




- 1Department of Microbiology, Institute for Water and Wetland Research, Radboud University Nijmegen, Nijmegen, Netherlands
- 2Department of Molecular Biology NCMLS, Radboud University Nijmegen, Nijmegen, Netherlands
- 3Department of Microbiology and Microbial Engineering, School of Life Sciences, Fudan University, Shanghai, China
- 4Department of Biotechnology, Delft University of Technology, Delft, Netherlands
Anaerobic ammonium-oxidizing (anammox) bacteria are key players in the global nitrogen cycle and responsible for significant global nitrogen loss. Moreover, the anammox process is widely implemented for nitrogen removal from wastewaters as a cost-effective and environment-friendly alternative to conventional nitrification-denitrification systems. Currently, five genera of anammox bacteria have been identified, together forming a deep-branching order in the Planctomycetes-Verrucomicrobium-Chlamydiae superphylum. Members of all genera have been detected in wastewater treatment plants and have been enriched in lab-scale bioreactors, but genome information is not yet available for all genera. Here we report the metagenomic analysis of a granular sludge anammox reactor dominated (∼50%) by “Candidatus Jettenia asiatica.” The metagenome was sequenced using both Illumina and 454 pyrosequencing. After de novo assembly 37,432 contigs with an average length of 571 nt were obtained. The contigs were then analyzed by BLASTx searches against the protein sequences of “Candidatus Kuenenia stuttgartiensis” and a set of 25 genes essential in anammox metabolism were detected. Additionally all reads were mapped to the genome of an anammox strain KSU-1 and de novo assembly was performed again using the reads that could be mapped on KSU-1. Using this approach, a gene encoding copper-containing nitrite reductase NirK was identified in the genome, instead of cytochrome cd1-type nitrite reductase (NirS, present in “Ca. Kuenenia stuttgartiensis” and “Ca. Scalindua profunda”). Finally, the community composition was investigated through MetaCluster analysis, 16S rRNA gene analysis and read mapping, which showed the presence of other important community members such as aerobic ammonia-oxidizing bacteria, methanogens, and the denitrifying methanotroph “Ca. Methylomirabilis oxyfera”, indicating a possible active methane and nitrogen cycle in the bioreactor under the prevailing operational conditions.
Introduction
Two microbial processes are responsible for the release of fixed nitrogen: denitrification and anaerobic ammonium oxidation (anammox). Anammox bacteria oxidize ammonia to dinitrogen gas under anaerobic conditions with nitrite as the electron acceptor. The anammox process is widely applied for wastewater treatment and has economical and environmental advantages over the conventional nitrogen removal processes nitrification and denitrification (Jetten et al., 1997; Van Dongen et al., 2001; Siegrist et al., 2008; Kartal et al., 2010). Furthermore, anammox bacteria are important players in the global nitrogen cycle, and widely distributed in various ecosystems (citations). It is now estimated that they contribute significantly to global nitrogen loss (Thamdrup and Dalsgaard, 2002; Kuypers et al., 2005; Hamersley et al., 2007; Humbert et al., 2010).
Until now five genera of anammox bacteria have been identified: “Candidatus Brocadia”, “Candidatus Kuenenia”, “Candidatus Scalindua”, “Candidatus Anammoxoglobus”, and “Candidatus Jettenia”, together forming a monophyletic order Brocadiales that ranches deeply in the phylum Planctomycetes (Jetten et al., 2010). Various aspects of the anammox bacteria, such as growth and metabolism, biochemistry and bio-energetics, cell biology, application, and environmental importance, have been addressed in various studies (reviewed in Jetten et al., 2009; van Niftrik and Jetten, 2012). Using environmental shotgun sequencing, which is used to retrieve microbial genomes or genomic fragments from complicated environmental samples, the first metagenome of an anammox bacterium was sequenced from an enrichment culture (∼75%) of “Candidatus Kuenenia stuttgartiensis” (hereafter: Kuenenia; Strous et al., 2006). Based on this metagenome, the central metabolic pathways of anammox bacteria were predicted and resolved (Strous et al., 2006; de Almeida et al., 2011; Kartal et al., 2011). Based on these findings, the anammox catabolism may be divided into three steps: the reduction of nitrite to NO catalyzed by a cd1 nitrite: nitric oxide oxidoreductase (NirS); the conversion of equimolar amounts of NO, and ammonium to hydrazine catalyzed by hydrazine synthase (HZS), and finally the oxidation of hydrazine to dinitrogen gas catalyzed by hydrazine dehydrogenase (HDH; de Almeida et al., 2011; Figure 1).
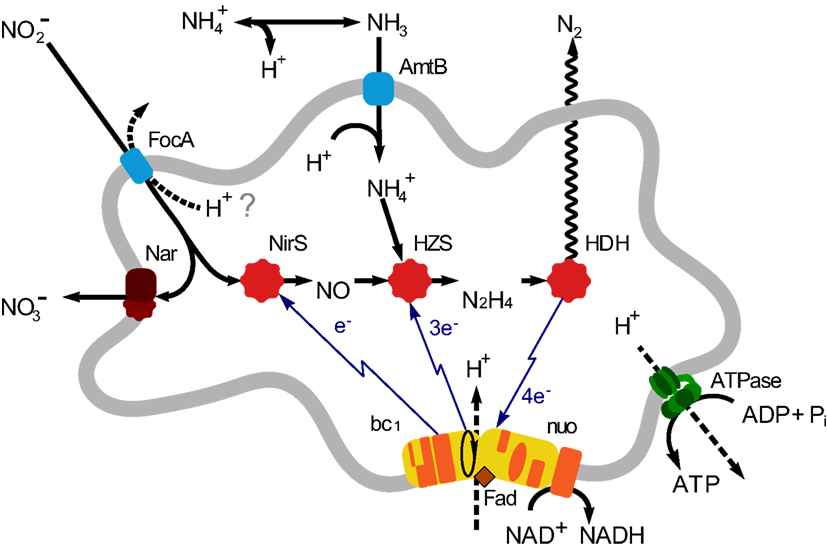
Figure 1. Overview of anammox metabolism in “Candidatus Jettenia asiatica”. AmtB, ammonium transport protein; FocA, Nitrite transporter; nitrite/formate transporter; NirS, Nitrite reductase; Nar, Nitrate reductase; HZS, Hydrazine synthesis; HDH, hydrazine dehydrogenase; bc1, cytochrome bc1 complex; Nuo, NADH ubiquinone oxidoreductase.
However, the metabolic processes described above and the proteins catalyzing these were predicted based on the genome of Kuenenia. Recently, the genome sequences of three more anammox species were reported; a marine species “Candidatus Scalindua profunda” (hereafter: Scalindua), a waste water species KSU-1, and a freshwater species “Candidatus Brocadia fulgida” (Hereafter: Brocadia) and all of them showed differences to Kuenenia (Gori et al., 2011; Hira et al., 2012; Van de Vossenberg et al., 2012). This indicated that genomic analyses of other anammox bacteria could contribute to a more comprehensive understanding of the metabolic features of these extraordinary microorganisms. To this end, a metagenome analysis was performed on an anammox enrichment dominated by Candidatus “Jettenia asiatica” (hereafter: Jettenia). Important anammox genes in the dataset were identified and compared to those in the genomes of Kuenenia and the anammox strain KSU-1 (Hira et al., 2012).
Materials and Methods
Metagenomic Sequencing and Assembly
Bulk community DNA was extracted from the granular sludge of an anaerobic bioreactor. The sludge was dominated by Jettenia (∼50%) as determined by 16S rRNA gene quantitative PCR assays (Quan et al., 2008). The community DNA was isolated and sequenced using Illumina and 454 pyrosesquencing, generating 16,296,896 reads with an average length of 33.6 nt and 162,543 reads with an average length of 215 nt respectively. De novo assembly was performed with the CLC genomics workbench (v. 5.1; CLCbio) using default setting (Word size: 22; Bubble size: 50; Minimum contig length: 200; Insertion: 3; Deletion cost: 2). After assembly, ∼33% of the reads were assembled into 37,432 contigs with an average length of 570 nt, about 1/3 of the contigs were longer than 500 nt and 2,549 contigs were longer than 1000 nt, the longest being 4.3 kb.
The contigs had a highly diverse GC content and coverage and many of them probably belonged to other community members rather than Jettenia. To select contigs which most likely originate from Jettenia, two approaches were used:
First, all contigs were used as query in a BLASTx search against the 4,663 proteins of Kuenenia using a cutoff of E-value < 1−10. After that, 12,850 contigs that had hits with Kuenenia proteins were retrieved and used as query in a new BLASTx search against NCBI protein database (nr database). Only contigs with best hits with Kuenenia and BLAST E-value < 1−10 and hit length >50 were considered as contigs highly likely to belong to Jettenia. A total of 1,187 contigs were obtained in this way. Although the remaining contigs did not have a best hit with Kuenenia, 150 contigs did not have any hit within the nr database and 2062 contigs have best hits with Kuenenia with E-value > 0.001 and 5779 between 0.001 and 1E−10. For these contigs it was difficult to decide if they belong to Jettenia. Therefore the following approach was also used: All 37,432 contigs were binned using the MetaCluster 3.0 software (Wang et al., 2012). The binning separated the contigs into two groups, containing 10,343 and 22,929 contigs, respectively. The GC content of the two groups was clearly different (Figure 2).
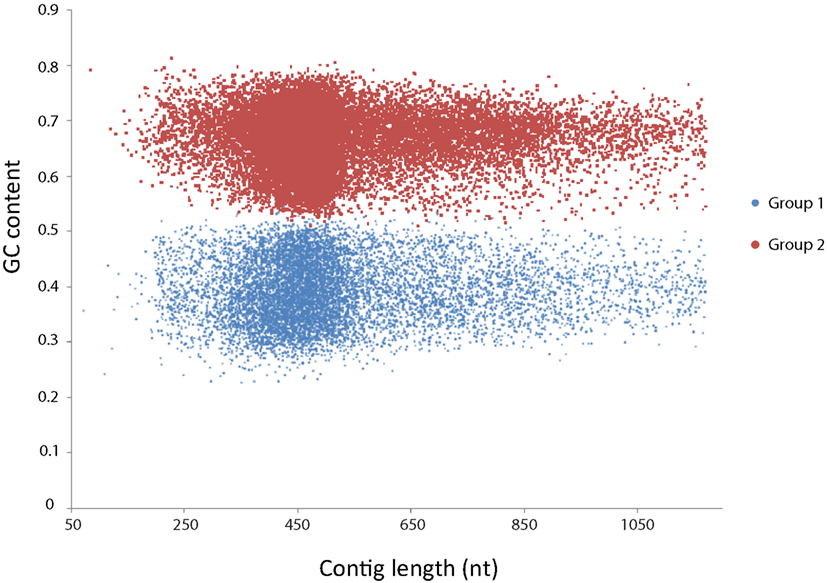
Figure 2. MetaCluster binning of the de novo assembly yielded two bins, clearly separated by GC content. Group 1 (blue) consisted of 10,343 contigs with an average GC content of 39.4%. Group 2 (red) consisted of 22,939 contigs with an average GC content of 66.2%.
Contigs belonging to group 1 had an average GC content of 39.4%, which was much lower than contigs of group 2 (66.9%). The average length and coverage of contigs in group 1 were higher than contigs belonging to group 2, suggesting that these contigs belonged to the dominant organism in the enrichment. Also, previously sequenced anammox bacteria all had GC contents close to 40% (Strous et al., 2006; Gori et al., 2011; Van de Vossenberg et al., 2012).
In addition, 28 important anammox genes involved in nitrogen metabolism and two genes encoding acetyl-CoA synthase of Kuenenia were selected as a reference protein dataset and gene products of these were downloaded from NCBI database. All 37,432 contigs were used as queries in a BLASTx search against this reference protein dataset with a cutoff E-value < 1−10. Contigs with best hits with the reference protein dataset were retrieved and used as queries again in a new BLASTx search against the NCBI nr database to ensure that they still were the best hit with the target Kuenenia proteins (Table 1). All contigs listed in Table 1, except contig270, contig5773, and contig10239, belonged to group 1 (10,343 contigs), which further strengthened the notion that contigs within this group were likely to belong to Jettenia.
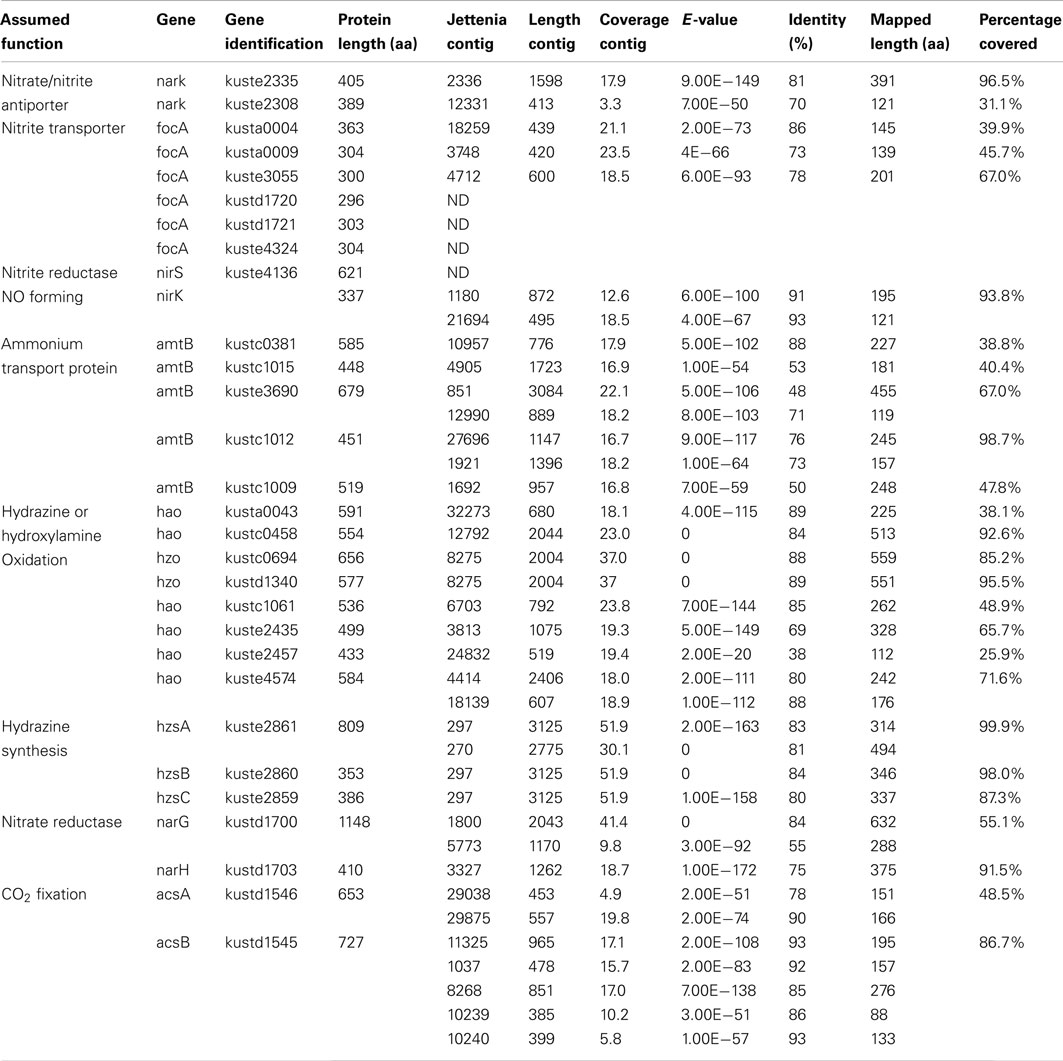
Table 1. Important anammox genes mapped by Jettenia contigs (First assembly).
Mapping Reads to KSU-1 Genome
Previous phylogenetic analysis based on 16S rRNA genes indicated that Jettenia was most closely related (98% similarity of 16S rRNA gene) to an anammox bacterium strain called KSU-1 (Quan et al., 2008; Viancelli et al., 2011). Additionally, the HZS subunit A (hzsA), a phylomarker for anammox bacteria, of Jettenia and KSU-1 shared 92.8% sequence identity on nucleotide level and 97.0% on amino acid level, which was much higher than any other pair of anammox bacteria (Harhangi et al., 2012). Therefore, in order to achieve a more efficient assembly, four large genome contigs of anammox strain KSU-1 were obtained from NCBI database (GenBank accession number: NZ_BAFH01000001-NZ_BAFH01000004) as reference sequences. All reads generated by both sequencing methods were then mapped to the reference sequences. In total, 76% of the reference sequences were mapped by 1,060,817 reads. The reads were then extracted and used for a new de novo assembly, which generated 3,209 contigs with an average length of 840 nt. The BLASTx search of Jettenia contigs against the Kuenenia reference protein dataset was performed again now using the 3,209 contigs generated by the new assembly as query (Table 2).
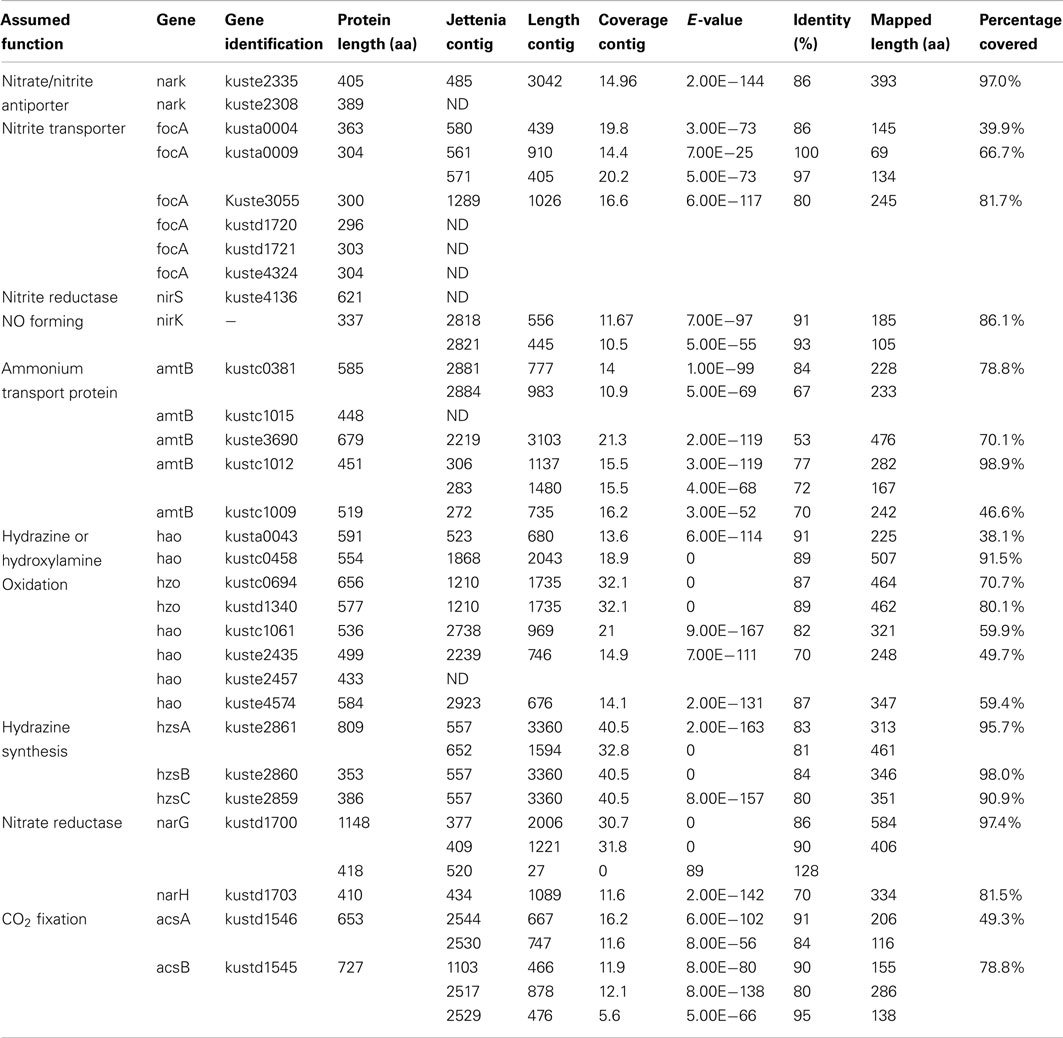
Table 2. Important anammox genes mapped by Jettenia contigs (Second assembly).
Phylogenetic Analyses
After the first assembly, the gene encoding the A subunit of HZS complex (hzsA) was recovered in two contigs (contig297–contig270). A Maximum likelihood tree of hzsA was then constructed using MEGA5 (Tamura et al., 2011), with reference sequences obtained from NCBI database (Figure 3).
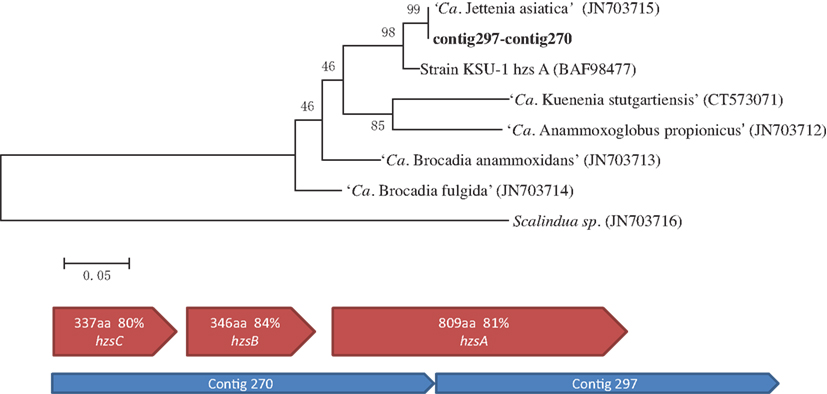
Figure 3. Maximum likelihood trees indicating the phylogenetic relationships within the Brocadiales based on hydrazine synthase subunit A (hzsA) protein sequences. Numbers at the nodes are percentages of bootstrap values based on 1000 re-samplings. NCBI accession number of reference sequences are shown in brackets (above). Similar organization of hydrazine synthase (hzsABC) genes cluster in Jettenia and Kuenenia. Blue pentagons represent Jettenia contigs contains HZS encoding genes, red pentagons indicating organization of Kuenenia hydrazine synthase gene cluster (below).
Also, the 16S rRNA gene database was downloaded from the Ribosomal Database Project (RDP; release 10, update 29; Cole et al., 2009) and a subset of cultured or identified microorganisms (339,774 sequences) was used as a reference dataset. All reads generated were mapped to this dataset in order to investigate the composition of the other community members besides Jettenia. Read mapping was also performed on the genomes of anaerobic methane oxidizer “Ca. Methylomirabilis. oxyfera” (FP565575) and ammonium-oxidizing bacteria Nitrosomonas eutropha (NC_008344.1) and Nitrosomonas europaea (NC004757.1) to investigate the presences of other important community members.
Results and Discussion
Genes for Anammox Metabolism Mapped by Jettenia Contigs
The BLASTx results showed that most important anammox genes of Kuenenia could be identified in the assembled Jettenia contigs. The hit lengths of some of these with the target proteins, however, were relatively short (Table 1). A new assembly using reads mapped to KSU-1 genome increased the mapped length of some genes significantly. For example, for kustc0381, the mapped length increased to 78.8% of full length when using contigs from the new assembly (Table 2). However, for some other genes such as kustc0694, kuste1009, and kuste2308, the mapped lengths slightly decreased when using contigs generated from second assembly, or could not be mapped at all (Table 2).
More sequence data with longer read length might lead to a more efficient assembly and would subsequently resolve this problem, but at this moment, we believe that the data presented here using the combination of contigs from two assemblies was sufficient to provide evidence that the predicted genes exist in the Jettenia genome.
Transportation of Nitrite/Nitrate and Ammonium
All anammox bacteria have to take up inorganic nitrogen substrates (ammonium, nitrite, and nitrate) from the environment, where they usually exist in limiting amounts. Furthermore, under oxygen-limited conditions, anammox bacteria may have to compete for ammonium with other community members such as heterotrophs and aerobic ammonium-oxidizing archaea (AOA) or bacteria (AOB). Under these conditions, there will also be competition for nitrite with nitrite-oxidizing bacteria (NOB; Fussel et al., 2012). Therefore, many genes encoding for nitrite, nitrate, and ammonium transport were present in the genomes of anammox bacteria most notably ammonium-transporter amtB, nitrate/nitrite antiporter narK, and formate/nitrite transporter focA.
Five copies of amtB were identified in the genome of Kuenenia (Strous et al., 2006). In the Jettenia metagenome, seven contigs were found to have best BLASTx hit with these five amtB proteins with an average identity of 66% (Table 1) on amino acid basis, which indicated that all five amtB genes were very likely also encoded by the genome of Jettenia. Six focA genes were found in Kuenenia genome, but only three of them (kusta0004, kusta0009, and kuste3055) could be partially retrieved from the metagenome of Jettenia (Tables 1 and 2). In the genome of Brocadia also only one gene encoding for focA protein could be identified which shared a 79% similarity with kusta0004 (Gori et al., 2011). However, in the genomes of Scalindua and KSU-1 multiple genes encoding focA were indentified (Van de Vossenberg et al., 2012). This might reflect the ability of this this marine anammox to thrive in nutrient limited conditions, such as marine oxygen-minimum zones (OMZ) where the concentration of inorganic nitrogen compounds are usually very low (Lam and Kuypers, 2011). On the other hand, less focA protein encoding genes in Brocadia and Jettenia genomes might indicate that those species possess other proteins which also function as nitrite transporter. In the genome of KSU-1, however, five formate/nitrite transport Two of the genes found in KSU-1 (ZP_10099012 and ZP_10100997), shared 82% identity and have best BLASTp hit with kuste3055. The other three (ZP_10098545, ZP_10098546, ZP_10098796) have best hits with kusta0009, kusta0004, and kustd1720, respectively.
For nitrate transport, two gene copies encoding a putative nitrate/nitrite antiporter narK (kuste2308, kuste2335) in the Kuenenia genome were partially mapped on Jettenia contigs (Tables 1 and 2).
Nitrite/Nitrate Conversion
Once transported, inorganic nitrogen substrates must be converted in several steps in the anammox metabolism, resulting in dinitrogen gas. First, nitrite is reduced to NO, this reaction could be catalyzed by a cytochrome cd1-type nitrite reductase nirS in Kuenenia (Strous et al., 2006). The nirS encoding genes were also identified the metagenome of Scalindua (Van de Vossenberg et al., 2012). The expression level of those genes analysed by transcriptomic and proteomic studies, however, were different: The Scalindua transcriptome data showed that nirS was highly expressed (Van de Vossenberg et al., 2012), but the nirS expression levels in Kuenenia were much lower (Kartal et al., 2011). In our Jettenia assembly, the nirS of Kuenenia (kuste4136) could not be mapped by any contig. BLASTx search also showed that none of the contigs has best hit with this gene. NirS also seemed to be absent from the metagenome of Brocadia fulgida (Gori et al., 2011). Again, the low sequencing depth could be an explanation for the absence of nirS gene. However, since at least one copy of all other important genes for anammox metabolism could be (partially) retrieved, the absence of nirS suggested that Jettenia was likely to employ another way to mediate the conversion of nitrite to NO. Very recently, the genome analysis of anammox strain KSU-1 showed that instead of nirS, a copper nitrite reductase nirK-homolog was detected. Biochemical studies confirmed that this nirK catalyzed the reduction of nitrite to NO (Hira et al., 2012). The nirK gene encodes a copper-containing nitrite reductase (CuNIR), which was previously identified in denitrifiers, but had not been detected in anammox bacteria. After mapping our Jettenia contigs to the KSU-1 assembled contigs, two Jettenia contigs (contig1180 and contig21694) covered almost the full length of nirK with only a gap of 21 amino acids. Together with the BLASTx search result (Tables 1 and 2), this strongly suggested that Jettenia also contained a nirK gene as an alternative for nirS to convert nitrite to NO.
To reduce nitrate to nitrite, a nitrate reductase complex is required, including narG and narH. In the Kuenenia genome, both genes were retrieved and together with other three genes formed a large gene cluster encoding a nitrate reductase complex (Strous et al., 2006). Large parts of Kuenenia narG and narH could be mapped by Jettenia contigs, which indicated both genes were present in the genome. In anammox bacteria, however, it is believed that narGH normally functions in the reverse way as a nitrite oxidoreductase nxrAB and would catalyze the oxidation of nitrite to nitrate (Jetten et al., 2009). Therefore, the narGH retrieved from Jettenia genome could also function in this way, similar to other anammox bacteria.
Hydrazine Metabolism and N2 Formation
NO, produced thorough nitrite reduction, and ammonium are are the direct precursors for the production of hydrazine (N2H4). HZS, encoded by gene cluster kuste2859-2861 (hzsCBA) in the Kuenenia genome (Kartal et al., 2011), catalyzes this reaction. The genes of the HZS cluster (kuste2859-2861) were the most highly expressed genes in both proteome and transcriptome data of Kuenenia (Kartal et al., 2011). In the transcriptomic and proteomic analyses of Scalindua, this was also observed (Van de Vossenberg et al., 2012). In the Brocadia metagenome, the coverage of contigs containing hzs genes was much higher than other contigs, which might be the result of multiple copies (Gori et al., 2011). Also here, this gene cluster could be perfectly mapped by two Jettenia contigs (contig297 and contig270) with identities between 80 and 84% (Table 1; Figure 3). Based on the BLASTx search, the two contigs should be concatenated together, but the metagenomic assembly failed to link them probably as a result of low coverage at the linking region. Moreover, the coverages of contig297 and 270 were 52 and 30-fold, respectively. This was much higher than the average coverage of contigs listed in Table 1, which was consistent with findings in other anammox genome assemblies (Gori et al., 2011).
Finally, an octaheme hydroxylamine oxidoreductase-like (hao-like) protein, HDH, catalyzes the oxidation of hydrazine to produce dinitrogen gas (Kartal et al., 2011). In the Kuenenia genome, at least 8 hao-like octaheme proteins were identified (Strous et al., 2006). Two of them, kustc0694, and kustd1340 were predicted to function as true HDHs. Both of them could be mapped by one same Jettenia contig, the contig8275 with high identities (Table 1), which is not surprising because the amino acid sequences of kustc0694 and kustd1340 are 96.4% identical.
Other hao-like proteins retrieved from Kuenenia genome could also be partially mapped by assembled Jettenia contigs, some of them, such as kustc0458, were nearly fully mapped (Tables 1 and 2). These hao-like proteins might be involved in some other reactions in the anammox metabolism, such as electron transfer, detoxification of potentially hazardous nitrogen compounds, dissimilatory nitrate/nitrite reduction to ammonia (DNRA) or ammonia formation (Campbell et al., 2009; Kartal et al., 2011; Van de Vossenberg et al., 2012).
Carbon Fixation
Previous studies indicated that the carbon fixation by anammox bacteria was accomplished through the acetyl-coenzyme A (CoA) pathway (Schouten et al., 2004), with reducing power required supplied by hydrazine oxidization (Kartal et al., 2011). A complete acetyl-CoA pathway was identified in the genome of Kuenenia (Strous et al., 2006). Two genes encoding the key enzyme in this pathway, the carbon monoxide dehydrogenase/acetyl-CoA synthase subunit alpha and beta (acsA and acsB) were retrieved with high identities to genes of Kuenenia in seven and five contigs by both assemblies, respectively (Tables 1 and 2), indicating Jettenia employed the same carbon fixation pathway as the previously studied anammox bacteria.
Other Important Community Members
Previous phylogenetic analyses of the same granule sample indicated a highly diverse community. Aerobic ammonia-oxidizing bacteria (AOB) were detected with a low abundance (Quan et al., 2008). In addition, the nutritional condition in the granule reactor would also allow the growth of other microorganisms such as methanogens and methane oxidizers or the recently reported denitrifying methanotroph “Ca. Methylomirabilis oxyfera” (Ettwig et al., 2010). The MetaCluster grouping result suggested that two thirds of the 37,432 assembled contigs were not originating from Jettenia but from other community members. Moreover, only ∼7% reads could be mapped to the KSU-1 genome. Therefore, it was likely that there were other members in the community with significant abundances. Analyses based on 16S rRNA genes and read mapping to reference genomes were performed to investigate the community diversity.
Mapping all sequence reads to a subset of 339,774 16S rRNA gene sequences of the RDP database confirmed a high abundance of Jettenia, with 15.6-fold coverage. A clone from a methanogenic archaeon (JN836400) was the second most abundant, with 4.6-fold coverage. Other abundant community members belonged to the Burkholderia (HQ222273) with 2.6-fold coverage and the Chloroflexi (HQ133206) with 2.1-fold coverage.
Since a methanogen was the second most abundant organism as detectable by 16S mapping, we decided to investigate this group of organisms in more detail. A mcrA gene, encoding the methyl coenzyme M reductase alpha subunit and a phylogenetic marker for methanogens, was detected on contig 9376 with a best hit to Methanothermobacter thermoautotrophicus (91% identity at amino acid level). Further, all contigs in group 2 were submitted to KEGG Automatic Annotation Server (KAAS; Moriya et al., 2007) for pathway mapping. A nearly complete methanogenesis pathway was retrieved with 82 contigs involved.
Since the granular sludge was originated from an anaerobic reactor system with ample supply of nitrite in the influent, the presence of methane could create a suitable niche for the anaerobic methanotroph “Ca. M. oxyfera”. Indeed mapping the reads to the “Ca. M. oxyfera” genome resulted in a mapping of 131,689 reads. Based on the amount of reads that could be mapped to KSU-1 (1,060,817), this resulted in an estimate of ∼6% abundance of “Ca. M. oxyfera” in the enrichment culture. Based on these findings we hypothesize that a full methane cycle is present in the enrichment.
As Nitrosomonas sp. were previously detected (Quan et al., 2008), we also performed a read mapping on the two available Nitrosomonas genomes; N. europaea and N. eutropha. 90,095 and 30,150 reads could be mapped to these genomes respectively, leading to an abundance estimate of ∼6% Nitrosomonas sp. in the enrichment. These findings suggest that a small oxygen gradient is present in the granular sludge, increasing the amount of nitrite available for Jettenia (Figure 4).
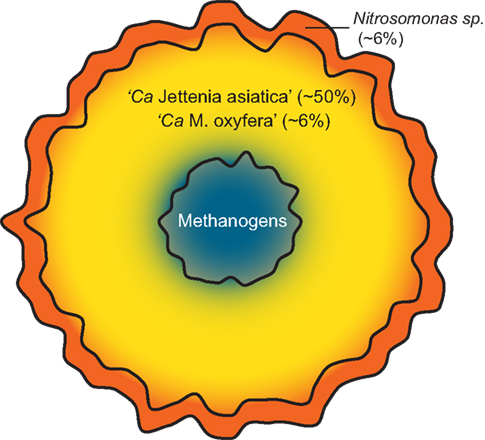
Figure 4. Schematic model of the community composition of the sludge granules. The microaerobic conditions in the outer layer of the granule (red) allow Nitrosomonas sp. to convert ammonium to nitrite. In the completely anaerobic interior of the granule (blue) methanogens form the last step in the anaerobic degradation of organic matter and produce methane. The methane formed allows coexistence of NC10 bacterium “Ca. Methylomirabilis oxyfera”, which oxidizes methane using nitrite as electron acceptor, with the dominant anammox bacterium “Ca. Jettenia asiatica” (yellow). Abundance of “Ca. Jettenia asiatica” is estimated based on qPCR (Quan et al., 2008). Abundance estimates of “Ca. Methylomirabilis oxyfera” and Nitrosomonas sp. are based on read mapping on the genomes of “Ca. Methylomirabilis oxyfera”, Nitrosomonas europaea and Nitrosomonas eutropha. No abundance estimate could be given for the methanogen community, since the Jettenia reference abundance was taken with Bacteria specific primers.
There were still more than 50% reads which could neither be mapped to any references mentioned above, nor be assembled into contigs. Additionally, less than 7% of the reads could be mapped to the genome of KSU-1. Since this organism is closely related to Jettenia, this leads to an abundance estimate that is much lower than previous reported (50%). On the other hand, 16S rRNA gene mapping confirmed the earlier qPCR analysis which indicated that Jettenia is the dominant bacterium in the enrichment. However, the 50% abundance of anammox bacteria was based on the 16S rRNA gene analyses with bacteria specific primers (Quan et al., 2008). Archaea, such as methanogens, were not included in the calculation and neither were Eukaryotes. However, Archaea were included in the 16S mapping and did not constitute a larger fraction than Jettenia. Thereby, Eukaryotes could affect the sequencing results significantly even when present at low abundance, because of their relatively large genomes. Therefore, we hypothesize that the majority of the reads obtained from the enrichment originated from a low abundance Eukaryotic population.
Concluding Remarks
Here we presented sequencing and metagenomic analysis of a community enriched for anammox bacterium “Candidatus Jettenia asiatica.” Although it was impossible to distil a high quality draft genome of this organism from the metagenome, we were able to identify all essential genes for anammox metabolism based on the comparison with other anammox bacteria such as Kuenenia and KSU-1. The identification of a copper-containing nitrite reductase NirK in Jettenia genome instead of cytochrome cd1-type nitrite reductase NirS indicated a flexible metabolism of anammox bacteria in terms of nitrite reduction to NO. However, the role of this enzyme, as well as the evolutionary relationship between Jettenia and other anammox bacteria still need to be studied.
Additionally, we could confirm earlier results on the presence of methanogens as important players in the side population and have shown clear indications of the presence of anaerobic methanotroph “Ca. M. oxyfera” in the culture. Together with anammox bacteria and aerobic ammonium oxidizers, these microorganisms formed a mutualistic community (Figure 4). Oxygen cannot penetrate into the very core of the granule, which could allow the growth of methanogens. The methanogens produce methane which is required for the anaerobic methanotroph. However, such a theory still needs further studies to confirm.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jack van de Vossenberg for the anammox scheme and Boran Kartal for critical discussions. Ziye Hu and M. S. M. Jetten were supported by ERC 232937 and DS by BE-Basic fp07-2.
References
Campbell, B. J., Smith, J. L., Hanson, T. E., Klotz, M. G., Stein, L. Y., Lee, C. K., et al. (2009). Adaptations to submarine hydrothermal environments exemplified by the genome of nautilia profundicola. PLoS Genet. 5, e1000362. doi:10.1371/journal.pgen.1000362
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., et al. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145.
de Almeida, N. M., Maalcke, W. J., Keltjens, J. T., Jetten, M. S. M., and Kartal, B. (2011). Proteins and protein complexes involved in the biochemical reactions of anaerobic ammonium-oxidizing bacteria. Biochem. Soc. Trans. 39, 303–308.
Ettwig, K. F., Butler, M. K., Le Paslier, D., Pelletier, E., Mangenot, S., Kuypers, M. M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548.
Fussel, J., Lam, P., Lavik, G., Jensen, M. M., Holtappels, M., Gunter, M., et al. (2012). Nitrite oxidation in the Namibian oxygen minimum zone. ISME J. 6, 1200–1209.
Gori, F., Tringe, S. G., Kartal, B., Marchiori, E., and Jetten, M. S. M. (2011). The metagenomic basis of anammox metabolism in Candidatus “Brocadia fulgida”. Biochem. Soc. Trans. 39, 1799–1804.
Hamersley, M. R., Lavik, G., Woebken, D., Rattray, J. E., Lam, P., Hopmans, E. C., et al. (2007). Anaerobic ammonium oxidation in the Peruvian oxygen minimum zone. Limnol. Oceanogr. 52, 923–933.
Harhangi, H. R., Le Roy, M., van Alen, T., Hu, B. L., Groen, J., Kartal, B., et al. (2012). Hydrazine synthase, a unique phylomarker with which to study the presence and biodiversity of anammox bacteria. Appl. Environ. Microbiol. 78, 752–758.
Hira, D., Toh, H., Migita, C., Okubo, H., Nishiyama, T., Hattori, M., et al. (2012). Anammox organism KSU-1 expresses a NirK-type copper-containing nitrite reductase instead of a NirS-type with cytochrome cd1. FEBS Letters 586, 1658–1663.
Humbert, S., Tarnawski, S., Fromin, N., Mallet, M.-P., Aragno, M., and Zopfi, J. (2010). Molecular detection of anammox bacteria in terrestrial ecosystems: distribution and diversity. ISME J. 4, 450–454.
Jetten, M. S. M., Op den Camp, H. J. M., Kuenen, J. G., and Strous, M. (2010). “Description of the order Brocadiales”, in Bergey’s Manual of Systematic Bacteriology: The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes, Vol. 4, eds N. R. Krieg, J. T. Staley, B. P. Hedlund, B. J. Paster, N. Ward, W. Ludwig, and W. B. Whitman (Heidelberg: Springer), 506–603.
Jetten, M. S. M., Horn, S. J., and vanLoosdrecht, M. C. M. (1997). Towards a more sustainable municipal wastewater treatment system. Water Sci. Technol. 35, 171–180.
Jetten, M. S. M., van Niftrik, L., Strous, M., Kartal, B., Keltjens, J. T., and Op den Camp, H. J. M. (2009). Biochemistry and molecular biology of anammox bacteria. Crit. Rev. Biochem. Mol. Biol. 44, 65–84.
Kartal, B., Kuenen, J. G., and van Loosdrecht, M. C. M. (2010). Sewage treatment with anammox. Science 328, 702–703.
Kartal, B., Maalcke, W. J., de Almeida, N. M., Cirpus, I., Gloerich, J., Geerts, W., et al. (2011). Molecular mechanism of anaerobic ammonium oxidation. Nature 479, U127–U159.
Kuypers, M. M. M., Lavik, G., Woebken, D., Schmid, M., Fuchs, B. M., Amann, R., Jorgensen, B. B., and Jetten, M. S. M. (2005). Massive nitrogen loss from the Benguela upwelling system through anaerobic ammonium oxidation. Proc. Natl. Acad. Sci. U.S.A. 102, 6478–6483.
Lam, P., and Kuypers, M. M. M. (2011). Microbial nitrogen cycling processes in oxygen minimum zones. Annu. Rev. Mar. Sci. 3, 317–345.
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185.
Quan, Z.-X., Rhee, S.-K., Zuo, J.-E., Yang, Y., Bae, J.-W., Park, J. R., Lee, S.-T., and Park, Y.-H. (2008). Diversity of ammonium-oxidizing bacteria in a granular sludge anaerobic ammonium-oxidizing (anammox) reactor. Environ. Microbiol. 10, 3130–3139.
Schouten, S., Strous, M., Kuypers, M. M. M., Rijpstra, W. I. C., Baas, M., Schubert, C. J., et al. (2004). Stable carbon isotopic fractionations associated with inorganic carbon fixation by anaerobic ammonium-oxidizing bacteria. Appl. Environ. Microbiol. 70, 3785–3788.
Siegrist, H., Salzgeber, D., Eugster, J., and Joss, A. (2008). Anammox brings WWTP closer to energy autarky due to increased biogas production and reduced aeration energy for N-removal. Water Sci. Technol. 57, 383–388.
Strous, M., Pelletier, E., Mangenot, S., Rattei, T., Lehner, A., Taylor, M. W., et al. (2006). Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature 440, 790–794.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739.
Thamdrup, B., and Dalsgaard, T. (2002). Production of N-2 through anaerobic ammonium oxidation coupled to nitrate reduction in marine sediments. Appl. Environ. Microbiol. 68, 1312–1318.
Van de Vossenberg, J., Woebken, D., Maalcke, W. J., Wessels, H. J. C. T., Dutilh, B. E., Kartal, B., et al. (2012). The metagenome of the marine anammox bacterium “Candidatus Scalindua profunda” illustrates the versatility of this globally important nitrogen cycle bacteriumem. Environ. Microbiol. doi:10.1111/j.1462-2920.2012.02774.x
Van Dongen, U., Jetten, M. S. M., and van Loosdrecht, M. C. M. (2001). The SHARON((R))-Anammox((R)) process for treatment of ammonium rich wastewater. Water Sci. Technol. 44, 153–160.
van Niftrik, L., and Jetten, M. S. M. (2012). Anaerobic ammonium-oxidizing bacteria: unique microorganisms with exceptional properties. Microbiol. Mol. Biol. Rev. 76, 585–596.
Viancelli, A., Kunz, A., Esteves, P. A., Bauermann, F. V., Furukawa, K., Fujii, T., et al. (2011). Bacterial biodiversity from an anaerobic up flow bioreactor with ANAMMOX Activity Inoculated with swine sludge. Braz. Arch. Biol. Technol. 54, 1035–1041.
Keywords: “Ca Jettenia asiatica”, metagenome, complex community
Citation: Hu Z, Speth DR, Francoijs K-J, Quan Z-X and Jetten MSM (2012) Metagenome analysis of a complex community reveals the metabolic blueprint of anammox bacterium “Candidatus Jettenia asiatica”. Front. Microbio. 3:366. doi: 10.3389/fmicb.2012.00366
Received: 15 June 2012; Accepted: 27 September 2012;
Published online: 29 October 2012.
Edited by:
Naomi L. Ward, University of Wyoming, USAReviewed by:
Frank Stewart, Georgia Institute of Technology, USAAlan J. Wolfe, Loyola University Chicago, USA
Copyright: © 2012 Hu, Speth, Francoijs, Quan and Jetten. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: M. S. M. Jetten, Department of Microbiology, Institute for Water and Wetland Research, Radboud Universiteit Nijmegen, Heyendaalseweg 135, 6525 AJ Nijmegen, Netherlands. e-mail: m.jetten@science.ru.nl