 Margarita Gomila
Margarita Gomila Arantxa Peña
Arantxa Peña Magdalena Mulet
Magdalena Mulet Jorge Lalucat
Jorge Lalucat Elena García-Valdés
Elena García-Valdés
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 18 March 2015
Sec. Evolutionary and Genomic Microbiology
Volume 6 - 2015 | https://doi.org/10.3389/fmicb.2015.00214
The genus Pseudomonas currently contains 144 species, making it the genus of Gram-negative bacteria that contains the largest number of species. Currently, multilocus sequence analysis (MLSA) is the preferred method for establishing the phylogeny between species and genera. Four partial gene sequences of housekeeping genes (16S rRNA, gyrB, rpoB, and rpoD) were obtained from 112 complete or draft genomes of strains related to the genus Pseudomonas that were available in databases. These genes were analyzed together with the corresponding sequences of 133 Pseudomonas type strains of validly published species to assess their correct phylogenetic assignations. We confirmed that 30% of the sequenced genomes of non-type strains were not correctly assigned at the species level in the accepted taxonomy of the genus and that 20% of the strains were not identified at the species level. Most of these strains had been isolated and classified several years ago, and their taxonomic status has not been updated by modern techniques. MLSA was also compared with indices based on the analysis of whole-genome sequences that have been proposed for species delineation, such as tetranucleotide usage patterns (TETRA), average nucleotide identity (ANIm, based on MUMmer and ANIb, based on BLAST) and genome-to-genome distance (GGDC). TETRA was useful for discriminating Pseudomonas from other genera, whereas ANIb and GGDC clearly separated strains of different species. ANIb showed the strongest correlation with MLSA. The correct species classification is a prerequisite for most diversity and evolutionary studies. This work highlights the necessity for complete genomic sequences of type strains to build a phylogenomic taxonomy and that all new genome sequences submitted to databases should be correctly assigned to species to avoid taxonomic inconsistencies.
The genus Pseudomonas is one of the most complex bacterial genera and is currently the genus of Gram-negative bacteria with the largest number of species; in fact, the number of species in the genus has increased every year (10 additional species in 2013 and six in 2014 through October). The current number of recognized and validly published species is 144, including 10 subspecies; these species are present in the List of Prokaryotic Names with Standing in Nomenclature (Parte, 2014).
The taxonomy of the genus has evolved simultaneously with the available methodologies since its first description. The genus Pseudomonas was described by Migula in 1894 according to the morphological characteristics of its members (Migula, 1900). For many years, the genus comprised many species that were not always well-characterized until the work of Stanier et al. (1966) in which the physiological and biochemical properties clearly established the taxonomical basis for the identification of the species. In 1984, the genus was revised, and a subdivision of five groups was implemented based on the DNA–DNA hybridisation (DDH) and rRNA-DNA hybridisation results (Palleroni, 1984). Later, the five groups were recognized as being associated with the class Proteobacteria (De Vos and De Ley, 1983; De Vos et al., 1985, 1989; De Ley, 1992); the members of the genus Pseudomonas “sensu stricto” were shown to belong to rRNA-DNA group I in the subclass Gammaproteobacteria. Since then, several authors have reviewed the taxonomic status of the genus Pseudomonas (Moore et al., 1996; Anzai et al., 2000; Peix et al., 2009; Mulet et al., 2012a). The approved list of bacterial names (Skerman et al., 1980) included 96 Pseudomonas species; however, only 31 of those species are considered true species in the genus Pseudomonas in the accepted taxonomy.
Although the 16S rRNA gene is the basic tool of the current bacterial classification system, it is known that closely related species of bacteria cannot be differentiated based on this gene. Therefore, over the past 10 years, other gene sequences have been used as phylogenetic molecular markers in taxonomic studies, such as atpD, gyrB, rpoB, recA, and rpoD (Yamamoto and Harayama, 1998; Hilario et al., 2004; Tayeb et al., 2005, 2008). Mulet and collaborators have demonstrated that the analysis of the sequences of four housekeeping genes (16S rRNA, gyrB, rpoB, and rpoD) in all known species of the genus clarified the phylogeny and greatly facilitated the identification of new strains (Mulet et al., 2012a; Sánchez et al., 2014). The multilocus sequence analysis (MLSA) approach based on the sequence analysis of the four housekeeping genes has proven reliable for species delineation and strain identification in Pseudomonas (Mulet et al., 2012b).
Whole-genome sequences can provide valuable information on the evolutionary and taxonomic relationships in bacteriology. In 2005, Coenye et al. (2005) published an article entitled “Toward a prokaryotic genomic taxonomy” and presented an overview of available approaches to assess the taxonomic relationships between prokaryotic species based on complete genome sequences. These genomic methods are delineated to substitute the experimental DDH by providing the possibility of creating accumulative databases of whole genome sequences. The digital methods used in the genome comparisons for the species delineation in several bacterial genera have been recently discussed by Li et al. (2015) and Colston et al. (2014). The methods tested in the present study, applied to strains in the genus Pseudomonas, were: tetranucleotide usage patterns (TETRA; Teeling et al., 2004), average nucleotide identity (ANIm and ANIb; Goris et al., 2007), and genome-to-genome distance (GGDC; Meier-Kolthoff et al., 2013).
Genome sequencing is expected to provide a relevant tool in bacterial taxonomy, and results obtained in the analysis of Pseudomonas species will assist in validating the proposed methods of comparison. Next-generation sequencing (NGS) currently provides an exponentially increasing number of whole genome sequences of bacterial strains, and the sequences are used frequently for comparative analyses from which phylogenetic or evolutionary conclusions are drawn. The correct strain assignation to a known species, however, is essential for correct conclusions. Many of the Pseudomonas strains under study were isolated and classified several years ago, and their correct taxonomic position is frequently dubious with the current taxonomic tools. The need for a taxonomical revision of several strains defined as different species of the genus Pseudomonas was evident when the strains were analyzed with a MLSA using the combined genes atpD, carA, recA, and 16S rRNA (Hilario et al., 2004) or using gyrB, rpoD, and 16S rRNA (Yamamoto et al., 2000; Mulet et al., 2010). In fact, Mulet et al. (2013) performed a taxonomical revision of P. putida strains based on a MLSA with the combined genes 16S rRNA, gyrB, and rpoD. Their results demonstrated that strains assigned to biovar A of the species were located in the P. putida group although not all belonged to the species P. putida. Biovar B strains were scattered among six subgroups of the P. fluorescens group and also belonged within the P. putida group.
Four partial gene sequences of housekeeping genes (16S rRNA, gyrB, rpoB, and rpoD) were obtained from 112 complete or draft genomes of strains related to the genus Pseudomonas that were available in databases until December 2012. These genes were analyzed together with the corresponding sequences of 133 Pseudomonas type strains of validly published species to assess their correct phylogenetic assignations. Because of the complex taxonomical relationships among species and pathovars in the P. syringae phylogenetic group, only six of the 63 available complete P. syringae genomes were considered. The P. syringae species complex has been studied at the intraspecies level in a recent publication with similar methodology (Marcelletti and Scortichini, 2014).
The main objectives of the present study were: (i) to infer the phylogeny and taxonomic affiliation of the 112 whole genome sequenced strains in the existing taxonomy of the genus Pseudomonas; (ii) to compare MLSA with the genome-based methods for species delineation (TETRA, ANIb, ANIm, and GGDC); and (iii) to compare the genome-based methods against each other.
A total of 253 Pseudomonas strains were analyzed in this study, comprising 112 complete or draft genomes of the Pseudomonas strains available in the databases and 141 strains of validly published Pseudomonas species (Mulet et al., 2012a). Those 141 taxonomically well-characterized strains included 133 Pseudomonas type strains, two subspecies of Pseudomonas chlororaphis (P. chlororaphis subsp. aurantiaca and P. chlororaphis subsp. aureofaciens) and Pseudomonas pseudoalcaligenes, the later synonym of Pseudomonas oleovorans subsp. oleovorans (Saha et al., 2010). In addition to the type strains, four taxonomically well-characterized strains of the Pseudomonas stutzeri phylogenetic group were also included: two strains of the species P. stutzeri (both members of the genomovar 1, ATCC 27951 and A15) and two strains of Pseudomonas balearica (LS401 and st101). “Pseudomonas alkylphenolia” JCM 16553 was also included although it has no standing in the nomenclature (Veeranagouda et al., 2011). The set of 112 genome sequences of Pseudomonas was retrieved from the Genbank database on 31st December of 2012. All complete and draft genomes not taxonomically identified as members of the P. syringae group were included in the analysis. Six genomes affiliated with the P. syringae group were also selected. Genomes that did not contain the full-length 16S rRNA, gyrB, rpoB, and rpoD genes sequences were removed from the dataset. The list of the 112 complete or draft genomes analyzed is shown in Supplementary Table 1.
The sequences of the 16S rRNA, gyrB, rpoB, and rpoD genes were extracted from each complete genome studied and were compared with the corresponding sequences of all species type strains described until 2012. The 16S rRNA, gyrB, rpoB, and rpoD gene sequences of the type strains were retrieved from our previous publications (Mulet et al., 2010, 2012a) and are available in the public National Centre for Biotechnology Information (NCBI) database. A series of individual trees was generated from the 16S rRNA, gyrB, rpoB, and rpoD partial gene sequences. Concatenated gene trees were constructed using the individual alignments in the following order: 16S rRNA (1309 nt), gyrB (803 nt), rpoD (791 nt), and rpoB (923 nt).
The alignments were conducted using a hierarchical method for multiple alignments implemented in the program CLUSTAL_X (Thompson et al., 1997). Automatically aligned sequences were checked manually. Similarities and evolutionary distances were calculated with programs implemented in PHYLIP (Phylogeny Inference Package, version 3.5c) (Felsenstein, 1981). Gene distances were calculated from nucleotide sequences using the Jukes-Cantor method (Jukes and Cantor, 1969), and dendrograms were generated using the neighbor-joining (NJ), minimum-evolution (ME), and maximum parsimony (MP) methods. A bootstrap analysis of 1000 replications was also performed. Values higher than 50% (from 1000) are indicated only at the groups or subgroups branching nodes of the corresponding trees. The topologies of the trees were visualized using the TreeView program (Page, 1996).
Among the Pseudomonas genomes, the correlation of the tetranucleotide signatures (TETRA), the average nucleotide identity (ANI) and the GGDC were calculated between pairwise genomic comparisons. The statistical calculations of the tetranucleotide frequencies (TETRA) (Teeling et al., 2004), the ANIb and the ANIm were calculated using the JSpecies software tool available at the webpage http://www.imedea.uib.es/jspecies. To calculate the ANI, the genomic sequence from one of the genomes in a pair (“the query”) was cut by the software into 1020 nucleotide consecutive fragments. The 1020 nt fragments were then used to search against the whole genomic sequence of the other genome in the pair (“the reference”) using the BLASTN algorithm (Altschul et al., 1997). The ANI between the query genome and the reference genome was calculated as the mean identity of all the BLASTN matches that showed more than 30% overall sequence identity over an alignable region of at least 70% of their length. The ANI was calculated based on the BLAST algorithm, ANIb (Altschul et al., 1997; Goris et al., 2007), and the MUMmer ultra-rapid aligning tool, ANIm (Kurtz et al., 2004). The recommended species cut-off was 95% for the ANIb and ANIm indices, and higher than 0.99 for the TETRA signature (Richter and Rosselló-Móra, 2009). The GGDC method functions based on the principle that two genomes are locally aligned using BLAST, which produces a set of high-scoring segment pairs (HSPs); the information in these HSPs is transformed to a single GGDC value using a specific distance formula that sets the species cut-off at 70% similarity. GGDC was calculated using the web service http://ggdc.dsmz.de (Meier-Kolthoff et al., 2013). GGDC 2.0 is an updated and enhanced version with improved DDH-prediction models and additional features such as confidence-interval estimation. The matrices obtained in our study for each parameter were used to generate a dendrogram using Permut Matrix software by applying an average linkage method (UPGMA hierarchical clustering) and Pearson's distance correlation (Caraux and Pinloche, 2005). The dendrograms were constructed using the average value of the duplicate analyses for each strain to assess topology coherence.
Parametric correlations based on the Pearson's product-moment coefficient and non-parametric correlations using the Spearman's rank correlation coefficient and Kendall tau rank correlation coefficient were calculated between all the whole-genome comparison results and the concatenated phylogenetic MLSA. Correlation analysis were performed using SPSS Plot v.11.0 software. Representations between the whole-genome comparison results and the concatenated phylogenetic MLSA were also graphed.
The phylogenetic analysis included the 141 reference strains of species that were validly described and used in a previous paper (Mulet et al., 2012a) combined with 112 Pseudomonas strains that had complete or draft genomes available in databases (Supplementary Table 1). A series of individual and concatenated phylogenetic trees from 16S rRNA, gyrB, rpoB, and rpoD partial gene alignments were generated. Individual dendrograms were generated using different methods, namely the NJ, MP, and ME methods. Their topologies were congruent (data not shown), as previously demonstrated by Mulet et al. (2010). Phylogenetic groups (G) and subgroups (SG) were defined by the length and branching order of the concatenated gene tree, as previously proposed (Mulet et al., 2010) and updated in 2012 (Mulet et al., 2012a). The name of the first species described in a group or subgroup was chosen to designate that group or subgroup. The resulting groups were supported by high bootstrap values.
A phylogenetic tree (Figure 1) was generated based on the concatenated sequences with a total length of 3711 nucleotides in the following order: 16S rRNA (1278 nt), gyrB (801 nt), rpoD (717 nt), and rpoB (915 nt). Phylogenetic assignation to a known species, group or subgroup of the 112 complete or draft genomes analyzed was congruent in all the trees (data not shown). Forty-eight of the 112 strains (42.8% of the genomes analyzed) were located in the same phylogenetic branch as the corresponding species type strain (or genomovar reference strains in the P. stutzeri species) with a similarity higher than 97%, which was the accepted species threshold (Mulet et al., 2010, 2012a), and their species assignations were considered correct. As observed in Table 1, 22 of the whole genome sequenced strains studied (20%) were only assigned to the genus and 34 strains (30%) were not correctly identified at the species level. For example, strain BBc6R8, which had been identified as P. fluorescens, was included in the P. gessardii phylogenetic subgroup, and P. fulva 12-X was closer to P. straminea than to the P. fulva type strain. The closest species type strain for the 112 complete or draft genomes analyzed based on the concatenated analysis of four genes is listed in Table 1 and in Supplementary Table 2. Thirty-seven of the genomes were less than 97% similar to the closest type strain and might be considered representatives of Pseudomonas species not yet described or have to be assigned to genomovars in the case of P. stutzeri.
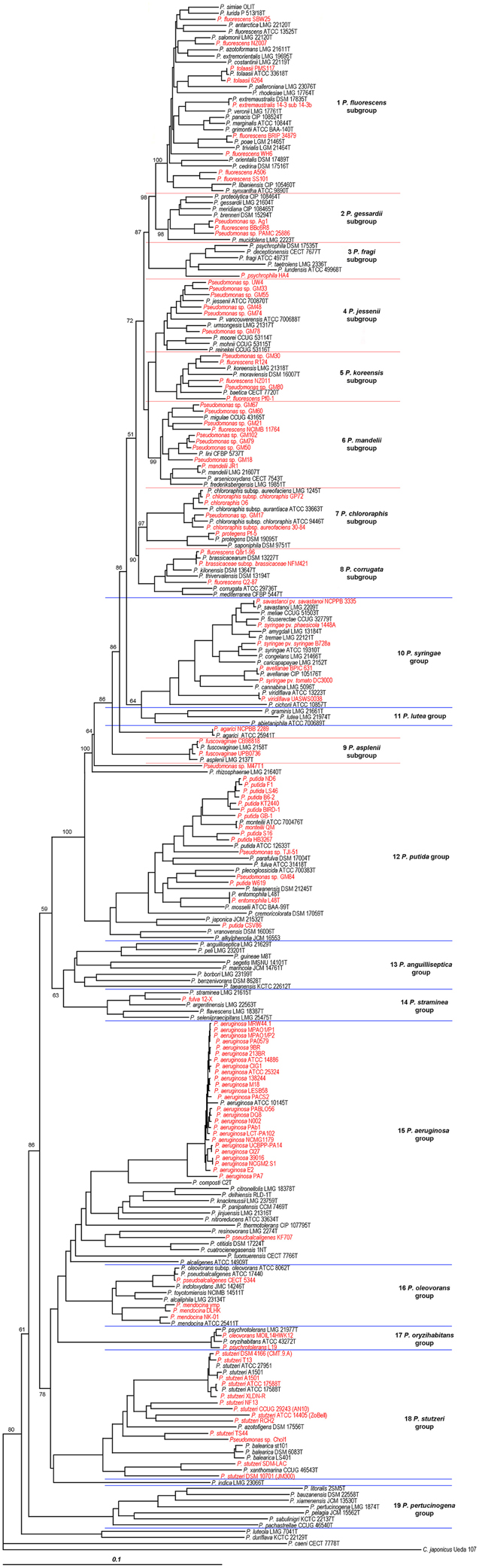
Figure 1. Phylogenetic tree of the 112 complete or draft genomes of strains related to the genus Pseudomonas and the 141 well described Pseudomonas strains used in this study based on the phylogenetic analysis of four concatenated genes (16S rRNA, gyrB, rpoB, and rpoD). The strains analyzed in this study whose genomes have been sequenced are labeled in red. Distance matrices were calculated by the Jukes-Cantor method. Dendrograms were generated by neighbor-joining. Cellvibrio japonicum Ueda107 was used as outgroup. The bar indicates sequence divergence. Percentage bootstrap values only of groups and subgroups higher than 50% of 1000 replicates are indicated at branching nodes.
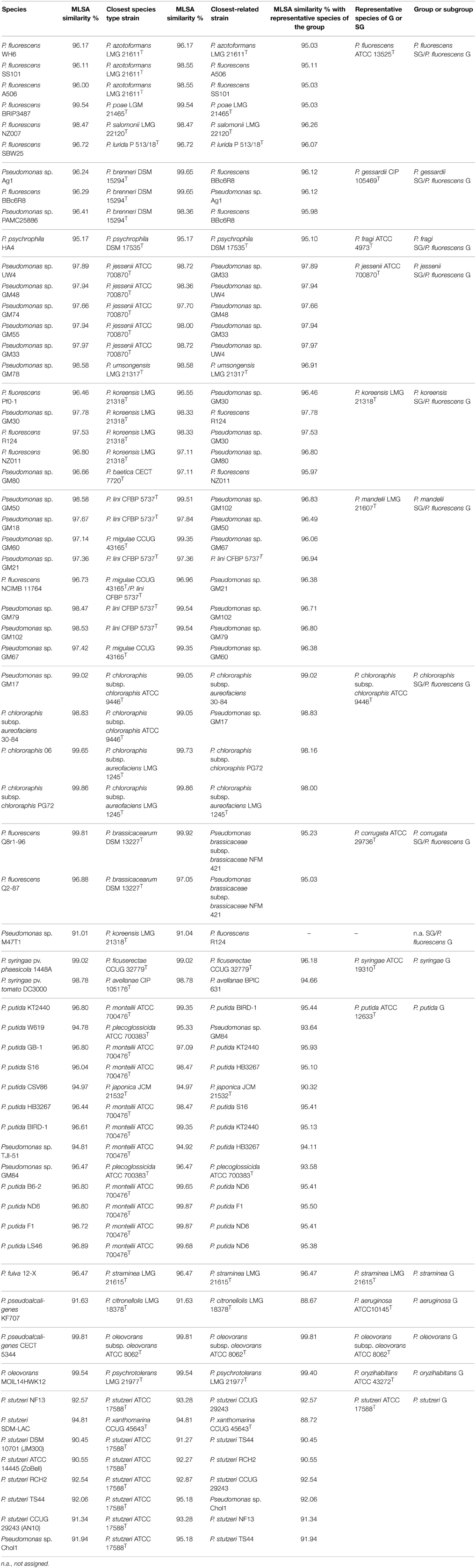
Table 1. Phylogenetic affiliation based on concatenated MLSA analysis for the 63 whole genome sequenced strains not assigned, or incorrectly assigned at the species level, including strains of P. stutzeri genomovars.
TETRA, average nucleotide identity based on MUMmer (ANIm) or BLAST (ANIb) and GGDC comparisons were calculated for the 112 genomes. In total, each final dataset consisted of 12,544 pairwise values, including the 112 pairwise comparisons of each genome with itself. Each square matrix obtained was transformed in a lower-triangular matrix using the average value of the duplicate analyses for each pair. The final dataset consisted of 6328 pairwise values. A dendrogram was generated for each matrix to assess their phylogenetic coherence (data not shown). The ANIb dendrogram showed the best topology congruence compared to the MLSA phylogenetic tree (Supplementary Figure 1).
Phylogenetic similarities in the analysis of the four concatenated genes were compared with all the indices calculated in the whole genome analyses. The results are plotted in Figure 2. The overall relationship between the MLSA analysis and the whole-genome analysis was found to be non-linear, which is consistent with previous studies performed using ANI and 16S rRNA gene sequence similarities (Konstantinidis and Tiedje, 2005a,b; Mulet et al., 2010; Kim et al., 2014). Correlation analyses between all the whole-genome comparison results and concatenated phylogenetic MLSA distances were also performed (Supplementary Table 3). Among the various methods proposed for substituting the experimental DDH high correlations were found between MLSA and ANIb (0.933 Spearman's rho and 0.917 Pearson coefficients) and between MLSA and GGDC (0.928 Spearman's rho and 0.766 Pearson's coefficients). The Pearson's correlation coefficients between ANIm and MLSA, GGDC and MLSA, and TETRA and MLSA were 0.838, 0.766, and 0.65, respectively. The ANIb values were selected and thoroughly analyzed as indicated in Figure 3. Square A (ANIb ≥ 95; MLSA ≥ 97, the threshold species delimitation by each index) included 315 pairwise comparisons of strains of the same species, such as P. aeruginosa, P. mendocina, and P. putida, subspecies of P. oleovorans, and P. stutzeri members of the same genomovar. Most of the values (5845) accumulated in square B (ANIb < 90%; MLSA < 97%) and corresponded to interspecies comparisons. In a few cases, strains in the pairwise comparisons located in square A were assigned to different species, which supported a non-correct species identification for at least one of the strains compared (i.e., the pair P. brassicacearum NFM 421 and P. fluorescens Q8r1 or the pair P. oleovorans MOIL14HWK12 and P. psychrotolerans L19). The transition between squares A and B (ANIb in the range of <95% and ≥90%) included only 56 (0.9%) pairwise comparisons of strains; nine of those comparisons had MLSA values <97% and 47 comparisons had MLSA values ≥97%. Twenty-four of the 56 pair-wise values were combinations between P. aeruginosa PA7 (considered a taxonomic outlier of the P. aeruginosa species; Roy et al., 2010) and the other genomes of the P. aeruginosa strains. The rest of the values (32) were distributed among a few strains in the P. chlororaphis SG (three strains, two comparisons), the P. fluorescens SG (two strains, one comparison), the P. gessardii SG (two strains, one comparison), the P. jessenii SG (five strains, nine comparisons), the P. mandelii SG (seven strains, seven comparisons), the P. putida G (seven strains, six comparisons) and the P. stutzeri SG (four strains, two comparisons). These last two pairs of P. stutzeri strains were clearly outliers in the plot: AN10/NF13 (MLSA 93% and ANIb 93%) and Chol1/TS44 (MLSA 95% and ANIb 92%). The four strains had been phylogenetically assigned to four different genomovars of P. stutzeri (Lalucat et al., 2006; Peña et al., 2013).
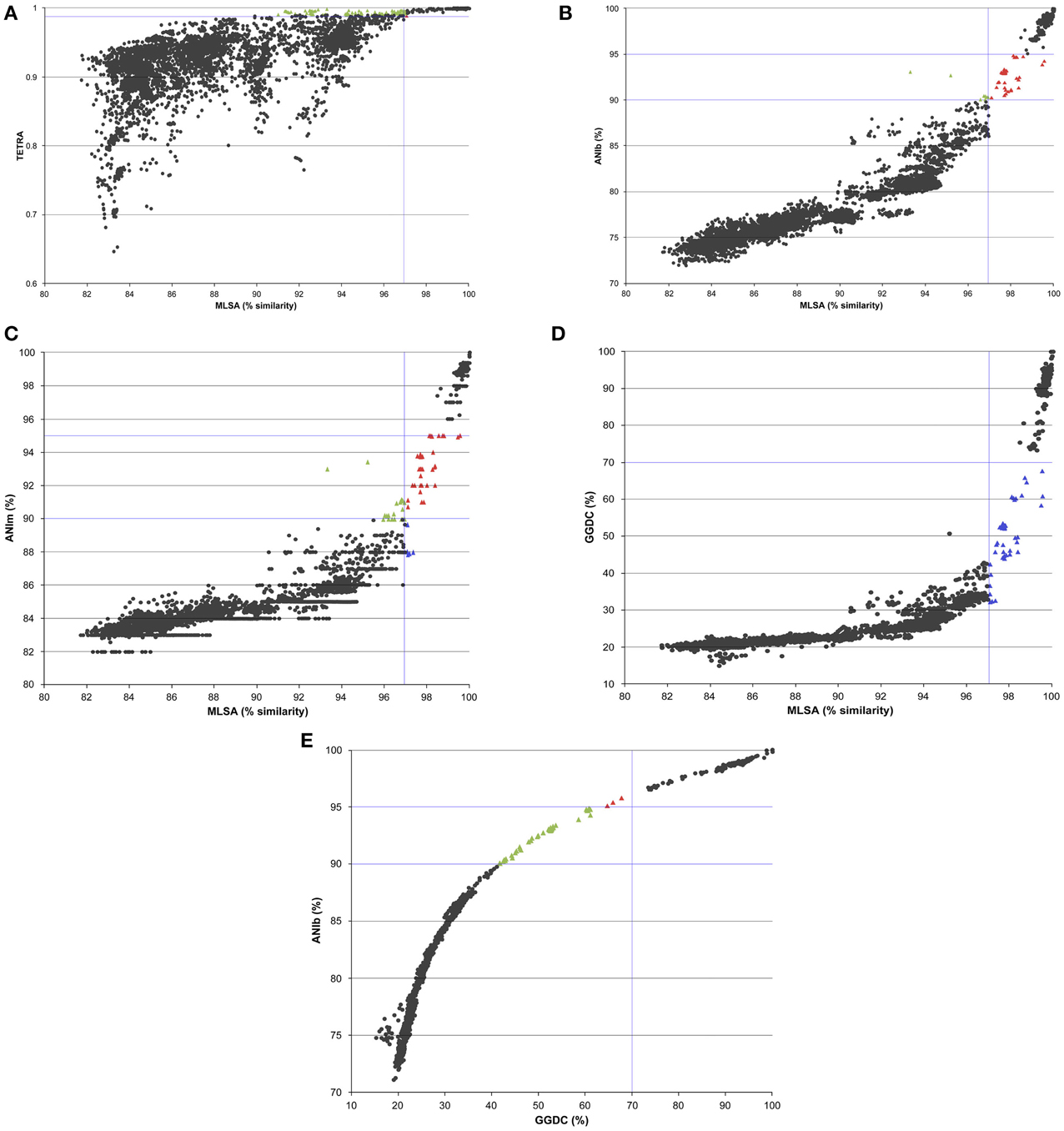
Figure 2. Graphs representing the relationship between TETRA (A), ANIb (B), ANIm (C), and GGDC (D) indices vs. MLSA sequence similarity for the genomes studied; (E) shows the relationship between ANIb and GGDC indices. Each dot represents a pairwise comparison; the genomic indices are plotted against the corresponding MLSA sequence similarity. TETRA signatures values in black circles indicate TETRA < 0.99 and MLSA < 97% and TETRA > 0.99 and MLSA > 97%; green triangles indicate TETRA > 0.99 and MLSA < 97%; and red triangles TETRA < 0.99 and MLSA > 97%. ANIb and ANIm black circles indicate genomic values <90% and MLSA < 97% and genomic values >95% and MLSA > 97%; green triangles genomic values between 90 and 95% and MLSA < 97%; red triangles genomic values between 90 and 95% and MLSA > 97%; and blue circles genomic values between 85 and 90% and MLSA > 97%. The values of GGDC < 70% and MLSA < 97% and GGDC > 70% and MLSA > 97% are indicated in black circles in GGDC plots; in green triangles are indicated values of GGDC < 70% and MLSA > 97%.
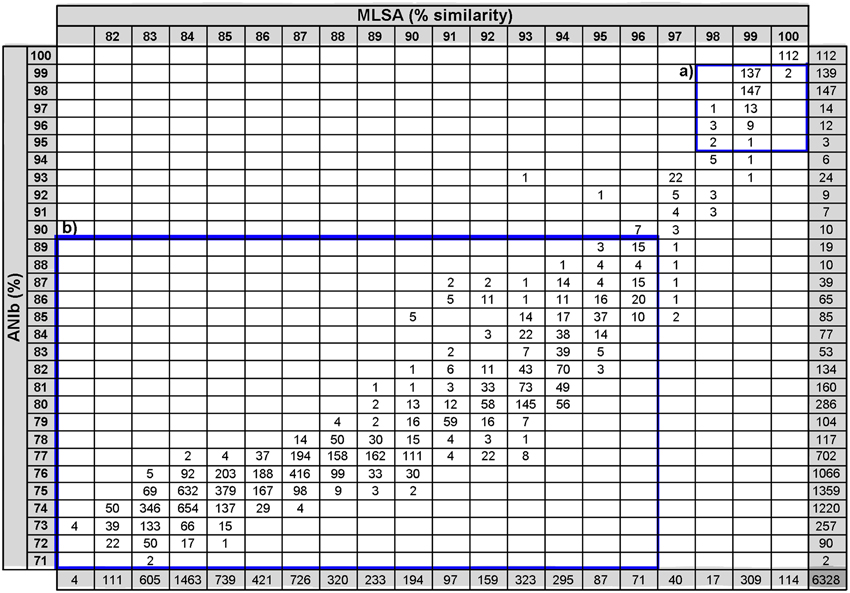
Figure 3. Association table between ANIb values and MLSA sequence similarities. The number of strain pairs is displayed in each category square. Square A indicates ANIb values ≥95% and MLSA ≥97%; square B indicates ANIb values <90% and MLSA values <97%.
Six pairwise values were located in the region of 97% MLSA similarity and below 90% ANIb and corresponded to two strains in the P. corrugata SG (classified as P. fluorescens Q2-87 and P. brassicacearum subsp. brassicacearum NFM421), three strains (P. fluorescens R124, P. fluorescens NZ011, and Pseudomonas sp. GM80) were in the P. koreensis SG, and four strains were in the P. mandelii SG (Pseudomonas sp. GM21, GM50, GM79, and GM102).
The graph representations for ANIm, ANIb, and GGDC plotted against MLSA (Figure 2) were very similar although the proposed species threshold for GGDC (70%) appeared to clearly discriminate the species boundaries because only a few pairs of strains (36 comparisons) were found between 50 and 70% GGDC distances, including combinations of the same strains as those detected in the ANIb/MLSA comparisons (strain PA7, strains in the P. chlororaphis SG and P. mandelii SG and four strains in the P. stutzeri G). In an attempt to find discontinuities in the graphs, the ANIb values were plotted against the GGDC values (Figure 2E). A high correlation of 0.940 was found between ANIb and GGDC, and a discontinuity might be observed in the region with 94–96% ANIb and 52–70% GGDC, in which only nine comparisons were detected and can be considered exceptions. Each pair of strains was closely related in the MLSA phylogenetic tree: P. avellanae 631/P. syringae DC300; Pseudomonas sp. GM50/GM102; Pseudomonas sp. GM33/P. putida UW4; P. chlororaphis strains; P. fluorescens 86/A506; and P. fluorescens A506/SS101.
Relationships between the MLSA and ANIb values were analyzed independently for the genomes of species in the P. aeruginosa, P. putida, and P. stutzeri phylogenetic groups (Figure 4). Correlation indices are shown in Supplementary Table 4. High Pearson's correlation coefficients between the MLSA and ANIb indices were detected among the strains in these three phylogenetic groups (i.e., 0.976, 0.983, and 0.971, respectively). In the three graphs, two different clusters were observed. In the P. aeruginosa group plot, a cluster at MLSA similarities higher than 98% and ANIb values higher than 98% was observed, and another cluster showed MLSA similarities of approximately 97.5% and ANIb values of approximately 93%. This last cluster of red-colored triangles in Figure 4A included all pairwise value combinations of P. aeruginosa PA7 with the other P. aeruginosa strains. In the P. putida and P. stutzeri graphs, the first cluster belonged to MLSA similarities higher than 97% and ANIb values higher than 95%, corresponding to intraspecies and intragenomovar comparisons, whereas a second cluster had MLSA similarities lower than 97% and ANIb values lower than 91%. Clear gaps were observed in the three plots.
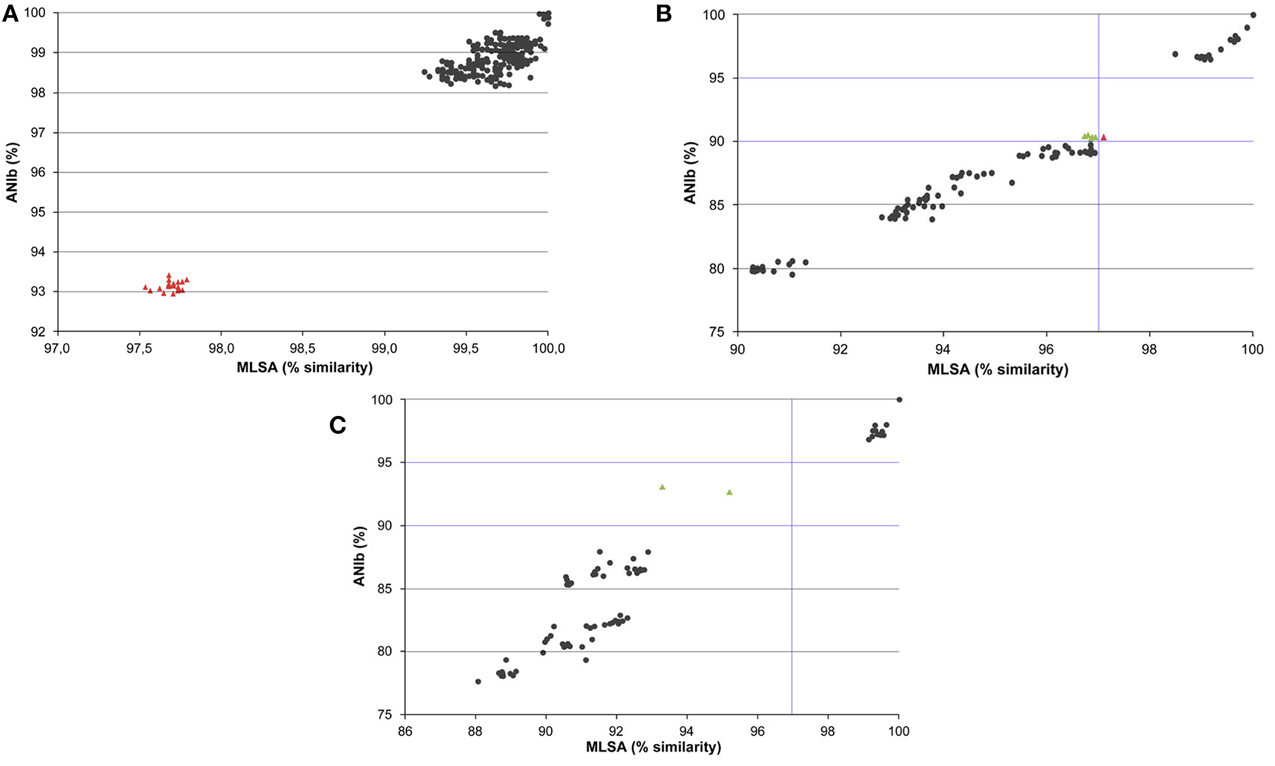
Figure 4. Graphs representing relationships between ANIb values and MLSA sequence similarities of pairwise comparisons of strains assigned to the P. aeruginosa (A), P. putida (B), and P. stutzeri (C) groups. Each dot represents pairwise values between the ANIb indices plotted against the corresponding MLSA sequence similarity. In the ANIb range of 90–95%, green triangles indicate pairwise comparisons with MLSA values lower than 97% and red triangles values between 97 and 98%; other values are indicated with black circles.
Bacterial species are considered groups of strains that are characterized by a certain degree of phenotypic consistency, by a significant degree (70%) of DNA–DNA hybridisation (DDH; Wayne et al., 1987) and by over 98.7–99% of 16S ribosomal RNA gene sequence similarity (Stackebrandt and Ebers, 2006). 16S rRNA gene sequences are highly conserved among strains of the same bacterial species and are frequently used to identify and classify microorganisms. Taxonomic classifications based only on the analysis of the 16S rRNA gene can create misclassifications in some instances, and the additional analysis of other housekeeping gene sequences should be performed for a correct phylogenetic affiliation. A threshold of 97% similarity in the MLSA study of four housekeeping genes (16S rRNA, gyrB, rpoD, and rpoB genes) has been proposed by Mulet et al. (2010), (2012a) for species differentiation in the genus Pseudomonas. The whole genome analysis of 112 Pseudomonas strains in the present study revealed that these genomes were in the range of 71–99% ANIb values, resulting in a continuous gradient of genetic relatedness, as shown in Figure 3. This type of continuous genetic gradient has been previously reported for the 10 strains studied by Konstantinidis et al. (2009) in the Shewanella genus. However, only a few strain comparisons were found in the ANIb region between 90 and 95% or in the GGDC region between 50 and 70%, which are below the species thresholds. The strains in these regions were classified as subspecies or genomovars within a species or were considered taxonomic outliers. The ANIb and GGDC values between these strains were consistent with the experimental DDH in the P. stutzeri group (Lalucat et al., 2006), the P. putida group (Regenhardt et al., 2002), and the P. aeruginosa group (Roy et al., 2010). These pairs of strains are always phylogenetically closely related, and extensive genetic exchange between each pair, or with other bacteria of the group or of other groups cannot be excluded. These strains can be considered different ecotypes of the same species or strains in the speciation process. A comparative study of their genomes must be conducted to confirm this possibility, which has been performed for the P. aeruginosa strain PA7. This strain is considered an outlier within the species, and its genome contains a similar number of genes as the other P. aeruginosa strains, but more than 1000 exclusive genes were found in PA7 compared with the P. aeruginosa strains PAO1, PA14, and LESB58 (Roy et al., 2010). This is a good example of the difficulty in obtaining taxonomic conclusions until the whole genome sequence of all type strains of the species in the group are available.
Intraspecies comparisons were characterized by ANIb values higher than 95%, which corresponds to MLSA values higher than 97% and GGDC values higher than 70%; these thresholds are considered the species boundaries by the three methods. However, the strains in the P. putida G assigned to the P. putida species were genomically very diverse, and some of those strains likely represent a new species, as has been proposed by Mulet et al. (2013) and more recently by Ohji et al. (2014).
P. aeruginosa, the type species of the genus, is phenotypically represented by homogeneous strains. Generally, there is no doubt in the identification of this species, and the strains studied were also very coherent in the genomic comparisons. However, this is not always true for other Pseudomonas species. The analysis of the 112 draft or complete genomes revealed that 63 strains (57%) were not assigned to species (22 strains) or were not correctly assigned (34 strains). These data were supported by the MLSA analysis and the whole-genome comparisons based on ANIb and GGDC. This result raised the question of which species those 56 genomes belonged to. The correct identification at the species level requires a polyphasic taxonomic study, but the MLSA tree, which included all the type strains, provides an adequate phylogenetic assignation to known species or the prediction of putative novel species. It is important to emphasize that incorrect identifications can lead to mistaken conclusions.
Several recent studies support our observation of wrong assignation to species of strains in the genus Pseudomonas. Duan and collaborators revealed that Pseudomonas sp. UW4 belongs to the fluorescens group, specifically the P. jessenii subgroup, and not to P. putida as previously proposed (Duan et al., 2013). Paulsen et al. (2005) published the complete genome of P. fluorescens Pf-5, and this strain was later reclassified as P. protegens Pf-5 (Ramette et al., 2011).
MLSA is the most convenient method nowadays for the assessment of the phylogenetic relationships among the species in the genus Pseudomonas until whole genome sequences of the type strains are available, and the best correlation with MLSA was found with ANIb in the study of the different digital whole genome comparisons tested. However, the use of GGDC was shown to be useful in species discrimination. As previously observed by other authors, most of the intraspecies ANIb values were found to be higher than 96%, which is within the range previously recommended for species delineation (Konstantinidis and Tiedje, 2005a,b) and corresponds to MLSA values higher than 97%, as proposed by Mulet et al. (2010).
In conclusion, because the resolution of the 16S rRNA tree was not sufficient to differentiate 63 genomes from other closely related Pseudomonas species, the classification of these bacteria should follow the phylogeny of the housekeeping genes until the whole genome sequence of the type strains of all Pseudomonas species is known. In recent years, thanks to NGS technologies, a remarkable increase in the number of sequenced genomes, drafts or complete, are available, but the correct assignation of the sequenced strains to the corresponding species with the accepted taxonomic tools is important before comparative analyses with other genomes can be performed. The need for the whole genome sequences of all the type strains, which are the only species references that are publicly available in culture collections, is evident. The project “Genomic Encyclopedia of Bacteria and Archaea: Sequencing a Myriad of Type Strains” (GEBA project) was initiated to address this problem (Kyrpides et al., 2014).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Financial support was obtained from the Spanish MINECO through projects CGL2011-24318 and Consolider CSD2009-00006, as well as funds for competitive research groups from the Government of the Balearic Islands (the last two funds with FEDER cofunding). MG and AP were supported by a postdoctoral contract from the University of the Balearic Islands.
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2015.00214/abstract
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, M., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Anzai, Y., Kim, H., Park, J., Wakabayashi, H., and Oyaizu, H. (2000). Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evol. Microbiol. 50, 1563–1589. doi: 10.1099/00207713-50-4-1563
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Caraux, G., and Pinloche, S. (2005). Permutmatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 21, 1280–1281. doi: 10.1093/bioinformatics/bti141
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coenye, T., Gevers, D., Van de Peer, Y., Vandamme, P., and Swings, J. (2005). Towards a prokaryotic genomic taxonomy. FEMS Microbiol. Rev. 29, 147–167. doi: 10.1016/j.femsre.2004.11.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Colston, S. M., Fullmer, M. S., Beka, L., Lamy, B., Gogarten, J. P., and Graf, J. (2014). Bioinformatic genome comparisons for taxonomic and phylogenetic assignments using Aeromonas as a test case. mBio 5:e02136-14. doi: 10.1128/mBio.02136-14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Ley, J. (1992). “The proteobacteria: ribosomal rna cistron similarities and bacterial taxonomy,” in The Prokaryotes, a Handbook on the Biology of Bacteria, Ecophysiology, Isolation, Identification and Applications, 2nd Edn., Vol. 2, eds A. Balows, H. G. Truper, M. Dworkin, W. Harder, and K. H. Schleifer (Berlin: Springer), 2111–2140.
De Vos, P., and De Ley, J. (1983). Intra- and intergeneric similarities of Pseudomonas and Xanthomonas ribosomal ribonucleic acid cistrons. Int. J. Syst. Bacteriol. 33, 487–509. doi: 10.1099/00207713-33-3-487
De Vos, P., Goor, M., Gillis, M., and De Ley, J. (1985). Ribosomal ribonucleic acid cistron similarities of phytopathogenic Pseudomonas species. Int. J. Syst. Bacteriol. 35, 169–184. doi: 10.1099/00207713-35-2-169
De Vos, P., Van Landschoot, A., Segers, P., Tytgat, R., Gillis, M., Bauwens, M., et al. (1989). Genotypic relationships and taxonomic localization of unclassified Pseudomonas and Pseudomonas-like strains by deoxyribonucleic acid: ribosomal ribonucleic acid hybridizations. Int. J. Syst. Bacteriol. 39, 35–49. doi: 10.1099/00207713-39-1-35
Duan, J., Jiang, W., Cheng, Z., Heikkila, J. J., and Glick, B. R. (2013). The complete genome sequence of the plant growth-promoting bacterium Pseudomonas sp. UW4. PLoS ONE 8:e58640. doi: 10.1371/journal.pone.0058640
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hilario, E., Buckley, T. R., and Young, J. M. (2004). Improved resolution on the phylogenetic relationships among Pseudomonas by the combined analysis of atpD, carA, recA and 16S rDNA. Antonie van Leeuwenhoek 86, 51–64. doi: 10.1023/B:ANTO.0000024910.57117.16
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jukes, T., and Cantor, C. (1969). “Evolution of protein molecules,” in Mammalian Protein Metabolism, ed H. N. Munro (New York, NY: Academic Press), 21–132.
Kim, M., Oh, H.-S., Park, S.-C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Konstantinidis, K. T., Serres, M. H., Romine, M. F., Rodrigues, J. L. M., Auchtung, J., McCue, L.-A., et al. (2009). Comparative systems biology across an evolutionary gradient within the Shewanella genus. Proc. Natl. Acad. Sci. U.S.A. 106, 15909–15914. doi: 10.1073/pnas.0902000106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Konstantinidis, K. T., and Tiedje, J. M. (2005a). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Konstantinidis, K. T., and Tiedje, J. M. (2005b). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5, R12. doi: 10.1186/gb-2004-5-2-r12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kyrpides, N. C., Hugenholtz, P., Eisen, J. A., Woyke, T., Göker, M., Parker, C. T., et al. (2014). Genomic encyclopedia of bacteria and archaea: sequencing a myriad of type strains. PLoS Biol. 12:e1001920. doi: 10.1371/journal.pbio.1001920
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lalucat, J., Bennasar, A., Bosch, R., García-Valdés, E., and Palleroni, N. J. (2006). Biology of Pseudomonas stutzeri. Microbiol. Mol. Biol. Rev. 70, 510–547. doi: 10.1128/MMBR.00047-05
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, X., Huang, Y., and Whitman, B. (2015). The relationship of the whole genome sequence identity to DNA hybridization varies between genera of prokaryotes. Antonie van Leeuwenhoek 107, 241–249. doi: 10.1007/s10482-014-0322-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marcelletti, S., and Scortichini, M. (2014). Definition of plant pathogenic Pseudomonas genomospecies of the Pseudomonas syringae complex through multiple comparative approaches. Phytopathology 104, 1274–1282. doi: 10.1094/PHYTO-12-13-0344-R
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Migula, W. (1900). System der Bakterien. Handbuck der Morphologie, Entwickelung-Geschichte und Systematik der Bakterien, Vol. 2. Jena: Verlag von Gustav Fischer. doi: 10.1099/00207713-37-4-463
Moore, E. R. B., Mua, M., Arnscheidt, A., Böttger, E. C., Hutson, R. A., Collins, M. D., et al. (1996). The determination and comparison of the 16S rDNA gene sequences of species of the genus Pseudomonas (sensu stricto) and estimation of the natural intrageneric relationships. Syst. Appl. Microbiol. 19, 476–492. doi: 10.1016/S0723-2020(96)80021-X
Mulet, M., García-Valdés, E., and Lalucat, J. (2013). Phylogenetic affiliation of Pseudomonas putida biovar A and B strains. Res. Microbiol. 164, 351–359. doi: 10.1016/j.resmic.2013.01.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mulet, M., Gomila, M., Lemaitre, B., Lalucat, J., and García-Valdés, E. (2012b). Taxonomic characterization of Pseudomonas strain L48 and formal proposal of Pseudomonas entomophila sp. nov. Syst. Appl. Microbiol. 35, 145–149. doi: 10.1016/j.syapm.2011.12.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mulet, M., Gomila, M., Scotta, C., Sánchez, D., Lalucat, J., and García-Valdés, E. (2012a). Concordance between whole-cell matrix-assisted laser-desorption/ionization time-of-flight mass spectrometry and multilocus sequence analysis approaches in species discrimination within the genus Pseudomonas. Syst. Appl. Microbiol. 35, 455–464. doi: 10.1016/j.syapm.2012.08.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mulet, M., Lalucat, J., and García-Valdés, E. (2010). DNA sequence-based analysis of the Pseudomonas species. Environ. Microbiol. 12, 1513–1530. doi: 10.1111/j.1462-2920.2010.02181.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ohji, S., Yamazoe, A., Hosoyama, A., Tsuchikane, K., Ezaki, T., and Fujita, N. (2014). The complete genome sequence of Pseudomonas putida NBRC 14164T confirms high intraspecies variation. Genome Announc. 2:e00029-14. doi: 10.1128/genomeA.00029-14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Page, R. D. M. (1996). TREEVIEW: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12, 357–358
Palleroni, N. J. (1984). “Genus I. Pseudomonas Migula 1894,” in Bergey's Manual of Systematic Bacteriology, Vol. 1, eds N. R. Krieg and J. G. Holt (Baltimore, MD: Williams & Wilkins), 141–199.
Parte, A. (2014). LPSN-list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 42, D613–D616. doi: 10.1093/nar/gkt1111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Paulsen, I. T., Press, C. M., Ravel, J., Kobayashi, D. Y., Myers, G. S., Mavrodi, D. V., et al. (2005). Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat. Biotechnol. 23, 873–878. doi: 10.1038/nbt1110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Peix, A., Ramírez-Bahena, M. H., and Velázquez, E. (2009). Historical evolution and current status of the taxonomy of genus Pseudomonas. Infec. Gen. Evol. 9, 1132–1147. doi: 10.1016/j.meegid.2009.08.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Peña, A., Busquets, A., Gomila, M., Mayol, J., Bosch, R., Nogales, B., et al. (2013). Draft genome of Pseudomonas stutzeri strain NF13, a nitrogen fixer isolated from the Galapagos rift hydrothermal vent. Genome Announc. 1:e0011313. doi: 10.1128/genomeA.00113-13
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramette, A., Frapolli, M., Fischer-Le Saux, M., Gruffaz, C., Meyer, J. M., Défago, G., et al. (2011). Pseudomonas protegens sp. nov., widespread plant-protecting bacteria producing the biocontrol compounds 2,4-diacetylphloroglucinol and pyoluteorin. Syst. Appl. Microbiol. 34, 180–188. doi: 10.1016/j.syapm.2010.10.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Regenhardt, D., Heuer, H., Heim, S., Fernandez, D. U., Strömpl, C., Moore, E. R., et al. (2002). Pedigree and taxonomic credentials of Pseudomonas putida strain KT2440. Environ. Microbiol. 4, 912–915. doi: 10.1046/j.1462-2920.2002.00368.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roy, P. H., Tetu, S. G., Larouche, A., Elbourne, L., Tremblay, S., Ren, Q., et al. (2010). Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PLoS ONE 5:e8842. doi: 10.1371/journal.pone.0008842
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Saha, R., Spröer, C., Bec, B., and Bagley, S. (2010). Pseudomonas oleovorans subsp. lubricantis subsp. nov., and reclassification of Pseudomonas pseudoalcaligenes ATCC 17440T as later synonym of Pseudomonas oleovorans ATCC 8062T. Curr. Microbiol. 60, 294–300. doi: 10.1007/s00284-009-9540-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sánchez, D., Mulet, M., Rodríguez, A. C., David, Z., Lalucat, J., and García-Valdés, E. (2014). Pseudomonas aestusnigri sp. nov., isolated from crude oil-contaminated intertidal sand samples after the Prestige oil spill. Syst. Appl. Microbiol. 37, 89–94. doi: 10.1016/j.syapm.2013.09.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Skerman, V. B. D., McGowan, V., and Sneath, P. H. A. (1980). Approved list of bacterial names. Int. J. Syst. Bacteriol. 30, 225–420. doi: 10.1099/00207713-30-1-225
Stackebrandt, E., and Ebers, J. (2006). Taxonomic parameters revisited: tarnished gold standards. Microbiol. Today 33, 152–155.
Stanier, R. Y., Palleroni, N. J., and Doudoroff, M. (1966). The aerobic Pseudomonads: a taxonomic study. J. Gen. Microbiol. 43, 159–271. doi: 10.1099/00221287-43-2-159
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tayeb, L., Ageron, E., Grimont, F., and Grimont, P. A. D. (2005). Molecular phylogeny of the genus Pseudomonas based on rpoB sequences and application for the identification of isolates. Res. Microbiol. 156, 763–773. doi: 10.1016/j.resmic.2005.02.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tayeb, L. A., Lefevre, M., Passet, V., Diancourt, L., Brisse, S., and Grimont, P. A. D. (2008). Comparative phylogenies of Burkholderia, Ralstonia, Comamonas, Brevundimonas and related organisms derived from rpoB, gyrB and rrs gene sequences. Res. Microbiol. 159, 169–177. doi: 10.1016/j.resmic.2007.12.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Teeling, H., Meyerdierks, A., Bauer, M., Amann, R., and Glöckner, F. O. (2004). Application of tetranucleotide frequencies for the assignment of genomic fragments. Environ. Microbiol. 6, 938–947. doi: 10.1111/j.1462-2920.2004.00624.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The Clustal X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Veeranagouda, Y., Lee, K., Cho, A. R., Cho, K., Anderson, E. M., and Lam, J. S. (2011). Ssg, a putative glycosyltransferase, functions in lipo- and exopolysaccharide biosynthesis and cell surface-related properties in Pseudomonas alkylphenolia. FEMS Microbiol. Lett. 315, 38–45. doi: 10.1111/j.1574-6968.2010.02172.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wayne, L. G., Brenner, D. J., Colwell, R. R., Grimont, P. A. D., Kandler, O., Krichevsky, M. I., et al. (1987). Report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Int. J. Syst. Bacteriol. 37, 463–464. doi: 10.1099/00207713-37-4-463
Yamamoto, S., and Harayama, S. (1998). Phylogenetic relationships of Pseudomonas putida strains deduced from the nucleotide sequences of gyrB, rpoD and 16S rRNA genes. Int. J. Syst. Bacteriol. 48, 813–819. doi: 10.1099/00207713-48-3-813
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamamoto, S., Kasai, H., Arnold, D. L., Jackson, R. W., Vivian, A., and Harayama, S. (2000). Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology 146, 2385–2394. doi: 10.1099/00207713-37-4-463
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Pseudomonas, genomics, multilocus sequence analysis, taxonomy, systematics
Citation: Gomila M, Peña A, Mulet M, Lalucat J and García-Valdés E (2015) Phylogenomics and systematics in Pseudomonas. Front. Microbiol. 6:214. doi: 10.3389/fmicb.2015.00214
Received: 03 December 2014; Accepted: 02 March 2015;
Published: 18 March 2015.
Edited by:
Frank T. Robb, University of Maryland, USAReviewed by:
Edward R. B. Moore, University of Gothenburg, SwedenCopyright © 2015 Gomila, Peña, Mulet, Lalucat and García-Valdés. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena García-Valdés, Department of Biology, Universitat de les Illes Balears, Crtra. Valldemossa km 7.5, 07122 Palma de Mallorca, SpainZWxlbmEuZ2FyY2lhdmFsZGVzQHVpYi5lcw==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.