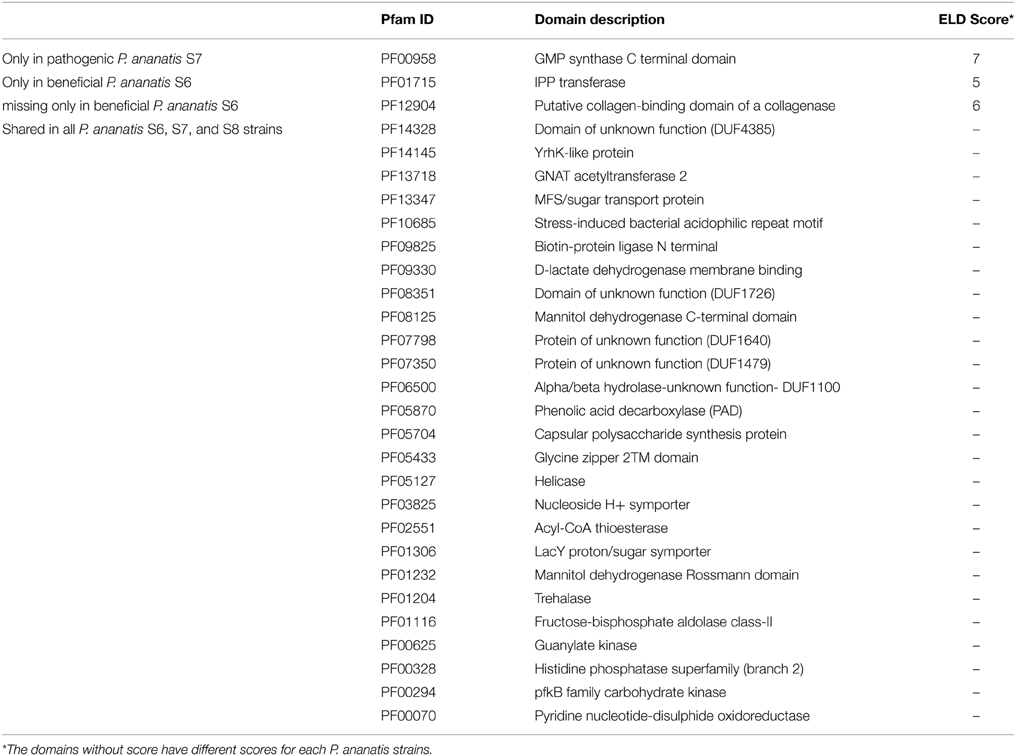
Table 6. Differences of eukaryotic-like protein domain (ELD) enrichment in P. ananatis strains of diverse phenotype.
The genus Pantoea comprises bacteria that are frequently associated with eukaryotic hosts such as plants but strains, even those belonging to the same species (such as P. ananatis), have different type of interactions with their host ranging from pathogenicity to mutualism (De Maayer et al., 2014). In our study we showed that genetically closely related P. ananatis strains with different effects on plant growth colonize maize seeds.
In our study, the maize seed endophyte P. ananatis S6 showed clear beneficial effects on maize growth, while strain S7 induced weak pathogenicity symptoms. P. ananatis S8 had hardly any effect and can be considered as commensal. The pan genome of eight P. ananatis genomes indicated as open pan genome that they can colonize and exploit several different environmental niches by De Maayer et al. (2014). As three P. ananatis strains (S6, S7, and S8) are also capable to colonize inside maize seeds and interact with their host, we can expect that the pan genome of these strains can be defined as open pan genome.
Our comparative analysis showed that an average of 85–87% of CDSs predicted for each individual strain of P. ananatis S6, S7, and S8 have orthologs encoded by the genomes of the other strains (P. ananatis AJ13355, P. ananatis LMG20103, P. ananatis LMG5342 and P. ananatis PA13). These results suggest that the core genomes of strains S6, S7, and S8 strains are highly conserved (Figure 1). Despite the overall high degree of similarity between the core genomes of the three maize seed endophytes, we found differences in transposase/integrases/phage related genes, type VI secretion system, and eukaryotic-like protein domains. Similarly, the analysis of the open pan-genome of eight sequenced genomes of P. ananatis indicated that between 89.3 and 95.7% of the proteins are common between all strains and they are important for metabolism and cellular processes (De Maayer et al., 2014).
Genes of the accessory genome of selected P. ananatis strains analyzed by De Maayer et al. (2014) encoded mainly poorly characterized proteins including transposases, integrases, and mobile genetic elements. The role of horizontal gene transfer in the diversification of P. ananatis strains was suggested (De Maayer et al., 2014). Similarly, phage related genes were reported to have a significant role in transferring pathogenicity factors to their bacterial host and thereby to affect bacterial evolution (Lima-Mendez et al., 2008). Due to the differences found in regard mobile genetic elements such as integrase genes, transposase genes and phage related genes, our study confirms a potential role of these elements in the diversification of related strains colonizing the same ecological niche. An over-representation of transposase genes and mobile elements also indicates the genomes' potential for acquisition of novel functions. The reduced number of mobile elements in P. ananatis S6 on the other hand could indicate high stability of its genome, implying good adaption to the habitat.
De Maayer et al. (2012a) proposed the Large Pantoea Plasmid (LLP-1) as genetic determinant of niche adaption and functional diversification of the genus Pantoea. All three maize seed endophytes S6, S7, and S8 contain a LLP-1 plasmid and no differences in LPP-1 related genes were found between the genomes of these strains and the core genome of P. ananatis. Our analysis revealed further that genes encoding the pigment biosyntetic (CrtEXYIBZ) and thiamine biosynthesis (ThiOSGF) proteins are present on the plasmid of P. ananatis S6, S7, and S8 (Supplementary Table 1). These genes are among those genes identified by De Maayer et al. (2014) to be specific for plant-associated bacteria (PAB) among P. ananatis. In addition, the core proteome of the maize seed P. ananatis strains contains PAB-specific CDs with prediction functions in metabolism and transport of carbohydrates, iron uptake and metabolism, and carbon, nitrogen and energy sources (De Maayer et al., 2014). In conclusion, our findings support the concept of functional diversification of the species P. ananatis proposed by De Maayer et al. (2014).
The T6SS is one of the most studied secretion system in P. ananatis (Coutinho and Venter, 2009; De Maayer et al., 2011; Shyntum et al., 2014). Three T6SS loci (T6SS-1, -2, and –3) have been described in P. ananatis strains, translocating effectors into the host plant (De Maayer et al., 2011; Shyntum et al., 2014). The T6SS-1 locus is found on the genomes of all P. ananatis strains, while T6SS-2 is restricted to pathogenic strains of P. ananatis. The presence of T6SS-1 and T6SS-2 in both pathogenic and non-pathogenic P. ananatis strains support the idea that the T6SS itself is not necessarily a determinant of pathogenicity and could play a role in competition against other microorganisms, fitness or niche adaptation (Weber et al., 2009; English et al., 2012; Shyntum et al., 2014). T6SS-3 was found to be mainly restricted to P. ananatis AJ13355, P. ananatis LMG 20103, and P. ananatis PA4 (De Maayer et al., 2014).
Beside the T6SS loci related genes VgrG and Hcp genes are present in the maize seed P. ananatis strains. The VgrG genes were found in S6, S7, and S8, whereas differences were seen in the presence of hemolysin co-regulated effector proteins (Hcp) between these three strains. A recent study showed that three hcp genes exist in P. ananatis strains comprising hcp-1, hcp-2 (having homologs in all sequenced strains of P. ananatis) and hcp-3 genes (found in P. ananatis PA13) (Shyntum et al., 2014). The hcp-3 gene is highly divergent from hcp-1, hcp-2, and the T6SS associated hcp genes (Shyntum et al., 2014). The plant-beneficial strain S6 has orthologs with all hcp genes identified in the orthologous gene families, while plant-pathogenic strain S7 has no orthologs for hcp genes. HcpC is presented in all P. ananatis strains but it is missing from S7 and S8 strains. This Hcp protein is located on the plasmid sequence of this strain. Paralogs of hcp influence bacterial motility, protease production and biofilm formation (Sha et al., 2013). A potential role of Hcp and VgrG proteins in host interaction is not described. As all hcp genes are present in other P. ananatis strains (ranging from pathogenic to saprophytic life style), the hcp genes in in the beneficial P. ananatis strain S6 might not be responsible for the differences in the phenotype of plant-microbe interaction of the three maize seed strains S6, S7, and S8.
The analysis of effector candidates containing eukaryotic-like protein domains (ELDs) revealed varying molecular repertoire in the genomes of the three maize seed P. ananatis strains. The plant-beneficial strain S6 carries a gene for a tRNA delta-isopentenylpyrophosphate (IPP) transferase domain which is not present in the strains S7 and S8. In E. coli this enzyme is involved in increasing spontaneous mutation frequency when cells need to adapt to environmental stress (Connolly and Winkler, 1989). Moreover, tRNA modifications mediated by tRNA delta-isopentenylpyrophosphate (IPP) transferase are required for virulence in Shigella flexneri by regulating posttranscriptional expression of the regulatory gene virF (Durand et al., 1997). The collagen-binding domain of bacterial collagenases is missing in the beneficial P. ananatis S6, although present in S7 and S8. This domain is a major component of the extracellular matrix (ECM) and plays a role in cell attachment, haemostasis, differentiation and bacterial adhesion in human and plant pathogens (Foster and Hook, 1998). Interestingly, in Yersinia enterocolitica it is a part of the pathogenic bacterial strategy for avoiding host response (Nummelin et al., 2004). The GMP synthase domain exclusively found in the pathogenic P. ananatis strain S7 is known to play an important role in cell-to-cell signaling in regulation of virulence in the plant pathogen Xanthomonas campestris (Ryan et al., 2006a). This domain is also involved in aggregative behavior, adhesion, biofilm formation, and the virulence of animal and plant pathogens (Ryan et al., 2006b). The role of these EDLs in the interaction of the three maize seed strains P. ananatis S7, S8, and S9 with maize plants remains unclear and merits further investigation.
Overall, our study showed that groups of bacterial endophytes with highly related genotypes but different phenotypes in terms of effects on host plants may exist in the same ecological niche. It can be expected that seed endophytes colonize, at least to a certain extent, plants derived from these seeds. Consequently, both, potential plant pathogens and mutualistic endophytes, may be together transmitted to the developing plant.
To predict the phenotype of plant-microbe interactions from traits manifested on the genome of bacteria is an attractive idea and would very much facilitate efforts in selecting microbial inoculants for improved plant production in sustainable agriculture. However, given the high genomic similarity between strains showing distinct phenotypes in regard to their interaction with plants, we conclude that plant pathogenicity and mutualism in P. ananatis may be based on rather subtle differences, e.g., on the expression of genes leading to plant defense reactions. Plant-bacteria interactions, irrespectively of whether pathogenic or beneficial must be considered as a multi-dimensional system and the expression of pathogenic or beneficial effects might depend on a multitude of parameters such as the plant/bacterial physiology, environmental conditions and/or a very fine-tuned interaction between bacterial elicitors and plant response.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants provided by the FWF (National Science Foundation, grant no. P22867-B16 and P26203-B22). The maize seeds of cultivars Kaleo and Mazurka used in this experiment were kindly provided by DOW AgroSciences, Neusiedl am See, Austria and cultivar DaSilvie was kindly provided by Saatbau Linz, Austria.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00440/abstract
Alikhan, N.-F., Petty, N., Ben Zakour, N., and Beatson, S. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Azad, H., Holmes, G., and Cooksey, D. (2000). A new leaf blotch disease of sudangrass caused by Pantoea ananas and Pantoea stewartii. Plant Dis. 84, 973–979. doi: 10.1094/PDIS.2000.84.9.973
Badger, J. H., and Olsen, G. J. (1999). CRITICA: coding region identification tool invoking comparative analysis. Mol. Biol. Evol. 16, 512–524. doi: 10.1093/oxfordjournals.molbev.a026133
Bashan, Y., Diab, S., and Okon, Y. (1982). Survival ofXanthomonas campestris pv.vesictoria in pepper seeds and roots in symptomless and dry leaves in non-host plants and in the soil. Plant Soil 68, 161–170. doi: 10.1007/BF02373702
Bordiec, S., Paquis, S., Lacroix, H., Dhondt, S., Ait Barka, E., Kauffmann, S., et al. (2011). Comparative analysis of defence responses induced by the endophytic plant growth-promoting rhizobacterium Burkholderia phytofirmans strain PsJN and the non-host bacterium Pseudomonas syringae pv. pisi in grapevine cell suspensions. J. Exp. Bot. 62, 595–603. doi: 10.1093/jxb/erq291
Choi, O., Lim, J. Y., Seo, Y. S., Hwang, I., and Kim, J. (2012). Complete genome sequence of the rice pathogen Pantoea ananatis strain PA13. J. Bacteriol. 194, 531. doi: 10.1128/JB.06450-11
Compant, S., Mitter, B., Colli-Mull, J. G., Gangl, H., and Sessitsch, A. (2011). Endophytes of grapevine flowers, berries, and seeds: identification of cultivable bacteria, comparison with other plant parts, and visualization of niches of colonization. Microb. Ecol. 62, 188–197. doi: 10.1007/s00248-011-9883-y
Connolly, D. M., and Winkler, M. E. (1989). Genetic and physiological relationships among the miaA gene, 2-methylthio-N6-(delta 2-isopentenyl)-adenosine tRNA modification, and spontaneous mutagenesis in Escherichia coli K-12. J. Bacteriol. 171, 3233–3246.
Cota, L., Costa, R., Silva, D., Parreira, D., Lana, U., and Casela, C. (2010). First report of pathogenicity of Pantoea ananatis in sorghum (Sorghum bicolor) in Brazil. Australas. Plant Dis. Notes 5, 120–122. doi: 10.1071/DN10044
Cother, E., Reinke, R., Mckenzie, C., Lanoiselet, V., and Noble, D. (2004). An unusual stem necrosis of rice caused by Pantoea ananas and the first record of this pathogen on rice in Australia. Australas. Plant Pathol. 33, 495–503. doi: 10.1071/AP04053
Coutinho, T. A., and Venter, S. N. (2009). Pantoea ananatis: an unconventional plant pathogen. Mol. Plant Pathol. 10, 325–335. doi: 10.1111/j.1364-3703.2009.00542.x
Darling, A., Mau, B., Blattner, F., and Perna, N. (2004). Mauve: multiple alignment of conserved genomic sequences with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679. doi: 10.1093/bioinformatics/btm009
De Maayer, P., Chan, W., Blom, J., Venter, S., Duffy, B., Smith, T., et al. (2012a). The large universal Pantoea plasmid LPP-1 plays a major role in biological and ecological diversification. BMC Genomics 13:625. doi: 10.1186/1471-2164-13-625
De Maayer, P., Chan, W., Rezzonico, F., Buhlmann, A., Venter, S., Blom, J., et al. (2012b). Complete genome sequence of clinical isolate Pantoea ananatis LMG5342. J. Bacteriol. 194, 1615–1616. doi: 10.1128/JB.06715-11
De Maayer, P., Chan, W., Venter, S., Toth, I., Birch, P., Joubert, F., et al. (2010). Genome sequence of Pantoea ananatis LMG20103, the causative agent of Eucalyptus blight and dieback. J. Bacteriol. 192, 2936–2937. doi: 10.1128/JB.00060-10
De Maayer, P., Chan, W. Y., Rubagotti, E., Venter, S. N., Toth, I. K., Birch, P. R., et al. (2014). Analysis of the Pantoea ananatis pan-genome reveals factors underlying its ability to colonize and interact with plant, insect and vertebrate hosts. BMC Genomics 15:404. doi: 10.1186/1471-2164-15-404
De Maayer, P., Venter, S., Kamber, T., Duffy, B., Coutinho, T., and Smits, T. (2011). Comparative genomics of the type VI secretion systems of Pantoea and Erwinia species reveals the presence of putative effector islands that may be translocated by the VgrG and Hcp proteins. BMC Genomics 12:e576. doi: 10.1186/1471-2164-12-576
Durand, J. M., Bjork, G. R., Kuwae, A., Yoshikawa, M., and Sasakawa, C. (1997). The modified nucleoside 2-methylthio-N6-isopentenyladenosine in tRNA of Shigella flexneri is required for expression of virulence genes. J. Bacteriol. 179, 5777–5782.
English, G., Trunk, K., Rao, V., Srikannathasan, V., Hunter, W., and Coulthurst, S. (2012). New secreted toxin and immunity proteins encoded within the Type VI secretion system gene cluster of Serratia marcescens. Mol. Microbiol. 86, 921–936. doi: 10.1111/mmi.12028
Foster, T. J., and Hook, M. (1998). Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 6, 484–488. doi: 10.1016/S0966-842X(98)01400-0
Garcia-Alcalde, F., Okonechnikov, K., Carbonell, J., Cruz, L. M., Gotz, S., Tarazona, S., et al. (2012). Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679. doi: 10.1093/bioinformatics/bts503
Gitaitis, R. D., and Gay, J. D. (1997). First report of a leaf blight, seed stalk rot, and bulb decay of onion by pantoea ananas in Georgia. Plant Dis. 81, 1096–1096. doi: 10.1094/PDIS.1997.81.9.1096C
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A., Eddy, S. R., and Bateman, A. (2005). Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124. doi: 10.1093/nar/gki081
Hara, Y., Kadotani, N., Izui, H., Katashkina, J., Kuvaeva, T., Andreeva, I., et al. (2012). The complete genome sequence of Pantoea ananatis AJ13355, an organism with great biotechnological potential. Appl. Microbiol. Biotechnol. 93, 331–341. doi: 10.1007/s00253-011-3713-5
Hardoim, P. R., Hardoim, C. C., Van Overbeek, L. S., and Van Elsas, J. D. (2012). Dynamics of seed-borne rice endophytes on early plant growth stages. PLoS ONE 7:e30438. doi: 10.1371/journal.pone.0030438
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jehl, M. A., Arnold, R., and Rattei, T. (2011). Effective–a database of predicted secreted bacterial proteins. Nucleic Acids Res. 39, D591–D595. doi: 10.1093/nar/gkq1154
Jensen, L. J., Julien, P., Kuhn, M., Von Mering, C., Muller, J., Doerks, T., et al. (2008). eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 36, D250–D254. doi: 10.1093/nar/gkm796
Johnston-Monje, D., and Raizada, M. N. (2011). Conservation and diversity of seed associated endophytes in Zea across boundaries of evolution, ethnography and ecology. PLoS ONE 6:e20396. doi: 10.1371/journal.pone.0020396
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, H. J., Lee, J. H., Kang, B. R., Rong, X., Mcspadden Gardener, B. B., Ji, H. J., et al. (2012). Draft genome sequence of Pantoea ananatis B1-9, a nonpathogenic plant growth-promoting bacterium. J. Bacteriol. 194, 729. doi: 10.1128/JB.06484-11
Kloepper, J. W., Mcinroy, J. A., Liu, K., and Hu, C.-H. (2013). Symptoms of fern distortion syndrome resulting from inoculation with opportunistic endophytic fluorescent Pseudomonas spp. PLoS ONE 8:e58531. doi: 10.1371/journal.pone.0058531
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lima-Mendez, G., Van Helden, J., Toussaint, A., and Leplae, R. (2008). Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics 24, 863–865. doi: 10.1093/bioinformatics/btn043
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 0955–0964. doi: 10.1093/nar/25.5.0955
Lukashin, A., and Borodovsky, M. (1998). GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115. doi: 10.1093/nar/26.4.1107
Mundt, J. O., and Hinkle, N. F. (1976). Bacteria within ovules and seeds. Appl. Environ. Microbiol. 32, 694–698.
Naveed, M., Mitter, B., Yousaf, S., Pastar, M., Afzal, M., and Sessitsch, A. (2014). The endophyte Enterobacter sp. FD17: a maize growth enhancer selected based on rigorous testing of plant beneficial traits and colonization characteristics. Biol. Fertil. Soils 50, 249–262. doi: 10.1007/s00374-013-0854-y
Nummelin, H., Merckel, M. C., Leo, J. C., Lankinen, H., Skurnik, M., and Goldman, A. (2004). The Yersinia adhesin YadA collagen-binding domain structure is a novel left-handed parallel β-roll. EMBO J. 23, 701–711. doi: 10.1038/sj.emboj.7600100
Paccola-Meirelles, L. D., Ferreira, A. S., Meirelles, W. F., Marriel, I. E., and Casela, C. R. (2001). Detection of a bacterium associated with a leaf spot disease of maize in Brazil. J. Phytopathol. 149, 275–279. doi: 10.1046/j.1439-0434.2001.00614.x
Pop, M., Phillippy, A., Delcher, A. L., and Salzberg, S. L. (2004). Comparative genome assembly. Brief. Bioinform. 5, 237–248. doi: 10.1093/bib/5.3.237
Reiter, B., and Sessitsch, A. (2006). Bacterial endophytes of the wildflower Crocus albiflorus analyzed by characterization of isolates and by a cultivation-independent approach. Can. J. Microbiol. 52, 140–149. doi: 10.1139/w05-109
Rijavec, T., Lapanje, A., Dermastia, M., and Rupnik, M. (2007). Isolation of bacterial endophytes from germinated maize kernels. Can. J. Microbiol. 53, 802–808. doi: 10.1139/W07-048
Ryan, R. P., Fouhy, Y., Lucey, J. F., Crossman, L. C., Spiro, S., He, Y.-W., et al. (2006a). Cell–cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc. Natl. Acad. Sci. U.S.A. 103, 6712–6717. doi: 10.1073/pnas.0600345103
Ryan, R. P., Fouhy, Y., Lucey, J. F., and Dow, J. M. (2006b). Cyclic Di-GMP signaling in bacteria: recent advances and new puzzles. J. Bacteriol. 188, 8327–8334. doi: 10.1128/JB.01079-06
Sayers, E. W., Barrett, T., Benson, D. A., Bolton, E., Bryant, S. H., Canese, K., et al. (2012). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 40, D13–D25. doi: 10.1093/nar/gkr1184
Serrano, F. (1928). Bacterial fruitlet brown-rot of pineapple in the Philippines. Phil. J. Sci. 36, 271–305.
Sha, J., Rosenzweig, J., Kozlova, E., Wang, S., Erova, T., Kirtley, M., et al. (2013). Evaluation of the roles played by Hcp and VgrG type 6 secretion system effectors in Aeromonas hydrophila SSU pathogenesis. Microbiology 159, 1120–1135. doi: 10.1099/mic.0.063495-0
Shyntum, D., Venter, S., Moleleki, L., Toth, I., and Coutinho, T. (2014). Comparative genomics of type VI secretion systems in strains of Pantoea ananatis from different environments. BMC Genomics 15:163. doi: 10.1186/1471-2164-15-163
Smits, T., Rezzonico, F., Kamber, T., Goesmann, A., Ishimaru, C., Stockwell, V., et al. (2010). Genome sequence of the biocontrol agent Pantoea vagans C9-1. J. Bacteriol. 192, 6486–6487. doi: 10.1128/JB.01122-10
Stall, R. E., Alexander, L. J., and Hall, C. B. (1969). Effect of tobacco mosaic virus and bacterial infections on occurrence of graywall of tomato (Erwinia ananas). Fla State Hort. Soc. Proc 81, 157–161.
Steel, R. G. D., Torrie, J. H., and Dicky, D. A. (1997). Principles and Procedures of Statistics: a Biometrical Approach, 3rd Edn. Singapore: McGraw Hill Book Int. Co.
Stothard, P., and Wishart, D. S. (2005). Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539. doi: 10.1093/bioinformatics/bti054
Uniprot consortium. (2009). The universal protein resource (UniProt) Nucleic Acids Res. 37, D169–D174. doi: 10.1093/nar/gkn664
Weber, B., Hasic, M., Chen, C., Wai, S., and Milton, D. (2009). Type VI secretion modulates quorum sensing and stress response in Vibrio anguillarum. Environ. Microbiol. 11, 3018–3028. doi: 10.1111/j.1462-2920.2009.02005.x
Wells, J., Sheng, W., Ceponis, M., and Chen, T. (1987). Isolation and characterization of strains of Erwinia ananas from honeydew melons. Phytopathol 77, 511–514. doi: 10.1094/Phyto-77-511
Wilson, D. (1995). Endophyte: the evolution of a term, and clarification of its use and definition. Oikos 73, 274–276. doi: 10.2307/3545919
Wu, M., and Scott, A. J. (2012). Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28, 1033–1034. doi: 10.1093/bioinformatics/bts079
Keywords: seed endophyte, Pantoea ananatis, comparative genomics, plant growth promotion
Citation: Sheibani-Tezerji R, Naveed M, Jehl M-A, Sessitsch A, Rattei T and Mitter B (2015) The genomes of closely related Pantoea ananatis maize seed endophytes having different effects on the host plant differ in secretion system genes and mobile genetic elements. Front. Microbiol. 6:440. doi: 10.3389/fmicb.2015.00440
Received: 30 January 2015; Accepted: 23 April 2015;
Published: 12 May 2015.
Edited by:
Anna Maria Pirttilä, University of Oulu, FinlandReviewed by:
Laila Pamela Partida-Martinez, Centro de Investigaciones Avanzadas del Instituto Politecnico Nacional, MexicoCopyright © 2015 Sheibani-Tezerji, Naveed, Jehl, Sessitsch, Rattei and Mitter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Rattei, Division of Computational Systems Biology, Department of Microbiology and Ecosystem Science, University of Vienna, Althanstrasse 14, 1090 Vienna, Austria,dGhvbWFzLnJhdHRlaUB1bml2aWUuYWMuYXQ=;
Birgit Mitter, Unit of Bioresources, Health and Environment Department, AIT Austrian Institute of Technology GmbH, Konrad Lorenz Strasse 24, 3430 Tulln, Austria,YmlyZ2l0Lm1pdHRlckBhaXQuYWMuYXQ=
†Present Address: Muhammad Naveed, Institute of Soil and Environmental Sciences, University of Agriculture Faisalabad, Faisalabad, Pakistan
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.