 Ashleigh Holmes
Ashleigh Holmes Louise Birse
Louise Birse Robert W. Jackson
Robert W. Jackson Nicola J. Holden
Nicola J. Holden
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Microbiol. , 10 June 2014
Sec. Plant Pathogen Interactions
Volume 5 - 2014 | https://doi.org/10.3389/fmicb.2014.00286
This article is part of the Research Topic Plants as alternative hosts for human and animal pathogens View all 11 articles
Analysis of microbial gene expression during host colonization provides valuable information on the nature of interaction, beneficial or pathogenic, and the adaptive processes involved. Isolation of bacterial mRNA for in planta analysis can be challenging where host nucleic acid may dominate the preparation, or inhibitory compounds affect downstream analysis, e.g., quantitative reverse transcriptase PCR (qPCR), microarray, or RNA-seq. The goal of this work was to optimize the isolation of bacterial mRNA of food-borne pathogens from living plants. Reported methods for recovery of phytopathogen-infected plant material, using hot phenol extraction and high concentration of bacterial inoculation or large amounts of infected tissues, were found to be inappropriate for plant roots inoculated with Escherichia coli O157:H7. The bacterial RNA yields were too low and increased plant material resulted in a dominance of plant RNA in the sample. To improve the yield of bacterial RNA and reduce the number of plants required, an optimized method was developed which combines bead beating with directed bacterial lysis using SDS and lysozyme. Inhibitory plant compounds, such as phenolics and polysaccharides, were counteracted with the addition of high-molecular-weight polyethylene glycol and hexadecyltrimethyl ammonium bromide. The new method increased the total yield of bacterial mRNA substantially and allowed assessment of gene expression by qPCR. This method can be applied to other bacterial species associated with plant roots, and also in the wider context of food safety.
The analysis of bacterial gene expression is important in the determination of how adaptation to different environments develops and informs on the roles of different genes during this process. Human pathogens are now recognized to interact with plants and use them as hosts, as a result of recent high-profile outbreaks from contaminated fruit and vegetables (Cooley et al., 2007; Buchholz et al., 2011). Adaptation of food-borne pathogens to secondary hosts has opened up new areas of research and investigation. Current research suggests that the interaction of human pathogens is more complex than previously perceived in that they can persist for long periods of time (reviewed in Brandl, 2006; Holden et al., 2009; Barak and Schroeder, 2012) and invoke an immune response from the plant (Thilmony et al., 2006; Schikora et al., 2008; Roy et al., 2013). Furthermore, the interactions include quite specific and targeted recognition of the plant host cells by the bacteria (Rossez et al., 2013). Important questions remain as to how the bacteria adapt to secondary hosts, for which analysis of bacterial gene expression is fundamental.
Bacterial gene expression is typically assessed using quantitative reverse transcriptase PCR (qPCR) or microarray techniques, and more recently RNA-seq (An et al., 2013), which require the isolation of high quality and quantity of mRNA. Bacterial mRNA has a short half-life and is less stable than eukaryotic mRNA transcripts, which have capped and polyadenylated RNA tails. Therefore, appropriate measures need to be in place to capture mRNA transcripts that may be inherently unstable, but nevertheless important for bacterial adaptation. In addition to challenges with bacterial mRNA stability, extractions from mixed samples bring additional considerations, not least for inhibitory compounds that may affect downstream analysis. Although there are a number of published techniques for the isolation of total RNA from bacteria-infected plant leaves (Schenk et al., 2008; Soto-Suarez et al., 2010; Fink et al., 2012; Goudeau et al., 2013), samples can still be dominated by plant RNA making it challenging to assess bacterial mRNA. In published reports, leaves were typically infected via infiltration, introducing a high bacterial inoculum into the sample. In other studies, total RNA was extracted from inoculated plant extracts (Mark et al., 2005; Hernandez-Morales et al., 2009; Kyle et al., 2010; Shidore et al., 2012), such as leaf lysates, which helped to reduce the dominance of plant RNA in the sample.
There are limited studies where bacterial expression has been assessed directly from the plant root system (roots and rhizosphere). However, roots are known to support relatively high densities of bacteria, providing a more favorable habitat than the phyllosphere. In general, published reports describe that a high number of plant roots is required to retrieve sufficient bacterial RNA (Matilla et al., 2007; Hou et al., 2012, 2013; Zyśko et al., 2012). Further to this, the application of techniques described for RNA purification from infected leaves cannot always be successfully applied to roots. Therefore, optimization of the RNA extraction methods is required to obtain sufficient quantity and quality of bacterial mRNA, coupled with a reduction in the amount of accompanying plant root RNA. We optimized the method for the food-borne pathogen Escherichia coli O157:H7, frequently associated food-borne outbreaks from consumption of contaminated spinach and lettuce. Although roots of these plants are not consumed, the pathogen can colonize this habitat successfully (Wright et al., 2013), from where it can contaminate the edible portion, either directly or through cross-contamination during processing.
E. coli O157:H7 isolate Sakai stx-negative (Hayashi et al., 2001) was routinely cultured overnight in Luria broth at 37°C, with aeration. For plant-infection assays, the bacteria were sub-cultured at 1:50 dilution into 15 ml rich-defined MOPs media supplemented with 0.2% glucose (RD MOPS glucose; Neidhardt et al., 1974), in a 200 ml Erlenmeyer flask and incubated at 18°C, with aeration for ~18 h. Bacterial cultures were diluted to OD600 of 0.2 (equivalent to ~2 × 108 cfu ml-1) in sterile phosphate buffered saline (PBS) prior to infecting plant roots.
Lettuce (Lactuca sativa) cultivar All Year Round or spinach (Spinacia oleracea) cultivar Amazon seeds (Sutton Seeds, UK) were soaked in sterile distilled water for 2 h before being surface sterilized in 2% calcium hypochlorite solution (10 ml) for 10 min. The seeds were then washed vigorously six times with sterile distilled water and germinated on distilled water agar (0.5% w/v) in the dark for 3–5 days, at ~22°C. Seedlings were transplanted into 175 ml hydroponic tubes (Greiner, Frickenhausen, Germany) containing autoclaved perlite and sterile 0.5× Murashige and Skoog (MS) medium (Sigma Aldrich, USA). Seedlings were grown in a cabinet with a light intensity of 150 μmol m2 s-1 (16 h photoperiod) for a further 21 days at 22°C. To assess bacterial numbers from infected plant material, the roots were washed with PBS to remove excess, non-adherent bacteria. The root sample was homogenized using mortar and pestle and serially diluted for viable counts on selective agar plates.
Total RNA was extracted from infected root samples either by the BPEX method (Schenk et al., 2008) or by the Bead/SDS/phenol method (Figure 1): a method which was adapted and optimized from Jahn et al. (2008) and Schenk et al. (2008).
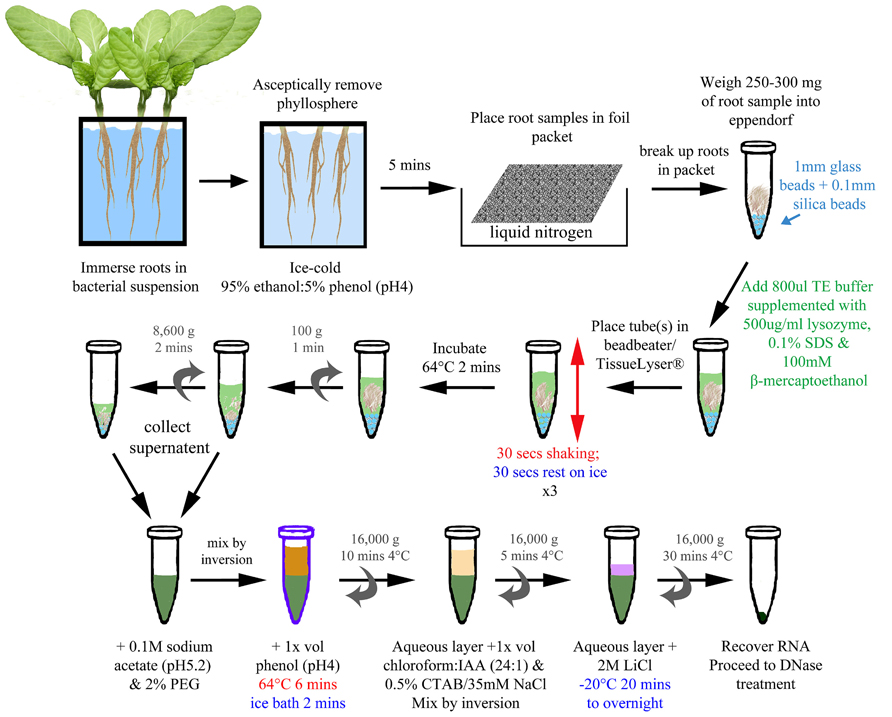
FIGURE 1. Bead/SDS/phenol method of RNA extraction from infected plant roots. A schematic of the optimized method (described in detail within Section “Material and Methods”). The final concentrations of reagents are indicated in the figure. Colored text has been used to highlight different stages and treatments (e.g., blue for addition of beads/cold; green for lysis; red for shaking/heat).
Sample preparation was common to both methods. Whole plants were gently removed from the hydroponic tubes and washed with sterile PBS to remove as much perlite as possible before bacterial infection. Plants were pooled into groups of 5 and the roots were submerged into 20–25 ml bacterial suspension (OD600 = 0.2 equivalent to ~ 5 × 107–1 × 108 cfu ml-1), and incubated at 18°C for 2 h. After incubation, the plants were removed from the bacterial suspension and all the tissue above the crown (shoots, leaves, and petioles) were aseptically removed with a sterile scalpel and the roots immediately immersed into 20 ml ice-cold, 95% ethanol: 5% phenol (pH 4.0). The sample was incubated on ice for 5 min and gently agitated on a vortex mixer to remove any excess and loosely attached bacteria. The root sample was placed into a foil packet and immediately stored in liquid nitrogen until all samples had been processed.
The BPEX method was originally developed to recover Pseudomonas syringae phytopathogen mRNA from infected plant material (Schenk et al., 2008), and tested for recovery of E. coli mRNA from infected roots. The main change from the published method was in the tissue type (root vs leaf disks), although the protocol is provided to allow a direct comparison with the optimized method. Infected plant tissue was ground to a fine powder in liquid nitrogen with a pre-cooled pestle and mortar. The sample (~1 g) was transferred to an Eppendorf containing 750 μl of bacteria plant extraction (BPEX) buffer [0.35 M glycine, 0.7 M NaCl, 2% (w/v) polyethylene glycol (PEG) 20000, 40 mM EDTA, 50 mM NaOH, 4% (w/v) SDS] supplemented with 100 mM β-mercaptoethanol just prior to use. The sample was mixed on a vortex mixer in the buffer prior to incubation at 95°C for 90 s with shaking. An equal volume (750 μl) of phenol/chloroform mix (5:1, pH 4.0) (Sigma, St. Louis, USA) was added and the sample shaken for 5 min to form an emulsion before centrifugation at 16,000 × g for 7 min. The upper phase was collected and added to an equal volume of phenol/chloroform mix (5:1, pH 4.0), shaken, and centrifuged again as the previous step. The upper phase was collected and added to an equal volume of phenol/chloroform/isoamyl alcohol (IAA) mix (25:24:1, pH 4.0) (Sigma, St. Louis, USA) shaken and centrifuged again as before. A volume of 495 μl of the upper phase was added to 550 μl chloroform/IAA mix (24:1) and overlayed with 55 μl pre-warmed (55°C) hexadecyltrimethyl ammonium bromide (CTAB)/NaCl (10% CTAB, 0.7 M NaCl) solution. The suspension was shaken and centrifuged as before. The upper phase was added to 1/4 vol 8 M LiCl solution, mixed by inversion and the RNA precipitated at –20°C for 30 min. To collect the RNA, the sample was centrifuged at 16,000 × g for 20 min at 4°C and the RNA pellet resuspended in 100 μl RNase-free water. The RNA was cleaned and DNase treated using RNeasy Plant Mini kit (Qiagen, Limburg, Netherlands) as per the manufacturer’s guidelines.
The optimized method is represented in Figure 1. The samples were removed from the liquid nitrogen, beaten with a spatula to break up the root into smaller pieces, and transferred into an 1.5 ml micro-centrifuge tube (DNase, RNase free: Ambion, Austin, USA) preloaded with ~250 mg mixture of 1 mm glass and 0.1 mm silica beads (ThermoScientific Ltd., Waltham, USA), from which the weight was determined. Cooled, sterile equipment was used throughout the process. The micro-centrifuge tube was then returned to liquid nitrogen for processing subsequent samples until all of the samples were collected. For the lysis step, 800 μl of Tris-EDTA (TE) buffer (10 mM Tris-HCl; 1 mM EDTA) supplemented with 500 μg ml-1 lysozyme, 0.1% SDS, and 100 mM β-mercaptoethanol was added to each root sample. The tubes were placed into TissueLyser (Qiagen, Limburg, Netherlands) and agitated for 30 s with a 30 s interval on ice for three cycles. After the last cycle, the samples were returned to ice before transfer to a heatblock set at 64° C for 2 min. To extract nucleic acids, the supernatant was collected and pooled after two centrifugation steps: first at 100 × g for 1 min to pellet the beads and large fragments of root, followed by a second at 8,600 × g for 2 min to compact the debris further, yielding more supernatant. One hundred millimolar sodium acetate (pH 5.2) and 2% (w/v) PEG 20000 was added to the supernatant and inverted to mix. An equal volume (~1 ml) of phenol (pH 4.0) was then added, mixed by inversion, and the sample incubated at 64°C for 6 min, with the tubes inverted every 40 s. The sample was transferred to an ice bath for 2 min before centrifugation at 16,000 × g for 10 min at 4°C. The upper aqueous layer was added to an equal volume of chloroform/IAA mix (24:1) and 1/20 volume of pre-warmed (55°C) CTAB/NaCl solution in a fresh micro-centrifuge tube. The sample was mixed by inversion and then centrifuged at 16,000 × g for 5 min at 4°C. The upper aqueous layer was added to 1/4 vol 8 M LiCl, mixed by inversion, and then incubated at –20°C for 20 min to overnight to precipitate the nucleic acid. The nucleic acid was recovered by centrifugation at 16,000 × g for 30 min at 4°C. The pellet was resuspended in 100 μl RNase-free water and the sample cleaned and DNase treated using the RNeasy Plant Mini kit (Qiagen, Limburg, Netherlands) as per the manufacturer’s instructions.
Total RNA concentration was determined using a NanoDrop (Wilmington, USA) spectrophotometer and the relative proportions of ribosomal RNA determined using a BioAnalyser 2100 (Agilent Technologies, Santa Clara, USA), for both methods.
cDNA was transcribed from 1 μg total RNA using Superscript II (Invitrogen, Carlsbad, USA) following the random primer protocol. A mixture of ten 11-mer oligonucleotide primers (Ea1 ± Ea10) at 100 nM (Fislage et al., 1997) designed specifically for Enterobacteriaceae mRNA was used. Quantitative reverse transcriptase PCR was carried out using specific primers for E. coli O157:H7 gyrB housekeeping gene (gyrB.RT.F = CATCAGAGAGGTCGGCTTCC; gyrB.RT.R = CATGGAGCGTCGTTATCCGA) using StepOnePlusTM real-time PCR system (Applied Biosystems, Life Technologies, Carlsbad, USA) and iTaqTM Universal SYBR® Green Supermix (Bio-Rad, Hercules, USA). Each 20 μl reaction volume was composed of iTaq SuperMix, 300 nM of forward and reverse primers and 1 μl of cDNA (diluted 1:4). The PCR program consisted of an initial denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s, and annealing and extension at 60°C for 1 min. Melt curve analysis was also performed with an initial denaturation at 95°C for 15 s, followed by annealing at 60°C for 1 min with an 0.3°C increase (step and hold) and final step of 95°C for 15 s. Data were collected from three technical replicates and from two biological replicates.
An optimized method was previously reported for the extraction of P. syringae mRNA from infiltrated plant leaves (Schenk et al., 2008). A buffer system was developed that yielded 75–125 μg of total RNA per 150–200 mg sample, from the equivalent of 20 infiltrated leaf disks (7 mm). We tested this protocol (termed the “BPEX” method) to determine if it could also be used to recover mRNA of a food-borne pathogen from infected roots. Our work focuses on the bacterial genes induced during the stages of colonization of lettuce and spinach roots, and as such the roots in our experiments are not infected via infiltration; rather the bacteria colonize the outer surface. This necessitated processing the entire root to recover sufficient RNA. Also, to obtain equivalent bacterial numbers to those reported, where each leaf disk was infiltrated with 1 × 108 cfu bacteria, at least 20 root samples were pooled for each extraction. In this method, the samples were ground to a fine powder in liquid nitrogen, prior to incubation in an extraction buffer (BPEX) coupled with cell lysis. RNA was purified using traditional acidic phenol extraction and LiCl-mediated precipitation.
Use of the BPEX method typically yielded 3.6–9.6 μg of total RNA from 250 mg E. coli O157:H7-infected lettuce roots. Analysis of the total RNA showed strong bands corresponding with 18S and 28S rRNA of lettuce (Figure 2A: Lanes 1, 2, 3) with only very faint bacterial RNA corresponding to 16S and 23S rRNA in mixed samples (Figure 2A; Lanes 1, 2, 3). Examination of the electropherogram trace showed that the bacterial rRNA peaks were considerably smaller than the plant rRNA peaks, e.g., BPEX sample 1 (Figure 2B). The pre-dominance of plant rRNA in the BPEX-prepared samples indicated that plant mRNA would also dominate over bacterial transcripts. Therefore, to increase the levels of bacterial mRNA with a concomitant decrease in plant-derived RNA, a modified method was designed. The aim was threefold: (i) to reduce the number of plants required per extraction; (ii) to reduce the carry-over of plant tissue in the sample; and (iii) incorporate a lysis step which targeted bacterial cells.
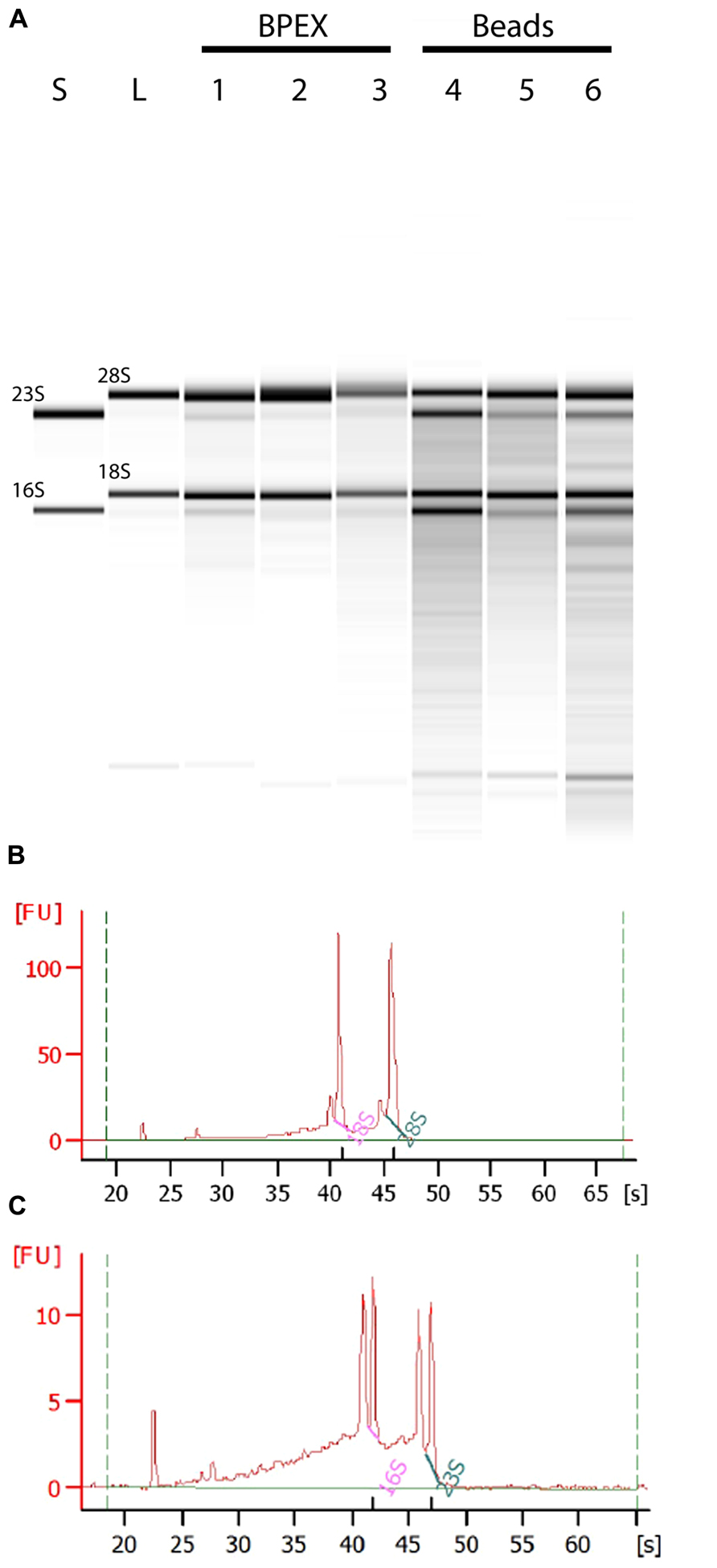
FIGURE 2. Total RNA from plant root extractions by BPEX and Bead/SDS/phenol methods. Intact plant roots were suspended in 20 ml of bacterial inoculum (OD600 of 0.2) for 2 h at 18°C. Total RNA was extracted using either the BPEX or the Bead/SDS/phenol method and RNA quality was assessed by spectroscopy on a Bioanalyser 2100 machine (Agilent). (A) A montage of an electropherograph of the total RNA levels following extraction of inoculated plant material. The lanes show RNA from an in vitro culture of E. coli O157:H7 isolate Sakai only (lane S); uninfected lettuce roots (lane L); E. coli O157:H7 Sakai infected lettuce roots (Lane 1, 306 ng μl-1; Lane 4, 119 ng μl-1; Lane 5, 195 ng μl-1); and E. coli O157:H7 Sakai infected spinach roots (Lane 2, 248 ng μl-1; Lane 3, 301 ng μl-1; Lane 6, 141 ng μl-1). Lanes 1, 2, and 3 show extraction using the BPEX method, and Lanes 4, 5, and 6 using the Bead method. The samples were run alongside a commercial RNA transcript ladder (0.2, 0.5, 1.0, 2.0, 4.0, and 6.0 kb, Agilent); the 16S and 23S (bacterial), and 18S and 28S (plant) rRNA bands are indicated. Electropherogram of E. coli O157:H7 Sakai infected lettuce roots extracted with (B) the BPEX method (sample derived from lane 1) or with (C) the novel Bead/SDS/phenol method (sample derived from lane 4). The uninfected spinach control was similar to lettuce (lane L) (not shown).
Physical disruption of tissues and cells using inert beads has been used for nucleic acid extraction from filamentous fungi (Leite et al., 2012), microalgae (Kim et al., 2012), soil and sludge samples (Griffiths et al., 2000), and in commercial kits. We postulated that this method of disruption may also aid in bacterial RNA extraction and used a combination of 1 mm glass beads and 0.1 mm silica beads for sample lysis. Since tissue can become over-heated during agitation, which leads to nucleic acid degradation, the samples were “rested” for 30 s on ice between the three treatment cycles.
Bacterial cells were specifically targeted for lysis in an attempt to reduce plant-derived nucleic acid contamination, with the inclusion of lysozyme in the extraction buffer. However, a combination of the hot SDS/phenol extraction protocol described previously (Jahn et al., 2008) with the bead beating step resulted in very low RNA yields; not greater than 0.5 μg. It was possible that contaminants or inhibitors from the plant material, such as polysaccharides, phenolic compounds, and secondary metabolites deleteriously affected RNA recovery. Therefore, the protocol was modified to include additional steps after the initial lysis and the incorporation of PEG and CTAB/NaCl treatments, followed by LiCl precipitation (Figure 1). The inclusion of high-molecular-weight PEG in the extraction protocol promotes the removal of polysaccharides and phenolic compounds from plant tissues that bind to or co-precipitate with RNA (Gehrig et al., 2000). CTAB is a detergent that acts to separate polysaccharides from nucleic acids (Chang et al., 1993; Jaakola et al., 2001). Lithium chloride is inefficient at precipitating DNA, proteins, or carbohydrates, and thus improves RNA yield and purity compared to other nucleic acid precipitation methods, i.e., ethanol and sodium acetate (Barlow et al., 1963; Cathala et al., 1983).
A combination of physical disruption using beads together with the chemical treatments (Figure 1) resulted in total RNA concentrations that averaged 5.1 μg (range from 1.5 to 8.9 μg) from 250 mg of infected root tissue. Assessment of the RNA showed a substantial increase in bacterial-derived 16S and 23S rRNA in bead-treated samples compared to BPEX samples (Figure 2A – compare lanes 4–6 with lanes 1–3). The optimized method increased the bacterial rRNA to levels that were equivalent or close to plant rRNA (Figure 2C).
Bacterial gene expression was quantified from the samples obtained using the optimized method, by carrying out qPCR. The gyrB gene was selected as a housekeeping gene that was expected to be expressed under the conditions tested and expression from the root-derived samples was compared to samples obtained from bacteria grown under in vitro conditions. The amount of gyrB transcript was found to be similar between both lettuce roots and in vitro samples (Table 1), indicating similar copy numbers and stable gyrB expression under in vitro conditions and in plant extracts. It is of note that the amount of detectable gyrB was lower in the infected spinach root extracts compared to lettuce roots (higher Ct value for gyrB).
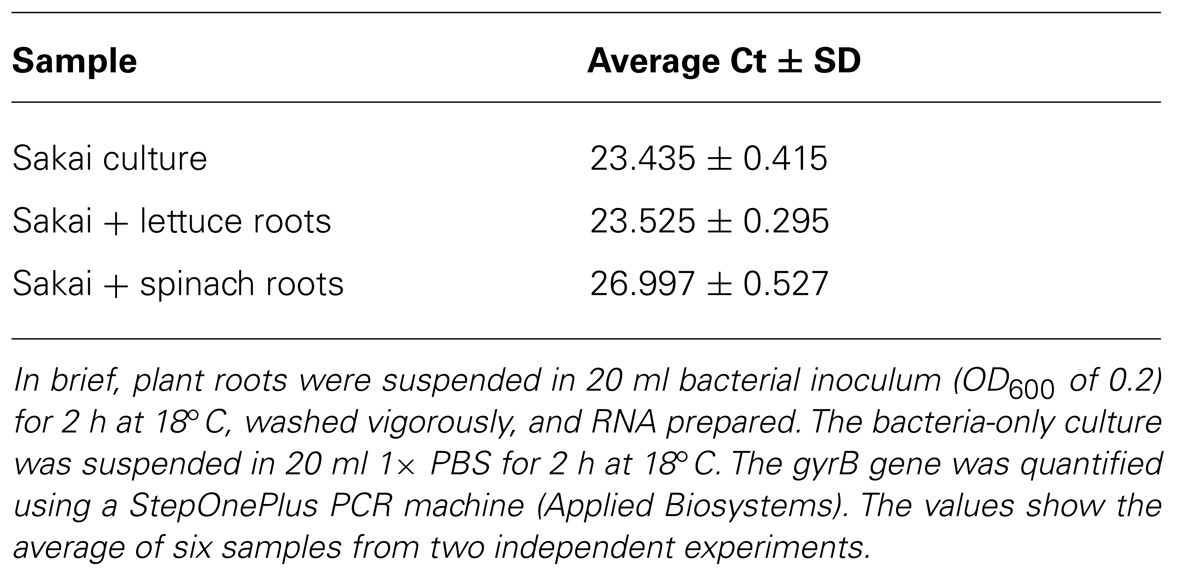
TABLE 1. Comparison of Ct values from E. coli O157:H7 isolate Sakai for an in vitro culture and for infected plant roots.
To quantify the bacteria present in the samples, the average number of E. coli O157:H7 (Sakai) cells present in a 250 mg root sample (typically from 7 to 8 roots) was determined. The experimental set-up was as described for RNA extraction, although the roots were washed with PBS instead of the initial step of mRNA preservation in the ice-cold phenol/ethanol mixture. On average, 5.6 × 107 cfu of E. coli O157:H7 (Sakai) were present in each lettuce sample. The values for spinach roots were similar, which represent between 10 and 15% of the initial inoculum used for infection. These figures are equivalent to the limit of bacterial gene detection reported by Schenk et al. (2008) of ~ 5 107 cfu/sample derived from 20 leaf disks.
The isolation of total RNA from plant material is well defined for leaves, but less so for studies involving the roots or rhizosphere. The majority of reports focus on analysis of gene expression for phytopathogens and the number of examples describing the extraction of food-borne pathogen RNA from plants is limited. Here, we describe optimization of a method that resulted in high-quality bacterial mRNA from the infected roots of fresh produce plants: lettuce and spinach.
Reported investigations of gene expression of enteric bacteria, such as Salmonella enterica (Goudeau et al., 2013), E. coli K-12 and E. coli O157:H7 (Kyle et al., 2010; Fink et al., 2012) used large quantities of plant material, e.g., 100 g of leaf tissue coupled with high concentrations of bacteria, approx. 108 cfu per sample. Samples were processed to separate bacteria from plant tissue using physical methods, such as a Stomacher (Kyle et al., 2010; Goudeau et al., 2013) or by shaking (Fink et al., 2012) followed by filtration to remove plant debris. RNA was prepared either using commercial kits (Kyle et al., 2010; Goudeau et al., 2013) or a hot-phenol method (Fink et al., 2012). Some of these studies investigated gene expression on post-harvest material, using pre-packaged commercial leaves rather than propagating plants, where it is possible to obtain the large amount of material required. In contrast, investigation of bacteria gene expression on living plant roots presents challenges in obtaining similar weights of material. A hydroponic system for high-throughput propagation of lettuce plants was described recently (Hou et al., 2012, 2013), where sterilized seeds were germinated in 96-well pipette tip boxes. Whole transcriptome analysis of E. coli was examined three days after inoculation with the plant roots, although the report does not state the weight of material required for RNA extraction. The main difference in our method was the amount of plant material required and the time at which the samples were taken. For example, our method allows for gene expression analysis after a short time of interaction, rather than after several days of colonization where the bacterial numbers are likely to have increased, dependent on the inoculation time and plant-bacterial system investigated.
The optimized method uses a combination of bead-beating, SDS lysis, phenol extraction, and CTAB purification to extract high-quality bacterial RNA from as little as 250–300 mg infected roots. Optimization has been carried out for two fresh produce plant species that have previously been associated with large-scale outbreaks of food-borne pathogens. It was interesting to note that the efficiency of recovery was lower for spinach root compared to lettuce, although the numbers of bacteria recovered from the plant tissue were similar. Since E. coli O157:H7 Sakai adheres to lettuce and spinach roots in comparable numbers (Wright et al., 2013), we anticipated similar levels of gyrB transcript. The reduction in level may be as a result of plant-associated inhibitors in spinach roots, or because of different plant-derived environmental cues acting on gyrB expression. Therefore, we suggest that the method needs to be validated if it is adopted for other plant species. Verification of the technique using qPCR shows that this method could be applied to food safety settings, e.g., for detection of food-borne pathogens in fresh produce. It may be possible to couple this technique to standard methods, which would enable examination of viability and expression of genes of interest, e.g., toxin genes. Furthermore, it facilitates analysis of bacterial gene expression in planta for not only food-borne pathogens but also other plant-associated bacteria, providing an insight into the adaptive processes that underpin host–microbe interactions.
Al authors were involved in the design of the work and interpretation of data; Ashleigh Holmes and Louise Birse were involved in data acquisition and analysis; Ashleigh Holmes, Louise Birse, and Nicola J. Holden drafted the work; all authors approved the final version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The work was supported by BBSRC grant BB/I014179/1 and Scottish Government RESAS funding. The authors thank Pete Headley and Jenny Morris (James Hutton Institute) for technical assistance; Sonia Humphris and the reviewers for helpful suggestions.
An, S.-Q., Febrer, M., Mccarthy, Y., Tang, D.-J., Clissold, L., Kaithakottil, G., et al. (2013). High-resolution transcriptional analysis of the regulatory influence of cell-to-cell signalling reveals novel genes that contribute to Xanthomonas phytopathogenesis. Mol. Microbiol. 88, 1058–1069. doi: 10.1111/mmi.12229
Barak, J. D., and Schroeder, B. K. (2012). Interrelationships of food safety and plant pathology: the life cycle of human pathogens on plants. Annu. Rev. Phytopathol. 50, 241–266. doi: 10.1146/annurev-phyto-081211-172936
Barlow, J. J., Mathias, A. P., Williamson, R., and Gammack, D. B. (1963). A simple method for the quantitative isolation of undegraded high molecular weight ribonucleic acid. Biochem. Biophys. Res. Commun. 13, 61–66. doi: 10.1016/0006-291X(63)90163-3
Brandl, M. T. (2006). Fitness of human enteric pathogens on plants and implications for food safety. Annu. Rev. Phytopathol. 44, 367–392. doi: 10.1146/annurev.phyto.44.070505.143359
Buchholz, U., Bernard, H., Werber, D., Bohmer, M. M., Remschmidt, C., Wilking, H., et al. (2011). German outbreak of Escherichia coli O104:H4 associated with sprouts. N. Engl. J. Med. 365, 1763–1770. doi: 10.1056/NEJMoa1106482
Cathala, G., Savouret, J. F., Mendez, B., West, B. L., Karin, M., Martial, J. A., et al. (1983). A method for isolation of intact, translationally active ribonucleic acid. DNA 2, 329–335. doi: 10.1089/dna.1983.2.329
Chang, S., Puryear, J., and Cairney, J. (1993). A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 11, 113–116. doi: 10.1007/BF02670468
Cooley, M., Carychao, D., Crawford-Miksza, L., Jay, M. T., Myers, C., Rose, C., et al. (2007). Incidence and tracking of Escherichia coli O157:H7 in a major produce production region in California. PLoS ONE 2:e1159. doi: 10.1371/journal.pone.0001159
Fink, R. C., Black, E. P., Hou, Z., Sugawara, M., Sadowsky, M. J., and Diez-Gonzalez, F. (2012). Transcriptional responses of Escherichia coli K-12 and O157:H7 associated with lettuce leaves. Appl. Environ. Microbiol. 78, 1752–1764. doi: 10.1128/aem.07454-11
Fislage, R., Berceanu, M., Humboldt, Y., Wendt, M., and Oberender, H. (1997). Primer design for a prokaryotic differential display RT-PCR. Nucl. Acids Res. 25, 1830–1835. doi: 10.1093/nar/25.9.1830
Gehrig, H. H., Winter, K., Cushman, J., Borland, A., and Taybi, T. (2000). An improved RNA isolation method for succulent plant species rich in polyphenols and polysaccharides. Plant Mol. Biol. Rep. 18, 369–376. doi: 10.1007/BF02825065
Goudeau, D. M., Parker, C. T., Zhou, Y., Sela, S., Kroupitski, Y., and Brandl, M. T. (2013). The Salmonella transcriptome in lettuce and cilantro soft rot reveals a niche overlap with the animal host intestine. Appl. Environ. Microbiol. 79, 250–262. doi: 10.1128/aem.02290-12
Griffiths, R. I., Whiteley, A. S., O’Donnell, A. G., and Bailey, M. J. (2000). Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl. Environ. Microbiol. 66, 5488–5491. doi: 10.1128/AEM.66.12.5488-5491.2000
Hayashi, T., Makino, K., Ohnishi, M., Kurokawa, K., Ishii, K., Yokoyama, K., et al. (2001). Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8, 11–22. doi: 10.1093/dnares/8.1.11
Hernandez-Morales, A., De La Torre-Zavala, S., Ibarra-Laclette, E., Hernandez-Flores, J., Jofre-Garfias, A., Martinez-Antonio, A., et al. (2009). Transcriptional profile of Pseudomonas syringae pv. phaseolicola NPS3121 in response to tissue extracts from a susceptible Phaseolus vulgaris L. cultivar. BMC Microbiol. 9:257. doi: 10.1186/1471-2180-9-257
Holden, N., Pritchard, L., and Toth, I. (2009). Colonization outwith the colon: plants as an alternative environmental reservoir for human pathogenic enterobacteria. FEMS Microbiol. Rev. 33, 689–703. doi: 10.1111/j.1574-6976.2008.00153.x
Hou, Z., Fink, R. C., Black, E., Sugawara, M., Zhang, Z., Diez-Gonzalez, F., et al. (2012). Gene expression profiling of Escherichia coli in response to interactions with the lettuce rhizosphere. J. Appl. Microbiol. 113, 1076–1086. doi: 10.1111/j.1365-2672.2012.05412.x
Hou, Z., Fink, R. C., Sugawara, M., Diez-Gonzalez, F., and Sadowsky, M. J. (2013). Transcriptional and functional responses of Escherichia coli O157:H7 growing in the lettuce rhizoplane. Food Microbiol. 35, 136–142. doi: 10.1016/j.fm.2013.03.002
Jaakola, L., Pirttila, A. M., Halonen, M., and Hohtola, A. (2001). Isolation of high quality RNA from bilberry (Vaccinium myrtillus L.) fruit. Mol. Biotechnol. 19, 201–203. doi: 10.1385/mb:19:2:201
Jahn, C. E., Charkowski, A. O., and Willis, D. K. (2008). Evaluation of isolation methods and RNA integrity for bacterial RNA quantitation. J. Microbiol. Methods 75, 318–324. doi: 10.1016/j.mimet.2008.07.004
Kim, B.-H., Ramanan, R., Cho, D.-H., Choi, G.-G., La, H.-J., Ahn, C.-Y., et al. (2012). Simple, rapid and cost-effective method for high quality nucleic acids extraction from different strains of Botryococcus braunii. PLoS ONE 7:e37770. doi: 10.1371/journal.pone.0037770
Kyle, J. L., Parker, C. T., Goudeau, D., and Brandl, M. T. (2010). Transcriptome analysis of Escherichia coli O157:H7 exposed to lysates of lettuce leaves. Appl. Environ. Microbiol. 76, 1375–1387. doi: 10.1128/AEM.02461-09
Leite, G. M., Magan, N., and Medina, A. (2012). Comparison of different bead-beating RNA extraction strategies: an optimized method for filamentous fungi. J. Microbiol. Methods 88, 413–418. doi: 10.1016/j.mimet.2012.01.011
Mark, G. L., Dow, J. M., Kiely, P. D., Higgins, H., Haynes, J., Baysse, C., et al. (2005). Transcriptome profiling of bacterial responses to root exudates identifies genes involved in microbe-plant interactions. Proc. Natl. Acad. Sci. U.S.A. 102, 17454–17459. doi: 10.1073/pnas.0506407102
Matilla, M. A., Espinosa-Urgel, M., Rodriguez-Herva, J. J., Ramos, J. L., and Ramos-Gonzalez, M. I. (2007). Genomic analysis reveals the major driving forces of bacterial life in the rhizosphere. Genome Biol. 8, R179. doi: 10.1186/gb-2007-8-9-r179
Neidhardt, F. C., Bloch, P. L., and Smith, D. F. (1974). Culture medium for enterobacteria. J. Bacteriol. 119, 736–747.
Rossez, Y., Holmes, A., Wolfson, E. B., Gally, D. L., Mahajan, A., Pedersen, H. L., et al. (2013). Flagella interact with ionic plant lipids to mediate adherence of pathogenic Escherichia coli to fresh produce plants. Environ. Microbiol. doi: 10.1111/1462-2920.12315 [Epub ahead of print].
Roy, D., Panchal, S., Rosa, B. A., and Melotto, M. (2013). Escherichia coli O157:H7 induces stronger plant immunity than Salmonella enterica Typhimurium SL1344. Phytopathology 103, 326–332. doi: 10.1094/PHYTO-09-12-0230-FI
Schenk, A., Weingart, H., and Ullrich, M. S. (2008). Extraction of high-quality bacterial RNA from infected leaf tissue for bacterial in planta gene expression analysis by multiplexed fluorescent Northern hybridization. Mol. Plant Pathol. 9, 227–235. doi: 10.1111/j.1364-3703.2007.00452.x
Schikora, A., Carreri, A., Charpentier, E., and Hirt, H. (2008). The dark side of the salad: Salmonella typhimurium overcomes the innate immune response of Arabidopsis thaliana and shows an endopathogenic lifestyle. PLoS ONE 3:e2279. doi: 10.1371/journal.pone.0002279
Shidore, T., Dinse, T., Öhrlein, J., Becker, A., and Reinhold-Hurek, B. (2012). Transcriptomic analysis of responses to exudates reveal genes required for rhizosphere competence of the endophyte Azoarcus sp. strain BH72. Environ. Microbiol. 14, 2775–2787. doi: 10.1111/j.1462-2920.2012.02777.x
Soto-Suarez, M., Bernal, D., Gonzalez, C., Szurek, B., Guyot, R., Tohme, J., et al. (2010). In planta gene expression analysis of Xanthomonas oryzae pathovar oryzae, African strain MAI1. BMC Microbiol. 10:170. doi: 10.1186/1471-2180-10-170
Thilmony, R., Underwood, W., and He, S. Y. (2006). Genome-wide transcriptional analysis of the Arabidopsis thaliana interaction with the plant pathogen Pseudomonas syringae pv. tomato DC3000 and the human pathogen Escherichia coli O157:H7. Plant J. 46, 34–53. doi: 10.1111/j.1365-313X.2006.02725.x
Wright, K. M., Chapman, S., Mcgeachy, K., Humphris, S., Campbell, E., Toth, I. K., et al. (2013). The endophytic lifestyle of Escherichia coli O157:H7: quantification and internal localization in roots. Phytopathology 103, 333–340. doi: 10.1094/PHYTO-08-12-0209-FI
Keywords: food-borne pathogens, lettuce, spinach, rhizosphere, mRNA isolation
Citation: Holmes A, Birse L, Jackson RW and Holden NJ (2014) An optimized method for the extraction of bacterial mRNA from plant roots infected with Escherichia coli O157:H7. Front. Microbiol. 5:286. doi: 10.3389/fmicb.2014.00286
Received: 24 March 2014; Paper pending published: 27 April 2014;
Accepted: 23 May 2014; Published online: 10 June 2014.
Edited by:
Adam Schikora, IPAZ, JLU Giessen, GermanyReviewed by:
Saul Burdman, The Hebrew University of Jerusalem, IsraelCopyright © 2014 Holmes, Birse, Jackson and Holden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola J. Holden, Cell and Molecular Sciences, The James Hutton Institute, Invergowrie, Dundee, DD2 5DA, UK e-mail:bmljb2xhLmhvbGRlbkBodXR0b24uYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.