 Naoko Kajitani1,2
Naoko Kajitani1,2
- 1 Laboratory of Mammalian Molecular Biology, Graduate School of Biostudies, Kyoto University, Kyoto, Japan
- 2 Laboratory of Gene Analysis, Department of Viral Oncology, Institute for Virus Research, Kyoto University, Kyoto, Japan
- 3 Department of Viral Oncology, Graduate School of Medicine, Kyoto University, Kyoto, Japan
Human papillomaviruses (HPVs) target the stratified epidermis, and can causes diseases ranging from benign condylomas to malignant tumors. Infections of HPVs in the genital tract are among the most common sexually transmitted diseases, and a major risk factor for cervical cancer. The virus targets epithelial cells in the basal layer of the epithelium, while progeny virions egress from terminally differentiated cells in the cornified layer, the surface layer of the epithelium. In infected basal cells, the virus maintains its genomic DNA at low-copy numbers, at which the viral productive lifecycle cannot proceed. Progression of the productive lifecycle requires differentiation of the host cell, indicating that there is tight crosstalk between viral replication and host differentiation programs. In this review, we discuss the regulation of the HPV lifecycle controlled by the differentiation program of the host cells.
Introduction
Human papillomavirus (HPV) infections of the anogenital organs are a very common “sexually transmitted disease (STD).” Although the incidence of cancer progression is low, a HPV infection is frequently detected in anogenital cancers. As for cervical cancer, HPV DNA is detected in more than 90% of cases. Approx. 5.5 million new cases of HPV infection are reported and there are c.a. 450,000 diagnoses of cervical cancer per year worldwide, leading to approximately 200,000 deaths each year, which ranks second among cancers in women (Parkin and Bray, 2006). HPV infections have also been associated with the head and neck squamous cell carcinomas (HNSCCs).
Human papillomavirus is categorized as a small virus containing DNA. More than 120 types of HPV have been identified and one-third of them target mucosal membranes, the remainder target the cutaneous membranes. Mucosa-tropic HPVs can be classified into two types based on their association with malignant carcinomas: a high-risk type (such as HPV type 16, 18, 31) and a low-risk type (such as HPV6 and 11; Howley, 1996). Prophylactic vaccines for HPV16 and18, Cervarix (GlaxoSmithKline), and for HPV6, 11, 16, and 18, Gardasil (Merck & Co.), have been developed recently and effectively prevent primary infections. They, however, cannot be used as therapeutic vaccines, indicating the importance of a Pap smear and the development of effective treatment strategies (Carter et al., 2011). In order to inhibit HPV-induced cancer, an understanding of the molecular basis of the infection and the characteristics of the infected lesions is important.
Genome Organization of HPV and Functions of Viral Proteins
Human papillomaviruses have a common gene organization (Figure 1): an early region encoding non-structural genes, the late region for structural genes, and a regulatory region (long control region: LCR).
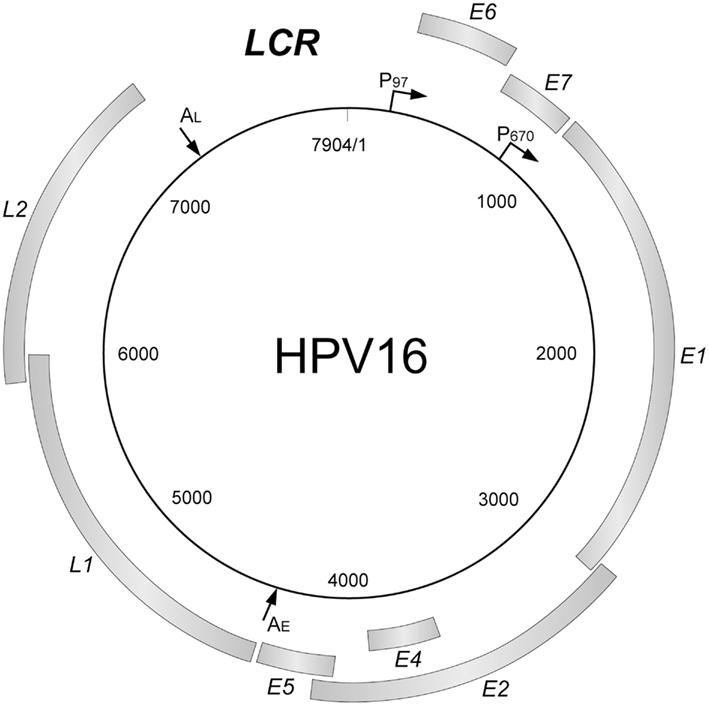
Figure 1. The genome organization of HPV16. The HPV genome has a circular double-stranded DNA structure. The viral genes are transcribed in a single direction (clockwise). There are genes coding for non-structural proteins (E1, E2, E4, E5, E6, and E7) and structural proteins (L1, L2), and a transcriptional control region (long control region; LCR). LCR contains a DNA replication origin and functions as the regulator for the DNA replication. The major promoters and polyadenylation signals are indicated (P97, P670, AE, AL).
The functions of each viral protein are summarized in Table 1. E1 and E2 are cooperatively involved in the initiation of viral DNA replication. E2 also functions as a transcriptional transactivator. E6 and E7 modulate the cell cycle control and contribute to viral genome maintenance (Frattini et al., 1996; Stubenrauch et al., 1998; Thomas et al., 1999). They also contribute to cancer development (Münger et al., 2004). Though E4 and E5 are speculated to modulate the productive phase of the HPV lifecycle, their biological roles remain unclear (Fehrmann et al., 2003; Genther et al., 2003; Nakahara et al., 2005; Wilson et al., 2005, 2007; Fang et al., 2006). Both L1 and L2 are capsid proteins.
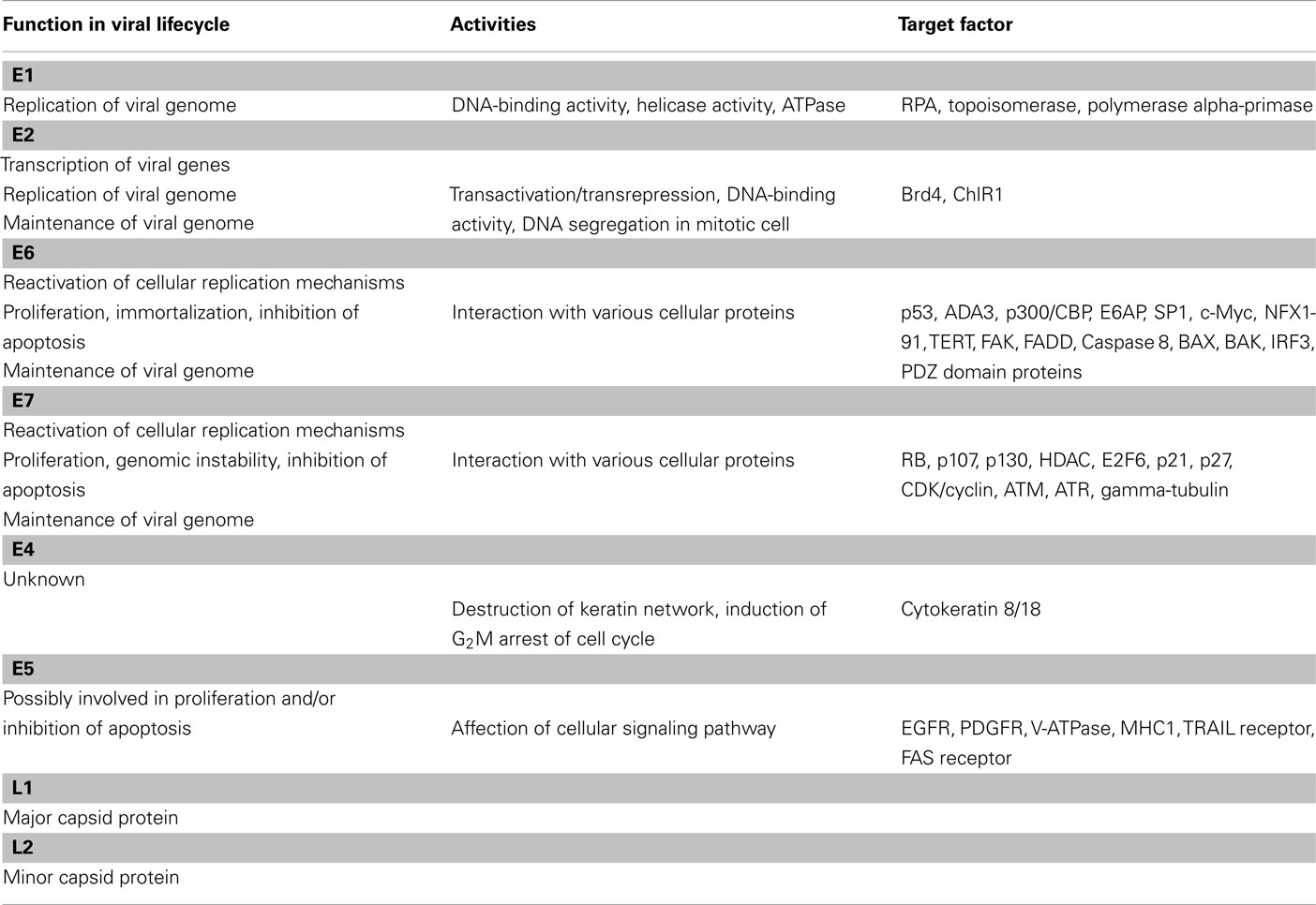
Table 1. Summary of HPV gene functions.
HPV Lifecycle
The target of a HPV infection is the stratified epithelium. In the normal stratified epithelium, the cell attached to the basal membrane (basal cell) is the only cell that has the potential to proliferate. The basal cell divides into a new basal cell and a daughter cell that is detached from the basal membrane, and the daughter cell launches its differentiation process. The daughter cells exit from the cell cycle and change their gene expression pattern, proceeding to terminal differentiation, then peel off from the epithelium (Jones et al., 2007). The lifecycle of HPV is tightly regulated by the differentiation program of the host cells (Figure 2). In this section, the differentiation-dependent lifecycle of HPV is briefly summarized.
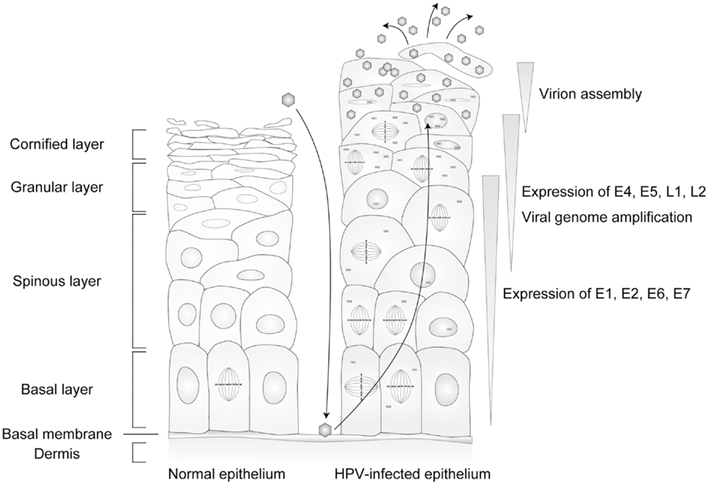
Figure 2. The lifecycle of human papillomaviruses (HPVs). HPVs infect specifically the cells in the basal layer of the stratified epithelium through lesions. Viral genomes are maintained as episomal DNA in the nuclei of infected cells. The viral lifecycle is strictly controlled by host cell differentiation, and the late lifecycle (productive lifecycle) occurs in upper layers of the epithelia that are terminally differentiated, and the progenitor virions are released from the cornified keratinocytes.
Entry of HPV into the Basal Cells of Stratified Epidermis
Human papillomavirus virions invade through damaged areas of the epithelium and infect the basal cells. Although the receptor for the HPV infection has not been fully characterized, the following model has been postulated; virions initially attach to the heparan sulfate proteoglycan (HSPG) on the basal membrane, and transfer to the receptor expressed on the keratinocytes moving on the basal membrane in the wound-healing process, then enter the cells (Kines et al., 2009).
Low-Level Expression of Viral Genes and Genome Maintenance in the Basal Layer
Following viral entry and uncoating, HPV genomic DNA is transported into the nucleus and maintained at a low-copy number in the basal cells (50 ∼ 100 copies per cell; in the basal layer, Figure 2; Moody and Laimins, 2010). Genome maintenance as episomal status is essential for the establishment of the early phase of the viral lifecycle (McBride et al., 2006).
Productive Replication of HPV in the Differentiated Cells
After leaving the basal membrane, the infected cells initiate the differentiation program. Because HPV does not encode DNA polymerase activity for viral genome replication, the host DNA replication machinery is required. However, the DNA replication activity is suppressed in the differentiated cells that exit from the cell division cycle. To ensure that the viral genome is replicated, HPV needs to reactivate cell division among the differentiation-initiated cells. E6 and E7 inactivate p53 and retinoblastoma protein (pRb), respectively, which enables the cells to maintain their DNA replication potential (Münger et al., 2004).
In the upper layers of the stratified epithelium (in the spinous layer, Figure 2), the expression of viral genes that are required for viral genome replication is markedly accelerated (Hummel et al., 1992; Ozbun and Meyers, 1997), inducing viral genome amplification to thousands of copies per cell (Bedell et al., 1991). Following the genome amplification, in the terminally differentiated cells, the synthesis of capsid proteins is triggered. The capsid proteins assemble into virions that encapsidate viral genomic DNA. The progenitor virions are released externally with peeled keratinocytes.
Differentiation-Dependent Control of HPV Lifecycle
The differentiation-dependent lifecycle of HPV is controlled of multiple levels, such as transcription, post-transcriptional processing, translation, and DNA replication. In the following sections, each regulatory mechanism is summarized.
Transcriptional Regulation of Viral Genes
Human papillomavirus has two major promoters, the early promoter and the late promoter. In HPV16, P97, and P670 have been identified as the early and late promoters, respectively (Figure 1). Transcriptional activity is mainly controlled by the LCR. A transcriptional enhancer is located within the LCR, with which various cellular transcription factors can associate (Figure 3).
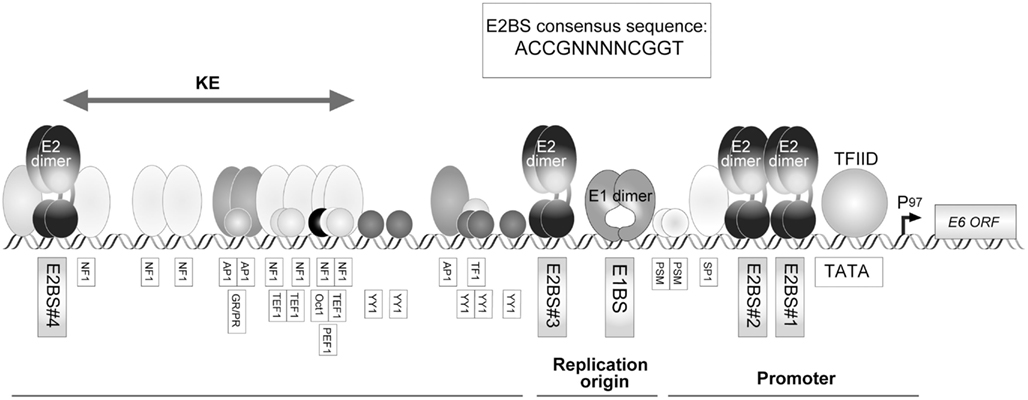
Figure 3. The structure of HPV16 LCR (region of the control of early promoter P97). The early promoter P97 and replication origin are located in LCR, which are regulated by various cellular factors. Activity of P97 is regulated by AP-1, NF1, SP1, TFIID, TF1, Oct-1, PSM, and the viral transcription factor E2. Four E2-binding sites (E2BS) have been identified in HPV16 LCR and the consensus sequence for E2BS is shown in an inset. A glucocorticoid receptor and progesterone receptor (GR/PR) recognition element was also identified in the LCR. The existence of a keratinocyte-specific enhancer (KE) has been proposed (Desaintes and Demeret, 1996).
The binding sites for the viral transcriptional regulator, E2, are found in HPV16 LCR. Viral gene expression is regulated by the occupancy status of the E2-binding sites (E2BSs; Figure 3), which is partly defined by the E2 expression level controlled by cellular differentiation status (Steger and Corbach, 1997; Hadaschik et al., 2003).
E2 functions in viral genome segregation by tethering the viral DNA to the mitotic chromatin, in which a cellular protein, bromodomain-containing protein 4 (Brd4), has been reported to be involved (McPhillips et al., 2006). Interaction between E2 and Brd4 is also required for the E2-mediated transcriptional activation and repression (McPhillips et al., 2006; Wu et al., 2012).
A ubiquitous transcription factor, Sp1 is a well-known regulator for HPV gene expression. The Sp1-binding site partially overlaps with one of the E2BSs (E2BS#2), and a TATA box element is located close to the promoter–proximal E2BS (E2BS#1; Figure 3). The binding of E2 to those E2BSs, therefore, interferes with the assembly of the transcriptional initiation complex, resulting in a suppression of E6/E7 expression that is governed by the early promoter activity (Tan et al., 1992). It was also reported that Sp1 altered the chromatin structure of HPV16 LCR, offering the accessibility of transcription factors (Stünkel and Bernard, 1999).
Transcriptional Control in the Undifferentiated Cells
Transcripts of viral early genes are expressed in the infected basal cells, which is essential for the viral DNA replication (Dürst et al., 1992). It was reported that a unique promoter, P14, was utilized for E1 expression and the E2BSs were considered as necessary for the P14 activity (Lace et al., 2008). The transcript initiated from P14 is a poly-cistronic mRNA containing E6, E7, and E1, in which the shunting in ribosomal scanning process enables the translation of E1 (Remm et al., 1999). The regulatory mechanism for E2 expression has not been clarified. The early promoter is used for E6 and E7 expression, in which several transcription factors, including AP-1, glucocorticoid receptor, NF1, Oct-1, Sp1, YY-1, and CDP, are involved (Figure 3; Desaintes and Demeret, 1996).
Transcription in the Differentiated Cells
The early promoter is activated in association with the differentiation process, increasing the E1/E2 expression (Hummel et al., 1992; Ozbun and Meyers, 1997). Although levels of E6 and E7 also increase with the early promoter’s activation, the E2 overexpressed in the upper layer is thought to suppress their transcription via the mechanism mentioned above. E6 and E7 are important in maintaining infected cells in an undifferentiated state, but terminal differentiation is required for the productive replication of HPV. The inhibition of E6/E7 expression by E2 might promote cellular differentiation, and the cells undergo terminal differentiation, which is suitable for the viral productive lifecycle.
AP-1, a heterodimer composed of Fos and Jun, is considered to be involved in the differentiation-dependent transcriptional control in keratinocytes; there are reports that the expression profiles of Fos and Jun family members were modified, and that the interaction between AP-1 and KRF-1, a keratinocyte-specific transcription factor (Mack and Laimins, 1991), was strengthened in the differentiation process (Desaintes and Demeret, 1996; Thierry, 2009). Several transcriptional factors were reported to be involved in the differentiation-dependent control of LCR function; EPOC-1/Skn-1a, C/EBP-α, -β, c-Myb, NF1, NFATx, Pax5, and WT1 (Desaintes and Demeret, 1996; Thierry, 2009).
The late promoter is specifically activated in the differentiated layers of epithelium. The late promoter activity is suppressed by CDP (CCAAT displacement protein) and YY-1, whose binding potential was reported to be decreased in differentiated keratinocytes (Ai et al., 1999, 2000). There was also a report that the expression ratio of a transcription factor, Sp1 and its antagonist, Sp3, was altered through the differentiation, which activated the late promoter activity (Apt et al., 1996). The binding of hSkn-1a and C/EBPα to the proximal region of the late promoter contributes to the control of the late promoter activity (Kukimoto and Kanda, 2001; Wooldridge and Laimins, 2008). The involvement of E7 in the regulation of the late promoter activity was also described (Bodily and Laimins, 2011; Bodily et al., 2011). It still remains necessary to clarify the regulatory mechanism for the late promoter in the differentiation of epithelial cells.
Methylation of the HPV Genome during the Cell Differentiation Process
HPV gene expression is controlled by the methylation of HPV genomic DNA. As E2BSs contain CpG dinucleotides (see inset in Figure 3), they can be modified by DNA methylation in the host cell. E2BSs are reported to be highly methylated in undifferentiated cells, inhibiting E2-binding, and demethylation at the E2BSs occurs in association with the cell differentiation (Kim et al., 2003; Vinokurova and von Knebel Doeberitz, 2011).
RNA Processing
For conversion of the gene expression profile from the early to late phase of viral replication, RNA processing is considered critical. The primary transcript of HPV encodes multiple viral genes, and precise RNA processing is essential to produce the mRNA for each viral gene at an appropriate stage of cell differentiation (Schwartz, 2008).
In the early phase of the viral lifecycle, the primary transcription initiated by the early promoter is terminated at the early poly(A) signal, AE (Figure 1), and the transcript is processed by using the early splicing signals, which produces the mRNAs encoding the viral early genes. In the differentiated cells, the transcripts for the late genes are expressed from the late promoter and utilize a late poly(A) signal, AL (Figure 1), and late splicing signals. The early and late splicing signals compete for the splicing factors, so their usages are generally exclusive.
Multiple splicing signals are found in the HPV genome, which are utilized for the expressions of various viral genes (Figure 4). These splicing signals can be categorized into three groups; early phase-specific signals (DS226, SA409, SA526, SA742 in HPV16), late phase-specific signals (SD 3632 and SA5639), and non-specific signals (SD880, SA2709, SA3358; Schwartz, 2008).
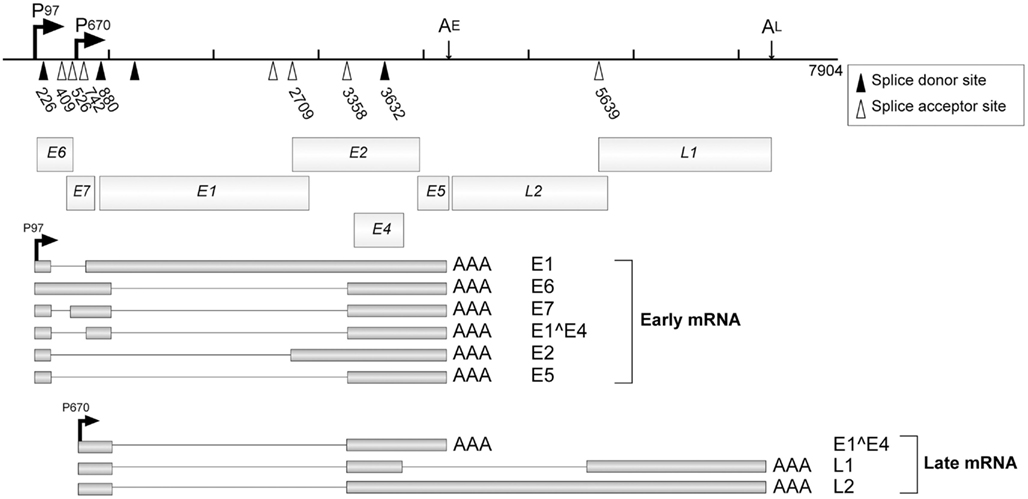
Figure 4. The splicing signals and the transcripts of HPV16. P97 and P670 are the early and late promoters, respectively, for HPV16. AE and AL indicate the early and late polyadenylation signals, respectively. Open triangles indicate splicing acceptors and filled triangles indicate splicing donors. Major mRNAs and their products are indicated. This figure is cited from a review (Schwartz, 2008).
Early splicing events have three major roles; regulation of the expression ratio of early genes, production of splicing variants of viral genes, and suppression of late gene expressions. The early splicing sites of HPV16, SD226, SA409, and SA526, are located in the E6 ORF, which enhances the translation efficiency of E7. Because the initiation codon for E6 is leaky and there are several splicing signals in the E6 ORF, various variants of E6 can be expressed. Those variants were reported to counteract the full-length E6, which might be important for the fine-tuning of E6 activities.
SA3358 is utilized in both the early and late phases of viral replication to produce HPV16 E1∧E4 mRNA. A strong splicing enhancer was identified downstream of SA3358, and it accelerated the polyadenylation at AE and suppressed the late gene expression in the early phase (Figure 5A). In undifferentiated cells, SA3358 competed with the late splicing signals for the splicing factors (SRp20, hnRNPL etc.), which might contribute to the suppression of late gene expression (Rush et al., 2005; Jia et al., 2009).
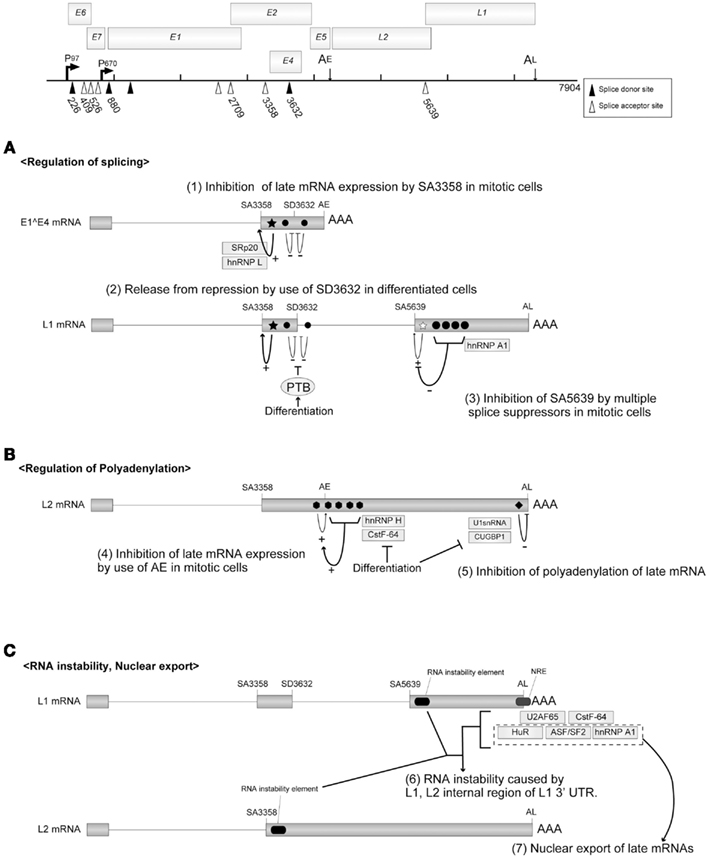
Figure 5. Regulatory mechanisms for mRNA processing. The HPV16 genome with the promoters, the poly(A) signals, and the splicing signals are shown at the top of the figure. Open triangles indicate splicing acceptors and filled triangles indicate splicing donors. (A) HPV16 splicing regulatory elements are indicated as filled stars (splicing enhancers) or filled circles (splicing silencers). (B) HPV16 polyadenylation regulatory elements are indicated as filled hexagons (polyadenylation stimulatory elements) and a filled rectangle (polyadenylation inhibitory element). (C) The regulatory elements for the instability and nuclear export of HPV16 late mRNA are indicated. This figure is cited from a review (Schwartz, 2008).
The late mRNAs are transcribed from the late promoter, and a major splicing event occurs between SD880 and SA3358 in HPV16. For the production of L1 mRNA, additional splicing between SD3632 and SA5639 is required. The mRNAs for L1 and L2 are poly-cistronic, and the mechanism for bypassing the upstream ATG remains to be explained.
SD3632 in HPV16 is used exclusively for late mRNA production. SD3362 is located close to SA3358 and AE, and the usage of SD3362 was suppressed by a splicing suppressor in dividing cells. It was indicated that the polypyrimidine tract-binding protein (PTB) interfered with the splicing suppressor in differentiated cells, which potentiated late gene expression (Figure 5A; Somberg et al., 2008).
A late phase-specific SA5639 in HPV16 was reported to be regulated by the cis-acting elements found in the L1 coding region (Zhao et al., 2004). A splicing enhancer was identified in the 3′ region of SA5639. Although the enhancer was activated in dividing cells, its function was hindered by multiple splicing suppressors located in the L1 coding region. It was reported that hnRNP A1 is associated with those multiple splicing suppressors (Figure 5A; Zhao et al., 2007).
To produce the late mRNAs, it is essential to bypass the polyadenylation at AE. Enhanced utilization of AE could, therefore, inhibit the late gene expression. The 5′ 400 nt of the HPV16 L2 ORF was reported to enhance the polyadenylation at AE, where multiple-G-motifs are located (Oberg et al., 2005). The hnRNP H interacted with the G-motifs and accelerated the polyadenylation at AE, and the expression of hnRNP H decreased as the cell differentiation progressed. CstF-64 was also reported to bind the L2 coding region of HPV31 and enhance the polyadenylation at AE, and the expression of CstF-64 diminished during the cell differentiation process (Figure 5B; Terhune et al., 2001).
Within the 3′UTR of the late mRNA (late UTR), a motif highly homologous to the U1snRNA was identified. It was reported that U1-70K, a component of U1snRNP, bound to the U1snRNA motif of BPV1 and interfered with the polyadenylation (Furth et al., 1994). Although U1-70K binding was not detected with HPV16, CUG binding protein 1 (CUGBP1) was reported to interact with the late UTR element of HPV16 and inhibit the polyadenylation process (Figure 5B; Goraczniak and Gunderson, 2008).
Control of Late mRNA Stability
There are RNA instability elements within the L1 and L2 coding mRNAs of HPV16, which function in undifferentiated cells (Mori et al., 2006), although the mechanism for RNA destabilization remains to be clarified (Sokolowski et al., 1998; Collier et al., 2002). There is a GU-rich negative regulatory element (NRE) in the late UTR of HPV16, which is a RNA instability element (Kennedy et al., 1991). Various factors, ASF/SF2, CstF064, U2AF65, hnRNPA1, and HuR, are reported to regulate the differentiation-dependent events of HPV replication through binding to the NRE (Figure 5C; Dietrich-Goetz et al., 1997; Koffa et al., 2000; Cheunim et al., 2008).
A highly U-rich region was located in the early UTR of HPV16 and reduced mRNA stability; a U-rich region is a signature feature of unstable mRNA (Jeon and Lambert, 1995; Barreau et al., 2005).
Nuclear Export of Late mRNAs
The L1 mRNA of HPV16 was retained in the nucleus in undifferentiated W12 epithelial cells (Koffa et al., 2000), suggesting that the nuclear export of late mRNAs was inhibited in the dividing cells. Although the factor(s) that mediates the nuclear export of late mRNAs has not been identified, candidates include HuR, hnRNA, and ASF/SF2, which are proteins shuttling between the nucleus and cytoplasm (Figure 5C; Carlsson and Schwartz, 2000; Koffa et al., 2000; McPhillips et al., 2004; Zhao et al., 2004).
Translational Control of Late Gene Expression
As the inhibitory mechanism for late gene expression, the involvement of translational inhibition was also reported. Translation efficiency was suppressed with in vitro translated RNA containing the late UTR of HPV1, suggesting the late UTR had the potential to inhibit the translation. For the inhibition, poly(A)-binding protein (PABP) was considered to be responsible (Wiklund et al., 2002). It was also reported that poly(C) binding protein 1 and 2 (PCBP-1 and -2) and hnRNP K bound to the L2 coding region of HPV16 mRNA and inhibited the in vitro translation (Collier et al., 1998). The rare codon usages found in L1 and L2 might also contribute to the inhibition of late gene translation (Gu et al., 2004). In terminally differentiated cells, the altered expression ratios of tRNA species could compensate for the inhibitory effect of the rare codon usages (Fang et al., 2007).
Regulation of Viral DNA Replication
E1 and E2 have essential roles in the HPV genome’s replication (Kadaja et al., 2009). E2 is a DNA-binding protein that recognizes E2-binding sites (E2BSs) located in the LCR (Figure 3). E2 has transcriptional transactivator activity, as well as the capacity to bind to the viral DNA replication factor E1. E1 has DNA helicase and ATPase activities and weak DNA-binding capacity. Through its interaction with E2, E1 is recruited to the replication origin (ori), which is essential for the initiation of viral DNA replication (Chiang et al., 1992a,b). E2 also contributes to the segregation of viral DNA in the cell division process by tethering the viral DNA to the host chromosome through interaction with Brd4 and/or ChlR1, both of which can bind to chromatin (McBride et al., 2006). Accurate segregation of the viral genome is essential to maintain the HPV infection in the basal cells, in which the copy number of the viral genome is very low.
The replication potential of E1 is regulated by its interaction with cellular proteins. p56, one of the interferon stimulated genes (ISGs), directly interacts with E1 and translocates it to the cytoplasm, which might contribute to the interferon-mediated inhibition of HPV replication (Terenzi et al., 2008). The interaction of WD repeat protein p80 (WDR80) with E1 is reported to be required for the efficient maintenance of the viral genome in undifferentiated keratinocytes (Côté-Martin et al., 2008).
As mentioned, the expression levels of E1 and E2 increase in association with the differentiation of the epithelial cells, which could be responsible for the vegetative genome amplification.
Recently, it was reported that E6 and E7 activated the ATM DNA damage pathway in differentiation-dependent manner, by which Chk2 was activated. Chk2 potentiated caspase-3 and -7, and the caspases in turn cleaved the E1 protein, which might be required for viral DNA amplification in the differentiated cells (Moody et al., 2007; Moody and Laimins, 2009).
Nuclear accumulation of E1 blocks cell cycle progression in early S-phase and triggers the activation of a DNA damage response (DDR) and of the ATM pathway (Fradet-Toucotte et al., 2011), and the activation of DDR might facilitate HPV DNA replication (Sakakibara et al., 2011). The nuclear accumulation of E1 is regulated by phosphorylation of the nuclear export signal (NES) found in E1 via Cyclin E/A-Cdk2 (Fradet-Toucotte et al., 2010).
DNA replication of HPV utilizes the replication mode with a “θ-structure” in undifferentiated cells, and the mode changes to “rolling circle replication” in differentiated cells, which enables the rapid synthesis of multiple copies of viral DNA. The molecular mechanism supporting the DNA replication in the differentiated cells, however, has not been fully explained (Flores and Lambert, 1997).
Involvement of Cellular Transcription Factors in Viral DNA Replication
It was reported that the binding of hSkn-1a to its recognition site proximal to the ori region was required for the viral genome replication of HPV16 (Kukimoto et al., 2008). In other HPV types, Sp1 and TATA box binding protein (TBP) inhibit viral genome replication, in which competition between E2 and Sp1 or TBP may be involved (Demeret et al., 1995; Hartley and Alexander, 2002). These transcription factors might alter the chromatin structure, which could inhibit the access of E1 to the origin (Demeret et al., 1995). The inhibition of STAT-1 expression by E6 and E7 was also reported to be important for viral genome amplification (Hong et al., 2011).
Virion Production
A report described that HSP70 was activated in response to the keratinocyte differentiation and co-localized with HPV31 L1 in the differentiated layer of epithelium (Song et al., 2010). It was reported that the disulfide bond among the HPV16 L1 pentamer was formed in a differentiation-dependent manner and had an important role in virion stability (Conway et al., 2011), indicating that virion production was regulated not only by the quantity of the late gene products but also by a differentiation-dependent mechanism.
The Modulation of Cell Proliferation/Differentiation by HPV Gene Products
As described above, HPV replication is strictly regulated by the differentiation program of the host cells. Conversely the HPV infections modulate the proliferation/differentiation status of the host cells, indicating tight communication between the virus and the host cell, which is required for completion of the viral replication.
Acceleration of Cell Proliferation and Inhibition of Cell Differentiation
The acceleration of cell proliferation and inhibition of differentiation are induced by the expression of E6 and E7 (Longworth and Laimins, 2004; Moody and Laimins, 2010). E7 inhibits the functions of the pocket protein family, activates the E2F-dependent promoter, and induces S-phase-specific gene expression (Moody and Laimins, 2010). E7 maintains Cyclin E or Cyclin A–CDK2 activity in differentiated cells by targeting p21 and p27, important regulators for growth arrest in the differentiation process. E6 mediates ubiquitination of p53 in association with E6AP, causing the proteasomal degradation of p53 (Moody and Laimins, 2010), and disturbs p53-mediated growth arrest. The association between E6 and histone acetyltransferases (HATs) might be also involved in the inhibition of p53 function (Moody and Laimins, 2010). E6 was reported to target cellular PDZ proteins, which might contribute to the immortalization of the infected cells (Thomas et al., 2008). E6 was reported to activate telomerase activity by inducing the overexpression of c-Myc and by modulating the expression of NFX1-91, which also contributed to the immortalization process (Gewin et al., 2004).
The functions of E6 and E7 in the activation of the DNA replication machinery of the host cell are required to ensure the viral genome’s replication in the differentiated cells (Thomas et al., 1999), resulting in the aberrant proliferation and the retarded differentiation of the host cells. With a transgenic mouse model expressing HPV16 E6 and/or E7 under the K14 promoter, dysplasia was observed at the stratified epidermis (Griep et al., 1993).
Although the normal differentiation of keratinocytes is not fully understood, reports describe the involvement of Notch, MAPK, NF-κB, p63, the AP2 family, C/EBP, IRF6, GRHL3, and KLF4 (Blanpain and Fuchs, 2009). There are also papers describing the contribution of c-Myc to the differentiation of epithelial stem cells, and differentiation-dependent demethylation at histone H3K27 (Blanpain and Fuchs, 2009). Recently, some of these factors were found to be associated with HPV gene products (Lathion et al., 2003; Chakrabarti et al., 2004; An et al., 2008; Melar-New and Laimins, 2010), and it is reasonable that these associations modify the cell differentiation program of the infected cells.
Inhibition of the Apoptotic Induction
The aberrant proliferation and/or DNA replication in the HPV-infected cells induce p53-dependent apoptotic cell death. The inactivation of pRb by E7 also potentiates the p53 activity. The p53-dependent apoptosis is counteracted by E6 activity (Garnett et al., 2006; Moody and Laimins, 2010). E6 induces the proteasomal degradation of p53. It has also been reported that E6 directly binds to p53 and inhibits its DNA-binding potential (Lechner and Laimins, 1994), and that E6 interacts with HDAC p300, ADA3, and/or CREB-binding protein (CBP), which destabilizes p53 (Patel et al., 1999; Zimmermann et al., 1999; Kumar et al., 2002). The HPV-infected cells also escaped from anoikis by the E6-mediated expression of FAK and the phosphorylation of paxillin, which activates FAK (McCormack et al., 1997). It was reported that interaction between E7 and p600 was involved in the inhibition of anoikis (Huh et al., 2005).
Several membrane-spanning death receptors have been identified, such as TNF receptor type 1 (TNFR1), FAS receptor, and TRAIL receptor. The high-risk type E6 was reported to interact with TNFR1 and suppress TNFα-induced apoptosis (Filippova et al., 2002). E6 is also known to interact with FADD and caspase8, which might inhibit the apoptosis mediated by FAS and TRAIL (Filippova et al., 2004; Garnett et al., 2006). In addition, E6 was reported to be associated with pro-apoptotic Bcl2 members, BAK and BAX, and to interfere with intrinsic apoptosis (Garnett et al., 2006). It was reported that E5 could inhibit FAS- and TRAIL-mediated apoptosis (Garnett et al., 2006). In addition, there was a paper that described the inhibitory effect of E7 on apoptosis, in which the upregulation of cellular inhibitor of apoptosis protein (c-IAP) by E7 was involved (Garnett et al., 2006).
The Modulation of miRNA Expression through Cell Differentiation
Recently, it was reported that HPV E6 and E7 induced the aberrant expression of tumor suppressive miRNAs (Zheng and Wang, 2011). E6 and E7 are known to target c-Myc, p53, and E2F, and these transcription factors are reported to be involved in the regulation of miRNA expression, so E6 and E7 could cause the uncoordinated expression of those miRNAs. E6 and E7 target p53 and pRb, respectively, and cause the unregulated expression of the miR-15/16 cluster, the miR-17-92 family, miR-21, miR-23b, miR-34a, and the miR-106b/93/25 cluster. Such aberrant expression was suspected to be involved in the development of cervical cancer. It was also reported that E6, E7, and E5 suppress the expression of miR-203 which participates in the differentiation of epithelial cells (McKenna et al., 2010; Greco et al., 2011). Through the suppression of miR-203, the expression level of p63 is upregulated in the differentiated cells, which might contribute to the genome amplification and the late gene expression in the upper layers of epithelium (Melar-New and Laimins, 2010).
Epigenetic Alteration Induced by the HPV Infection
There was a report describing the epigenetic alteration of cells mediated by HPV gene expression (Hsu et al., 2011; Hyland et al., 2011; McLaughlin-Drubin et al., 2011; Zheng and Wang, 2011). The epigenetic alteration induced by the HPV infection was considered to modify the miRNA expression pattern, which might change the cell differentiation program. Although there is a possibility that some epigenetic alteration occurs also in the normal cell differentiation process, there has been no report related to it.
Conclusion
Human papillomavirus suppresses its replication to a “maintenance level” or “latent infection mode” in the basal cells, and maintains the DNA synthesis potential of the infected cells detached from the basal membrane to maintain viral genome replication. In terminally differentiated cells, a tremendous level of genome amplification and late gene expression takes place. After completion of virion assembly, the virions are released externally with the cornified cells (the regulation of the differentiation-dependent lifecycle of HPV is summarized in Figure 6). One of the reasons for this unique lifecycle of HPV is escape from the immune-surveillance system (Bodily and Laimins, 2011). Because both L1 and L2 could have immunogenicity, their expressions should be suppressed until the infected cells move to the upper layer of the epithelium, where the host immune-surveillance system does not well function. The hyperproliferation induced by E6 and E7 is required for viral genome amplification and contributes to the augmentation of progeny virion production by expanding the pool of the infected cells.
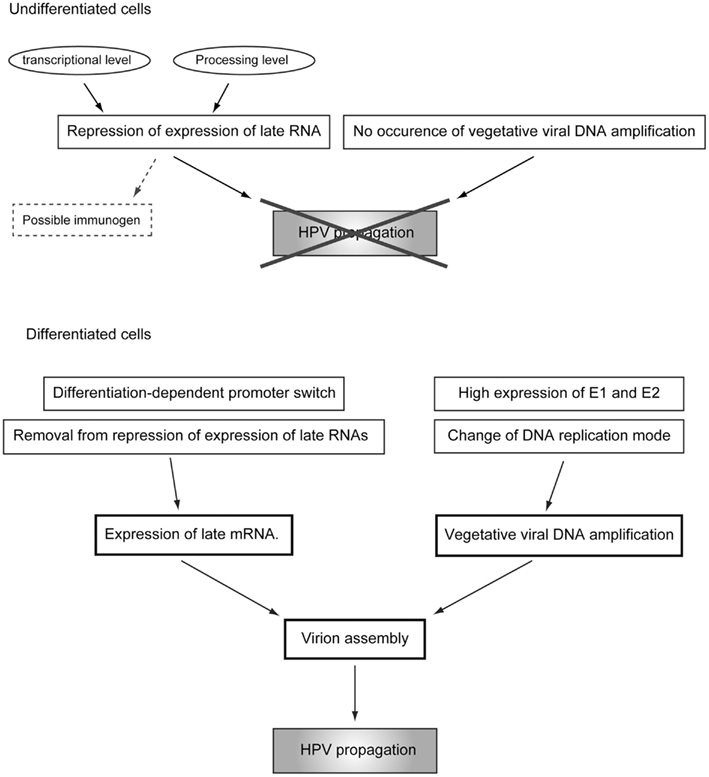
Figure 6. Regulatory mechanisms of the differentiation-dependent lifecycle of HPV.
Tight communication between the virus and the host cell is a unique character of HPV biology, and raises the possibility of using HPV as a probe to investigate the development of the stratified epithelium. In this review, we did not describe the details of the cancer progression induced by HPV infections. HPV-induced cancer is a good model for “multi-step carcinogenesis,” and the study of HPV biology provides novel insights into cancer development.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The research was supported in part by a Grant-in-Aid for Cancer Research from the Ministry of Health Labor and Welfare of Japan and a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (H. Sakai).
References
Ai, W., Narahari, J., and Roman, A. (2000). Yin yang 1 negatively regulates the differentiation-specific E1 promoter of human papillomavirus type 6. J. Virol. 74, 5198–5205.
Ai, W., Toussaint, E., and Roman, A. (1999). CCAAT displacement protein binds to and negatively regulates human papillomavirus type 6 E6, E7, and E1 promoters. J. Virol. 73, 4220–4229.
An, J., Mo, D., Liu, H., Veena, M. S., Srivatsan, E. S., Massoumi, R., and Rettig, M. B. (2008). Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-kappaB activation. Cancer Cell 14, 394–407.
Apt, D., Watts, R. M., Suske, G., and Bernard, H. U. (1996). High Sp1/Sp3 ratios in epithelial cells during epithelial differentiation and cellular transformation correlate with the activation of the HPV-16 promoter. Virology 224, 281–291.
Barreau, C., Paillard, L., and Osborne, H. B. (2005). AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 33, 7138–7150.
Bedell, M. A., Hudson, J. B., Golub, T. R., Turyk, M. E., Hosken, M., Wilbanks, G. D., and Laimins, L. A. (1991). Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J. Virol. 65, 2254–2260.
Blanpain, C., and Fuchs, E. (2009). Epidermal homeostasis: a balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 10, 207–217.
Bodily, J., and Laimins, L. A. (2011). Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol. 19, 33–39.
Bodily, J. M., Mehta, K. P., Cruz, L., Meyers, C., and Laimins, L. A. (2011). The E7 open reading frame acts in cis and in trans to mediate differentiation-dependent activities in the human papillomavirus type 16 life cycle. J. Virol. 85, 8852–8862.
Carlsson, A., and Schwartz, S. (2000). Inhibitory activity of the human papillomavirus type 1 AU-rich element correlates inversely with the levels of the elav-like HuR protein in the cell cytoplasm. Arch. Virol. 145, 491–503.
Carter, J. R., Ding, Z., and Rose, B. R. (2011). HPV infection and cervical disease: a review. Aust. N. Z. J. Obstet. Gynaecol. 51, 103–108.
Chakrabarti, O., Veeraraghavalu, K., Tergaonkar, V., Liu, Y., Androphy, E. J., Stanley, M. A., and Krishna, S. (2004). Human papillomavirus type 16 E6 amino acid 83 variants enhance E6-mediated MAPK signaling and differentially regulate tumorigenesis by notch signaling and oncogenic Ras. J. Virol. 78, 5934–5945.
Cheunim, T., Zhang, J., Milligan, S. G., McPhillips, M. G., and Graham, S. V. (2008). The alternative splicing factor hnRNP A1 is up-regulated during virus-infected epithelial cell differentiation and binds the human papillomavirus type 16 late regulatory element. Virus Res. 131, 189–198.
Chiang, C. M., Dong, G., Broker, T. R., and Chow, L. T. (1992a). Control of human papillomavirus type 11 origin of replication by the E2 family of transcription regulatory proteins. J. Virol. 66, 5224–5231.
Chiang, C. M., Ustav, M., Stenlund, A., Ho, T. F., Broker, T. R., and Chow, L. T. (1992b). Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc. Natl. Acad. Sci. U.S.A. 89, 5799–5803.
Collier, B., Goobar-Larsson, L., Sokolowski, M., and Schwartz, S. (1998). Translational inhibition in vitro of human papillomavirus type 16 L2 mRNA mediated through interaction with heterogeneous ribonucleoprotein K and poly(rC)-binding proteins 1 and 2. J. Biol. Chem. 273, 22648–22656.
Collier, B., Oberg, D., Zhao, X., and Schwartz, S. (2002). Specific inactivation of inhibitory sequences in the 5′ end of the human papillomavirus type 16 L1 open reading frame results in production of high levels of L1 proteins in human epithelial cells. J. Virol. 76, 2739–2752.
Conway, M. J., Cruz, L., Alam, S., Christensen, N. D., and Meyers, C. (2011). Differentiation-dependent interpentameric disulfide bond stabilizes native human papillomavirus type 16. PLoS ONE 6, e22427. doi:10.1371/journal.pone.0022427
Côté-Martin, A., Moody, C., Fradet-Turcotte, A., D’Abramo, C. M., Lehoux, M., Joubert, S., Poirier, G. G., Coulombe, B., Laimins, L. A., and Archambault, J. (2008). Human papillomavirus E1 helicase interacts with the WD repeat protein p80 to promote maintenance of the viral genome in keratinocytes. J. Virol. 82, 1271–1283.
Demeret, C., Le Moal, M., Yaniv, M., and Thierry, F. (1995). Control of HPV 18 DNA replication by cellular and viral transcription factors. Nucleic Acids Res. 23, 4777–4784.
Desaintes, C., and Demeret, C. (1996). Control of papillomavirus DNA replication and transcription. Semin. Cancer Biol. 7, 339–347.
Dietrich-Goetz, W., Kennedy, I. M., Levins, B., Stanley, M. A., and Clements, J. B. (1997). A cellular 65-kDa protein recognizes the negative regulatory element of human papillomavirus late mRNA. Proc. Natl. Acad. Sci. U.S.A. 94, 163–168.
Dürst, M., Glitz, D., Schneider, A., and zur Hausen, H. (1992). Human papillomavirus type 16 (HPV 16) gene expression and DNA replication in cervical neoplasia: analysis by in situ hybridization. Virology 189, 132–140.
Fang, L., Budgeon, L. R., Doorbar, J., Briggs, E. R., and Howett, M. K. (2006). The human papillomavirus type 11 E1/¥E4 protein is not essential for viral genome amplification. Virology 351, 271–279.
Fang, N. X., Gu, W., Ding, J., Saunders, N. A., Frazer, I. H., and Zhao, K. N. (2007). Calcium enhances mouse keratinocyte differentiation in vitro to differentially regulate expression of papillomavirus authentic and codon modified L1 genes. Virology 365, 187–197.
Fehrmann, F., Klumpp, D. J., and Laimins, L. A. (2003). Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 77, 2819–2831.
Filippova, M., Parkhurst, L., and Duerksen-Hughes, P. J. (2004). The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J. Biol. Chem. 279, 25729–25744.
Filippova, M., Song, H., Connolly, J. L., Dermody, T. S., and Duerksen-Hughes, P. J. (2002). The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J. Biol. Chem. 277, 21730–21739.
Flores, E. R., and Lambert, P. F. (1997). Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 71, 7167–7179.
Fradet-Toucotte, A., Bergeron-Labrecque, F., Moody, C. A., Lehoux, M., Laimins, L. A., and Archambault, J. (2011). Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 85, 8996–9012.
Fradet-Toucotte, A., Moody, C. A., Laimins, L. A., and Archambault, J. (2010). Nuclear export of human papillomavirus type 31 E1 is regulated by cdk2 phosphorylation and required for viral genome maintenance. J. Virol. 84, 11747–11760.
Frattini, M. G., Lim, H. B., and Laimins, L. A. (1996). In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc. Natl. Acad. Sci. U.S.A. 93, 3062–3067.
Furth, P. A., Choe, W. T., Rex, J. H., Byrne, J. C., and Baker, C. C. (1994). Sequences homologous to 5′ splice sites are required for the inhibitory activity of papillomavirus late 3′ untranslated regions. Mol. Cell. Biol. 14, 5278–5289.
Garnett, T. O., Filippova, M., and Duerksen-Hughes, P. J. (2006). Accelerated degradation of FADD and procaspase 8 in cells expressing human papillomavirus 16 E6 impairs TRAIL-mediated apoptosis. Cell Death Differ. 13, 1915–1926.
Genther, S. M., Sterling, S., Duensing, S., Münger, K., Sattler, C., and Lambert, P. F. (2003). Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol. 77, 2832–2842.
Gewin, L., Myers, H., Kiyono, T., and Galloway, D. A. (2004). Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes Dev. 18, 2269–2282.
Goraczniak, R., and Gunderson, S. I. (2008). The regulatory element in the 3′-untranslated region of human papillomavirus 16 inhibits expression by binding CUG-binding protein 1. J. Biol. Chem. 283, 2286–2296.
Greco, D., Kivi, N., Qian, K., Leivonen, S. K., Auvinen, P., and Auvinen, E. (2011). Human papillomavirus 16 E5 modulates the expression of host microRNAs. PLoS ONE 6, e21646. doi:10.1371/journal.pone.0021646
Griep, A. E., Herber, R., Jeon, S., Lohse, J. K., Dubielzig, R. R., and Lambert, P. F. (1993). Tumorigenicity by human papillomavirus type 16 E6 and E7 in transgenic mice correlates with alterations in epithelial cell growth and differentiation. J. Virol. 67, 1373–1384.
Gu, W., Li, M., Zhao, W. M., Fang, N. X., Bu, S., Frazer, I. H., and Zhao, K. N. (2004). tRNASer(CGA) differentially regulates expression of wild-type and codon-modified papillomavirus L1 genes. Nucleic Acids Res. 32, 4448–4461.
Hadaschik, D., Hinterkeuser, K., Oldak, M., Pfister, H. J., and Smola-Hess, S. (2003). The papillomavirus E2 protein binds to and synergizes with C/EBP factors involved in keratinocyte differentiation. J. Virol. 77, 5253–5265.
Hartley, K. A., and Alexander, K. A. (2002). Human TATA binding protein inhibits human papillomavirus type 11 DNA replication by antagonizing E1-E2 protein complex formation on the viral origin of replication. J. Virol. 76, 5014–5023.
Hong, S., Mehta, K. P., and Laimins, L. A. (2011). Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J. Virol. 85, 9486–9494.
Howley, M. P. (1996). “Papillomavirinae: the viruses and their replication,” in Fields Virology, 3rd Edn, eds B. N. Fields, D. M. Knipe, and P. M. Howley (Philadelphia: Lippincott Williams and Wilkins), 2045–2076.
Hsu, C. H., Peng, K. L., Jhang, H. C., Lin, C. H., Wu, S. Y., Chiang, C. M., Lee, S. C., Yu, W. C., and Juan, L. J. (2011). The HPV E6 oncoprotein targets histone methyltransferases for modulating specific gene transcription. Oncogene. doi:10.1038/onc.2011.415. [Epub ahead of print].
Huh, K. W., DeMasi, J., Ogawa, H., Nakatani, Y., Howley, P. M., and Münger, K. (2005). Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc. Natl. Acad. Sci. U.S.A. 102, 11492–11497.
Hummel, M., Hudson, J. B., and Laimins, L. A. (1992). Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 66, 6070–6080.
Hyland, P. L., McDade, S. S., McCloskey, R., Dickson, G. J., Arthur, K., McCance, D. J., and Patel, D. (2011). Evidence for alteration of EZH2, BMI1, and KDM6A and epigenetic reprogramming in human papillomavirus type 16 E6/E7-expressing keratinocytes. J. Virol. 85, 10999–11006.
Jeon, S., and Lambert, P. F. (1995). Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 92, 1654–1658.
Jia, R., Liu, X., Tao, M., Kruhlak, M., Guo, M., Meyers, C., Baker, C. C., and Zheng, Z. M. (2009). Control of the papillomavirus early-to late switch by differentially expressed SRp20. J. Virol. 83, 167–180.
Jones, P. H., Simons, B. D., and Watt, F. M. (2007). Sic transit gloria: farewell to the epidermal transit amplifying cell? Cell Stem Cell 1, 371–381.
Kadaja, M., Isok-Paas, H., Laos, T., Ustav, E., and Ustav, M. (2009). Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 5, e1000397. doi:10.1371/journal.ppat.1000397
Kennedy, I. M., Haddow, J. K., and Clements, J. B. (1991). A negative regulatory element in the human papillomavirus type 16 genome acts at the level of late mRNA stability. J. Virol. 65, 2093–2097.
Kim, K., Garner-Hamrick, P. A., Fisher, C., Lee, D., and Lambert, P. F. (2003). Methylation patterns of papillomavirus DNA, its influence on E2 function, and implications in viral infection. J. Virol. 77, 12450–12459.
Kines, R. C., Thompson, C. D., Lowy, D. R., Schiller, J. T., and Day, P. M. (2009). The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. U.S.A. 106, 20458–20463.
Koffa, M. D., Graham, S. V., Takagaki, Y., Manley, J. L., and Clements, J. B. (2000). The human papillomavirus type 16 negative regulatory RNA element interacts with three proteins that act at different posttranscriptional levels. Proc. Natl. Acad. Sci. U.S.A. 97, 4677–4682.
Kukimoto, I., and Kanda, T. (2001). Displacement of YY1 by differentiation-specific transcription factor hSkn-1a activates the P(670) promoter of human papillomavirus type 16. J. Virol. 75, 9302–9311.
Kukimoto, I., Mori, S., Sato, H., Takeuchi, T., and Kanda, T. (2008). Transcription factor human Skn-1a enhances replication of human papillomavirus DNA through the direct binding to two sites near the viral replication origin. FEBS J. 275, 3123–3135.
Kumar, A., Zhao, Y., Meng, G., Zeng, M., Srinivasan, S., Delmolino, L. M., Gao, Q., Dimri, G., Weber, G. F., Wazer, D. E., Band, H., and Band, V. (2002). Human papillomavirus oncoprotein E6 inactivates the transcriptional coactivator human ADA3. Mol. Cell. Biol. 22, 5801–5812.
Lace, M. J., Anson, J. R., Turek, L. P., and Haugen, T. H. (2008). Functional mapping of the human papillomavirus type 16 E1 cistron. J. Virol. 82, 10724–10734.
Lathion, S., Schaper, J., Beard, P., and Raj, K. (2003). Notch1 can contribute to viral-induced transformation of primary human keratinocytes. Cancer Res. 63, 8687–8694.
Lechner, M. S., and Laimins, L. A. (1994). Inhibition of p53 DNA binding by human papillomavirus E6 proteins. J. Virol. 68, 4262–4273.
Longworth, M. S., and Laimins, L. A. (2004). Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 68, 362–372.
Mack, D. H., and Laimins, L. A. (1991). A keratinocyte-specific transcription factor, KRF-1, interacts with AP-1 to activate expression of human papillomavirus type 18 in squamous epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 88, 9102–9106.
McBride, A. A., Oliveila, J. G., and McPhillips, M. G. (2006). Partitioning viral genomes in mitosis: same idea, different targets. Cell Cycle 5, 1499–1502.
McCormack, S. J., Brazinski, S. E., Moore, J. L., Werness, B. A., and Goldstein, D. J. (1997). Activation of the focal adhesion kinase signal transduction pathway in cervical carcinoma cell lines and human genital epithelial cells immortalized with human papillomavirus type 18. Oncogene 15, 265–274.
McKenna, D. J., McDade, S. S., Patel, D., and McCance, D. J. (2010). MicroRNA 203 expression in keratinocytes is dependent on regulation of p53 levels by E6. J. Virol. 85, 10644–10652.
McLaughlin-Drubin, M. E., Crum, C. P., and Münger, K. (2011). Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. U.S.A. 108, 2130–2135.
McPhillips, M. G., Oliveira, J. G., Spindler, J. E., Mitra, R., and McBride, A. A. (2006). Brd4 is required for E2-mediated transcriptional activation but not genome partitioning of all papillomavirus. J. Virol. 80, 9530–9543.
McPhillips, M. G., Veerapraditsin, T., Cumming, S. A., Karali, D., Milligan, S. G., Boner, W., Morgan, I. M., and Graham, S. V. (2004). SF2/ASF binds the human papillomavirus type 16 late RNA control element and is regulated during differentiation of virus-infected epithelial cells. J. Virol. 78, 10598–10605. doi:10.1371/journal.ppat.1000605
Melar-New, M., and Laimins, L. A. (2010). Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J. Virol. 84, 5212–5221.
Moody, C. A., Fradet-Turcotte, A., Archambault, J., and Laimins, L. A. (2007). Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc. Natl. Acad. Sci. U.S.A. 104, 19541–19546.
Moody, C. A., and Laimins, L. A. (2009). Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 5, e1000605.
Moody, C. A., and Laimins, L. A. (2010). Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer 10, 550–560.
Mori, S., Ozaki, S., Yasugi, T., Yoshikawa, H., Taketani, Y., and Kanda, T. (2006). Inhibitory cis-element-mediated decay of human papillomavirus type 16 L1-transcript in undifferentiated cells. Mol. Cell. Biochem. 288, 47–57.
Münger, K., Baldwin, A., Edwards, K. M., Hayakawa, H., Nguyen, C. L., Owens, M., Grace, M., and Huh, K. (2004). Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 78, 11451–11460.
Nakahara, T., Peh, W. L., Doorbar, J., Lee, D., and Lambert, P. F. (2005). Human papillomavirus type 16 E1circumflexE4 contributes to multiple facets of the papillomavirus life cycle. J. Virol. 79, 13150–13165.
Oberg, D., Fay, J., Lambkin, H., and Schwartz, S. (2005). A downstream polyadenylation element in human papillomavirus type 16 L2 encodes multiple GGG motifs and interacts with hnRNP H. J. Virol. 79, 9254–9269.
Ozbun, M. A., and Meyers, C. (1997). Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J. Virol. 71, 5161–5172.
Parkin, D. M., and Bray, F. (2006). Chapter 2: the burden of HPV-related cancers. Vaccine 24(Suppl. 3), S3/11–S3/25.
Patel, D., Huang, S. M., Baglia, L. A., and McCance, D. J. (1999). The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 18, 5061–5072.
Remm, M., Remm, A., and Ustav, M. (1999). Human papillomavirus type 18 E1 protein is translated from polycistronic mRNA by a discontinuous scanning mechanism. J. Virol. 73, 3062–3070.
Rush, M., Zhao, X., and Schwartz, S. (2005). A splicing enhancer in the E4 coding region of human papillomavirus type 16 is required for early mRNA splicing and polyadenylation as well as inhibition of premature late gene expression. J. Virol. 79, 12002–12015.
Sakakibara, N., Mitra, R., and McBride, A. A. (2011). The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 85, 8981–8995.
Sokolowski, M., Tan, W., Jellne, M., and Schwartz, S. (1998). mRNA instability elements in the human papillomavirus type 16 L2 coding region. J. Virol. 72, 1504–1515.
Somberg, M., Zhao, X., Fröhlich, M., Evander, M., and Schwartz, S. (2008). Polypyrimidine tract binding protein induces human papillomavirus type 16 late gene expression by interfering with splicing inhibitory elements at the major late 5′ splice site, SD3632. J. Virol. 82, 3665–3678.
Song, H., Moseley, P. L., Lowe, S. L., and Ozbun, M. A. (2010). Inducible heat shock protein 70 enhances HPV31 viral genome replication and virion production during the differentiation-dependent life cycle in human keratinocytes. Virus Res. 147, 113–122.
Steger, G., and Corbach, S. (1997). Dose-dependent regulation of the early promoter of human papillomavirus type 18 by the viral E2 protein. J. Virol. 71, 50–58.
Stubenrauch, F., Lim, H. B., and Laimins, L. A. (1998). Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J. Virol. 72, 1071–1077.
Stünkel, W., and Bernard, H. U. (1999). The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J. Virol. 73, 1918–1930.
Tan, S. H., Gloss, B., and Bernard, H. U. (1992). During negative regulation of the human papillomavirus-16 E6 promoter, the viral E2 protein can displace Sp1 from a proximal promoter element. Nucleic Acids Res. 20, 251–256.
Terenzi, F., Saikia, P., and Sen, G. C. (2008). Interferon-induced protein, p56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J. 27, 3311–3321.
Terhune, S. S., Hubert, W. G., Thomas, J. T., and Laimins, L. A. (2001). Early polyadenylation signals of human papillomavirus type 31 negatively regulate capsid gene expression. J. Virol. 75, 8147–8157.
Thierry, F. (2009). Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma. Virology 384, 375–379.
Thomas, J. T., Hubert, W. G., Ruesch, M. N., and Laimins, L. A. (1999). Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. U.S.A. 96, 8449–8454.
Thomas, M., Narayan, N., Pim, D., Tomaic, V., Massimi, P., Nagasaka, K., Kranjec, C., Gammoh, N., and Banks, L. (2008). Human papillomaviruses, cervical cancer and cell polarity. Oncogene 27, 7018–7030.
Vinokurova, S., and von Knebel Doeberitz, M. (2011). Differential methylation of the HPV 16 upstream regulatory region during epithelial differentiation and neoplastic transformation. PLoS ONE 6, e24451. doi:10.1371/journal.pone.0024451
Wiklund, L., Sokolowski, M., Carlsson, A., Rush, M., and Schwartz, S. (2002). Inhibition of translation by UAUUUAU and UAUUUUUAU motifs of the AU-rich RNA instability element in the HPV-1 late 3′ untranslated region. J. Biol. Chem. 277, 40462–40471.
Wilson, R., Fehrmann, F., and Laimins, L. A. (2005). Role of the E1–E4 protein in the differentiation-dependent life cycle of human papillomavirus type 31. J. Virol. 79, 6732–6740.
Wilson, R., Ryan, G. B., Knight, G. L., Laimins, L. A., and Roberts, S. (2007). The full-length E1E4 protein of human papillomavirus type 18 modulates differentiation-dependent viral DNA amplification and late gene expression. Virology 362, 453–460.
Wooldridge, T. R., and Laimins, L. A. (2008). Regulation of human papillomavirus type 31 gene expression during the differentiation-dependent lifecycle through histone modifications and transcription factor binding. Virology 374, 371–380.
Wu, S.-Y., Lee, A.-Y., Hou, S. Y., Kemper, J. K., Erdjument-Bromage, H., Tempst, P., and Chiang, C.-M. (2012). Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 20, 2383–2396.
Zhao, X., Fay, J., Lambkin, H., and Schwartz, S. (2007). Identification of a 17-nucleotide splicing enhancer in HPV-16 L1 that counteracts the effect of multiple hnRNP A1-binding splicing silencers. Virology 369, 351–363.
Zhao, X., Rush, M., and Schwartz, S. (2004). Identification of an hnRNP A1-dependent splicing silencer in the human papillomavirus type 16 L1 coding region that prevents premature expression of the late L1 gene. J. Virol. 78, 10888–10905.
Zheng, Z. M., and Wang, X. (2011). Regulation of cellular miRNA expression by human papillomaviruses. Biochim. Biophys. Acta 1809, 668–677.
Keywords: HPV, differentiation, epithelial cell, keratinocyte
Citation: Kajitani N, Satsuka A, Kawate A and Sakai H (2012) Productive lifecycle of human papillomaviruses that depends upon squamous epithelial differentiation. Front. Microbio. 3:152. doi: 10.3389/fmicb.2012.00152
Received: 28 December 2011; Accepted: 02 April 2012;
Published online: 24 April 2012.
Edited by:
Akio Adachi, The University of Tokushima Graduate School, JapanReviewed by:
Iwao Kukimoto, National Institute of Infectious Diseases, JapanSeiichiro Mori, National Institute of Infectious Diseases, Japan
Copyright: © 2012 Kajitani, Satsuka, Kawate and Sakai. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hiroyuki Sakai, Laboratory of Gene Analysis, Department of Viral Oncology, Institute for Virus Research, Kyoto University, Kyoto 606-8507, Japan. e-mail:aHNha2FpQHZpcnVzLmt5b3RvLXUuYWMuanA=