
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 13 June 2012
Sec. Evolutionary and Genomic Microbiology
Volume 3 - 2012 | https://doi.org/10.3389/fmicb.2012.00214
This article is part of the Research Topic Recent advances in genomic and genetic studies in the Archaea View all 12 articles
 Michaela Wagner Marleen van Wolferen Alexander Wagner Kerstin Lassak Benjamin H. Meyer Julia Reimann
Michaela Wagner Marleen van Wolferen Alexander Wagner Kerstin Lassak Benjamin H. Meyer Julia Reimann Sonja-Verena Albers*
Sonja-Verena Albers*For reverse genetic approaches inactivation or selective modification of genes are required to elucidate their putative function. Sulfolobus acidocaldarius is a thermoacidophilic Crenarchaeon which grows optimally at 76°C and pH 3. As many antibiotics do not withstand these conditions the development of a genetic system in this organism is dependent on auxotrophies. Therefore we constructed a pyrE deletion mutant of S. acidocaldarius wild type strain DSM639 missing 322 bp called MW001. Using this strain as the starting point, we describe here different methods using single as well as double crossover events to obtain markerless deletion mutants, tag genes genomically and ectopically integrate foreign DNA into MW001. These methods enable us to construct single, double, and triple deletions strains that can still be complemented with the pRN1 based expression vector. Taken together we have developed a versatile and robust genetic tool box for the crenarchaeote S. acidocaldarius that will promote the study of unknown gene functions in this organism and makes it a suitable host for synthetic biology approaches.
In contrast to the bacterial domain, research on Archaea has long been hampered by the absence of genetic tools to study gene functions in vivo. One major reason for this was that archaea are resistant to most commonly used antibiotics that are used in microbial genetics as selectable markers (Leigh et al., 2011). These compounds very often target the peptidoglycan synthesis which is absent in most archaea (Albers and Meyer, 2011) or the antibiotics are readily degraded at the conditions at which various archaea live.
Genetic systems were described for haloarchaea and methanogens only in the late 1990s, respectively, and have constantly improved since then (Leigh et al., 2011). For another euryarchaeon Thermococcus kodakariaensis an effective gene deletion system was established by Sato et al. (2003, 2005) for which also a complementary expression vector has been optimized (Santangelo et al., 2008). The most recent system was developed for Pyrocococus furiosus. Hence, nowadays genetic toolboxes exist for a variety of euryarchaeota (Waege et al., 2010).
In the kingdom crenarchaeota, only for organisms of the order Sulfolobales genetic systems have been developed. Sulfolobales are thermoacidophilic microorganisms that grow optimally at temperatures around 80°C and pH values between 2.5 and 3.5. Since the first description of Sulfolobus acidocaldarius by Brock et al. (1972), members of the Sulfolobales have developed into model organisms for studying DNA transcription, replication, translation, DNA repair, RNA processing, and cell division. Moreover, most Sulfolobales possess the non-phosphorylated Entner–Douderoff pathway and its regulation upon temperature shifts was the topic of a systems biology approach (Albers et al., 2009). This initiative has led to the development of standard operating procedures for omics approaches in Sulfolobales and should facilitate the exchange and comparability of obtained data (Zaparty et al., 2009).
The first shuttle vectors and a gene deletion method in S. solfataricus were based on the β-galactosidase, LacS, as a selection marker (Worthington et al., 2003; Aucelli et al., 2006; Berkner et al., 2007) and this method was successfully employed in a number of studies (Schelert et al., 2004, 2006; Szabo et al., 2007; Zolghadr et al., 2007; Frols et al., 2008; Maaty et al., 2009). However, as the selection is based on minimal media containing only lactose as a carbon source, this method is quite tedious as Sulfolobus species do not grow well on sugar minimal media. In some archaeal genetic systems the complementation of uracil auxotrophic mutants by the pyrEF genes is used on uracil free media to obtain gene deletion mutants. This marker also enables for the use of 5-fluoroorotic acid (5-FOA), which can be employed as counter selection marker for constructing markerless deletion mutants. For S. islandicus a system for obtaining unmarked deletion mutants relying on the pyrEF selection including a shuttle vector has been established (Deng et al., 2009; Peng et al., 2009) and successfully used in different studies (Zhang et al., 2010; Gudbergsdottir et al., 2011). Other Sulfolobus shuttle vectors that rely on pyrEF selection were constructed based on the virus SSV1 and the plasmid pRN1 (Jonuscheit et al., 2003; Berkner et al., 2007). For S. islandicus it has furthermore been shown recently that the drug simvastatin can be used to select for the presence of plasmids in a host that overexpresses the hmg gene, encoding the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA; Zheng et al., 2012).
Recently, we employed pyrEF as a selectable marker to obtain insertional deletion mutants in S. acidocaldarius and moreover for persistence of a shuttle vector with an inducible promoter for expression (Wagner et al., 2009; Berkner et al., 2010). It was earlier demonstrated that S. acidocaldarius can efficiently recombine primers down to a length of 14 bp into its genome (Grogan and Stengel, 2008). Taking advantage of this ability Grogan and colleagues showed very recently that short flanking regions of only 40–50 bp incorporated into the 5’ end of primers led to site-specific integration of the obtained PCR products containing pyrE as a selectable marker into the S. acidocaldarius genome (Sakofsky et al., 2011). This method has been used in high-through put screens especially in Saccharomyces cerevisiae and opens these possibilities now for S. acidocaldarius. However, these methods lead to the consumption of the only available marker and therefore abolish the possibility of complementation in trans and also the construction of double or triple gene mutants.
Therefore we established different efficient methods to obtain unmarked deletion mutants in S. acidocaldarius relying on uracil auxotrophy as a selectable marker using the pop in/pop out method. These methods were employed for genomically tagging of genes enabling pull down experiments for studies on protein complexes. In addition, an ABC transporter of S. solfataricus was ectopically inserted into the upsE locus of S. acidocaldarius. Moreover, we present the use of a copper inducible promoter for homologous and heterologous production of proteins. Summarizing, we present here a versatile and complete genetic toolbox for S. acidocaldarius.
Several different auxotrophic mutants of S. acidocaldarius were published before. However, quite a few of these were obtained by UV mutagenesis (Grogan, 1991). Therefore the possibility existed that these mutants would also contain secondary mutations in their genomes. For that reason a new directed S. acidocaldarius pyrE deletion mutant was constructed. This mutant, dubbed MW001, was obtained by transforming the S. acidocaldarius wild type strain DSM639 with a PCR product that contained 930–1500 bp of the up- and downstream flanking regions of the pyrE gene (saci1597), which would delete the full length pyrE gene by homologous recombination. After transformation the cells were streaked on gelrite plates containing uracil and 5-FOA. Surprisingly, none of the obtained colonies exhibited the expected complete deletion of the pyrE gene, but only a deletion of 322 bp (91–412 bp). This phenomenon happened repeatedly and this particular deletion has also been isolated before (Grogan and Hansen, 2003). The deletion of the 322 bp in pyrE in MW001 was confirmed by PCR (Figure 1B) and sequencing. Growth of MW001 in medium containing NZ-amine medium was only possible upon the addition of uracil and was completely restored at a concentration of 10 μg/ml uracil (Figure 1A).
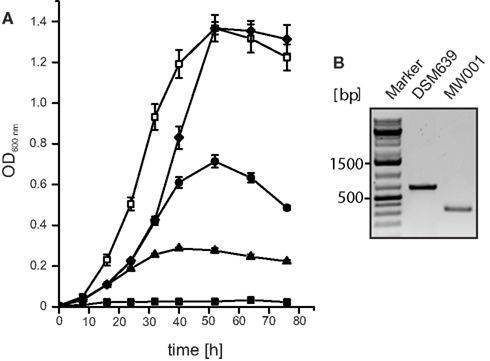
Figure 1. Construction of the uracil auxotrophic strain MW001. (A) Growth curves of MW001 in medium containing 0.2% NZ-amine and 0 (closed rectangles), 1 (closed triangles), 2.5 (closed circles), 5 (closed diamonds), 10 μg/ml (open rectangles) uracil. (B) PCR analysis of the wild type S. acidocaldarius strain DSM 639 and MW001 using primers 914/915 that amplify 663 bp of the full length pyrE and 340 bp from the deletion mutant pyrE.
We employed three different methods to obtain in-frame unmarked deletion mutants in S. acidocaldarius MW001. The first method is based on the classical “pop in/pop out” scheme using a single crossover recombination step (see Figure 2A). We designed pSVA406 for this purpose, which contained the pyrEF cassette of S. solfataricus (pyrEFSSO) and a multiple cloning site upstream of it, which was used to insert the approximately 500 bp long up- and downstream flanking regions of the gene of interest. The obtained deletion mutant plasmid was methylated and 100 ng plasmid DNA was electroporated into MW001. Cells were streaked on first selection plates that contained no uracil to select for the cells that would integrate the plasmid into their genome via a single crossover (Figure 2A). To enforce the “pop out” by a second single crossover recombination event, cells were then streaked on second selection plates that contained 5-FOA as only pyrEF- cells are resistant to 5-FOA. This second single crossover recombination step will either produce the wild type situation or the expected deletion mutant (Figure 2A). The method is here illustrated by deleting upsE (saci1494). In the last screening step we obtained in 50% of the colonies the wild type situation and 50% of analysed colonies we got the expected in-frame deletion mutants (Figure 3A). As upsE is part of the UV inducible pili operon in S. acidocaldarius which encodes proteins that after UV induction mediate cellular aggregation (Ajon et al., 2011), we confirmed that the obtained ΔupsE strain could indeed not aggregate anymore upon UV treatment (Figure 3B). The correctness of the obtained deletion mutants was confirmed by sequencing of PCR products that were achieved by using primers that were at least 200 bp up and downstream located to the primers used to construct the flanking regions for the deletion plasmid. This deletion method has been successfully used to produce single gene deletion mutants (Ellen et al., 2011; Meyer et al., 2011), and also double and triple deletion mutants as the marker cassette can be reused as often as wanted (Henche et al., 2012; Lassak et al., 2012). These mutants have also been successfully complemented by expression vectors.
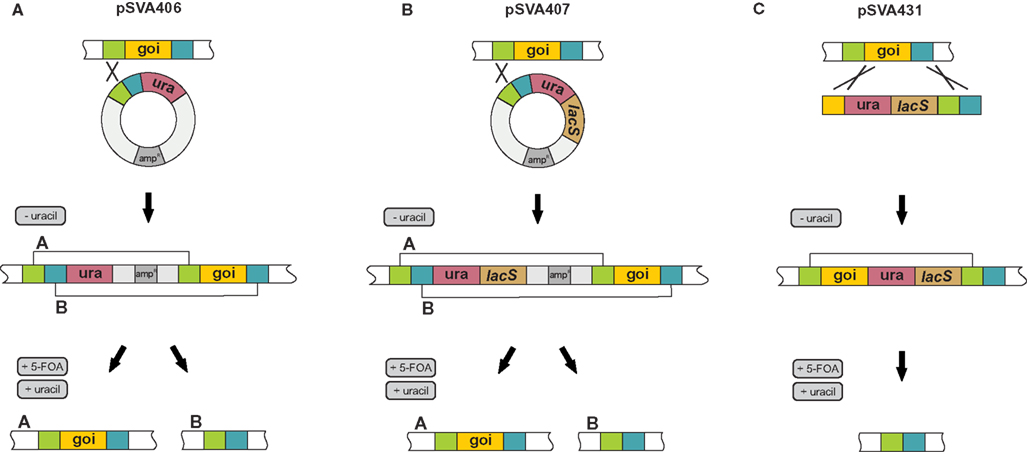
Figure 2. Depiction of the different methods to obtain markerless in-frame mutants. (A) Plasmid pSVA406 integrates via single crossover into the genome and by addition of 5-FOA it can be selected for a second single crossover event that leads either to the wild type situation or the aimed add deletion mutant. (B) By using pSVA407 deletion mutants can be obtained via the same method as employed by pSVA406, only that pSVA407 contains the lacS gene in the selection cassette that allows for the easy identification of integrants at the first selection level. (C) pSVA431 allows for double crossover insertion via the sequence of the gene and a downstream region of the gene of interest of the selection cassette. Thereby positives clones should be staining blue. Upon selection on 5-FOA plates the selection cassette will be excised and positive deletion mutants should be white. Goi, gene of interest; ura, pyrEFsso cassette; amp, ampicillin resistance cassette for selection in E.coli.
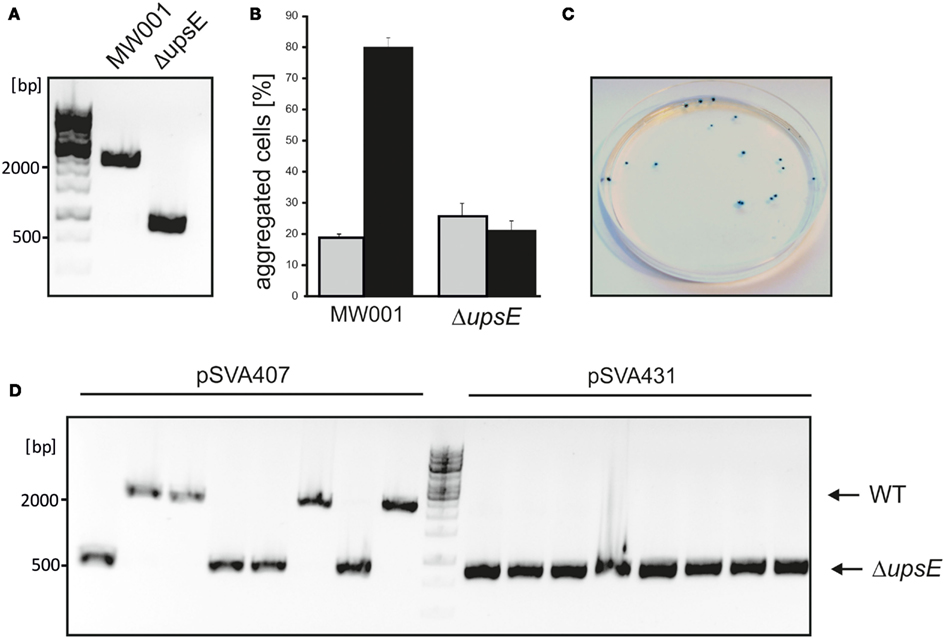
Figure 3. UpsE deletion mutants obtained by three different methods. (A) PCR to exemplify the difference in PCR product size obtained by primers 2073/2015 on either MW001 or any of the ΔupsE mutants. (B) As upsE is part of the pili operon that leads to aggregation of UV treated S. acidocaldarius cells (Ajon et al., 2011), an aggregation assay was performed with all the ΔupsE deletion mutants that were either obtained using pSVA406, pSVA407, or pSVA431. A typical result of an UV induced aggregation assay is shown. Aggregation of wild type and deletion mutant were scored before (gray bar) or after UV treatment (black bar). (C) First selection plate colonies from a pSVA407 transformation was sprayed with X-gal. (D) A typical result of a mutant PCR screen with primers 2073/2015 using either plasmid pSVA406 or pSVA431 to obtain the deletion mutant.
To improve the deletion mutant procedure we introduced the reporter gene lacS that encodes a β-galactosidase from S. solfataricus into pSVA406 yielding the deletion mutant plasmid pSVA407 (Figure 2B). Cells expressing lacS turn blue when sprayed with substrates like X-gal (5-bromo-4-chloro-indolyl-β-D-galactopyranoside), therefore integrants could be easily detected on the first selection plate. Figure 3C shows an example of such a plate, also demonstrating that most of the growing colonies were lacS positives.
Furthermore, we developed a third method which yields markerless deletion mutants via double crossover events. The deletion mutant plasmid pSVA431 consists of the pyrEFSSO marker cassette together with lacS reporter cassette that are flanked by two different multiple cloning sites. The first multiple cloning site harbors an approximately 500 bp part of the gene of interest and in the second multiple cloning site the up and downstream flanking regions were placed consecutively (Figure 2C). This cassette was transformed as a linear fragment into MW001. As the absence of uracil on the first selection plates selects for colonies containing the pyrEFSSO cassette, cells will only be able to form colonies when they integrate the linear fragment with double crossover via the matching part of the gene and the downstream flanking part of the gene (Figure 2C). Therefore all obtained colonies on first selection plates by means of this method turned blue after X-gal spraying (data not shown). When these strains were streaked on second selection plates containing 5-FOA and uracil, only the cells that loop the pyrEFSSO cassette and the lacS gene via a single crossover of the upstream flanking region out, can form colonies. Consequently, all white colonies that grow on second selection plates are deletion mutants (Figure 3D, right half of the gel). One further advantage of this method is that the genotype of integrants still harbors the functional full length gene of interest. If via this technique no colonies can be obtained on second selection plates or if the obtained colonies stay blue after X-gal spraying (which would mean that point mutations in the pyrEFSSO cassette occurred), the target gene can be considered essential for growth under these conditions.
To demonstrate that we can introduce ectopically DNA sequences into the S. acidocaldarius genome, we introduced the glucose transporter of S. solfataricus (sso2847-sso2850; glcS,T,U, and V, respectively, see Figure 4A; Albers et al., 1999) into the upsE locus (saci1494) of MW001. To this end plasmid pSVA445 was constructed containing the whole glucose transporter under the control of the maltose inducible promoter of malE (saci1665; Berkner et al., 2010) and the flanking regions that matched the sequence of upsE. pSVA445 was methylated and transformed linearized into MW001. Cells were plated on first selection plates. Colonies that had integrated pSVA445 were streaked on second selection plates. Obtained colonies were screened for positive clones that would have integrated the glucose transporter cassette of 4700 bp (glc transporter with promoter; 4450 bp only transporter; 6220 bp glc transporter together with pyrEF marker; data not shown) under the control of the maltose promoter. Successful production of the glucose transporter from the S. acidocaldarius genome was tested by analyzing wild type cells and the insertion mutant by western blot analysis using antibodies against GlcV, encoded by the last gene of the glucose transporter operon. As shown in Figure 4B, GlcV was detected in the membrane fraction of the insertion mutant, showing that the mal promoter upstream of glcS was sufficient to drive expression of the whole ABC transporter operon.
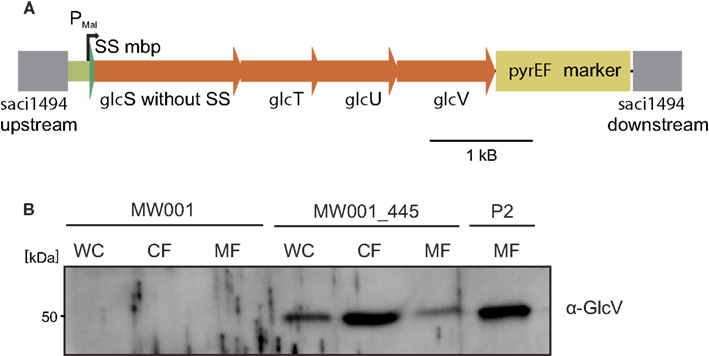
Figure 4. Insertion and expression of the S. solfataricus ABC glucose transporter in the S. acidocaldarius genome. (A) Depiction of the genomic integration of the glucose ABC transport operon of S. solfataricus in the upsE locus in the S. acidocaldarius genome. The signal peptide of GlcS, the glusoce binding protein, was exchanged by the MalE signal peptide from S. acidocaldarius. GlcTU are the predicted permase, whereas GlcV is the ATP binding protein in the glucose ABC transporter. (B) Western blot with specific GlcV antibodies on MW001, MW001_445, and S. solfataricus P2 cells. WC, whole cells; CF, cytoplasmic fraction; MF, membrane fraction.
To enable tag based affinity purification of homologously produced proteins and identification of native protein complexes, we used the “pSVA406” method (Figure 2A) to add tags to genes of interest in the genomic context that encoded for, e.g., His- or One-Strep-tags. Two examples will be discussed here, the purification of AglB (Saci_1274), a multiple spanning membrane protein, and of Saci_1210, a cytosolic protein. AglB was tagged in the genome by constructing plasmid pSVA1252 that contained the C-terminal part of aglB in which the stop codon was exchanged by a One-Strep-tag coding sequence and the downstream part of aglB. Using the pop in/pop out method the tag was inserted into the genome of MW001 and correct insertion was verified by sequencing MW001 saci1274::saci1274-One-Strep. This strain was grown and cells processed as described in Section “Materials and Methods.” Solubilized membrane proteins were subjected to Strep-tag affinity chromatography and the samples were analyzed by SDS-PAGE and immunoblotted with Streptactin antibodies (Figures 5A,B). The purified AglB-One-Strep appeared fuzzy, most probably as it is glycosylated, but its identity was confirmed by mass spectrometry. Two other proteins were co-purified that were identified as Saci_0260 and Saci_0262, subunits of the pyruvate carboxylase. These two proteins seem to be biotinylated proteins that bind to the chromatography material. The same proteins have been observed during Strep affinity purifications from cell extracts from S. solfataricus (Albers et al., 2006). Binding of these two proteins can be avoided by the addition of avidin to the cytoplasmic fraction.
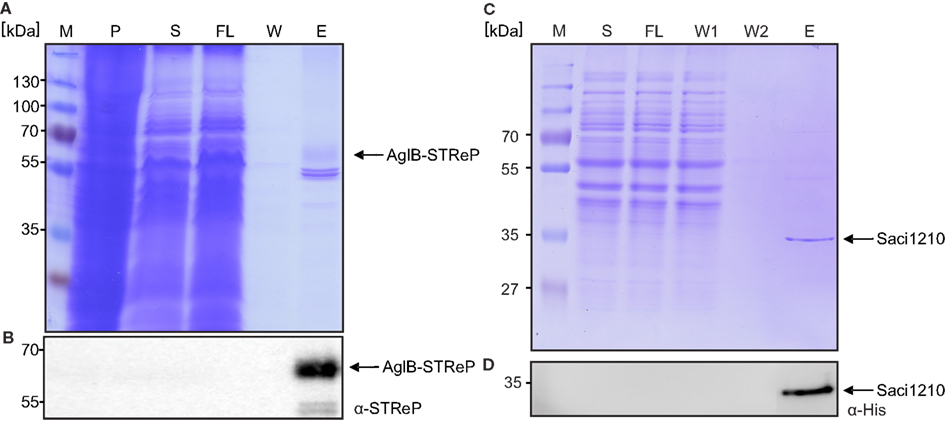
Figure 5. Purification of the genomic c-terminal Strep-Saci1274 fusion protein. (A) Commassie stained SDS-PAGE and (B) immuno blot with STRep-tag antibodies of the purification of Strep-Saci1274 fusion protein from S. acidocaldarius cells using Streptactin affinity material. (C) Commassie SDS-PAGE and an immune blot with a-His-tag antibodies (D) of the purification of Saci1210 which was genomically tagged using His_SELECT material. M, marker; P, insoluble membrane pellet; S, solubilized membrane proteins; FL, flow through; W, wash fraction; E, elution fraction.
In contrast to AglB, Saci_1210 was tagged with a tandem tag containing a Strep and a 6xHis-tag. This enabled us to test purification by either Strep- or His affinity chromatography using the same strain. Strep affinity chromatography of Saci_1210-Strep/His resulted in the same two contamination bands seen also in the AglB purification and no detectable Saci_1210 (data not shown). His-tag affinity chromatography resulted in highly pure Saci_1210-Strep/His in one step (Figures 5C,D). However, in contrast to S. solfataricus S. acidocaldarius exhibits a 14.7 kDa hypothetical protein, Saci_0386, that contains a natural stretch of six histidine residues at the C-terminus and therefore this protein co-purifies during His affinity chromatography (not shown in this blot). Concluding, His-tag affinity chromatography seems to be better suited for isolation of proteins from S. acidocaldarius as Strep-tag isolation leads to the co purification of two very prominent contaminating proteins of around 50 kD.
Several promoters have already been tested for efficient production of proteins in S. acidocaldarius of which the maltose inducible promoter of the maltose binding protein (saci1165) turned out to be the most reliable one (Berkner et al., 2010; Meyer et al., 2011; Henche et al., 2012; Lassak et al., 2012). Still this promoter exhibits quite some basic expression even under non-induced conditions. As a possible alternative we examined the copper inducible promoter of the copper inducible copper resistance cassette (saci0874–saci0872) that encodes CopT, the regulator of the operon, CopM, a metallochaperon, and CopA, a P-type copper ATPase (Ettema et al., 2006; Figure 6A). On the one hand CopT was described to be a negative regulator (Ettema et al., 2006) and on the other hand in a recent study (Villafane et al., 2011) CopT (called CopR in that publication) was described to be an activator of the cop operon. Therefore, the mal promoter of pCmalLacS was replaced with the copMA promoter region together with copT (highlighted in Figure 6A by a gray line, plasmids are schematically shown in Figure 6B). Hence, LacS expression would be driven by the copMA promoter (Figure 6A). The regulatory gene copT was included to ensure correct repression/activation of the copMA promoter.
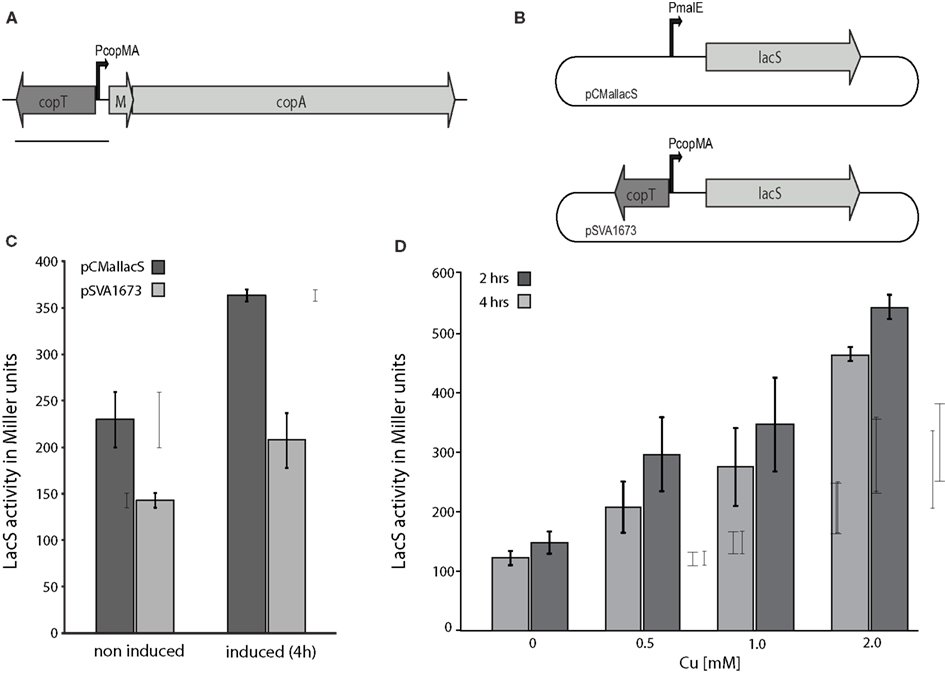
Figure 6. Comparison of copper inducible promoter and maltose inducible promoter for expression in MW001. (A) Operon structure of the copper resistance cluster of S. acidocaldarius. Black line indicates the part of the operon that was included into pSVA1673. (B) Schematic representation of the plasmids pCMallacS and pSVA1673 used in (C,D). (C) Direct comparison of LacS expression under control of Pmal (pCMallacS; dark gray) or PcopMA (pSVA1673; light gray) in MW001 cells under non-induced and induced conditions (0.4% maltose or 1 mM Cu, respectively). (D) Specific LacS activity by induction of PcopMA with increasing amounts of Cu in the medium. For each concentration the LacS activity was measured after 2 (light gray) and 4 h (dark gray) of induction.
The pSVA1673 was transformed into MW001 which was grown in medium supplemented with a minimal trace element solution to ensure very low levels of copper. The basic levels of expression were half of that obtained by the mal promoter under comparable conditions (Figure 6C). However, the induction of the promoter was only 1.4 fold after 4 h at a copper concentration of 1 mM whereas the maltose promoter was induced up to 1.8 fold. The copper promoter could be induced gradually by varying the amount of copper present in the medium (Figure 6D). Copper concentrations higher than 2 mM were not suitable because of cell lysis.
Detailed in vivo studies were long hampered in archaea as the development of genetic systems was very often obstructed by the absence of plasmids or selection markers. However, in the last few years enormous progress has been made and for most of the archaeal model systems genetic tools now exist (Leigh et al., 2011). Especially in the Sulfolobales genetic systems have been developed. For S. solfataricus and S. islandicus, respectively (Worthington et al., 2003; Albers and Driessen, 2008; She et al., 2009) these are proven to be successful as work based on them has been published.
Here we present a complete and versatile genetic system for S. acidocaldarius. Our system advances the system developed by Grogan that is based on marker insertion into the target gene (Sakofsky et al., 2011), to a system in which the selection marker can be reused. This therefore enables us to construct multiple allele mutants (single, double, and triple), specifically mutate genes in the genomic background or tag proteins genomically and complement obtained mutants by using an expression vector. In this manuscript we showed exemplary three different methods that can be employed to obtain markerless deletion mutants. We have used the same method for different projects in which we deleted target genes, constructed single, double, and triple mutants and complemented these using the pCmal based vector system (Berkner et al., 2007; Ajon et al., 2011; Meyer et al., 2011; Henche et al., 2012; Lassak et al., 2012).
We have also experienced that some genes could not be deleted by the pop in/pop out method, but we were able to obtain a deletion mutant by the PCR based insertion method (Sakofsky et al., 2011). This might be due to regulatory elements that might be disturbed by the insertion method. Therefore both methods can and should be used complementary.
We demonstrated that we can genomically tag genes in cis which will facilitate protein complex isolation and testing protein truncates or point mutations in their original genomic neighborhood. For affinity purification of such tagged proteins, His-tag affinity chromatography in our experience yielded better purification results, which could of course be due to a better accessible tag as the His-tag was located at the outmost C-terminus of the protein whereas the Strep-tag might have been occluded. However, His-tag affinity purification only yields one co purifying impurity. In the future other tags as, e.g., FLAG and HA tag will be tested to expand the genetic tool box.
Moreover, we introduced a ∼5 kB ABC transporter operon from S. solfataricus into the S. acidocaldarius genome and could demonstrate that the last gene was transcribed and translated correctly. This opens the option of integrating large sequences into the S. acidocaldarius genome and therefore streamlines its application in synthetic biology approaches.
For the expression in S. acidocaldarius we also tested the copper inducible copMA promoter, which showed decreased basic expression levels in comparison to the maltose inducible promoter. Therefore the copMA promoter might be useful for production of toxic proteins in S. acidocaldarius, especially regarding that the expression levels can be tightly titrated by the amount of copper added to the medium.
Concluding, we present here a very robust and versatile genetic tool box for S. acidocaldarius that has already proven to be widely applicable and very useful in delineating gene functions in this organism. We are currently working on the development of additional selective markers and the optimization of the expression vector.
Sulfolobus acidocaldarius DSM 639, MW001, and all constructed deletion mutants were aerobically grown in Brock media (Brock et al., 1972) with a pH of 3 at 76°C. The media were supplemented with 0.1% (w/v) tryptone or with 0.1% (w/v) N-Z-Amine and 0.2% dextrine. The growth of the cells was monitored by measurement of the optical density at 600 nm.
For pouring Sulfolobus plates a two times concentrated Brock media was supplemented with 6 mM CaCl2 and 20 mM MgCl2. For first selection plates 0.2% NZ-Amine (Fluka) and 0.4% dextrin, for second selection plates 0.2% tryptone, 0.4% dextrin, 200 μg/ml 5-FOA, and 20 μg/ml uracil was added to the two times concentrated solution and prewarmed to 75°C. This solution was mixed with an equal volume of fresh boiling 1.4% Gelrite solution (Carl Roth, Karlsruhe, Germany) and poured in 40 ml portions into petri dishes (150 × 20 mm, Sarstedt, Nümbrecht, Germany).
To obtain an uracil auxotrophic mutant of S. acidocaldarius DSM 639 deletion plasmid pSVA402 was constructed. To this end the up- and downstream regions of pyrE (saci1597) were amplified using primers 390/916 and 917/920, respectively. An overlap extension PCR was performed with both PCR products and the resulting pyrE deletion fragment was blunt end cloned into pGEM-T Easy (Promega, Mannheim, Germany) yielding pSVA402.
For the construction of the deletion plasmid pSVA406 the pyrEF (sso0615–sso0616) cassette of S. solfataricus together with its own promoter region was amplified using primers 938 and 939 (primer sequences available in the Appendix). This marker cassette was cloned into the MluI NsiI site of the recirculated pGEM-T Easy vector. Primer 938 contained additional BamHI and AvrII sites to expand the multiple cloning site of the resulting plasmid.
For construction of the deletion plasmid pSVA407 the lacS (sso3019) gene of S. solfataricus, encoding a β-galactosidase, was amplified with primers 933 and 940 and fused to the maltose inducible promoter of the maltose binding protein (saci1165) of S. acidocaldarius, which was amplified with primers 937 and 932, via overlap extension PCR. The resulting reporter cassette was cloned into the NsiI site of pSVA407. A correctly oriented clone was identified by restriction analysis.
The deletion plasmid pSVA431 was constructed by amplifying the pyrEF (sso0615–sso0616) and lacS (sso3019) cassette of S. solfataricus from pSVA407 using primer 939 and 1072. The resulting marker – reporter cassette was cloned into the pGEM-T Easy vector. The pSVA431 exhibited two multiple cloning sites up and downstream to the marker – reporter cassette, respectively.
For the construction of the upsE (saci1949) deletion plasmids via single crossover event the upstream region of upsE was amplified with the primers 2010 and 2011 and the downstream region was amplified with the primers 2012 and 2013. With both PCR products an overlap extension PCR was performed and cloned into the ApaI BamHI sites of pSVA406 and pSVA407 leading to the plasmids pSVA1804 and pSVA447. For the construction of the upsE deletion plasmid via double crossover the upsE gene was amplified with primers 532 and 639. The resulting PCR product was cloned into the BamHI and PstI site of the MCSI of pSVA431. A PCR was performed on pSVA1804 with the outer primers 2264 and 2265 to introduce different restriction sites and cloned into the NcoI and KpnI site of MCSII.
For the construction of the ectopic integration plasmid of the S. solfataricus glucose transporter into S. acidocaldarius pSVA445 the upsE (saci1949) upstream region was amplified using primers 2256 and 2257. The promoter region together with the signal sequence of the maltose binding protein (saci1165) was amplified employing primers 2258 and 986. glcS (sso2847) without signal sequence was amplified using the primers 987 and 988. glcT (sso2848), glcU (sso2849), and glcV (sso2850) were amplified in a single PCR with the primers 989 and 2259. The pyrEF marker cassette of S. solfataricus was amplified with the primers 2260 and 2261 and for the amplification of the upsE downstream region the primers 2262 and 2263 were used. All six PCR products were mixed together to perform an overlap extension PCR. The resulting PCR product was blunt end cloned into pGEM-T Easy following the standard protocol yielding pSVA445.
To simplify the purification of saci1274 by affinity chromatography, a One-Strep-tag was fused to this gene in the genome. For the construction of the insertion plasmid 900–1000 bp fragments of the up- and downstream regions of saci1274 were PCR amplified. For the upstream region the forward primer introduced an ApaI restriction site at the 5′end, whereas the reversed primer was designed to incorporate the One-Strep-tag sequence in front of the Stop codon (primers 1818 and 1819, respectively). For the downstream fragment the forward primer 1820 was designed to incorporate the complementary strand of the One-Strep-Tag, while the reverse primer 1821 contained a BamHI restriction site. Both fragments were fused via an overlapping PCR and the amplified PCR fragment was digested with ApaI and BamHI and ligated into plasmid pSVA407 yielding plasmid pSVA1224. The correct sequence was confirmed by sequencing. To increase the accessibility of the One-Strep-tag a longer linker sequence was added leading to the One-Strep sequence that was synthesized with a flanking 5′ AccI and 3′ PstI restriction site (sequence present in Figure A1 in Appendix). The product was digested with AccI and PstI and ligated into the plasmid pSVA1224, predigested with the same restriction enzymes, yielding the plasmid pSVA1252. Transformation and selection of the mutant strain was performed as described above.
To obtain genomically His-tagged saci1210 primers 1583 and 3106 were used to amplify the gene together with the C-terminal tag (Strep/His-tag). For the downstream region amplification with the primers 3107 and 1578 resulted in a PCR product with overlapping regions to the gene together with the tag. The overlap extension PCR product was cloned into the NcoI, BamHI site of pSVA406 yielding plasmid pSVA1097.
For the construction of copper inducible expression plasmid the promoter region of copM (saci0873) was amplified together with the predicted regulator copT (saci0874) with the primers 3428 and 3429. The resulting PCR product was cloned into the SacII, NcoI site of pCMalLacS which led to an exchange of the maltose inducible promoter of the maltose binding protein with the copper inducible promoter together with its regulator yielding pSVA1673.
Sulfolobus acidocaldarius MW001 were grown in 50 ml Brock media supplemented with 0.1% NZ-Amine, 0.2% sucrose, and 20 μg/ml uracil and adjusted to pH 3 with sulfuric acid. When the culture reached an OD600 nm of 0.5–0.7 (exponential growth phase) an aliquot was transferred to 400 ml fresh medium and harvested at an OD600 nm of 0.2–0.3. The culture was cooled down on ice, then centrifuged for 15 min at 4000 g and washed twice with 250 ml ice cold 20 mM sucrose. The pellet was resuspended with ice cold 20 mM sucrose to a theoretical final OD600 nm of 10 and aliquotted in 50 μl portions. The aliquots were directly used for transformation or frozen at −80°C without using liquid nitrogen for storage.
Prior to transformation into S. acidocaldarius, suicide- and shuttle plasmids were methylated to prevent restriction by the SuaI restriction enzyme. For that purpose E. coli ER1821 (New England Biolabs) bearing the additional plasmid pM.EsaBC4I (New England Biolabs, Frankfurt am Main, Germany) was transformed with plasmid DNAs.
Methylated deletion plasmids were electroporated in electro-competent wild type cells MW001 using a Gene Pulser Xcell (BioRad, München, Germany) with a constant time protocol with input parameters 1.5 kV, 25 μF, 600 Ω in 1 mm cuvettes. Before plating on uracil lacking and NZ-Amine containing plates, cells were regenerated for 30 min at 75°C in two-fold recovery solution (1% sucrose, 20 mM β-alanine, 1.5 mM malate buffer, pH 4.5, 10 mM MgSO4). The plates were sealed in plastic bags to avoid drying-out and incubated for around seven days at 75°C.
Single colonies appearing on plates were analyzed by colony PCR. To that end, single colonies were picked and lysed in 30 μl 0.2 M NaOH and the solution neutralized with 70 μl 0.2 M Tris pH 7.8. To amplify the genomic region of interest 0.5 μl lysate was used in 30 μl PCR reactions using Phusion High-Fidelity polymerase with Phusion HF buffer and monitored on an agarose gel.
Integration of pSVA407 or pSVA431 constructs could be visualized by X-Gal spraying of cells using a 25 mg/ml X-Gal stock solution in DMF diluted 1:5 with water. The 5 mg/ml X-Gal solution was sprayed on plates when single colonies appeared after around one week and the plates were directly put back into the 75°C incubator for 30 min. Transformants on first selection plates and point mutants on second selection plates turned blue while deletion mutants on second selection plates stayed white.
Ten liter culture of the strain MW001 Saci1274::Saci1274-One-Strep was grown in Brock medium until an OD of 0.8. Cells were harvested by centrifugation (3000 g; 4°C; 20 min). The cell pellet was resuspended in 40 ml buffer A (100 mM NaCl, 100 mM Tris HCl, 1 mM EDTA, pH 8) and lysed by a 20 min sonification with an intensity of 60% and an interval 20 s (Bandelin Sonopuls). Unbroken cells were removed by a low spin centrifugation 3000 g at 4°C for 20 min. The supernatant was centrifuged at 120,000 g at 4°C for 45 min and the membrane pellet resuspended in 12 ml of buffer A. 6 ml of the membrane fraction solubilized at 42°C under shaking condition in 30 ml Buffer S (2% n-dodecyl beta-D-maltoside (DDM), 100 mM NaCl, 100 mM Tris HCl, 1 mM EDTA, pH 8) supplemented with PMSF. Undissolved membranes were pelleted by an ultracentrifugation (120,000 g; 4°C for 45 min). The supernatant was added twice to a 0.8 ml STREP-Tag column (Strep-Tactin® Superflow®, IBA, Goettingen, Germany). Before loading the column was equilibrated with 10× column volume of buffer E (0.05% DDM, 100 mM NaCl, 100 mM Tris HCl, 1 mM EDTA, pH 8) and after the loading of the supernatant washed with 15× column volumes of the same buffer. The fusion protein was eluted with the buffer E containing 1 mM desthiobiotin.
Purification of Saci-Strep/His was performed with the difference that the cytoplasm was used for purification. For His affinity chromatography His-SELECT material from Sigma was used.
From each purification step 30 μl samples were loaded on a 11% SDS-PAGE and run at 100 V. Transfer to a PVDF membrane and blotting were performed as commonly done. The generated chemifluorescence of the Precision StrepTactin-AP Conjugate antibody (Biorad) or His-AP (Abcam, Cambridge, UK) was measured in a Fujifilm LAS-4000 Luminescent image analyzer (Fujifilm, Duesseldorf, Germany).
For the promoter activity assay pSVA1673 and pCmalLacS were transformed into MW001. Single colonies containing pCmalLacs were inoculated in Brock Medium with 0.1% NZ-Amine and 0.2% sucrose whereas pSVA1673 containing cells were grown in Brock medium supplemented with 0.2% maltose and 0.2% xylose. Moreover, for these cells the trace element solution only contained Na2B4O7 and MnCl2. For induction, 0.4% maltose or 1 mM CuSO4 were added to the medium, respectively.
Promoter activity was analysed by measuring the cleavage of o-nitrophenyl-β-D-galactopyranosid (ONPG) by LacS of S. solfataricus as described in Lassak et al. (2012). All given strains were assayed in biological as well as technical triplicates. The production of ONP was measured at 410 nm over a period of 4 h at 42°C using an Infinite 200 luminometer (Tecan, Maennedorf, Switzerland). Miller units were calculated as described previously (Lassak et al., 2012). With respect to the temperature optimum of LacS a conversion factor FT = 7 was determined and included in the final equation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Michaela Wagner, Marleen van Wolferen, and Sonja-Verena Albers were supported by intramural funds of the Max Planck Society. Alexander Wagner received funding from the BMBF in the network of ExpresSyS (0315586D). Kerstin Lassak was supported by the International Max Planck Research School (IMPRS).
Ajon, M., Frols, S., van Wolferen, M., Stoecker, K., Teichmann, D., Driessen, A. J., Grogan, D. W., Albers, S. V., and Schleper, C. (2011). UV-inducible DNA exchange in hyperthermophilic archaea mediated by type IV pili. Mol. Microbiol. 82, 807–817.
Albers, S. V., Birkeland, N. K., Driessen, A. J., Gertig, S., Haferkamp, P., Klenk, H. P., Kouril, T., Manica, A., Pham, T. K., Ruoff, P., Schleper, C., Schomburg, D., Sharkey, K. J., Siebers, B., Sierocinski, P., Steuer, R., van der Oost, J., Westerhoff, H. V., Wieloch, P., Wright, P. C., and Zaparty, M. (2009). SulfoSYS (Sulfolobus Systems Biology): towards a silicon cell model for the central carbohydrate metabolism of the archaeon Sulfolobus solfataricus under temperature variation. Biochem. Soc. Trans. 37, 58–64.
Albers, S. V., and Driessen, A. J. M. (2008). Conditions for gene disruption by homologous recombination of exogenous DNA into the Sulfolobus solfataricus genome. Archaea 2, 145–149.
Albers, S. V., Elferink, M. G., Charlebois, R. L., Sensen, C. W., Driessen, A. J., and Konings, W. N. (1999). Glucose transport in the extremely thermoacidophilic Sulfolobus solfataricus involves a high-affinity membrane-integrated binding protein. J. Bacteriol. 181, 4285–4291.
Albers, S. V., Jonuscheit, M., Dinkelaker, S., Urich, T., Kletzin, A., Tampe, R., Driessen, A. J. M., and Schleper, C. (2006). Production of recombinant and tagged proteins in the hyperthermophilic archaeon Sulfolobus solfataricus. Appl. Environ. Microbiol. 72, 102–111.
Albers, S. V., and Meyer, B. H. (2011). The archaeal cell envelope. Nat. Rev. Microbiol. 9, 414–426.
Aucelli, T., Contursi, P., Girfoglio, M., Rossi, M., and Cannio, R. (2006). A spreadable, non-integrative and high copy number shuttle vector for Sulfolobus solfataricus based on the genetic element pSSVx from Sulfolobus islandicus. Nucleic Acids Res. 34, e114.
Berkner, S., Grogan, D., Albers, S. V., and Lipps, G. (2007). Small multicopy, non-integrative shuttle vectors based on the plasmid pRN1 for Sulfolobus acidocaldarius and Sulfolobus solfataricus, model organisms of the (cren-)archaea. Nucleic Acids Res. 35, e88.
Berkner, S., Wlodkowski, A., Albers, S. V., and Lipps, G. (2010). Inducible and constitutive promoters for genetic systems in Sulfolobus acidocaldarius. Extremophiles. 14, 249–259.
Brock, T. D., Brock, K. M., Belly, R. T., and Weiss, R. L. (1972). Sulfolobus: a new genus of sulfur-oxidizing bacteria living at low pH and high temperature. Arch. Mikrobiol. 84, 54–68.
Deng, L., Zhu, H., Chen, Z., Liang, Y. X., and She, Q. (2009). Unmarked gene deletion and host-vector system for the hyperthermophilic crenarchaeon Sulfolobus islandicus. Extremophiles 13, 735–746.
Ellen, A. F., Rohulya, O. V., Fusetti, F., Wagner, M., Albers, S. V., and Driessen, A. J. (2011). The sulfolobicin genes of Sulfolobus acidocaldarius encode novel antimicrobial proteins. J. Bacteriol. 193, 4380–4387.
Ettema, T. J., Brinkman, A. B., Lamers, P. P., Kornet, N. G., de Vos, W. M., and van der Oost, J. (2006). Molecular characterization of a conserved archaeal copper resistance (cop) gene cluster and its copper-responsive regulator in Sulfolobus solfataricus P2. Microbiology 152, 1969–1979.
Frols, S., Ajon, M., Wagner, M., Teichmann, D., Zolghadr, B., Folea, M., Boekema, E. J., Driessen, A. J., Schleper, C., and Albers, S. V. (2008). UV-inducible cellular aggregation of the hyperthermophilic archaeon Sulfolobus solfataricus is mediated by pili formation. Mol. Microbiol. 70, 938–952.
Grogan, D. W. (1991). Selectable mutant phenotypes of the extremely thermophilic archaebacterium Sulfolobus acidocaldarius. J. Bacteriol. 173, 7725–7727.
Grogan, D. W., and Hansen, J. E. (2003). Molecular characteristics of spontaneous deletions in the hyperthermophilic archaeon Sulfolobus acidocaldarius. J. Bacteriol. 185, 1266–1272.
Grogan, D. W., and Stengel, K. R. (2008). Recombination of synthetic oligonucleotides with prokaryotic chromosomes: substrate requirements of the Escherichia coli/lambdaRed and Sulfolobus acidocaldarius recombination systems. Mol. Microbiol. 69, 1255–1265.
Gudbergsdottir, S., Deng, L., Chen, Z., Jensen, J. V., Jensen, L. R., She, Q., and Garrett, R. A. (2011). Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol. Microbiol. 79, 35–49.
Henche, A. L., Koerdt, A., Ghosh, A., and Albers, S. V. (2012). Influence of cell surface structures on crenarchaeal biofilm formation using a thermostable green fluorescent protein. Environ. Microbiol. 14, 779–793.
Jonuscheit, M., Martusewitsch, E., Stedman, K. M., and Schleper, C. (2003). A reporter gene system for the hyperthermophilic archaeon Sulfolobus solfataricus based on a selectable and integrative shuttle vector. Mol. Microbiol. 48, 1241–1252.
Lassak, K., Neiner, T., Ghosh, A., Klingl, A., Wirth, R., and Albers, S. V. (2012). Molecular analysis of the crenarchaeal flagellum. Mol. Microbiol. 83, 110–124.
Leigh, J. A., Albers, S. V., Atomi, H., and Allers, T. (2011). Model organisms for genetics in the domain Archaea: Methanogens, Halophiles, Thermococcales and Sulfolobales. FEMS Microbiol. Rev. 35, 577–608.
Maaty, W. S., Wiedenheft, B., Tarlykov, P., Schaff, N., Heinemann, J., Robison-Cox, J., Valenzuela, J., Dougherty, A., Blum, P., Lawrence, C. M., Douglas, T., Young, M. J., and Bothner, B. (2009). Something old, something new, something borrowed; how the thermoacidophilic archaeon Sulfolobus solfataricus responds to oxidative stress. PLoS ONE 4, e6964. doi:10.1371/journal.pone.0006964
Meyer, B. H., Zolghadr, B., Peyfoon, E., Pabst, M., Panico, M., Morris, H. R., Haslam, S. M., Messner, P., Schaffer, C., Dell, A., and Albers, S. V. (2011). Sulfoquinovose synthase – an important enzyme in the N-glycosylation pathway of Sulfolobus acidocaldarius. Mol. Microbiol. 82, 1150–1163.
Peng, N., Xia, Q., Chen, Z., Liang, Y. X., and She, Q. (2009). An upstream activation element exerting differential transcriptional activation on an archaeal promoter. Mol. Microbiol. 74, 928–939.
Sakofsky, C. J., Runck, L. A., and Grogan, D. W. (2011). Sulfolobus mutants, generated via PCR products, which lack putative enzymes of UV photoproduct repair. Archaea 2011, 864015.
Santangelo, T. J., Cubonova, L., and Reeve, J. N. (2008). Shuttle vector expression in Thermococcus kodakaraensis: contributions of cis elements to protein synthesis in a hyperthermophilic archaeon. Appl. Environ. Microbiol. 74, 3099–3104.
Sato, T., Fukui, T., Atomi, H., and Imanaka, T. (2003). Targeted gene disruption by homologous recombination in the hyperthermophilic archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 185, 210–220.
Sato, T., Fukui, T., Atomi, H., and Imanaka, T. (2005). Improved and versatile transformation system allowing multiple genetic manipulations of the hyperthermophilic archaeon Thermococcus kodakaraensis. Appl. Environ. Microbiol. 71, 3889–3899.
Schelert, J., Dixit, V., Hoang, V., Simbahan, J., Drozda, M., and Blum, P. (2004). Occurrence and characterization of mercury resistance in the hyperthermophilic archaeon Sulfolobus solfataricus by use of gene disruption. J. Bacteriol. 186, 427–437.
Schelert, J., Drozda, M., Dixit, V., Dillman, A., and Blum, P. (2006). Regulation of mercury resistance in the crenarchaeote Sulfolobus solfataricus. J. Bacteriol. 188, 7141–7150.
She, Q., Zhang, C., Deng, L., Peng, N., Chen, Z., and Liang, Y. X. (2009). Genetic analyses in the hyperthermophilic archaeon Sulfolobus islandicus. Biochem. Soc. Trans. 37, 92–96.
Szabo, Z., Sani, M., Groeneveld, M., Zolghadr, B., Schelert, J., Albers, S. V., Blum, P., Boekema, E. J., and Driessen, A. J. (2007). Flagellar motility and structure in the hyperthermoacidophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 189, 4305–4309.
Villafane, A., Voskoboynik, Y., Ruhl, I., Sannino, D., Maezato, Y., Blum, P., and Bini, E. (2011). CopR of Sulfolobus solfataricus represents a novel class of archaeal-specific copper-responsive activators of transcription. Microbiology 157, 2808–2817.
Waege, I., Schmid, G., Thumann, S., Thomm, M., and Hausner, W. (2010). Shuttle vector-based transformation system for Pyrococcus furiosus. Appl. Environ. Microbiol. 76, 3308–3313.
Wagner, M., Berkner, S., Ajon, M., Driessen, A. J., Lipps, G., and Albers, S. V. (2009). Expanding and understanding the genetic toolbox of the hyperthermophilic genus Sulfolobus. Biochem. Soc. Trans. 37, 97–101.
Worthington, P., Hoang, V., Perez-Pomares, F., and Blum, P. (2003). Targeted disruption of the alpha-amylase gene in the hyperthermophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 185, 482–488.
Zaparty, M., Esser, D., Gertig, S., Haferkamp, P., Kouril, T., Manica, A., Pham, T. K., Reimann, J., Schreiber, K., Sierocinski, P., Teichmann, D., van Wolferen, M., von Jan, M., Wieloch, P., Albers, S. V., Driessen, A. J., Klenk, H. P., Schleper, C., Schomburg, D., van der Oost, J., Wright, P. C., and Siebers, B. (2009). “Hot standards” for the thermoacidophilic archaeon Sulfolobus solfataricus. Extremophiles 14, 119–142.
Zhang, C., Guo, L., Deng, L., Wu, Y., Liang, Y., Huang, L., and She, Q. (2010). Revealing the essentiality of multiple archaeal pcna genes using a mutant propagation assay based on an improved knockout method. Microbiology 156, 3386–3397.
Zheng, T., Huang, Q., Zhang, C., Ni, J., She, Q., and Shen, Y. (2012). Development of a simvastatin selection marker for a hyperthermophilic acidophile, Sulfolobus islandicus. Appl. Environ. Microbiol. 78, 568–574.
Zolghadr, B., Weber, S., Szabo, Z., Driessen, A. J., and Albers, S. V. (2007). Identification of a system required for the functional surface localization of sugar binding proteins with class III signal peptides in Sulfolobus solfataricus. Mol. Microbiol. 64, 795–806.
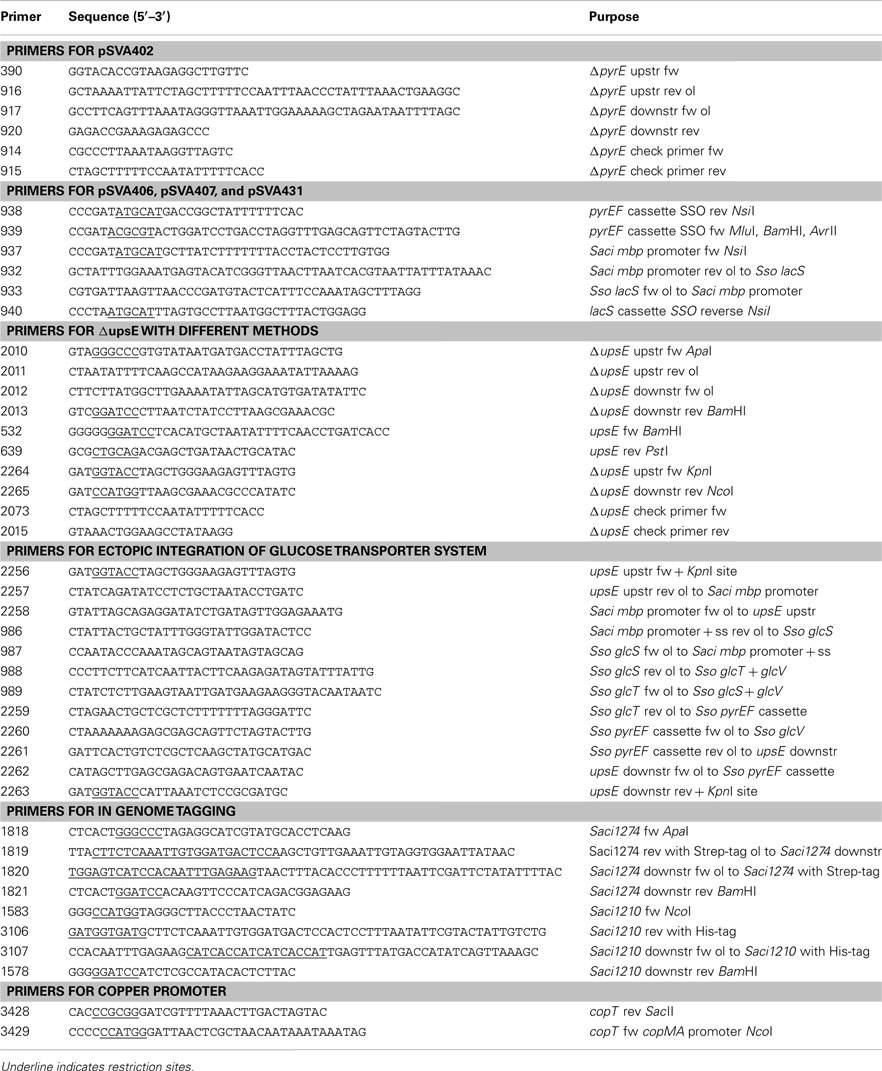
Table A1. Primers used in this study.
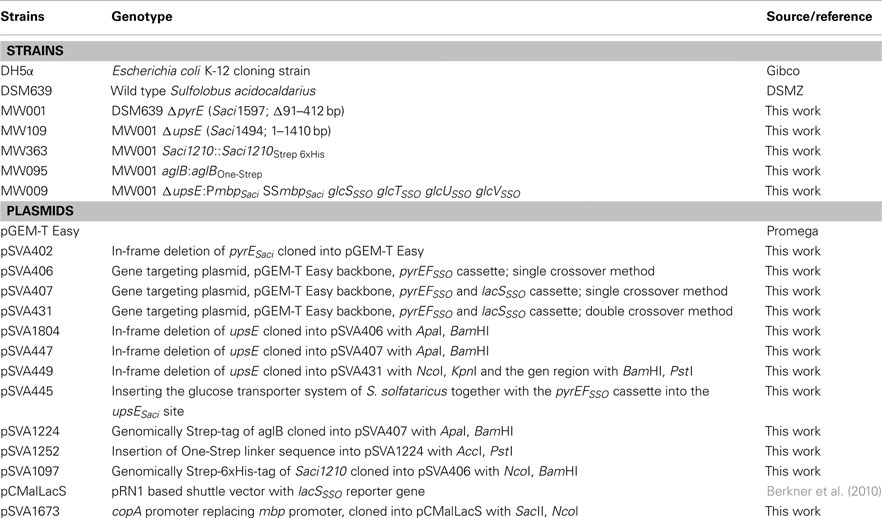
Table A2. Strains and plasmids used in this study.

Figure A1. Codon optimized One-STrEP-tag sequence.
Keywords: archaea, Sulfolobus, genetics, deletion mutant, expression system, in-frame deletion
Citation: Wagner M, van Wolferen M, Wagner A, Lassak K, Meyer BH, Reimann J and Albers S-V (2012) Versatile genetic tool box for the crenarchaeote Sulfolobus acidocaldarius. Front. Microbio. 3:214. doi: 10.3389/fmicb.2012.00214
Received: 06 March 2012; Paper pending published: 09 May 2012;
Accepted: 24 May 2012; Published online: 13 June 2012.
Edited by:
Zvi Kelman, University of Maryland, USACopyright: © 2012 Wagner, van Wolferen, Wagner, Lassak, Meyer, Reimann and Albers. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Sonja-Verena Albers, Molecular Biology of Archaea, Max Planck Institute for terrestrial Microbiology, Karl-von-Frisch-Strasse 10, 35043 Marburg, Germany. e-mail:YWxiZXJzQG1waS1tYXJidXJnLm1wZy5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.