 Jia-Rong Jheng
Jia-Rong Jheng Jin-Yuan Ho
Jin-Yuan Ho Jim-Tong Horng
Jim-Tong Horng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 05 August 2014
Sec. Virology
Volume 5 - 2014 | https://doi.org/10.3389/fmicb.2014.00388
This article is part of the Research Topic The unfolded protein response in virus infections. View all 11 articles
Endoplasmic reticulum (ER) stress is a general term for representing the pathway by which various stimuli affect ER functions. ER stress induces the evolutionarily conserved signaling pathways, called the unfolded protein response (UPR), which compromises the stimulus and then determines whether the cell survives or dies. In recent years, ongoing research has suggested that these pathways may be linked to the autophagic response, which plays a key role in the cell's response to various stressors. Autophagy performs a self-digestion function, and its activation protects cells against certain pathogens. However, the link between the UPR and autophagy may be more complicated. These two systems may act dependently, or the induction of one system may interfere with the other. Experimental studies have found that different viruses modulate these mechanisms to allow them to escape the host immune response or, worse, to exploit the host's defense to their advantage; thus, this topic is a critical area in antiviral research. In this review, we summarize the current knowledge about how RNA viruses, including influenza virus, poliovirus, coxsackievirus, enterovirus 71, Japanese encephalitis virus, hepatitis C virus, and dengue virus, regulate these processes. We also discuss recent discoveries and how these will produce novel strategies for antiviral treatment.
The endoplasmic reticulum (ER) is a eukaryotic organelle in which an array of cell functions takes place. These include the transportation of cellular materials, provision of increased surface area for cellular reactions, and the production of proteins, steroids, and lipids. The ER may be overloaded with molecular chaperones, folding enzymes, and massive protein products during normal processes, such as in the differentiation of B lymphocytes into antibody-secreting plasma cells (Shaffer et al., 2004; Ma et al., 2010) or in highly specialized cells for secretion (Harding and Ron, 2002). In addition, dysfunction of the ER, known as ER stress, results from pathogenic stress signals, such as hypoxia (Koumenis, 2006), ER–Ca2+ depletion, viral infections, or agents that affect Ca2+ balance (i.e., thapsigargin), protein glycosylation (i.e., tunicamycin), and ER–Golgi vesicular transport (i.e., brefeldin A), which lead to accumulation of misfolded and unfolded proteins (Kaufman, 1999). To reduce the adverse effects of accumulating misfolded or unfolded proteins, the cell operates an adaptive response known as the unfolded protein response (UPR) to reduce the load of newly synthesized proteins within the ER and eliminate inappropriately folded proteins through upregulation of ER chaperone expression. In addition, proteins that fail to correctly fold are then deployed to the distal secretory pathway from the ER by the ER-associated protein degradation (ERAD) pathway of the UPR (Hampton, 2000; Yoshida et al., 2003).
There are two ERAD models for protein degradation: ubiquitin-proteasome ERAD, designated as ERAD (I), and autophagy-lysosome ERAD, designated as ERAD (II) (Fujita et al., 2007; Korolchuk et al., 2010). Both models depend on retrotranslocation of ERAD substrates from the ER back to the cytoplasm with the help of the Cdc48p–p97 complex. Most soluble misfolded proteins are cleared through the ubiquitin-proteasome system, which involves action of a cascade of three canonical ubiquitin enzymes: E1 ubiquitin-activating enzyme initiates the reaction by using ATP to covalently activate and then conjugate the ubiquitin to an E2 ubiquitin-conjugating enzyme. Ubiquitin is then transferred from the ubiquitin-charged E2 to the lysine residue of a specific target or a growing ubiquitin chain by E3 ubiquitin ligase, which results in a multiubiquitin chain-tagged substrate. Proteins that are ubiquitinated with K48-linked chains are specifically recognized by the 26S proteasome and subjected to degradation (Hershko et al., 1983). In contrast, ERAD (II) degrades both soluble and insoluble misfolded protein aggregates in autolysosome. Autophagy receptors and adaptors, called p62/SQSTM1, NBR1, HDAC6, and ALFY, bind to proteins with K63-specific monoubiquitination or polyubiquitin chains and then guide them to the concave side of developing autophagosomes (Behrends and Fulda, 2012). Notably, p62 also recognizes K48 polyubiquitin-tagged proteins for autophagic clearance upon proteasome dysfunction. In addition to the protective role of UPR, prolonged and/or excess ER stress typically activates caspase-12, an ER-resident caspase, leading to UPR-mediated cell death (Szegezdi et al., 2006).
Basal autophagy plays a key role in maintaining cellular homeostasis through eliminating unwanted proteins and damaged organelles by cellular self-digestion in the lysosome to fulfill the demand for the building blocks required for cell survival (Levine and Klionsky, 2004; Shintani and Klionsky, 2004). Recently, the study of autophagy regulation has grown in different research areas, including regulation of cancer development and progression (Mahoney et al., 2013a), lipid metabolism (Singh et al., 2009), degenerative diseases (Wang et al., 2006), and the control of viral pathogenesis (Jackson et al., 2005). The first step of autophagy relies on the formation of an isolation membrane at the so-called preautophagosomal site (PAS) where a system of evolutionarily conserved proteins (Atg proteins) comes together. Recent reports have revealed that the ER serves as a subcellular platform for autophagy initiation (Axe et al., 2008). The elongation of the initial autophagic membrane requires continued processing by two ubiquitin-like protein-conjugation systems, the Atg12 and LC3 systems, which modify the autophagy proteins, Atg5 and Atg8/LC3, respectively (Geng and Klionsky, 2008). The autophagosome then fuses with endosomal and/or lysosomal vesicles to create an autolysosome, where digestion of intracellular components occurs (Eskelinen, 2005). In addition, autophagy can be induced by various physiological and pathological conditions such as nutrient deprivation, oxidative stress, and pathogen infections. The live-or-dead signal is modulated by UPR and autophagy and several lines of evidence suggest there is communication between these two pathways (Bernales et al., 2006; Ogata et al., 2006; Yorimitsu et al., 2006; Salazar et al., 2009); thus, it is believed that these two pathways could be a therapeutic target in certain circumstances (Figure 1). Herein, we review recent findings, focusing on the regulation of the UPR and autophagy involved in RNA virus infection as a new antiviral strategy.
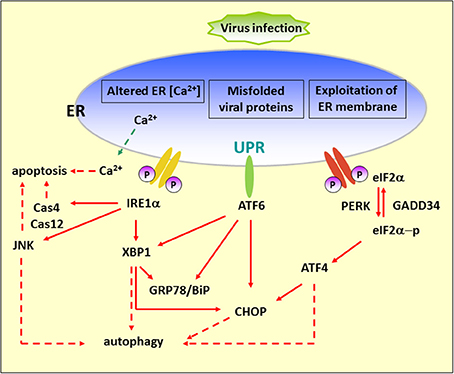
Figure 1. Diagram of the UPR arms and their connection to autophagy. Alteration of ER functions results from stress signals by RNA virus infection, by the exploitation of ER membrane for viral replication, rapid accumulation of viral proteins, imbalance of calcium concentration by viroporin, and the sabotage or depletion of ER membrane for viral release. This leads to the accumulation of misfolded and unfolded proteins, which triggers ER stress. To alleviate this adverse effect, the cells operate an adaptive UPR to reduce the load of the newly synthesized proteins in the ER by activating the PERK–eIF2α branch and eliminating inappropriate protein accumulation by upregulating ER chaperone proteins through IRE1 and ATF6 branches. In addition, the incurable misfolded proteins undergo retrotranslocation from the ER into cytosol for degradation by an ERAD mechanism. ER stress can contribute to autophagy via activation of JNK, XBP1, CHOP, and ATF4. Red dash arrows indicate the final outcome of the activated pathways, such as apoptosis and autophagy, caused by viral infection. Red solid arrows indicate the UPR pathways.
Viral virulence is determined by successful entrance, replication in the host cell, and release of mature virion. During the life cycle, ER stress may arise from the exploitation of the ER membrane, accumulation of misfolded proteins, imbalance of calcium concentration by viroporin, and the sabotage or depletion of the ER membrane during virion release. Details of viral effects are given as follows.
Many positive-strand RNA viruses cause the rearrangement of host intracellular membrane compartments that house replication complexes. ER, trans-Golgi, or lysosomes are the likely origin of virally induced membranes (Miller and Krijnse-Locker, 2008; Korolchuk et al., 2010). Upon poliovirus (PV) and coxsackievirus B3 (CVB3) infection, clusters of vesicles have been considered to derive from ER, although other cellular compartment marker proteins also colocalized with viral nonstructural proteins (Schlegel et al., 1996; Van Kuppeveld et al., 1997). Consistent with these findings, our previous study indicates that enterovirus 71 (EV71) nonstructural 2C protein, which participates in viral replication, is associated with the ER membrane through direct interaction with ER membrane protein reticulon 3 (RTN3), which is required and sufficient for immediate early virus replication and translation (Tang et al., 2007). In the RTN3 siRNA knockdown cells, synthesis of the 2C protein was ablated. However, in the RTN3 rescue cell line 2A3, the synthesis of viral protein and RNA was restored. Moreover, the interactions between RTN3 and two EV71 2C homologs of PV and CVA16 have been confirmed (Tang et al., 2007). Immunofluorescence studies reveal that replication of Flaviviruses dengue virus (DENV) and hepatitis C virus (HCV) may take place on perinuclear ER membranes (El-Hage and Luo, 2003). DENV2 nonstructural protein 2 (NS2A) is a 22-kDa hydrophobic protein containing five integral transmembrane segments that span the ER membrane. Functional analysis reveals that NS2A involves both DENV RNA synthesis and virion assembly/maturation (Xie et al., 2013). Furthermore, DENV infection induces ROCK-dependent vimentin rearrangement and subsequent ER redistribution (Lei et al., 2013). In addition, the HCV ER integral membrane protein, NS4B, is responsible for rearranging the ER membrane and inducing the formation of new ER-derived membrane structures, and this is possibly negatively regulated by RTN3-NS4B interaction (Lundin et al., 2003; Wu et al., 2014).
The N-glycosylation pathway in the ER modifies a mass of proteins at the asparagine residue of the consensus sequence Asn-X-Ser/Thr, where X is any amino acid except Pro (Kornfeld and Kornfeld, 1985; Gavel and Von Heijne, 1990). The modification influences protein folding and attributes various functional properties to the protein. Thus, interference with host protein glycosylation by viral proteins competing for the modification process may cause ER stress.
Viruses, including influenza A virus (IAV), hepatitis virus, and Japanese encephalitis virus (JEV), use this host cell process to enhance viral pathogenesis through facilitating folding and trafficking, affecting receptor interaction, and modulating host immune responses (Tatu et al., 1995; Dubuisson and Rice, 1996; Zai et al., 2013). Hemagglutinin (HA) of IAV is a type I transmembrane glycoprotein that determines viral antigenicity. Throughout the glycosylation process, HA rapidly associates with calnexin in a monoglucosylated form. Once folded, the HA monomers dissociate from calnexin and assemble into trimeric structures in the ER or in the intermediate compartment (Tatu et al., 1995). HCV envelope glycoproteins E1 and E2 have been shown to cooperate for the formation of a functional noncovalent heterodimer (Dubuisson et al., 1994; Dubuisson and Rice, 1996). Based on studies of HCV pseudoparticles, coexpression of both envelope glycoproteins has been shown to be necessary to produce infectious pseudoparticles (Bartosch et al., 2003). Glycosylation also occurs in JEV and WNV proteins, namely the precursor of membrane protein (prM), the envelope protein (E), and the nonstructural protein NS1, which affects the efficiency of virus release and infection (Hanna et al., 2005; Zai et al., 2013).
Typically, viroporins are composed by integral membrane proteins to form a hydrophilic pore, which targets different cellular compartments and ions, thus affecting various viral functions (Nieva et al., 2012). For example, IAV M2 reduces the acidity of vesicular compartments to trigger virus uncoating. It is also required for viral assembly and release. In the case of ER-targeting viroporins, rotavirus-encoded NSP4 modifies the calcium homeostasis by enhancing the calcium permeability of the ER membrane. This may be associated with virus-induced cell death and subsequent release of NSP4, which in turn causes activation of the phospholipase C-IP3 cascade in neighboring noninfected cells and is responsible for viral pathogenesis (Tian et al., 1995, 1996; Dong et al., 1997). On the other hand, 2B proteins of picornaviruses also participate in the remodeling of membrane structures and the formation of replication complexes (De Jong et al., 2008). Among them, CBV3 2B, PV1, and rhinovirus 2B are present at the membranes of the ER and Golgi complex and are responsible for the release of Ca2+ and H+ from these organelles.
Rotavirus studies propose that the double-layered particle (DLP)–VP4–NSP4 complex breaches the ER membrane and penetrates into the ER. The viral capsid protein, VP7, re-envelopes the immature particle (DLP) after removal of the ER membrane and NSP4, and forms the infectious triple-layered particle (Tian et al., 1996; Trask et al., 2012).
The induction of individual branches or part of the UPR by viruses was reported previously. Viruses have also evolved different means to modulate the arms of the UPR, which consequently expanded both the temporal and spatial superiority for virus replication or completion of the life cycle.
It has been reported that viruses regulate the host translational machinery to promote viral protein synthesis by inhibiting the synthesis of proteins involved in host immune responses. In enteroviruses, 2A and 3C proteases target translation factors such as eIF4GI and poly(A)-binding protein (PABP) to impede host translation (Lloyd, 2006). Moreover, modulation of the integrated stress response (ISR), which is determined by phosphorylation of eIF2α to attenuate cellular translation, is another strategy for promoting virulence (Figure 2) (Sonenberg and Hinnebusch, 2009). Four eIF2α kinases have been identified: heme-regulated inhibitor (HRI), which is a response to heme deficiency (Chen, 2007); double-stranded RNA-dependent protein kinase (PKR), which is induced by interferon (IFN) and activated by double-stranded RNA (dsRNA) during viral infection (Meurs et al., 1990); general control nonderepressible-2 (GCN2), which is activated by serum and amino acid deprivation (Harding et al., 2000); and finally, PKR-like ER kinase (PERK or PEK), which is activated by unfolded proteins in the ER (Ron, 2002).
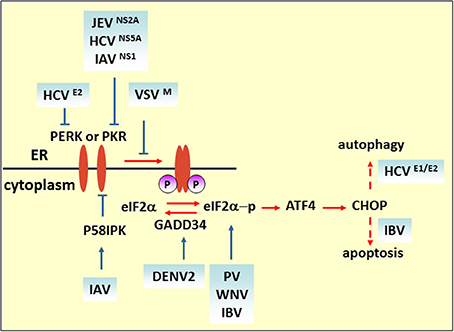
Figure 2. eIF2 pathway under viral infection. The M protein of VSV, the E2 and NS5A proteins of HCV, and NS2A of JEV counteract the phosphorylation of eIF2α for viral replication. Blue solid arrows indicate the direct target of the virus or viral proteins. IAV also targets eIF2α by inducing P58IPK, a cellular inhibitor of PERK and PKR. IBV upregulates eIF2α–ATF4–CHOP-mediated apoptosis to benefit viral replication.
Some researchers consider that eIF2α phosphorylation plays a role in hampering viral protein synthesis. For example, upon VSV infection, the induction of activated PERK only correlates with eIF2α phosphorylation at the later stage of infection. In MEF cells carrying a phosphorylation-insensitive eIF2α S51A variant, viral protein synthesis increased compared with a wild-type control, indicating that eIF2 phosphorylation is inhibitory to viral protein synthesis. As demonstrated by matrix (M) protein mutant virus (rM51RM), a viral protein (M protein) is involved in counteracting the antiviral response of the phosphorylation of eIF2α (Connor and Lyles, 2005). Like VSV infection, Chikungunya virus (CHIKV) induces PERK activation but delays eIF2α phosphorylation. The expression of CHIKV NSP4, which is the RNA-dependent-RNA polymerase, contributed to suppression of eIF2α phosphorylation, thus ensuring translation of viral proteins (Rathore et al., 2013). Furthermore, viruses containing type I or type II internal ribosomal entry sites (IRESs), such as PV, foot-and-mouth disease virus, mengovirus and EMCV, require many canonical translation initiation factors for initial replication (Beales et al., 2003; Sarnow, 2003). It is reported that PV switches translation mode from an eIF2-dependent to an eIF2-independent one during the course of infection to ensure efficient proliferation. Furthermore, studies have shown that the C terminal of the eIF5B fragment, cleavage by 3C proteases, and proteolytic activity of 2Apro can stimulate virus IRES translation of enteroviruses (De Breyne et al., 2008; Redondo et al., 2011). Interestingly, it is reported that phosphorylation of eIF2α is required for activation of IRES during cell differentiation (Gerlitz et al., 2002). Thus, whether the phosphorylation level of eIF2α positively correlates with IRES-dependent viral mRNA translational efficiency remains to be determined. Some viruses regulate the eIF2α pathway by interfering with the activation of eIF2α kinases. HCV NS5A protein, containing an IFN sensitivity-determining region (ISDR), interferes with PKR activity by binding to a PKR dimerization domain (PKR residues 244–296) (Gale et al., 1998), while HCV E2 protein binds to PERK and inhibits downstream eIF2α phosphorylation by acting as a pseudosubstrate (Pavio et al., 2003). Interestingly, it is reported that NS5A stimulates eIF2α phosphorylation in the absence of PKR, implying that NS5A may activate other eIF-2α kinases to regulate eIF2α phosphorylation (Tardif et al., 2002). Overexpression of HCV NS2 induces eIF2α phosphorylation (Von Dem Bussche et al., 2010). Taken together, these studies indicate that HCV proteins modulate eIF2α pathway in a complex way, and the effect of regulation on virus replication cannot be established unequivocally. The N-terminal region of NS2A of JEV contains a sequence that is highly similar to HCV NS5A ISDR and also inhibits PKR-induced eIF2α phosphorylation (Tu et al., 2012). DENV2 infection triggers and then suppresses PERK-mediated eIF2α phosphorylation by elevating the expression of growth arrest and DNA damage-inducible protein-34 (GADD34), which acts together with phosphatase 1 (PP1) to dephosphorylate eIF2α-P (Pena and Harris, 2011). Influenza virus nonstructural protein NS1 interferes with dsRNA binding to PKR, and the infection also induces and activates P58IPK, a cellular inhibitor of PKR and PERK. Both strategies deployed by NS1 and P58IPK prevent PKR dimerization and autophosphorylation, which limits eIF2α phosphorylation (Lee et al., 1992; Lu et al., 1995; Yan et al., 2002).
In some circumstances, such as when the host immune system specifically recognizes foreign viruses and kills them with cytotoxic T lymphocytes, or when cell death is directly induced in virus infected cells to prevent completion of the replication cycle, apoptotic cell death is considered to be a host strategy for fighting against viral infections. ATF4 is a transcriptional activator of the ISR, which is involved in the expression of ISR target genes such as c/EBP homologous protein (CHOP) and GADD34 (Ma and Hendershot, 2003). CHOP was originally identified as a transcriptional factor eliciting ER stress-induced apoptosis. In cells subjected to West Nile virus (WNV) infection, eIF2α phosphorylation and CHOP-mediated apoptosis were induced. Both viral protein expression level and virus titer are increased in CHOP-deficient cells (Medigeshi et al., 2007). On the other hand, a virus may induce apoptosis to facilitate replication or the spread of viral progeny. It is reported that coronavirus infectious bronchitis virus (IBV) upregulates eIF2α–ATF4–CHOP signaling in infected cells and that it relies on PERK or PKR activation. Knockdown of CHOP reduces IBV-induced apoptosis through activation of the extracellular signal-related kinase (ERK). Viral protein expression level is moderately suppressed in CHOP-knockdown cells, which suggests that upregulation of CHOP-mediated apoptosis during IBV infection probably promotes virus replication (Liao et al., 2013).
In addition to regulation of cell death, it is reported that HCV induces the expression of CHOP at mRNA and protein levels and is correlated with autophagy induction; knockdown of CHOP not only increases HCV PAMP-mediated innate immune activation, but also elevates its inhibitory effect on virus replication (Ke and Chen, 2011). However, upstream CHOP induction is a matter of debate. Overexpression of HCV E1 and/or E2 induces the expression of CHOP in a PERK-dependent manner (Chan and Egan, 2005); while upon HCV infection, CHOP protein is upregulated by PERK, activating transcription factor (ATF6), and inositol-requiring transmembrane kinase/endonuclease 1 (IRE1) collectively.
ATF6 is a type 2 transmembrane protein of 670 amino acids and is constitutively expressed as a 90-kDa protein (p90ATF6). Its C-terminal region is located in the ER, whereas the N-terminal region is located on the cytosolic side (Figure 3). Upon ER stress, ATF6 is cleaved to an N-terminal 50-kDa protein (p50ATF6) sequentially by the Golgi site-1 and site-2 proteases (S1P and S2P) (Ye et al., 2000). Nuclear translocation of p50ATF6, as a transcription factor, activates expression of ER stress and ERAD genes including ER chaperones, CHOP (aka GADD153), EDEM1, and X-box-binding protein 1 (XBP1) by targeting the cis-acting ER stress response element (ERSE) (CCAAT-N9-CCACG) and UPR element (UPRE) (GATGACGTG(T/G) NNN(A/T)T), although ATF6 has a much higher affinity for ERSE (Yoshida et al., 1998). In addition to directly regulating gene expression, ATF6 also modulates the innate immune response. Under subtilase cytotoxin (SubAB) treatment, cleavage and degradation of GRP78/BiP leads to activation of the AKT–NF-κB pathway through ATF6 activation (Yamazaki et al., 2009). Based on its pivotal role of connecting the arms of the UPR and converging the UPR and immune response, many viruses preferentially regulate ATF6 pathways to benefit replication. In WNV strain Kunjin (WNVKUN)-infected cells, expression of ATF6-target genes increases, but viral production decreases in ATF6 knockout MEF cells. Moreover, in ATF6 knockout MEF cells, phosphorylation of eIF2α, downstream CHOP activity, and Jak–STAT1 phosphorylation induced by IFNα are upregulated upon infection, which implies that virus-induced ATF6 activation is a prosurvival mechanism required for replication and inhibition of the antiviral signaling pathway (Ambrose and Mackenzie, 2013). However, it is still unclear whether WNVKUN NS4A and NS4B, potent inducers of the UPR, inhibit IFNα-induced Jak–STAT signaling in an ATF6-dependent manner (Ambrose and Mackenzie, 2011). Other Flavivirus infections, including HCV, JEV, and DENV2, also induce cleavage of ATF6, nuclear translocalization of ATF6 and increases in chaperone proteins expression. In HCV replication, silencing of ATF6 reduces HCV intracellular mRNA levels (Ke and Chen, 2011). However, in JEV-infected cells, knockdown of the ATF6-targeted gene, GRP78, by siRNA did not affect JEV viral RNA replication, although it did impair virus assembly or release. In sucrose gradient, mature JEV viruses that do not cofractionate with GPR78 displayed a significant decrease in viral infectivity, indicating that JEV acts with GPR78 to promote its infectivity (Wu et al., 2011). Notably, DENV2 triggers ATF6 signaling in a cell-type-specific manner. In A549 cells, nuclear-localized ATF6 was observed (Umareddy et al., 2007); however, no activating events can be detected in human fibrosarcoma 2fTGH cells, therefore, GPR78 upregulation may be mediated in an ATF6-independent fashion (Pena and Harris, 2011). This cell-type-specific regulation of ATF6, also observed in IAV infection, p50ATF6, and its target gene ERp57/GRP58 expression (Roberson et al., 2012), has been shown to increase in murine primary tracheal epithelial cells infected with influenza A/PR/8/34, which is known to be involved in influenza virus HA protein folding (Solda et al., 2006). Knockdown of ERp57 abrogates viral progeny production. However, ATF6 activity is not induced in infected human tracheobronchial epithelial (HTBE) cells (Hassan et al., 2012).
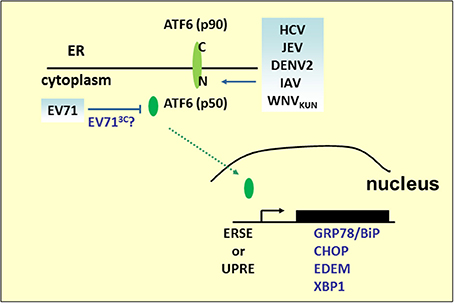
Figure 3. ATF6 pathway under viral infection. Many RNA viruses activate the UPR pathway by cleaving ATF6 to release the p50 fragment. The N-terminal p50 with transcription activity enters the nucleus to activate the expression of ER stress and ERAD genes, such as GRP78/BiP, CHOP, XBP1, or EDEM. However, the p50 fragment was not detected in the EV71 infection.
Although ATF6-mediated transcriptional activation is an ongoing research field, another role for ATF6 in virus infection has emerged. We have previously demonstrated that EV71 infection results in the decline of p90ATF6, while the GRP78 promoter containing classical ERSE sites responsive to p50ATF6 in EV71-infected cells was not activated (Jheng et al., 2010). Indeed, two potential 3C cleavage sites (glutamine–glycine; QG) located at adjacent amino acids 511–512 and 516–517 near the C terminus of p90ATF6 were computationally predicted. It would be interesting to investigate the role of viral 3C in the regulation of ATF6, for its possible contribution in manipulating virus infection.
IRE1 is an ER-localized type I transmembrane protein containing an ER luminal dimerization domain and cytosolic kinase and RNase domains (Mori et al., 1993; Sidrauski and Walter, 1997). During ER stress, accumulation of unfolded proteins in the ER stimulates IRE1 oligomerization and autophosphorylation (Figure 1). Its endoribonuclease activity initiates an unconventional splicing of the XBP1 mRNA, excising a 26-nt sequence and shifts the reading frame to produce a functional isoform XBP1(S), which contains a C-terminal transactivation domain absent from the unspliced form, XBP1(U). XBP1(S) then translocates to the nucleus where it induces expression of target genes containing UPRE or ERSE. These target genes are involved in ERAD, chaperone protein production, and ER membrane biosynthesis (Shamu and Walter, 1996; Friedlander et al., 2000).
Studies of the IRE1 signaling pathway demonstrate its significant role in virus infection (Figure 4). HCV glycoprotein E2 is an example of a virus-derived ERAD substrate. HCV infection activates the IRE1–XBP1–EDEM pathway, where EDEM1 and EDEM3, but not EDEM2, interact with HCV E2 to accelerate its degradation. Either knockdown of EDEMs or treating cells with kifunensine (KIF), a potent inhibitor of ER mannosidase, interferes with the binding of EDEMs with SEL1L, a component of ERAD complex, stabilizes E2 expression, and enhances virus replication and viral particle production. However, there is no interaction between EDEM proteins and the JEV envelope protein and abolishing the ERAD pathway by KIF does not affect JEV production (Saeed et al., 2011). The results emphasize the pivotal role of the ERAD pathway in the life cycle of specific viruses. Interestingly, UPRE reporter activity or ERAD of misfolded null Hong Kong α-antitrypsin is reduced in cells carrying HCV replicons, which lack structural proteins, even though upstream XBP1 splicing occurs (Tardif et al., 2004). This implies that HCV structural proteins play a key role in XBP1-mediated UPRE activation, and this is supported by a related study demonstrating that HCV E1 and/or E2 activates the XBP1–ERAD pathway (Chan and Egan, 2005). Furthermore, the IRE1 signaling pathway also participates in viral protein retrotranslocation. Hepatitis E virus (HEV) ORF2 is an N-linked glycoprotein which is cotranslationally translocated into the ER while a significant fraction of it is also observed in the cytoplasm. Based on the results of tunicamycin and KIF treatment, it is believed that glycosylation and ERAD are essential for ORF2 retrotranslocation from the ER to the cytoplasm (Surjit et al., 2007). However, no ubiquitination of ORF2 can be observed, and retrotranslocated ORF2 protein was stable in the cytoplasm when the cells were treated with proteasome inhibitor MG132, which suggests that ERAD is required for ORF2 access to the cytoplasm. Microarray analysis reveals that ORF2 overexpression causes upregulation of Hsp70B, Hsp72, and Hsp40. Hsp72 is an antiapoptotic heat shock protein that directly interacts with ORF2 (John et al., 2011). It is reported that expression of Hsp72 enhances XBP1 mRNA splicing and protects cells from ER stress-induced apoptosis by association with IRE1 (Gupta et al., 2010). Thus, further investigation is needed to examine the correlations of ORF2, IRE1, and Hsp72 in HEV replication.
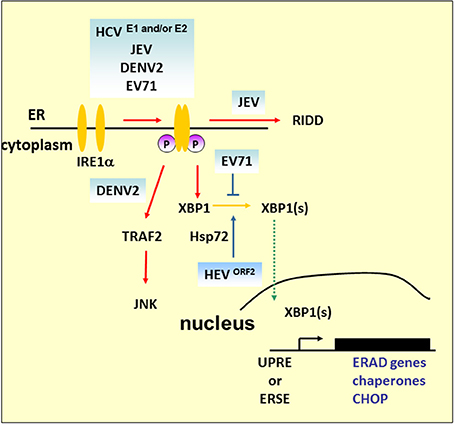
Figure 4. IRE1 pathway under viral infection. In addition to mediating Xbp1 mRNA splicing, studies demonstrated that Ire1 activates RIDD to promote the degradation of mRNAs encoding ER-targeted proteins to reduce the load of ER client proteins during ER stress. The mammalian IRE1–TRAF2–JNK pathway, independent of XBP1 splicing, may lead to the activation of apoptosis after prolonged ER stress. HCV and its structural proteins E1 and E2 play an important role in the activation of the IRE1–XBP1–ERAD pathway. Overexpression of ORF2 of HEV can upregulate antiapoptotic protein Hsp72 to activate XBP1 splicing. However, further study is required to determine whether HEV infection can activate XBP1 via Hsp72. DENV2 infection activates CHOP and GADD34 expression downstream of IRE1–XBP1 signaling. However, apoptosis activation by JNK, but not CHOP, is essential for DENV2 infection.
Under harsh ER stress, the activation of IRE1–XBP1 can also lead to the induction and expression of CHOP. DENV2 infection induces CHOP, and GADD34 expression is a downstream event of IRE1–XBP1 signaling. Of note is that induction of CHOP does not lead to apoptosis markers such as decreased expression of Bcl-2 or proteolytic cleavage of pro-caspase-9, pro-caspase-3, or PARP, which indicates a role beyond guiding cell death in infected cells (Pena and Harris, 2011). Indeed, it has been reported that CHOP exhibits protective effects against radiation-induced apoptosis or has a role in autophagy induction (Mayerhofer and Kodym, 2003; Ke and Chen, 2011). Another ER stress-induced cell death that relies on the IRE1–TRAF2 pathway is implicated in JNK activation (see Figure 4). The role of this pathway is emphasized by DENV2 infection; silencing of IRE1 decreases the virus titer, but the viral progeny output is not affected by silencing of XBP1 (Pena and Harris, 2011). However, JNK pathway inhibitors diminished virus yield significantly, which suggests that activation of JNK is essential for DENV2 infection (Ceballos-Olvera et al., 2010). Our previous findings also demonstrated that EV71 phosphorylates IRE1, but inhibits the expression of XBP1. The overexpression of XBP1 in cells appeared to inhibit viral entry, and therefore reduce viral RNA and viral particle formation (Jheng et al., 2012). As previous studies have reported that picornavirus infections induce JNK activation (Kim et al., 2004; Peng et al., 2014), further detailed studies of the IRE1-JNK activation in EV71 infection would extend our understanding of the contributions of IRE1-JNK in the virus life cycle.
IRE1 has also been linked to the mediation of the selective degradation of a subset of ER-localized mRNAs in a process known as regulated IRE1-dependent degradation (RIDD) (Hollien and Weissman, 2006). Mutation or removal of the signal sequences in targeted mRNAs prevents their decay (Kimmig et al., 2012). However, it has been observed that Drosophila mRNA Smt3, a homolog of a small ubiquitin-like modifier (aka SUMO), lacks any ER-targeting sequence, and is a noncanonical RIDD target, which implies that unknown specific features other than ER localization are involved in defining the RIDD substrates (Moore et al., 2013). RIDD has been suggested to play adaptive roles by reducing protein translocation load, such as decrease of proinsulin expression in pancreatic beta-cells faced with chronic high glucose, and protecting liver cells from acetaminophen-induced hepatotoxicity (Lipson et al., 2008; Hur et al., 2012). Alternatively, RIDD has also been suggested to play destructive roles under unmitigated ER stress because continued degradation of mRNAs encoding secretory cargo proteins and proteins involved in ER-resident protein folding occurs.
In addition to IRE1–XBP1 activation, JEV also induces activation of the RIDD cleavage pathway (Bhattacharyya et al., 2014). The addition of STF083010, a specific inhibitor of IRE1 RNase activity, to infected cells decreases the Tg-induced Xbp1 splicing and potential RIDD target transcripts. It also decreases viral protein expression as well as mature progeny formation, but does not affect viral RNA synthesis, which indicates that JEV viral RNA is not a substrate of RIDD, and RIDD activation is beneficial for viral infectivity.
It is not clear whether other viral infections trigger RIDD. To extrapolate from the study of HCV, HCV replicons activate the phosphorylation of IRE1 but impede XBP1 activation (Tardif et al., 2004). Depletion of IRE1 attenuates replicon translation, which implies that RIDD may enhance viral protein synthesis. Thus, the study of HCV replicon may have potential for deciphering the role of RIDD in HCV infection because it could uncouple XBP1 signaling from IRE1 activation.
Autophagy is a vesicular process that results in the degradation of the sequestered component, which can then be recycled by the cell. In mammalian cells, a complete autophagy includes the following four steps. (1) Induction. Induction is initiated by activation of the Unc-51-like kinase 1 (ULK1) complex. The ULK1 complex contains ULK1, focal adhesion kinase (FAK)-family-interacting protein of 200 kD (FIP200), Atg13 and Atg101 (Mizushima, 2010). ULK1 complex activity would be, at least, modulated by mTORC1, Akt, and AMPK (Inoki et al., 2003; Bach et al., 2011; Egan et al., 2011; Kim et al., 2011). mTORC1 is a serine/threonine kinase complex, which phosphorylates ULK1 and Atg13 and also inhibits autophagy. Akt and AMP-activated protein kinase (AMPK) phosphorylate TSC2 at different residues, which results in the GTP hydrolysis of Rheb and indirectly antagonizes the mTORC1 signaling pathway. Recently, the combination of bioinformatic and proteomic approaches has identified ULK1 as a direct target of AMPK and as involved in autophagy induction. (2) Vesicle nucleation. The Beclin1–PI3KC3 complex, generating PI3P, is essential for recruitment of PI3P effectors including DFCP1, WIPIs upstream of Atg proteins and lipids recruitment to the PAS, which is required for autophagosome construction (Proikas-Cezanne et al., 2004; Axe et al., 2008). Importantly, the activity of the Beclin1–PI3KC3 complex depends on its subunit composition. Complexes containing Atg14-like protein (ATG14L or Barkor) or ultraviolet irradiation resistance-associated gene (UVRAG) activate autophagy (Itakura et al., 2008); nevertheless, the RUN domain and cysteine-rich domain containing Beclin 1-interacting protein (Rubicon) act as negative regulators of autophagy (Matsunaga et al., 2009). (3) Vesicle expansion and completion. The cytosolic form of LC3 (LC3-I) is cleaved by the cysteine protease Atg4, followed by conjugation with phosphatidylethanolamine (PE) assisted by the Atg12–Atg5–Atg16L complex, which functions as an E3–like enzyme. LC3-PE leads to PAS expansion, and cytosolic cargos are then enclosed into double membrane vesicles called autophagosomes (Geng and Klionsky, 2008). (4) Autophagosome maturation. An autophagosome matures into an autolysosome by sequential fusion with endosomes and with lysosomes, the contents of which are degraded by hydrolases therein. It is reported that autolysosome formation is related to UVRAG and expression of lysosomal-associated membrane protein 2 (Lamp-2) (Liang et al., 2008; Fortunato et al., 2009).
Previous studies suggest that autophagy may be an important antiviral defense mechanism (Talloczy et al., 2006; Orvedahl et al., 2010); however, the role of autophagy in virus infection is complicated and may have opposite consequences for the viral pathogenesis. Many viruses manipulate autophagy for their own benefit by the following mechanisms.
The exploitation of autophagy has been identified in many RNA viruses including PV, CVB3, JEV, and HCV (Jackson et al., 2005; Wong et al., 2008; Tanida et al., 2009; Ke and Chen, 2011; Li et al., 2012). Increased amounts of autophagosomes, as well as colocalization of the autophagy marker protein LC3 and viral protein, were observed in virus-infected cells. In addition, cells treated with an autophagy inhibitor, or transfected with siRNA specifically obstructed autophagic processes, which reduced virus replication or virus titer. For example, in PV infection, virus yield was correlated with the induction of autophagy. Treating cells with siRNA targeting LC3 or Atg12 to block autophagy leads to reduced virus yield (Jackson et al., 2005). In addition, based on the topology of a double membrane compartment, digestion of the inner membrane under the autolysosome formation would allow efficient fusion of the autophagosomal membrane with the cytoplasm membrane. Thus, an emerging concept is that autophagy may also involve the nonlytic release of cytoplasm under autophagosome maturation, namely autophagic exit without lysis (AWOL), which may participate in the release of PV (Kirkegaard and Jackson, 2005; Taylor et al., 2009).
Virus-induced uncompleted autophagy was reported for CVB3-, rotavirus-, and IVA- infected cells (Gannage et al., 2009; Kemball et al., 2010; Alirezaei et al., 2012; Crawford et al., 2012). In CVB3-infected pancreatic acinar cells, an increase in the number of double-membraned autophagy-like vesicles was observed upon infection. However, the accumulation of autophagy substrate p62 and the formation of large autophagy-related structures named megaphagosomes indicate that CVB3 blocks a later stage of the autophagic pathway (Kemball et al., 2010). Further results highlight the impact of autophagy on CVB3 RNA replication and translation (Alirezaei et al., 2012). It was reported that rotavirus NSP4 viroporin initiates autophagy to transport viral proteins to sites of virus replication for assembly of mature particles, which involves an increase of cytoplasmic calcium and subsequent activation of the CaMKK-β–AMPK pathway. Rotavirus also interferes with autophagy maturation; however, the mechanism is still unknown (Crawford et al., 2012). Accumulated studies reveal that M2, HA, and NS1 proteins of IAV are involved in the induction of autophagy, while only M2 has been identified as playing a critical role in impeding fusion of autophagosomes with lysosomes (Gannage et al., 2009; Sun et al., 2012; Zhirnov and Klenk, 2013).
Autophagy-mediated immune responses that benefit virus replication have been reported in VSV, HCV, DENV, and JEV (Jounai et al., 2007; Ke and Chen, 2011; Jin et al., 2013). In VSV infection, the Atg5–Atg12 conjugate targets RIG-I/MDA5–MAVS-dependent type I IFN production by directly interacting with the MAVS and RIG-I, and negatively regulates MAVS-mediated NF-κB and type I IFN promoters, and permits VSV replication. Furthermore, through an unknown mechanism, HCV- or DENV-induced complete autophagy negatively regulates type I IFN production and promotes HCV replication (Ke and Chen, 2011). Recently, research about JEV has shown that in autophagy-impaired cells, virus infection induces aggregates of MAVS and activation of IFN regulatory factor 3 (IRF3), markers for activation of innate immune responses, which suggests that autophagy-mediated immune responses are required for viral replication (Jin et al., 2013).
As ER proliferation, which paradoxically commits the cell to cell death or survival, is observed both in UPR and autophagy, it is reasonable to propose a possible link between UPR pathways and the autophagic response. Indeed, many UPR-related transcription factors manage Atg expression (Table 1). As demonstrated previously, yeasts with mutations in the GCN2-signaling pathway are defective in starvation-induced autophagy. GCN4, which undertakes GCN2-dependent transcriptional activation, is essential for autophagy induction (Talloczy et al., 2002). Recently, results of multiple genetic models showed that the PERK–eIF2α–ATF4 pathway affects cMyc-dependent tumorigenesis by evoking cytoprotective autophagy; while pharmacologic or genetic inhibition of autophagy resulted in enhanced Myc-dependent apoptosis (Hart et al., 2012). Thus, UPR inhibition could provide new targets for the treatment of malignancies, characterized by cMyc overexpression. In addition, IRE1 also mediates autophagy in Huntington's disease under ER stress. Clearance of mutant huntingtin aggregates through autophagic flux was impaired via IRE1–TRAF2 signaling, which results in neuronal cytotoxicity (Lee et al., 2012). Although studies on UPR autophagy mainly focus on the regulation of eIF2α kinase and IRE1, transcriptional regulation of autophagic genes by ATF6 and SREBP2, a membrane-bound transcription factor activated through proteolytic processing upon ER stress, was noticed recently (Ogata et al., 2006; Seo et al., 2011; Gade et al., 2012). Death-associated protein kinase 1 (DAPK1), a positive mediator of IFN-regulated growth suppressor, is principally regulated by transcription factor C/EBP-β, one of the genes that increases expression during ER stress (Chen et al., 2004). DAPK1 promotes autophagy by phosphorylating Beclin 1, and therefore dissociating it from autophagy negative regulator Bcl2. An investigation found that activated ATF6 could directly interact with C/EBP-β carrying an ERK1/2 target site; this heterodimer then coacts to activate the DAPK1 promoter, which in turn induces autophagy. Additionally, XBP1, a downstream target of ATF6, is essential for C/EBP-β expression (Chen et al., 2004). The role of SREBP2 in autophagy was disclosed through gene ontology analysis (Seo et al., 2011). Further study shows that SREBP-2 activates autophagy gene expression, such as LC3B, ATG4B, and ATG4D, accompanied by increased LC3 puncta formation, while SREBP-2 deficiency obtains an opposite result.
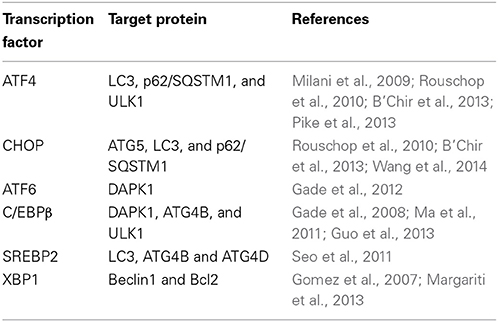
Table 1. Exploitation of autophagy by modulation of UPR transcription factors.
In virus infection, HCV is a well-documented model illustrating UPR autophagy regulation. Induction of UPR and incomplete autophagy was observed in cells transfected with HCV JFH1 RNA. Cells treated with siRNA targeting PERK, IRE1, and ATF6 showed a suppression of LC3 conversion and a decrease of HCV RNA replication (Sir et al., 2008). In the HCV infection system, HCV induces complete autophagy and CHOP plays a leading role in UPR autophagy signaling (Ke and Chen, 2011). Further efforts to decipher how HCV activates autophagy revealed that PERK–eIF2α–ATF4 and ATF6 pathways activated CHOP expression in HCV core protein-transfected cells where the core protein had not been demonstrated to induce ER stress previously. Moreover, HCV core protein may promote ATG12 and LC3 protein expression through transcriptional control by ATF4 and CHOP, respectively (Wang et al., 2014).
Recent studies suggest that completed autophagy induced by CHIKV infection is mediated by the independent induction of the endoplasmic reticulum and oxidative stress pathways. Knockdown of IRE1 or treated cells with the ROS inhibitor N-acetyl-l-cysteine inhibits formation of autophagosomes as well as the conversion of LC3-I to LC3-II. Moreover, an additive inhibitory effect on autophagosome formation was observed in infected cells silenced for IRE1mRNA and treated with N-acetyl-l-cysteine (Joubert et al., 2012).
Because UPR and autophagy play a role in viral pathogenesis, the regulation of UPR and autophagy may be an important strategy for the future development of new therapeutic approaches to combat viruses. For example, we have demonstrated that overexpression of GRP78 to relieve ER stress decreases EV71 replication (Jheng et al., 2010). Thus, agents such as GRP78/BiP inducer X (BIX) (Kudo et al., 2008) or chemical chaperone, tauroursodeoxycholic acid (TUDCA) (Ozcan et al., 2006), will be potentially useful in the treatment of EV71 (Table 2).
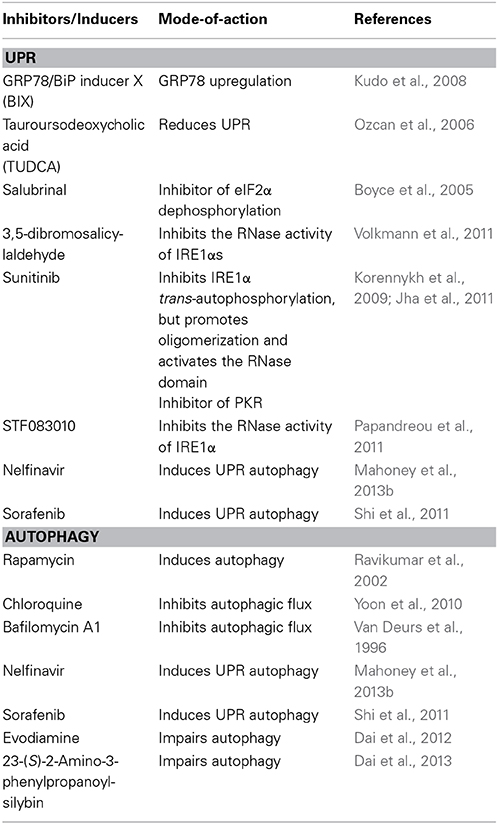
Table 2. Compounds affecting UPR and autophagy.
There are other established strategies to inhibit viruses by modulating UPR target eIF2α phosphorylation or IRE1, e.g., salubrinal is a small molecule that prevents dephosphorylation of eIF2α and 3,5-dibromosalicylaldehyde, an IRE1 inhibitor, may cause restriction of IVA (Boyce et al., 2005; Volkmann et al., 2011).
There is emerging evidence that pharmacological agents that directly activate or deactivate autophagy influence virus replication. Evodiamine and 3-(S)-2-amino-3-phenylpropanoyl-silybin have been identified as anti-IVA agents aimed at multiple processes of autophagy (Dai et al., 2012, 2013). Additionally, chloroquine-suppressed HCV replication has been proved (Mizui et al., 2010).
Because UPR and autophagy are closely related, combination treatment may show a synergistic effect of their application, which was demonstrated in cancer research. The combination of nelfinavir (which induces UPR autophagy) and chloroquine enhances cytotoxicity against cancer cells (Mahoney et al., 2013b); therefore, the use of combination treatment with improved efficacy and decreased toxicity represents a promising strategy to fight viruses.
Although UPR autophagy has been discussed in many research areas, its integrated response to virus infection is only now beginning to emerge. It needs to be experimentally proven whether virus-induced autophagy is associated with UPR. Furthermore, given what we know about the various means that viruses use to modulate UPR or autophagy to advantage their own virulence, the development of specific inducers or inhibitors for these molecules is one of the major challenges in this field.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are in debt to all previous and current laboratory members for their contributions and involvement in the study of EV71-induced ER stress. This work was supported by the National Science Council [NSC- 102-2325-B-182-016] and Chang Gung Memorial Hospital [CMRPD1B0422 and CMRPD1A0163].
Alirezaei, M., Flynn, C. T., Wood, M. R., and Whitton, J. L. (2012). Pancreatic acinar cell-specific autophagy disruption reduces coxsackievirus replication and pathogenesis in vivo. Cell Host Microbe 11, 298–305. doi: 10.1016/j.chom.2012.01.014
Ambrose, R. L., and Mackenzie, J. M. (2011). West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J. Virol. 85, 2723–2732. doi: 10.1128/JVI.02050-10
Ambrose, R. L., and Mackenzie, J. M. (2013). ATF6 signaling is required for efficient West Nile virus replication by promoting cell survival and inhibition of innate immune responses. J. Virol. 87, 2206–2214. doi: 10.1128/JVI.02097-12
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., et al. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701. doi: 10.1083/jcb.200803137
Bach, M., Larance, M., James, D. E., and Ramm, G. (2011). The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 440, 283–291. doi: 10.1042/BJ20101894
Bartosch, B., Dubuisson, J., and Cosset, F. L. (2003). Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197, 633–642. doi: 10.1084/jem.20021756
B'Chir, W., Maurin, A. C., Carraro, V., Averous, J., Jousse, C., Muranishi, Y., et al. (2013). The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699. doi: 10.1093/nar/gkt563
Beales, L. P., Holzenburg, A., and Rowlands, D. J. (2003). Viral internal ribosome entry site structures segregate into two distinct morphologies. J. Virol. 77, 6574–6579. doi: 10.1128/JVI.77.11.6574-6579.2003
Behrends, C., and Fulda, S. (2012). Receptor proteins in selective autophagy. Int. J. Cell Biol. 2012:673290. doi: 10.1155/2012/673290
Bernales, S., McDonald, K. L., and Walter, P. (2006). Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4:e423. doi: 10.1371/journal.pbio.0040423
Bhattacharyya, S., Sen, U., and Vrati, S. (2014). Regulated IRE1-dependent decay pathway is activated during Japanese encephalitis virus-induced unfolded protein response and benefits viral replication. J. Gen. Virol. 95, 71–79. doi: 10.1099/vir.0.057265-0
Boyce, M., Bryant, K. F., Jousse, C., Long, K., Harding, H. P., Scheuner, D., et al. (2005). A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307, 935–939. doi: 10.1126/science.1101902
Ceballos-Olvera, I., Chavez-Salinas, S., Medina, F., Ludert, J. E., and Del Angel, R. M. (2010). JNK phosphorylation, induced during dengue virus infection, is important for viral infection and requires the presence of cholesterol. Virology 396, 30–36. doi: 10.1016/j.virol.2009.10.019
Chan, S. W., and Egan, P. A. (2005). Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J. 19, 1510–1512. doi: 10.1096/fj.04-3455fje
Chen, C., Dudenhausen, E. E., Pan, Y. X., Zhong, C., and Kilberg, M. S. (2004). Human CCAAT/enhancer-binding protein beta gene expression is activated by endoplasmic reticulum stress through an unfolded protein response element downstream of the protein coding sequence. J. Biol. Chem. 279, 27948–27956. doi: 10.1074/jbc.M313920200
Chen, J. J. (2007). Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood 109, 2693–2699. doi: 10.1182/blood-2006-08-041830
Connor, J. H., and Lyles, D. S. (2005). Inhibition of host and viral translation during vesicular stomatitis virus infection. eIF2 is responsible for the inhibition of viral but not host translation. J. Biol. Chem. 280, 13512–13519. doi: 10.1074/jbc.M501156200
Crawford, S. E., Hyser, J. M., Utama, B., and Estes, M. K. (2012). Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-beta signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. U.S.A. 109, E3405–E3413. doi: 10.1073/pnas.1216539109
Dai, J. P., Li, W. Z., Zhao, X. F., Wang, G. F., Yang, J. C., Zhang, L., et al. (2012). A drug screening method based on the autophagy pathway and studies of the mechanism of evodiamine against influenza A virus. PLoS ONE 7:e42706. doi: 10.1371/journal.pone.0042706
Dai, J. P., Wu, L. Q., Li, R., Zhao, X. F., Wan, Q. Y., Chen, X. X., et al. (2013). Identification of 23-(s)-2-amino-3-phenylpropanoyl-silybin as an antiviral agent for influenza a virus infection in vitro and in vivo. Antimicrob. Agents Chemother. 57, 4433–4443. doi: 10.1128/AAC.00759-13
De Breyne, S., Bonderoff, J. M., Chumakov, K. M., Lloyd, R. E., and Hellen, C. U. (2008). Cleavage of eukaryotic initiation factor eIF5B by enterovirus 3C proteases. Virology 378, 118–122. doi: 10.1016/j.virol.2008.05.019
De Jong, A. S., De Mattia, F., Van Dommelen, M. M., Lanke, K., Melchers, W. J., Willems, P. H., et al. (2008). Functional analysis of picornavirus 2B proteins: effects on calcium homeostasis and intracellular protein trafficking. J. Virol. 82, 3782–3790. doi: 10.1128/JVI.02076-07
Dong, Y., Zeng, C. Q., Ball, J. M., Estes, M. K., and Morris, A. P. (1997). The rotavirus enterotoxin NSP4 mobilizes intracellular calcium in human intestinal cells by stimulating phospholipase C-mediated inositol 1,4,5-trisphosphate production. Proc. Natl. Acad. Sci. U.S.A. 94, 3960–3965. doi: 10.1073/pnas.94.8.3960
Dubuisson, J., Hsu, H. H., Cheung, R. C., Greenberg, H. B., Russell, D. G., and Rice, C. M. (1994). Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia and Sindbis viruses. J. Virol. 68, 6147–6160.
Dubuisson, J., and Rice, C. M. (1996). Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J. Virol. 70, 778–786.
Egan, D., Kim, J., Shaw, R. J., and Guan, K. L. (2011). The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7, 643–644. doi: 10.4161/auto.7.6.15123
El-Hage, N., and Luo, G. (2003). Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral proteins and RNA. J. Gen. Virol. 84, 2761–2769. doi: 10.1099/vir.0.19305-0
Eskelinen, E. L. (2005). Maturation of autophagic vacuoles in Mammalian cells. Autophagy 1, 1–10. doi: 10.4161/auto.1.1.1270
Fortunato, F., Burgers, H., Bergmann, F., Rieger, P., Buchler, M. W., Kroemer, G., et al. (2009). Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology 137, 350–360, 360.e351–355. doi: 10.1053/j.gastro.2009.04.003
Friedlander, R., Jarosch, E., Urban, J., Volkwein, C., and Sommer, T. (2000). A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2, 379–384. doi: 10.1038/35017001
Fujita, E., Kouroku, Y., Isoai, A., Kumagai, H., Misutani, A., Matsuda, C., et al. (2007). Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 16, 618–629. doi: 10.1093/hmg/ddm002
Gade, P., Ramachandran, G., Maachani, U. B., Rizzo, M. A., Okada, T., Prywes, R., et al. (2012). An IFN-gamma-stimulated ATF6-C/EBP-beta-signaling pathway critical for the expression of Death Associated Protein Kinase 1 and induction of autophagy. Proc. Natl. Acad. Sci. U.S.A. 109, 10316–10321. doi: 10.1073/pnas.1119273109
Gade, P., Roy, S. K., Li, H., Nallar, S. C., and Kalvakolanu, D. V. (2008). Critical role for transcription factor C/EBP-beta in regulating the expression of death-associated protein kinase 1. Mol. Cell. Biol. 28, 2528–2548. doi: 10.1128/MCB.00784-07
Gale, M. Jr., Blakely, C. M., Kwieciszewski, B., Tan, S. L., Dossett, M., Tang, N. M., et al. (1998). Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol. Cell. Biol. 18, 5208–5218.
Gannage, M., Dormann, D., Albrecht, R., Dengjel, J., Torossi, T., Ramer, P. C., et al. (2009). Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380. doi: 10.1016/j.chom.2009.09.005
Gavel, Y., and Von Heijne, G. (1990). Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 3, 433–442. doi: 10.1093/protein/3.5.433
Geng, J., and Klionsky, D. J. (2008). The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 9, 859–864. doi: 10.1038/embor.2008.163
Gerlitz, G., Jagus, R., and Elroy-Stein, O. (2002). Phosphorylation of initiation factor-2 alpha is required for activation of internal translation initiation during cell differentiation. Eur. J. Biochem. 269, 2810–2819. doi: 10.1046/j.1432-1033.2002.02974.x
Gomez, B. P., Riggins, R. B., Shajahan, A. N., Klimach, U., Wang, A., Crawford, A. C., et al. (2007). Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J. 21, 4013–4027. doi: 10.1096/fj.06-7990com
Guo, L., Huang, J. X., Liu, Y., Li, X., Zhou, S. R., Qian, S. W., et al. (2013). Transactivation of Atg4b by C/EBPbeta promotes autophagy to facilitate adipogenesis. Mol. Cell. Biol. 33, 3180–3190. doi: 10.1128/MCB.00193-13
Gupta, S., Deepti, A., Deegan, S., Lisbona, F., Hetz, C., and Samali, A. (2010). HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction. PLoS Biol. 8:e1000410. doi: 10.1371/journal.pbio.1000410
Hampton, R. Y. (2000). ER stress response: getting the UPR hand on misfolded proteins. Curr. Biol. 10, R518–R521. doi: 10.1016/S0960-9822(00)00583-2
Hanna, S. L., Pierson, T. C., Sanchez, M. D., Ahmed, A. A., Murtadha, M. M., and Doms, R. W. (2005). N-linked glycosylation of west nile virus envelope proteins influences particle assembly and infectivity. J. Virol. 79, 13262–13274. doi: 10.1128/JVI.79.21.13262-13274.2005
Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., et al. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. doi: 10.1016/S1097-2765(00)00108-8
Harding, H. P., and Ron, D. (2002). Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 51(Suppl. 3), S455–S461. doi: 10.2337/diabetes.51.2007.S455
Hart, L. S., Cunningham, J. T., Datta, T., Dey, S., Tameire, F., Lehman, S. L., et al. (2012). ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Invest. 122, 4621–4634. doi: 10.1172/JCI62973
Hassan, I. H., Zhang, M. S., Powers, L. S., Shao, J. Q., Baltrusaitis, J., Rutkowski, D. T., et al. (2012). Influenza A viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (IRE1) stress pathway. J. Biol. Chem. 287, 4679–4689. doi: 10.1074/jbc.M111.284695
Hershko, A., Heller, H., Elias, S., and Ciechanover, A. (1983). Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 258, 8206–8214.
Hollien, J., and Weissman, J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107. doi: 10.1126/science.1129631
Hur, K. Y., So, J. S., Ruda, V., Frank-Kamenetsky, M., Fitzgerald, K., Koteliansky, V., et al. (2012). IRE1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J. Exp. Med. 209, 307–318. doi: 10.1084/jem.20111298
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/S0092-8674(03)00929-2
Itakura, E., Kishi, C., Inoue, K., and Mizushima, N. (2008). Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 19, 5360–5372. doi: 10.1091/mbc.E08-01-0080
Jackson, W. T., Giddings, T. H. Jr., Taylor, M. P., Mulinyawe, S., Rabinovitch, M., Kopito, R. R., et al. (2005). Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. doi: 10.1371/journal.pbio.0030156
Jha, B. K., Polyakova, I., Kessler, P., Dong, B., Dickerman, B., Sen, G. C., et al. (2011). Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. J. Biol. Chem. 286, 26319–26326. doi: 10.1074/jbc.M111.253443
Jheng, J. R., Lau, K. S., Tang, W. F., Wu, M. S., and Horng, J. T. (2010). Endoplasmic reticulum stress is induced and modulated by enterovirus 71. Cell. Microbiol. 12, 796–813. doi: 10.1111/j.1462-5822.2010.01434.x
Jheng, J. R., Lin, C. Y., Horng, J. T., and Lau, K. S. (2012). Inhibition of enterovirus 71 entry by transcription factor XBP1. Biochem. Biophys. Res. Commun. 420, 882–887. doi: 10.1016/j.bbrc.2012.03.094
Jin, R., Zhu, W., Cao, S., Chen, R., Jin, H., Liu, Y., et al. (2013). Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE 8:e52909. doi: 10.1371/journal.pone.0052909
John, L., Thomas, S., Herchenroder, O., Putzer, B. M., and Schaefer, S. (2011). Hepatitis E virus ORF2 protein activates the pro-apoptotic gene CHOP and anti-apoptotic heat shock proteins. PLoS ONE 6:e25378. doi: 10.1371/journal.pone.0025378
Joubert, P. E., Werneke, S. W., De La Calle, C., Guivel-Benhassine, F., Giodini, A., Peduto, L., et al. (2012). Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 209, 1029–1047. doi: 10.1084/jem.20110996
Jounai, N., Takeshita, F., Kobiyama, K., Sawano, A., Miyawaki, A., Xin, K. Q., et al. (2007). The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. U.S.A. 104, 14050–14055. doi: 10.1073/pnas.0704014104
Kaufman, R. J. (1999). Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 13, 1211–1233. doi: 10.1101/gad.13.10.1211
Ke, P. Y., and Chen, S. S. (2011). Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 121, 37–56. doi: 10.1172/JCI41474
Kemball, C. C., Alirezaei, M., Flynn, C. T., Wood, M. R., Harkins, S., Kiosses, W. B., et al. (2010). Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 84, 12110–12124. doi: 10.1128/JVI.01417-10
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim, S. M., Park, J. H., Chung, S. K., Kim, J. Y., Hwang, H. Y., Chung, K. C., et al. (2004). Coxsackievirus B3 infection induces cyr61 activation via JNK to mediate cell death. J. Virol. 78, 13479–13488. doi: 10.1128/JVI.78.24.13479-13488.2004
Kimmig, P., Diaz, M., Zheng, J., Williams, C. C., Lang, A., Aragon, T., et al. (2012). The unfolded protein response in fission yeast modulates stability of select mRNAs to maintain protein homeostasis. Elife 1:e00048. doi: 10.7554/eLife.00048
Kirkegaard, K., and Jackson, W. T. (2005). Topology of double-membraned vesicles and the opportunity for non-lytic release of cytoplasm. Autophagy 1, 182–184. doi: 10.4161/auto.1.3.2065
Korennykh, A. V., Egea, P. F., Korostelev, A. A., Finer-Moore, J., Zhang, C., Shokat, K. M., et al. (2009). The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693. doi: 10.1038/nature07661
Kornfeld, R., and Kornfeld, S. (1985). Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 54, 631–664. doi: 10.1146/annurev.bi.54.070185.003215
Korolchuk, V. I., Menzies, F. M., and Rubinsztein, D. C. (2010). Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 584, 1393–1398. doi: 10.1016/j.febslet.2009.12.047
Koumenis, C. (2006). ER stress, hypoxia tolerance and tumor progression. Curr. Mol. Med. 6, 55–69. doi: 10.2174/156652406775574604
Kudo, T., Kanemoto, S., Hara, H., Morimoto, N., Morihara, T., Kimura, R., et al. (2008). A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 15, 364–375. doi: 10.1038/sj.cdd.4402276
Lee, H., Noh, J. Y., Oh, Y., Kim, Y., Chang, J. W., Chung, C. W., et al. (2012). IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 21, 101–114. doi: 10.1093/hmg/ddr445
Lee, T. G., Tomita, J., Hovanessian, A. G., and Katze, M. G. (1992). Characterization and regulation of the 58,000-dalton cellular inhibitor of the interferon-induced, dsRNA-activated protein kinase. J. Biol. Chem. 267, 14238–14243.
Lei, S., Tian, Y. P., Xiao, W. D., Li, S., Rao, X. C., Zhang, J. L., et al. (2013). ROCK is involved in vimentin phosphorylation and rearrangement induced by dengue virus. Cell Biochem. Biophys. 67, 1333–1342. doi: 10.1007/s12013-013-9665-x
Levine, B., and Klionsky, D. J. (2004). Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477. doi: 10.1016/S1534-5807(04)00099-1
Li, J. K., Liang, J. J., Liao, C. L., and Lin, Y. L. (2012). Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect. 14, 159–168. doi: 10.1016/j.micinf.2011.09.001
Liang, C., Lee, J. S., Inn, K. S., Gack, M. U., Li, Q., Roberts, E. A., et al. (2008). Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 10, 776–787. doi: 10.1038/ncb1740
Liao, Y., Fung, T. S., Huang, M., Fang, S. G., Zhong, Y., and Liu, D. X. (2013). Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J. Virol. 87, 8124–8134. doi: 10.1128/JVI.00626-13
Lipson, K. L., Ghosh, R., and Urano, F. (2008). The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PLoS ONE 3:e1648. doi: 10.1371/journal.pone.0001648
Lloyd, R. E. (2006). Translational control by viral proteinases. Virus Res. 119, 76–88. doi: 10.1016/j.virusres.2005.10.016
Lu, Y., Wambach, M., Katze, M. G., and Krug, R. M. (1995). Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology 214, 222–228. doi: 10.1006/viro.1995.9937
Lundin, M., Monne, M., Widell, A., Von Heijne, G., and Persson, M. A. (2003). Topology of the membrane-associated hepatitis C virus protein NS4B. J. Virol. 77, 5428–5438. doi: 10.1128/JVI.77.9.5428-5438.2003
Ma, D., Panda, S., and Lin, J. D. (2011). Temporal orchestration of circadian autophagy rhythm by C/EBPbeta. EMBO J. 30, 4642–4651. doi: 10.1038/emboj.2011.322
Ma, Y., and Hendershot, L. M. (2003). Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 278, 34864–34873. doi: 10.1074/jbc.M301107200
Ma, Y., Shimizu, Y., Mann, M. J., Jin, Y., and Hendershot, L. M. (2010). Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress Chaperones 15, 281–293. doi: 10.1007/s12192-009-0142-9
Mahoney, E., Byrd, J. C., and Johnson, A. J. (2013a). Autophagy and ER stress play an essential role in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Autophagy 9, 434–435. doi: 10.4161/auto.23027
Mahoney, E., Maddocks, K., Flynn, J., Jones, J., Cole, S. L., Zhang, X., et al. (2013b). Identification of endoplasmic reticulum stress-inducing agents by antagonizing autophagy: a new potential strategy for identification of anti-cancer therapeutics in B-cell malignancies. Leuk. Lymphoma 54, 2685–2692. doi: 10.3109/10428194.2013.781168
Margariti, A., Li, H., Chen, T., Martin, D., Vizcay-Barrena, G., Alam, S., et al. (2013). XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J. Biol. Chem. 288, 859–872. doi: 10.1074/jbc.M112.412783
Matsunaga, K., Saitoh, T., Tabata, K., Omori, H., Satoh, T., Kurotori, N., et al. (2009). Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 11, 385–396. doi: 10.1038/ncb1846
Mayerhofer, T., and Kodym, R. (2003). Gadd153 restores resistance to radiation-induced apoptosis after thiol depletion. Biochem. Biophys. Res. Commun. 310, 115–120. doi: 10.1016/j.bbrc.2003.08.130
Medigeshi, G. R., Lancaster, A. M., Hirsch, A. J., Briese, T., Lipkin, W. I., Defilippis, V., et al. (2007). West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 81, 10849–10860. doi: 10.1128/JVI.01151-07
Meurs, E., Chong, K., Galabru, J., Thomas, N. S., Kerr, I. M., Williams, B. R., et al. (1990). Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 62, 379–390. doi: 10.1016/0092-8674(90)90374-N
Milani, M., Rzymski, T., Mellor, H. R., Pike, L., Bottini, A., Generali, D., et al. (2009). The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 69, 4415–4423. doi: 10.1158/0008-5472.CAN-08-2839
Miller, S., and Krijnse-Locker, J. (2008). Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6, 363–374. doi: 10.1038/nrmicro1890
Mizui, T., Yamashina, S., Tanida, I., Takei, Y., Ueno, T., Sakamoto, N., et al. (2010). Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J. Gastroenterol. 45, 195–203. doi: 10.1007/s00535-009-0132-9
Mizushima, N. (2010). The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 22, 132–139. doi: 10.1016/j.ceb.2009.12.004
Moore, K. A., Plant, J. J., Gaddam, D., Craft, J., and Hollien, J. (2013). Regulation of sumo mRNA during endoplasmic reticulum stress. PLoS ONE 8:e75723. doi: 10.1371/journal.pone.0075723
Mori, K., Ma, W., Gething, M. J., and Sambrook, J. (1993). A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756. doi: 10.1016/0092-8674(93)90521-Q
Nieva, J. L., Madan, V., and Carrasco, L. (2012). Viroporins: structure and biological functions. Nat. Rev. Microbiol. 10, 563–574. doi: 10.1038/nrmicro2820
Ogata, M., Hino, S., Saito, A., Morikawa, K., Kondo, S., Kanemoto, S., et al. (2006). Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220–9231. doi: 10.1128/MCB.01453-06
Orvedahl, A., Macpherson, S., Sumpter, R. Jr., Talloczy, Z., Zou, Z., and Levine, B. (2010). Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7, 115–127. doi: 10.1016/j.chom.2010.01.007
Ozcan, U., Yilmaz, E., Ozcan, L., Furuhashi, M., Vaillancourt, E., Smith, R. O., et al. (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140. doi: 10.1126/science.1128294
Papandreou, I., Denko, N. C., Olson, M., Van Melckebeke, H., Lust, S., Tam, A., et al. (2011). Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117, 1311–1314. doi: 10.1182/blood-2010-08-303099
Pavio, N., Romano, P. R., Graczyk, T. M., Feinstone, S. M., and Taylor, D. R. (2003). Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2alpha kinase PERK. J. Virol. 77, 3578–3585. doi: 10.1128/JVI.77.6.3578-3585.2003
Pena, J., and Harris, E. (2011). Dengue virus modulates the unfolded protein response in a time-dependent manner. J. Biol. Chem. 286, 14226–14236. doi: 10.1074/jbc.M111.222703
Peng, H., Shi, M., Zhang, L., Li, Y., Sun, J., Zhang, L., et al. (2014). Activation of JNK1/2 and p38 MAPK signaling pathways promotes enterovirus 71 infection in immature dendritic cells. BMC Microbiol. 14:147. doi: 10.1186/1471-2180-14-147
Pike, L. R., Singleton, D. C., Buffa, F., Abramczyk, O., Phadwal, K., Li, J. L., et al. (2013). Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem. J. 449, 389–400. doi: 10.1042/BJ20120972
Proikas-Cezanne, T., Waddell, S., Gaugel, A., Frickey, T., Lupas, A., and Nordheim, A. (2004). WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 23, 9314–9325. doi: 10.1038/sj.onc.1208331
Rathore, A. P., Ng, M. L., and Vasudevan, S. G. (2013). Differential unfolded protein response during Chikungunya and Sindbis virus infection: CHIKV nsP4 suppresses eIF2alpha phosphorylation. Virol. J. 10:36. doi: 10.1186/1743-422X-10-36
Ravikumar, B., Duden, R., and Rubinsztein, D. C. (2002). Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117. doi: 10.1093/hmg/11.9.1107
Redondo, N., Sanz, M. A., Welnowska, E., and Carrasco, L. (2011). Translation without eIF2 promoted by poliovirus 2A protease. PLoS ONE 6:e25699. doi: 10.1371/journal.pone.0025699
Roberson, E. C., Tully, J. E., Guala, A. S., Reiss, J. N., Godburn, K. E., Pociask, D. A., et al. (2012). Influenza induces endoplasmic reticulum stress, caspase-12-dependent apoptosis, and c-Jun N-terminal kinase-mediated transforming growth factor-beta release in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 46, 573–581. doi: 10.1165/rcmb.2010-0460OC
Ron, D. (2002). Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 110, 1383–1388. doi: 10.1172/JCI16784
Rouschop, K. M., Van Den Beucken, T., Dubois, L., Niessen, H., Bussink, J., Savelkouls, K., et al. (2010). The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Invest. 120, 127–141. doi: 10.1172/JCI40027
Saeed, M., Suzuki, R., Watanabe, N., Masaki, T., Tomonaga, M., Muhammad, A., et al. (2011). Role of the endoplasmic reticulum-associated degradation (ERAD) pathway in degradation of hepatitis C virus envelope proteins and production of virus particles. J. Biol. Chem. 286, 37264–37273. doi: 10.1074/jbc.M111.259085
Salazar, M., Carracedo, A., Salanueva, I. J., Hernandez-Tiedra, S., Lorente, M., Egia, A., et al. (2009). Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Invest. 119, 1359–1372. doi: 10.1172/JCI37948
Sarnow, P. (2003). Viral internal ribosome entry site elements: novel ribosome-RNA complexes and roles in viral pathogenesis. J. Virol. 77, 2801–2806. doi: 10.1128/JVI.77.5.2801-2806.2003
Schlegel, A., Giddings, T. H. Jr., Ladinsky, M. S., and Kirkegaard, K. (1996). Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 70, 6576–6588.
Seo, Y. K., Jeon, T. I., Chong, H. K., Biesinger, J., Xie, X., and Osborne, T. F. (2011). Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab. 13, 367–375. doi: 10.1016/j.cmet.2011.03.005
Shaffer, A. L., Shapiro-Shelef, M., Iwakoshi, N. N., Lee, A. H., Qian, S. B., Zhao, H., et al. (2004). XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21, 81–93. doi: 10.1016/j.immuni.2004.06.010
Shamu, C. E., and Walter, P. (1996). Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 15, 3028–3039.
Shi, Y. H., Ding, Z. B., Zhou, J., Hui, B., Shi, G. M., Ke, A. W., et al. (2011). Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 7, 1159–1172. doi: 10.4161/auto.7.10.16818
Shintani, T., and Klionsky, D. J. (2004). Autophagy in health and disease: a double-edged sword. Science 306, 990–995. doi: 10.1126/science.1099993
Sidrauski, C., and Walter, P. (1997). The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039. doi: 10.1016/S0092-8674(00)80369-4
Singh, R., Kaushik, S., Wang, Y., Xiang, Y., Novak, I., Komatsu, M., et al. (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. doi: 10.1038/nature07976
Sir, D., Chen, W. L., Choi, J., Wakita, T., Yen, T. S., and Ou, J. H. (2008). Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48, 1054–1061. doi: 10.1002/hep.22464
Solda, T., Garbi, N., Hammerling, G. J., and Molinari, M. (2006). Consequences of ERp57 deletion on oxidative folding of obligate and facultative clients of the calnexin cycle. J. Biol. Chem. 281, 6219–6226. doi: 10.1074/jbc.M513595200
Sonenberg, N., and Hinnebusch, A. G. (2009). Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745. doi: 10.1016/j.cell.2009.01.042
Sun, Y., Li, C., Shu, Y., Ju, X., Zou, Z., Wang, H., et al. (2012). Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci. Signal. 5, ra16. doi: 10.1126/scisignal.2001931
Surjit, M., Jameel, S., and Lal, S. K. (2007). Cytoplasmic localization of the ORF2 protein of hepatitis E virus is dependent on its ability to undergo retrotranslocation from the endoplasmic reticulum. J. Virol. 81, 3339–3345. doi: 10.1128/JVI.02039-06
Szegezdi, E., Logue, S. E., Gorman, A. M., and Samali, A. (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885. doi: 10.1038/sj.embor.7400779
Talloczy, Z., Jiang, W., Virgin, H. W. T., Leib, D. A., Scheuner, D., Kaufman, R. J., et al. (2002). Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 99, 190–195. doi: 10.1073/pnas.012485299
Talloczy, Z., Virgin, H. W. T., and Levine, B. (2006). PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2, 24–29. doi: 10.4161/auto.2176
Tang, W. F., Yang, S. Y., Wu, B. W., Jheng, J. R., Chen, Y. L., Shih, C. H., et al. (2007). Reticulon 3 binds the 2C protein of enterovirus 71 and is required for viral replication. J. Biol. Chem. 282, 5888–5898. doi: 10.1074/jbc.M611145200
Tanida, I., Fukasawa, M., Ueno, T., Kominami, E., Wakita, T., and Hanada, K. (2009). Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 5, 937–945. doi: 10.4161/auto.5.7.9243
Tardif, K. D., Mori, K., Kaufman, R. J., and Siddiqui, A. (2004). Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 279, 17158–17164. doi: 10.1074/jbc.M312144200
Tardif, K. D., Mori, K., and Siddiqui, A. (2002). Hepatitis C virus subgenomic replicons induce endoplasmic reticulum stress activating an intracellular signaling pathway. J. Virol. 76, 7453–7459. doi: 10.1128/JVI.76.15.7453-7459.2002
Tatu, U., Hammond, C., and Helenius, A. (1995). Folding and oligomerization of influenza hemagglutinin in the ER and the intermediate compartment. EMBO J. 14, 1340–1348.
Taylor, M. P., Burgon, T. B., Kirkegaard, K., and Jackson, W. T. (2009). Role of microtubules in extracellular release of poliovirus. J. Virol. 83, 6599–6609. doi: 10.1128/JVI.01819-08
Tian, P., Ball, J. M., Zeng, C. Q., and Estes, M. K. (1996). The rotavirus nonstructural glycoprotein NSP4 possesses membrane destabilization activity. J. Virol. 70, 6973–6981.
Tian, P., Estes, M. K., Hu, Y., Ball, J. M., Zeng, C. Q., and Schilling, W. P. (1995). The rotavirus nonstructural glycoprotein NSP4 mobilizes Ca2+ from the endoplasmic reticulum. J. Virol. 69, 5763–5772.
Trask, S. D., McDonald, S. M., and Patton, J. T. (2012). Structural insights into the coupling of virion assembly and rotavirus replication. Nat. Rev. Microbiol. 10, 165–177. doi: 10.1038/nrmicro2673
Tu, Y. C., Yu, C. Y., Liang, J. J., Lin, E., Liao, C. L., and Lin, Y. L. (2012). Blocking double-stranded RNA-activated protein kinase PKR by Japanese encephalitis virus nonstructural protein 2A. J. Virol. 86, 10347–10358. doi: 10.1128/JVI.00525-12
Umareddy, I., Pluquet, O., Wang, Q. Y., Vasudevan, S. G., Chevet, E., and Gu, F. (2007). Dengue virus serotype infection specifies the activation of the unfolded protein response. Virol. J. 4:91. doi: 10.1186/1743-422X-4-91
Van Deurs, B., Holm, P. K., and Sandvig, K. (1996). Inhibition of the vacuolar H(+)-ATPase with bafilomycin reduces delivery of internalized molecules from mature multivesicular endosomes to lysosomes in HEp-2 cells. Eur. J. Cell Biol. 69, 343–350.
Van Kuppeveld, F. J., Hoenderop, J. G., Smeets, R. L., Willems, P. H., Dijkman, H. B., Galama, J. M., et al. (1997). Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 16, 3519–3532. doi: 10.1093/emboj/16.12.3519
Volkmann, K., Lucas, J. L., Vuga, D., Wang, X., Brumm, D., Stiles, C., et al. (2011). Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 286, 12743–12755. doi: 10.1074/jbc.M110.199737
Von Dem Bussche, A., Machida, R., Li, K., Loevinsohn, G., Khander, A., Wang, J., et al. (2010). Hepatitis C virus NS2 protein triggers endoplasmic reticulum stress and suppresses its own viral replication. J. Hepatol. 53, 797–804. doi: 10.1016/j.jhep.2010.05.022
Wang, J., Kang, R., Huang, H., Xi, X., Wang, B., and Zhao, Z. (2014). Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 10, 766–784. doi: 10.4161/auto.27954
Wang, Q. J., Ding, Y., Kohtz, D. S., Mizushima, N., Cristea, I. M., Rout, M. P., et al. (2006). Induction of autophagy in axonal dystrophy and degeneration. J. Neurosci. 26, 8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006
Wong, J., Zhang, J., Si, X., Gao, G., Mao, I., McManus, B. M., et al. (2008). Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82, 9143–9153. doi: 10.1128/JVI.00641-08
Wu, M. J., Ke, P. Y., Hsu, J. T., Yeh, C. T., and Horng, J. T. (2014). Reticulon 3 interacts with NS4B of the hepatitis C virus and negatively regulates viral replication by disrupting NS4B self-interaction. Cell. Microbiol. doi: 10.1111/cmi.12318. [Epub ahead of print].
Wu, Y. P., Chang, C. M., Hung, C. Y., Tsai, M. C., Schuyler, S. C., and Wang, R. Y. (2011). Japanese encephalitis virus co-opts the ER-stress response protein GRP78 for viral infectivity. Virol. J. 8:128. doi: 10.1186/1743-422X-8-128
Xie, X., Gayen, S., Kang, C., Yuan, Z., and Shi, P. Y. (2013). Membrane topology and function of dengue virus NS2A protein. J. Virol. 87, 4609–4622. doi: 10.1128/JVI.02424-12
Yamazaki, H., Hiramatsu, N., Hayakawa, K., Tagawa, Y., Okamura, M., Ogata, R., et al. (2009). Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J. Immunol. 183, 1480–1487. doi: 10.4049/jimmunol.0900017
Yan, W., Frank, C. L., Korth, M. J., Sopher, B. L., Novoa, I., Ron, D., et al. (2002). Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. U.S.A. 99, 15920–15925. doi: 10.1073/pnas.252341799
Ye, J., Rawson, R. B., Komuro, R., Chen, X., Dave, U. P., Prywes, R., et al. (2000). ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364. doi: 10.1016/S1097-2765(00)00133-7
Yoon, Y. H., Cho, K. S., Hwang, J. J., Lee, S. J., Choi, J. A., and Koh, J. Y. (2010). Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest. Ophthalmol. Vis. Sci. 51, 6030–6037. doi: 10.1167/iovs.10-5278
Yorimitsu, T., Nair, U., Yang, Z., and Klionsky, D. J. (2006). Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 281, 30299–30304. doi: 10.1074/jbc.M607007200
Yoshida, H., Haze, K., Yanagi, H., Yura, T., and Mori, K. (1998). Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749. doi: 10.1074/jbc.273.50.33741
Yoshida, H., Matsui, T., Hosokawa, N., Kaufman, R. J., Nagata, K., and Mori, K. (2003). A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 4, 265–271. doi: 10.1016/S1534-5807(03)00022-4
Zai, J., Mei, L., Wang, C., Cao, S., Fu, Z. F., Chen, H., et al. (2013). N-glycosylation of the premembrane protein of Japanese encephalitis virus is critical for folding of the envelope protein and assembly of virus-like particles. Acta Virol. 57, 27–33. doi: 10.4149/av_2013_01_27
Keywords: ATF6, eIF2α, enterovirus 71, ER stress, IRE1, unfolded protein response
Citation: Jheng J-R, Ho J-Y and Horng J-T (2014) ER stress, autophagy, and RNA viruses. Front. Microbiol. 5:388. doi: 10.3389/fmicb.2014.00388
Received: 02 May 2014; Accepted: 11 July 2014;
Published online: 05 August 2014.
Edited by:
Shiu-Wan Chan, The University of Manchester, UKReviewed by:
Youichi Suzuki, Duke-NUS Graduate Medical School, SingaporeCopyright © 2014 Jheng, Ho and Horng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jim-Tong Horng, Department of Biochemistry and Molecular Biology, College of Medicine, Chang Gung University, First Medical Building, 259 Wen-Hua First Road, Kweishan, Taoyuan 333, Taiwan e-mail:amltdG9uZ0BtYWlsLmNndS5lZHUudHc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.