 Romain Talon
Romain Talon Nicolas Coquelle
Nicolas Coquelle Dominique Madern
Dominique Madern Eric Girard
Eric Girard
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 February 2014
Sec. Extreme Microbiology
Volume 5 - 2014 | https://doi.org/10.3389/fmicb.2014.00066
This article is part of the Research Topic Proceedings of Halophiles 2013: The international congress on halophilic microorganisms View all 27 articles
Protein-solvent interactions govern the behaviors of proteins isolated from extreme halophiles. In this work, we compared the solvent envelopes of two orthologous tetrameric malate dehydrogenases (MalDHs) from halophilic and non-halophilic bacteria. The crystal structure of the MalDH from the non-halophilic bacterium Chloroflexus aurantiacus (Ca MalDH) solved, de novo, at 1.7 Å resolution exhibits numerous water molecules in its solvation shell. We observed that a large number of these water molecules are arranged in pentagonal polygons in the first hydration shell of Ca MalDH. Some of them are clustered in large networks, which cover non-polar amino acid surface. The crystal structure of MalDH from the extreme halophilic bacterium Salinibacter ruber (Sr) solved at 1.55 Å resolution shows that its surface is strongly enriched in acidic amino acids. The structural comparison of these two models is the first direct observation of the relative impact of acidic surface enrichment on the water structure organization between a halophilic protein and its non-adapted counterpart. The data show that surface acidic amino acids disrupt pentagonal water networks in the hydration shell. These crystallographic observations are discussed with respect to halophilic protein behaviors in solution
Salinibacter ruber (Sr) is a halophilic bacterium that was isolated from saltern crystallizer ponds in Spain (Antón et al., 2002). In contrast to most bacterial species that equilibrate osmotic pressure with compatible solute, S. ruber accumulates high KCl concentration within its cytoplasm, an adaptive strategy similar to that of haloarchaea (Halobacteriaceae) (Oren, 2002). S. ruber genome sequence has revealed some interesting characteristics related to haloadaption: numerous lateral gene transfers from haloarchaea and a mean pI-value of 5.2 of its whole proteome (Mongodin et al., 2005). This proteomic pI shift toward low values, which is typical in haloarchaea, is the consequence of an enrichment of Asp and Glu residues and is considered an adaptive signature of proteins facing high salt concentration (Oren, 2013). However this explanation has been recently challenged by the characterization of a bacterium (Halorodospira) that does not accumulate high KCl concentration in its cytoplasm and has nonetheless a high acidic proteome (Deole et al., 2013). Among the few cytoplasmic enzymes isolated from S. ruber (Bonete et al., 2003; Madern and Zaccai, 2004), the tetrameric malate dehydrogenase (MalDH) remains the most extensively characterized, at the biochemical and structural level (Coquelle et al., 2010). As observed for non-halophilic counterparts, this halophilic enzyme does not require salt to maintain its conformational stability. However, the Sr MalDH structure revealed an acidic amino acids enriched surface, typical to that observed for a halophilic enzyme, which is responsible for a favorable change of solubility in high concentration of salts (Coquelle et al., 2010). According to the solvation-stabilization model for halophilic protein (Madern et al., 2000; reviewed in Zaccai, 2013), high salt concentrations exert a major selective pressure through a strong impact on protein solubility. In order to compete against this deleterious effect of salts, halophilic proteins stay highly soluble by maintaining a solvation envelope composition as close as possible as the composition of the bulk. This model is based on biophysical measurements that have shown that a halophilic protein recruits a solvation envelope of high ionic concentration (Costenaro et al., 2002; Ebel et al., 2002). In the solvation-stabilization model, surface acidic amino acids are suggested to be responsible for this particular solvent organization. Even if several structure of halophilic protein have been solved (Frolow et al., 1996; Richard et al., 2000; Bieger et al., 2003; Irimia et al., 2003; Zeth et al., 2004; Besir et al., 2005; Britton et al., 2006; Winter et al., 2009; Yamamura et al., 2009; Wende et al., 2010; Bracken et al., 2011), attempts to describe how the solvation shell of a halophilic protein interacts with acidic residues using X-rays crystallography is still a challenge.
In our follow-up crystallographic study on Sr MalDH (Coquelle et al., 2010); we determined the direct effect on solvent organization due to its acidic surface, by using a comparison with a non-halophilic counterpart. For this purpose, we solved de novo the crystal structure of the non-halophilic Chloroflexus aurantiacus (Ca) MalDH at 1.7 Å resolution. It allowed the determination of a hydration shell consisting in 945 water molecules, which cluster themselves in large networks of structured water through pentameric/hexameric polygons. Direct and indirect effects of acidic amino acids substitutions, avoiding the formation of structured water in Sr MalDH are described here through the comparison with Ca MalDH. The data are analyzed with respect to the solvation-stabilization model for halophilic protein. In particular, we underline that difference in hydration-solvation characteristics should always be kept in mind while analyzing the solvation layer of a halophilic protein, using X-ray crystallography, or any other techniques.
Ca MalDH overexpression was done accordingly to Dalhus et al. (2002). The cells were lysed by sonication in a 50 mM Tris-HCl buffered at pH 7. The crude extract was incubated for half an hour at 70°C and centrifugated for 15 min at 17,000 g. The soluble portion of the extract was loaded on a Q sepharose column equilibrated in 50 mM Tris-HCl buffer at pH 7. The protein was eluted using a linear gradient of 0–1 M NaCl. Fractions containing Ca MalDH were extensively dialyzed against 50 mM potassium phosphate buffer (pH 7) and deposited on a hydroxyapatite column equilibrated with the same buffer. The enzyme was eluted with a linear gradient of 50–1000 mM ammonium phosphate. The active fractions were pooled and concentrated by centrifugation using an Amicon PM30. They were deposited on a Sephacryl S300 gel filtration column (1 × 100 cm) and then eluted using an isocratic buffer of 50 mM Tris-HCl buffered at pH 7. The purified fractions were concentrated at 20 mg/ml and stored at 4°C.
Crystallization was performed by vapor diffusion using the hanging-drop method at 293 K. Native Ca MalDH crystals (≈500 × 400 × 400 μm3) were grown within 2 days by mixing 1.5 μL of 20 mg· mL−1 protein solution and 1.5 μL of 4–14% PEG 400, 100 mM sodium acetate buffer at pH 4.6 and 40 mM cadmium acetate reservoir solution. Ca MalDH derivative crystals were obtained by a 10 s soaking of a native crystal in a 2.0 μL solution equivalent to the mother liquor containing 100 mM of GdHPDO3A lanthanide complex (Girard et al., 2003). Then the crystal was quickly back-soaked in 2.0 μL of the corresponding reservoir solution without the lanthanide complex.
Prior to data collection, native and derivative crystals were cryo-cooled in liquid nitrogen using mother liquor containing 25% PEG 400 as cryo-protectant.
Gd-derivative data were collected on a Nonius FR591 X-Ray home source (1.541 Å). Native data were collected on the FIP-BM30A beamline at the ESRF (Grenoble, France) with the X-ray beam wavelength set to 0.979 Å. Diffraction frames were integrated using the program XDS (Kabsch, 2010) and the integrated intensities were scaled and merged using the CCP4 programs SCALA and TRUNCATE (Winn et al., 2011) respectively. A summary of the processing statistics is given in Table 1.
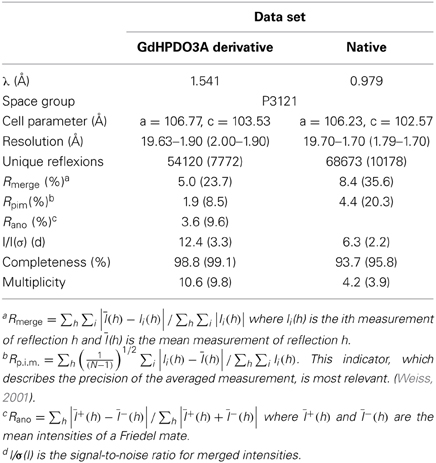
Table 1. Data collection and processing statistics.
Ca MalDH crystals belong to the P3121 space group with one A-D dimer per asymmetric unit leading to a solvent content of 49.5%.
Ca MalDH structure was determined de novo by the SIRAS (Single Isomorphous Replacement with Anomalous Scattering) method. As shown in Table 1, the high value of Rano clearly indicated the presence of GdHPDO3A complex binding sites, which was then confirmed by inspection of the anomalous Patterson map. Gadolinium positions were determined within the asymmetric unit using the program SHELXD (Sheldrick, 2010). Heavy-atom refinement and initial phasing were performed using the program SHARP (Bricogne et al., 2003). Phases from SHARP were improved by density modification using the CCP4 program DM (Cowtan and Main, 1996) leading to figures of merit of 0.235 and 0.793 after SHARP and density modification respectively. Automatic model building was performed with the program BUCCANEER (Cowtan, 2006) leading to an initial model consisting in 552 over the expected 618 A-D dimer residues.
The model was manually completed and improved in COOT (Emsley et al., 2010) prior to refinement with PHENIX (Adams et al., 2010). This model was then optimized through iterative rounds of refinement and model building. At the end stages of the refinement, TLS was used with TLS-groups determined with the TLSMD server (Brünger, 1992; Painter and Merritt, 2006a,b). The 1.7 Å resolution Ca MalDH final model consists in the complete (N-terminus, C-terminus and catalytic loop) residues sequence for each monomer of the Ca MalDH A-D dimer. The analysis of this final model (Table 2) showed no residues in disallowed regions of the Ramachandran plot (99.7% in preferred regions).

Table 2. Refinement statistics and model quality of the Ca MalDH structure.
In order to precisely assign the 945 water molecules in the model, we allowed the PHENIX program to automatically build solvent molecule up to 5.0 Å above the protein surface, with a distance of 1.7–3.0 Å between two water molecules or between a water molecules and the coordinated residue and only if the 1.0 σ contored 2Fo−Fc electron density map was interpretable. At the end, each water molecule was manually verified in COOT.
All the figures were made by using the Pymol program: The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC. All electrostatic calculations were performed using the Pymol plugin for APBS (Baker et al., 2001).
The structure of Ca MalDH enzyme was determined at 1.7 Å resolution using SIRAS phasing. The asymmetric unit contains a dimer, the physiological tetramer being generated by the crystal symmetry operators of the P3121 space group (Figure 1). This dimer delineated A-D will serve as the reference for all comparisons through this study. Our Ca MalDH model (4BGT) does not present major fold difference compared to the previously deposited (PDB accession code: 1GUY) structure (Dalhus et al., 2002), as confirmed by a root-mean-square deviation (RMSD) value of 0.42 Å for 594 A-D dimer superimposed residues. Moreover, the mobile loop (residues 83–89, following the linear numbering of 4BGT) covering the catalytic site, as well as the residues of the N- and C- termini have been modeled in each monomer of this new Ca MalDH structure. The detailed analysis of Ca MalDH fold and stabilization mechanism based on the 1GUY model has previously been published (Dalhus et al., 2002), and thus will not be further described in this study. The striking new feature in our model is the incredibly large number of modeled water molecules, i.e., 945 for the dimer A-D, which allows a detailed analysis of water organization.
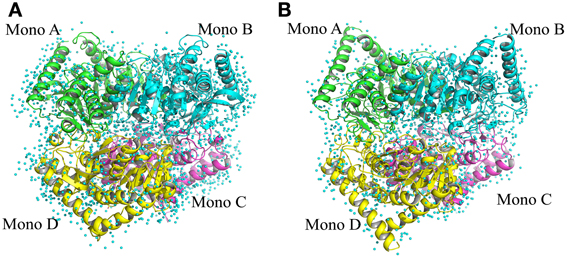
Figure 1. Ribbon drawing of tetrameric MalDH. (A) Chloroflexus aurantiacus MalDH. (B) Salinibacter ruber MalDH. Monomers are represented in four different colors. The water molecules are displayed by small blue spheres.
Sr MalDH shares more than 72% of sequence similarity with its non-halophilic counterpart Ca MalDH. The Sr MalDH model was obtained at a resolution of 1.55 Å, and also contains a large number of water molecules: 680 for the equivalent Ca MalDH A-D dimer (Coquelle et al., 2010). The overall structural similarity between one monomer of Sr and Ca MalDHs led to a RMSD of about 0.6 Å for 258 superimposed Cα.
Therefore, these two structures of excellent resolution, with a large number of water molecules in their solvent layers, provide a unique combination to finely compare the water organization at their surface.
A detailed analysis of the geometry of the 945 water molecules surrounding the dimeric Ca MalDH model (distance and angle) was performed and is presented in Table 3. It is outside the scope of this study to describe in great details both the geometry and interactions with the protein of all these water molecules. The role of water molecules in the folding process and stabilization of proteins has been well described in a work based on a larger set of proteins (Matsuoka and Nakasako, 2009). The most interesting feature of the water molecules in Ca MalDH structure is that 28% of them are organized in polygons (pentagons or hexagons), which can form extended clusters (Figure 2). These polygons are only observed at the surface of apolar residues. Geometrical properties of these polygons (Table 3) are in good agreement with those determined from a large statistical study using high-resolution structures (Lee and Kim, 2009).
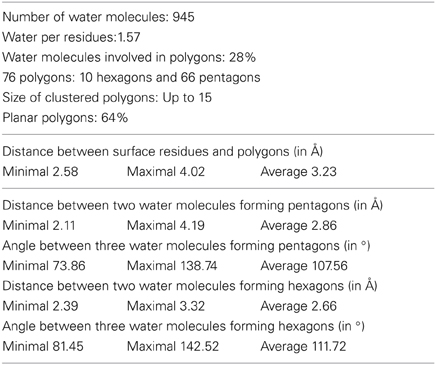
Table 3. Water statistics for dimer AD of Ca MalDH.
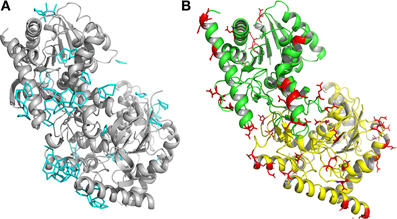
Figure 2. (A) Ribbon drawing of monomers A and D of Ca MalDH. The water polygons are represented in blue lines. (B) Ribbon drawing of monomer A (green) and monomer D (yellow) of Sr MalDH. The surface acidic amino acid is shown in red.
Based on Ca MalDH water analysis, a careful inspection of the halophilic Sr MalDH hydration layer at the surface of the protein was performed to detect any water polygon. Even though 43% of Sr MalDH water molecules were considered to be superimposable with those from Ca MalDH (using a cut off distance of 1.5 Å), no polygons were observed at the surface of the halophilic MalDH. However, 14 water molecules lie in the catalytic pocket of Sr MalDH, all of which are conserved in Ca MalDH. Five are organized as a pentagon, the only one observed in Sr MalDH (Figure 3). In Ca MalDH, the same water pentagon is present, but the catalytic pocket of Ca MalDH contains an extra water molecule, which closes a second pentagon in the catalytic pocket, adjacent to the first one (Figure 3A). A black arrow indicates the missing water molecule in Sr MalDH (Figure 3B).
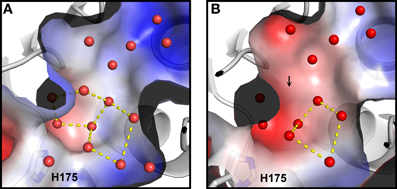
Figure 3. Close up views of the catalytic pocket. Electrostatic surface representation of Ca MalDH (A) and Sr MalDH (B). Water molecules are shown in small red spheres. Dashed lines cultured in yellow delineates the polygons. The catalytic histidine (H175) is indicated. Numbering of amino acids corresponds to linear numbering of Ca MalDH.
We therefore decided to have a closer look at surface regions where polygons are present in Ca MalDH to figure out the reasons why none are observed in Sr MalDH.
As mentioned, large networks of connected water polygons are present in Ca MalDH (Figure 2A). An example of such network is shown in Figure 4A. This network is anchored between helices α1G-α1G and αH and is made up of five pentagons and one hexagon. In the same protein region, no water polygon is observed in Sr MalDH (Figure 4B), which possesses four extra negative charges compared to Ca MalDH, due to substitutions at positions 199, 203, 283, and 285. These substitutions led to important electrostatic surface changes, with a highly negative one for Sr MalDH compared to the apolar surface of Ca MalDH (Figures 4C,D). The lateral chain of acidic residues D287 in Sr MalDH is orientated in such a conformation that the Sr MalDH hydration pattern is modified when compared to that of Ca MalDH. The data suggest that the replacement of non-polar amino acid residues by acidic amino acid in a halophilic protein modifies properties of the hydration shell. Around apolar surfaces of the non-halophilic MalDH, water molecules cannot form direct hydrogen bonds with the protein, and thus organize themselves as polygons with their nearest stable water neighbors. Acidic amino acids enrichment in these regions of Sr MalDH surface favors direct hydrogen bonding with water and therefore prevents polygons formation.
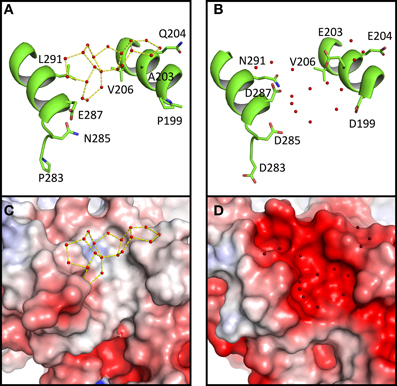
Figure 4. Close up views of Ca MalDH (A) and Sr MalDH (B). Water molecules are shown in small red spheres. Dashed lines coloured in yellow delineates the polygons. Important amino acids are represented in sticks. Electrostatic surface representation of Ca MalDH (C) and Sr MalDH (D).
We also observe that water polygons formation is hampered in halophilic Sr MalDH, not only by direct acidic amino acid substitution but also by the side chain reorganization of conserved residues, as illustrated in Figure 5. In Sr MalDH compared to Ca MalDH, two acidic amino acids are observed at position 158 and 204. Glutamate at position 158 induces a direct perturbation of water pentagon P1, as previously observed. But Glutamate 204 promotes an interaction with R201 side chain, which moved to a new position that hinders appropriate hydrogen bonding geometries requested for the formation of water polygon P2 (Figure 5).
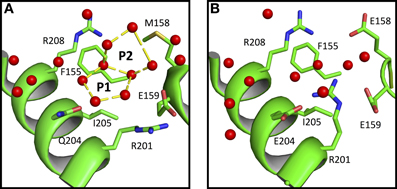
Figure 5. Close up view of Ca MalDH (A) and Sr MalDH (B). Water molecules are in red spheres. Linear numbering as in Ca MalDH.
These two examples clearly illustrate the key influence of acidic amino acid enrichment in halophilic protein on the water organization at their surface; either through direct impacts or via conformational rearrangements of surrounding residues. This leads to the destabilization of almost all water polygons observed in the non-halophilic protein structure.
This study presents for the first time a detailed analysis of the water organization at the surface of a halophilic protein and its non-halophilic counterpart. Both crystal structures were obtained at high resolution (better than 1.7 Å) and display similar crystallographic quality. The comparison of these hydration envelopes shows the effect of surface composition changes on the hydration shell structure. In the structure of Ca MalDH, we observed a large amount of stable water polygons. These specific water arrangements were first observed in the crystal structure of Crambin (Teeter, 1984). It has been analyzed that these water are not the results of crystallization process and are likely due to intrinsic interaction mode with the local hydrophobic surface of proteins (Nakasako, 1999, 2004). Water organization observed in Ca MalDH is in good agreement as apolar surface prevent direct hydrogen bonding of water molecules with the protein and favors polygonal structures. Acidic residues substitutions at the surface of Sr MalDH promote hydrogen bonding between the solvent and the protein. In particular, we observed that the changes in water structure organization in Sr MalDH are not only due to direct effects but also to long-range effects of amino acid substitutions. The latter is an indirect consequence of amino acids substitutions, selected to increase the Sr MalDH enzymatic activity at high salt concentration as analyzed in our previous work (Coquelle et al., 2010). Indeed, these changes modify the local dynamics of the protein surface, which should impact the dynamical properties of the nearest hydration water molecules, as previously observed (Nakasako et al., 2001).
At this stage, it is important to remind the concept of solvation/hydration of proteins.
This is an important issue that should be discussed. Because of the chemical properties of the protein surface, the solvent composition at the vicinity of a given protein surface is different from the bulk. In a simple binary system containing water and protein without any cosolvents, such as salt or other macromolecular solutes, a hydration shell surrounds the protein. In the presence of high concentration of additional compounds such as salts, sugars, precipitating agents etc., the protein solution should be described as a ternary system in which the protein is enveloped by a solvation shell. The thermodynamics of proteins in the three-component system is well understood in terms of preferential binding parameters (Von Hippel and Schleich, 1969; Inoue and Timasheff, 1972; Arakawa and Timasheff, 1982; Zaccai and Eisenberg, 1990; Timasheff, 1991; reviewed in Zaccai, 2013). In conditions that maintain protein solubility, the chemical potential of the solvation shell and the bulk are equilibrated (Figure 6). In salting-out conditions that favor protein aggregation and crystallization, the equilibrium is strongly perturbed because the small solutes are excluded from the solvation shell (Tardieu et al., 2002). In this case the solvation shell looks like a hydration shell.
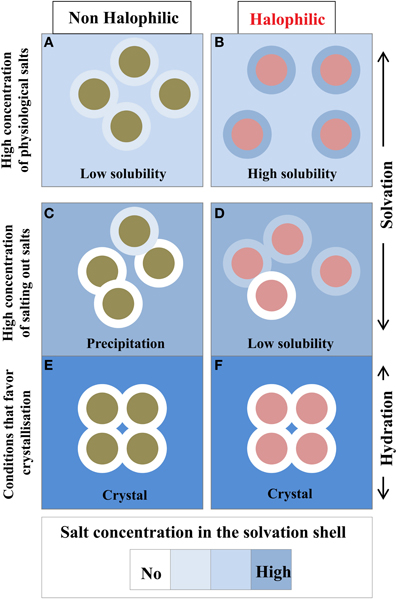
Figure 6. Compositional changes of the solvation layer between non-halophilic and halophilic proteins. Filled circles either represent non-halophilic (Green) or halophilic (Red) proteins with their solvation shell (external circle). Solubility measurements are taken from Coquelle et al. (2010). With the halophilic protein, due to its acidic enriched surface, the dominant inter particular effect is repulsive and its solvation shell composition is similar to the bulk solvent (B). These two effects strongly favor high solubility even in high physiological salt concentration. In equivalent conditions the non-halophilic protein solubility is reduced (A). In non-physiological conditions, i.e., in the presence of salting out salts (C,D) or additives that promote crystallization (E,F), the solvation shell of each enzyme starts to be depleted in salt. This situation impacts the solubility of each enzyme to diverse extent and could promote precipitation. In crystal conditions, the solvation envelope is a hydration shell, which contains none or very few ions.
Cytoplasmic protein isolated from extreme halophilic prokaryotes that use the KCl-in adaptive strategy, such as S. ruber or the Halobacteriaceae, maintain a high solubility at molar concentration of various salt (Coquelle et al., 2010). In the case of the tetrameric MalDH from Haloarcula marismortui, the measurements of the preferential binding parameters have shown that the enzyme obey the general thermodynamics rules of the three components system (Costenaro et al., 2002; Ebel et al., 2002): In salting out conditions, the solvation envelope of Hm MalDH is strongly depleted in salt and it looks like a hydration shell; such behaviors is equivalent to the situation encountered with a non-halophilic protein. However, in high concentration of various physiological salts, it has been measured that Hm MalDH preferential binding parameters depend on salt type, demonstrating that the composition of its solvation shell varies (Costenaro et al., 2002; Ebel et al., 2002). In these physiological salts, Hm MalDH solvation envelope is enriched in salt, reflecting its halophilic adaptation. Consequently, as the chemical potential of the solvation layer and the bulk solvent are close, Hm MalDH remains highly soluble at high salt concentration. We determined that Sr MalDH remains highly soluble in high concentration of physiological salts (Coquelle et al., 2010). Based on the observation made on Hm MalDH, this suggests that Sr MalDH solvation layer should also be enriched with salts.
The relationship between an increase in protein solubility and the shift toward negatively charged protein surfaces is not restricted to halophilic protein (Trevino et al., 2007). The favorable effect of acidic residues on protein solubility has been highlighted by an elegant thermodynamical work based on seven non-halophilic proteins, which displayed pIs ranging from 3.5 to 8 (Kramer et al., 2012). In the case of halophilic proteins, it has been demonstrated that their high negative charge density maintains a weak repulsive protein-protein interactions in high salt concentration (Costenaro et al., 2002; Ebel et al., 2002). Theoretically, this repulsive effect between macromolecules of same net charge could also be induced by positively charged amino acids. However, calculation of the solvent-accessible areas of the side-chain components between negatively and positively charged residues unravel their relative efficiency on solubility. Compared to positively charged residues, the favorable effect of acidic residues is due to the lower hydrophobic solvent exposed surface of their side chain (Britton et al., 1998).
Our comparative study of the hydration shell of Ca and Sr MalDHs sheds light on the close relation between solubility, acidic residue enrichment and solvation.
First, conclusions from the solvent properties analysis using X-ray crystallography should be drawn with cautiousness. Our observation confirms that salting-out conditions deplete the solvation layer in salts. Indeed, the additives used for crystal growth shift the conditions toward salt depletion in the solvation envelope. Consequently, even if several halophilic proteins have been crystallized in the presence of high concentrations of physiological salts, one should take precaution when discussing the role of the solvent layer as obtained from X-ray structures.
Second, the favorable change in solubility of halophilic proteins is driven by their protein surface enrichment in acidic residues, which plays a dual role. Indeed, our study shows that acidic residues, through their carboxyl groups that are known to form strong hydrogen bonds, can organize the solvation shell by direct as well as indirect interactions. They are therefore good candidates for interactions with hydrated salt ions as proposed by Zaccai (2013). Moreover, they promote slightly repulsive inter-particular interactions between each protein molecule, favoring solubility.
Recent data have suggested that acidic enrichment, considered as an adaptive signature of halophilic proteins, could also be due to genetic drift (Deole et al., 2013). Whatever the precise evolutionary mechanism responsible for the peculiar composition of protein isolated from halophilic microorganisms, our work helps to understand that acidic acid enrichment was an appropriate evolutionary innovation in the case of microorganisms that accumulates high concentration of KCl in their cytoplasm to maintain their turgor pressure in highly salted environment. Such enrichment allows halophilic proteins to compete against aggregation via their ability to reorganize protein-solvent interactions.
The role of acidic amino acids substitution on the solvent organization, highlighted in the present work, has to be completed by further studies involving enzymes from halophilic organisms that used different strategies to cope with high concentration of salts.
Dominique Madern and Eric Girard designed research, Romain Talon, Nicolas Coquelle, Dominique Madern, and Eric Girard performed research, Romain Talon, Nicolas Coquelle, Dominique Madern, and Eric Girard were involved in data analysis. Romain Talon, Nicolas Coquelle, Dominique Madern, and Eric Girard wrote the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by the Agence Nationale de la Recherche Grants “Ln23” ANR-13-BS07-0007-02. Romain Talon and Eric Girard also thank scientists of the FIP-BM30A beamline at the European Synchrotron radiation Facility (ESRF) for their help.
Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. doi: 10.1107/S0907444909052925
Antón, J., Oren, A., Benlloch, S., Rodríguez-Valera, F., Amann, R., and Rosselló-Mora, R. (2002). Salinibacter ruber gen. nov., sp. nov., a novel, extremely halophilic member of the Bacteria from saltern crystallizer ponds. Int. J. Syst. Evol. Microbiol. 52, 485–491. doi: 10.1186/1746-1448-4-15
Arakawa, T., and Timasheff, S. N. (1982). Preferential interactions of proteins with salts in concentrated solutions. Biochemistry 21, 6545–6552. doi: 10.1021/bi00268a034
Baker, N. A., Sept, D., Joseph, S., Holst, M. J., and McCammon, J. A. (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041. doi: 10.1073/pnas.181342398
Besir, H., Zeth, K., Bracher, A., Heider, U., Ishibashi, M., Tokunaga, M., et al. (2005). Structure of a halophilic nucleoside diphosphate kinase from Halobacterium salinarum. FEBS Lett. 579, 6595–6600. doi: 10.1016/j.febslet.2005.10.052
Bieger, B., Essen, L. O., and Oesterhelt, D. (2003). Crystal structure of halophilic dodecin. A novel dodecameric flavin binding protein from Halobacterium salinarum. Structure 4, 375–385. doi: 10.1016/S0969-2126(03)00048-0
Bonete, M. J., Pérez-Pomares, F., Diaz, S., Ferrer, J., and Oren, A. (2003). Occurrence of two different glutamate dehydrogenase activities in the halophilic bacterium Salinibacter ruber. FEMS Microbiol. Lett. 226, 181–186. doi: 10.1016/S0378-1097(03)00592-5
Bracken, C. D., Neighbor, A. M., Lamlenn, K. K., Thomas, G. C., Schubert, H. L., Whitby, F. G., et al. (2011). Crystal structures of a halophilic archaeal malate synthase from Haloferax volcanii and comparisons with isoforms A and G. BMC Struct. Biol. 11:23. doi: 10.1186/1472-6807-11-23
Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M., and Paciorek, W. (2003). Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 59, 2023–2030. doi: 10.1107/S0907444903017694
Britton, K. L., Baker, P. J., Fisher, M., Ruzheinikov, S., Gilmour, D. J., Bonete, M. J. et al. (2006). Analysis of protein solvent interactions in glucose dehydrogenase from the extreme halophile Haloferax mediterranei. Proc. Natl. Acad. Sci. U.S.A. 103, 4846–4851. doi: 10.1073/pnas.0508854103
Britton, K. L., Stillman, T. J., Yip, K. S., Forterre, P., Engel, P. C., and Rice, D. W. (1998). Insights into the molecular basis of salt tolerance from the study of glutamate dehydrogenase from Halobacterium salinarum. J. Biol. Chem. 273, 9023–9030. doi: 10.1074/jbc.273.15.9023
Brünger, A. T. (1992). Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. doi: 10.1038/355472a0
Coquelle, N., Talon, R., Juers, D. H., Girard, E., Kahn, R., and Madern, D. (2010). Gradual adaptive changes of a protein facing high salt concentrations. J. Mol. Biol. 404, 493–505. doi: 10.1016/j.jmb.2010.09.055
Costenaro, L., Zaccai, G., and Ebel, C. (2002). Link between protein–solvent and weak protein–protein interactions gives insight into halophilic adaptation. Biochemistry 41, 13245–13252. doi: 10.1021/bi025830z
Cowtan, K. D. (2006). The Buccaneer software for automated model building. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011. doi: 10.1107/S0907444906022116
Cowtan, K. D., and Main, P. (1996). Phase combination and cross validation in iterated density modification calculations. Acta Crystallogr. D Biol. Crystallogr. 52, 43–48. doi: 10.1107/S090744499500761X
Dalhus, B., Saarinen, M., Sauer, U. H., Eklund, P., Johansson, K., Karlsson, A., et al. (2002). Structural basis for thermophilic protein stability: structures of thermophilic and mesophilic malate dehydrogenases. J. Mol. Biol. 318, 707–721. doi: 10.1016/S0022-2836(02)00050-5
Deole, R., Challacombe, J., Raiford, D. W., and Hoff, W. D. (2013). An extremely halophilic proteobacterium combines a highly acidic proteome with a low cytoplasmic potassium content. J. Biol. Chem. 288, 581–588. doi: 10.1074/jbc.M112.420505
Ebel, C., Costenaro, L., Pascu, M., Faou, P., Kernel, B., Proust-De Martin, F., et al. (2002). Solvent interactions of halophilic malate dehydrogenase. Biochemistry 41, 13234–13244. doi: 10.1021/bi0258290
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. D. (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. doi: 10.1107/S0907444910007493
Frolow, F., Harel, M., Sussman, J. L., Mevarech, M., and Shoham, M. (1996). Insights into protein adaptation to a saturated salt environment from the crystal structure of a halophilic 2Fe-2S ferredoxin. Nat. Struct. Biol. 3, 452–458. doi: 10.1038/nsb0596-452
Girard, E., Stelter, M., Anelli, P. L., Vicat, J., and Kahn, R. (2003). A new class of gadolinium complexes employed to obtain high-phasing-power heavy-atom derivatives: results from SAD experiments with hen egg-white lysozyme and urate oxidase from Aspergillus flavus. Acta Crystallogr. D Biol. Crystallogr. 59, 118–126. doi: 10.1107/S0907444902020176
Inoue, H., and Timasheff, S. N. (1972). Preferential and absolute interactions of solvent components with proteins in mixed solvent systems. Biopolymers 11, 737–743. doi: 10.1002/bip.1972.360110402
Irimia, A., Ebel, C., Madern, D., Richard, S. B., Cosenza, L. W., Zaccai, G., et al. (2003). The Oligomeric states of Haloarcula marismortui malate dehydrogenase are modulated by solvent components as shown by crystallographic and biochemical studies. J. Mol. Biol. 325, 859–873. doi: 10.1016/S0022-2836(02)01450-X
Kabsch, E. (2010). XDS. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132. doi: 10.1107/S0907444909047337
Kramer, R. M., Shende, V. R., Motl, N., Pace, C. N., and Scholtz, J. M. (2012). Toward a molecular understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophys J. 102, 1907–1915. doi: 10.1016/j.bpj.2012.01.060
Lee, J., and Kim, S. H. (2009). Water polygons in high-resolution protein crystal structures. Protein Sci. 18, 1370–1376. doi: 10.1002/pro.162
Madern, D., Ebel, C., and Zaccai, G. (2000). Halophilic adaptation of enzymes. Extremophiles 4, 91–98. doi: 10.1007/s007920050142
Madern, D., and Zaccai, G. (2004). Molecular adaptation: the malate dehydrogenase from the extreme halophilic bacterium Salinibacter ruber behaves like a non-halophilic protein. Biochimie 86, 295–303. doi: 10.1016/j.biochi.2004.04.004
Matsuoka, D., and Nakasako, M. (2009). Probability distributions of hydration water molecules around polar protein atoms obtained by a database analysis. J. Phys. Chem. B 113, 11274–11292. doi: 10.1021/jp902459n
Mongodin, E. F., Nelson, K. E., Daugherty, S., DeBoy, R. T., Wister, J., Khouri, H., et al. (2005). The genome of Salinibacter ruber: convergence and gene exchange among hyperhalophilic bacteria and archaea. Proc. Natl. Acad. Sci. U.S.A. 102 18147–18152. doi: 10.1073/pnas.0509073102
Nakasako, M. (1999). Large-scale networks of hydration water molecules around bovine beta-trypsin revealed by cryogenic X-ray crystal structure analysis. J. Mol. Biol. 289, 547–564. doi: 10.1006/jmbi.1999.2795
Nakasako, M. (2004). Water-protein interactions from high-resolution protein crystallography. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 1191–1204. doi: 10.1098/rstb.2004.1498
Nakasako, M., Fujisawa, T., Adachi, S., Kudo, T., and Higuchi, S. (2001). Large-scale domain movements and hydration structure changes in the active-site cleft of unligated glutamate dehydrogenase from Thermococcus profundus studied by cryogenic X-ray crystal structure analysis and small-angle X-ray scattering. Biochemistry 40, 3069–3079. doi: 10.1021/bi002482x
Oren, A. (2002). Halophilic Microorganisms and their Environments. Dordrecht: Kluwer Scientific Publishers. doi: 10.1007/0-306-48053-0
Oren, A. (2013). Life at high salt concentrations, intracellular KCl concentrations, and acidic proteomes. Front. Microbiol. 4:315. doi: 10.3389/fmicb2013.00315
Painter, J., and Merritt, E. A. (2006a). Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D. Biol. Crystallogr. 62, 439–450. doi: 10.1107/S0907444906005270
Painter, J., and Merritt, E. A. (2006b). TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 39, 109–111. doi: 10.1107/s0021889805038987
Richard, S. B., Madern, D., Garcin, E., and Zaccai, G. (2000). Halophilic adaptation: novel solvent protein interactions observed in the 2.9 and 2.6 Å resolution structures of the wild type and a mutant of malate dehydrogenase from Haloarcula marismortui. Biochemistry 39, 992–1000. doi: 10.1021/bi991001a
Sheldrick, G. M. (2010). Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485. doi: 10.1107/S0907444909038360
Tardieu, A., Bonneté, F., Finet, D., and Vivares, D. (2002). Understanding salt or PEG induced attractive interactions to crystallize biological macromolecules. Acta Crystallogr. D Biol. Crystallogr. 58, 1549–1553. doi: 10.1107/S0907444902014439
Teeter, M. M. (1984). Water structure of a hydrophobic protein at atomic resolution: pentagon rings of water molecules in crystals of crambin. Proc. Natl. Acad. Sci. U.S.A. 81, 6014–6018. doi: 10.1073/pnas.81.19.6014
Timasheff, S. N. (1991). Solvent effect on protein stability. Curr. Opin. Struct. Biol. 2, 35–39. doi: 10.1016/0959-440X(92)90173-5
Trevino, S. R., Scholtz, J. M., and Pace, C. N. (2007). Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favourably than the other hydrophilic amino acids in RNase Sa. J. Mol. Biol. 366, 449–446. doi: 10.1016/j.jmb.2006.10.026
von Hippel, P. H., and Schleich, T. W. (1969). “The effects of neutral salts on the structure and conformational stability of macromolecules in solution,” in Biological Macromolecules, Vol. II, eds G. D. Fasman and S. N. Timasheff (New York, NY: Marcel Dekker Inc.), 417–584.
Weiss, M. S. (2001). Global indicators of X-ray data quality. J. Appl. Crystallogr. 34, 130–135. doi: 10.1107/S0021889800018227
Wende, A., Johansson, P., Vollrath, R., Dyall-Smith, M., Oesterhelt, D., and Grininger, M. (2010). Structural and biochemical characterization of a halophilic archaeal alkaline phosphatase. J. Mol. Biol. 400, 52–62. doi: 10.1016/j.jmb.2010.04.057
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta. Crystallogr. D Biol. Crystallogr. 67, 235–242. doi: 10.1107/S0907444910045749
Winter, J. A., Christofi, P., Morroll, S., and Bunting, K. A. (2009). The crystal structure of Haloferax volcanii proliferating cell nuclear antigen reveals unique surface charge characteristics due to halophilic adaptation. BMC Struct. Biol. 9:55 doi: 10.1186/1472-6807-9-55
Yamamura, A., Ichimura, T., Kamekura, M., Mizuki, T., Usami, R., Makino, T., et al. (2009). Molecular mechanism of distinct salt-dependent enzyme activity of two halophilic nucleoside diphosphate kinases. Biophys. J. 96, 4692–700. doi: 10.1016/j.bpj.2009.03.012
Zaccai, G. (2013). Hydration shells with a pinch of salt. Biopolymers 4, 233–238. doi: 10.1002/bip.22154
Zaccai, G., and Eisenberg, H. (1990). Halophilic proteins and the influence of solvent on protein stabilization. Trends Biochem. Sci. 15, 333–337. doi: 10.1016/0968-0004(90)90068-M
Keywords: halophilic, solvation, hydration, water pentagon, malate dehydrogenase, acidic proteins, adaptation, salinibacter
Citation: Talon R, Coquelle N, Madern D and Girard E (2014) An experimental point of view on hydration/solvation in halophilic proteins. Front. Microbiol. 5:66. doi: 10.3389/fmicb.2014.00066
Received: 30 December 2013; Accepted: 04 February 2014;
Published online: 21 February 2014.
Edited by:
R. Thane Papke, University of Connecticut, USAReviewed by:
Aharon Oren, The Hebrew University of Jerusalem, IsraelCopyright © 2014 Talon, Coquelle, Madern and Girard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominique Madern and Eric Girard, Equipe ELMA, Institut de Biologie Structurale, 6 Rue Jules Horowitz, 38000 Grenoble, France e-mail:ZG9taW5pcXVlLm1hZGVybkBpYnMuZnI=;ZXJpYy5naXJhcmRAaWJzLmZy
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.