
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 17 April 2012
Sec. Evolutionary and Genomic Microbiology
Volume 3 - 2012 | https://doi.org/10.3389/fmicb.2012.00132
 Stephen M. Techtmann1,2
Stephen M. Techtmann1,2 Alexander V. Lebedinsky3
Alexander V. Lebedinsky3 Albert S. Colman4
Albert S. Colman4 Tatyana G. Sokolova3 Tanja Woyke5 Lynne Goodwin5
Tatyana G. Sokolova3 Tanja Woyke5 Lynne Goodwin5 Frank T. Robb1,2*
Frank T. Robb1,2*Carbon monoxide (CO) is commonly known as a toxic gas, yet both cultivation studies and emerging genome sequences of bacteria and archaea establish that CO is a widely utilized microbial growth substrate. In this study, we determined the prevalence of anaerobic carbon monoxide dehydrogenases ([Ni,Fe]-CODHs) in currently available genomic sequence databases. Currently, 185 out of 2887, or 6% of sequenced bacterial and archaeal genomes possess at least one gene encoding [Ni,Fe]-CODH, the key enzyme for anaerobic CO utilization. Many genomes encode multiple copies of [Ni,Fe]-CODH genes whose functions and regulation are correlated with their associated gene clusters. The phylogenetic analysis of this extended protein family revealed six distinct clades; many clades consisted of [Ni,Fe]-CODHs that were encoded by microbes from disparate phylogenetic lineages, based on 16S rRNA sequences, and widely ranging physiology. To more clearly define if the branching patterns observed in the [Ni,Fe]-CODH trees are due to functional conservation vs. evolutionary lineage, the genomic context of the [Ni,Fe]-CODH gene clusters was examined, and superimposed on the phylogenetic trees. On the whole, there was a correlation between genomic contexts and the tree topology, but several functionally similar [Ni,Fe]-CODHs were found in different clades. In addition, some distantly related organisms have similar [Ni,Fe]-CODH genes. Thermosinus carboxydivorans was used to observe horizontal gene transfer (HGT) of [Ni,Fe]-CODH gene clusters by applying Kullback–Leibler divergence analysis methods. Divergent tetranucleotide frequency and codon usage showed that the gene cluster of T. carboxydivorans that encodes a [Ni,Fe]-CODH and an energy-converting hydrogenase is dissimilar to its whole genome but is similar to the genome of the phylogenetically distant Firmicute, Carboxydothermus hydrogenoformans. These results imply that T carboxydivorans acquired this gene cluster via HGT from a relative of C. hydrogenoformans.
Carbon monoxide (CO) is most commonly known as a potent human toxin. Alternatively, CO can provide an energy and carbon source in both anaerobic and aerobic microbes (Uffen, 1976; King and Weber, 2007; Sokolova et al., 2009; Techtmann et al., 2009). This ability to utilize CO relies upon enzymes known as carbon monoxide dehydrogenases (CODHs, EC 1.2.99.2; Ferry, 1995; Seravalli et al., 1995, 1997; Ragsdale, 2008). CO oxidation is performed by aerobic and anaerobic species using CODHs in different classes (Ragsdale, 2004; King and Weber, 2007). While the overall reaction catalyzed by both aerobic and anaerobic species is the same (CO + H2O → CO2 + 2H+ + 2e−), major differences result from the different terminal electron acceptors of the pathway. For aerobic species this reaction is catalyzed by the aerobic CODH (CoxSML) complex (Hugendieck and Meyer, 1992; Schubel et al., 1995). The Cox-type CODH contains a highly conserved Mo based active site (Meyer and Schlegel, 1983; Meyer and Rajagopalan, 1984; Meyer et al., 1993; Dobbek et al., 1999; Gnida et al., 2003). In the case of the aerobic Cox-type CODH, the electrons generated from oxidation of CO are transferred to oxygen or, in some cases, nitrate as the final electron acceptor (King, 2003, 2006; King and Weber, 2007). Anaerobic CODHs are distinct from Cox CODHs and have a Ni–Fe active site (Dobbek et al., 2001, 2004; Svetlitchnyi et al., 2001, 2004). The anaerobic CODH generates electrons from the oxidation of CO and transfers them to a variety of acceptors, allowing CO to be the source of reducing equivalents for various pathways including sulfate reduction, acetogenesis, methanogenesis, hydrogenogenesis, and metal reduction, and it can also reduce CO2 to CO used further for acetate synthesis by the Wood–Ljungdahl pathway (Gonzalez and Robb, 2000; Svetlitchnyi et al., 2001, 2004; Ragsdale, 2004, 2008; Wu et al., 2005; Sokolova et al., 2009).
The extreme thermophile Carboxydothermus hydrogenoformans is a prototype anaerobic carboxydotroph with five distinct anaerobic CODHs. C. hydrogenoformans can grow efficiently on CO as sole carbon and energy source, confirming that CO can fuel divergent pathways (Wu et al., 2005). The presence of multiple anaerobic CODHs encoded by a single organism requires that these homologs arose either by duplication of an ancestral CODH gene in the same lineage or else have been acquired by horizontal gene transfer (HGT) between separate lineages.
In this study we identified [Ni,Fe]-CODH genes in the rapidly expanding database of microbial genomes in order to understand the mechanisms of evolution and dispersion of [Ni,Fe]-CODHs. A comprehensive phylogenetic analysis was designed to observe if CODHs are found in distinct clades. Examination of the genomic context of each of the [Ni,Fe]-CODHs provided insights into the functions of [Ni,Fe]-CODH gene clusters. We then used these functional assignments to determine whether differences in inferred function of [Ni,Fe]-CODHs explain the pattern of phylogenetic divergence. In addition, in-depth statistical analysis was performed on two gene clusters that catalyze similar functions. By examining operons with the same function we control for differences that have arisen based on functional diversification. The two gene clusters examined were from the prototype thermophilic hydrogenogen – C. hydrogenoformans – and a thermophilic metal reducing hydrogenogen – Thermosinus carboxydivorans. These organisms were chosen based on the following criteria. (1) They are both thermophilic, thus controlling for selection due to adaptations to differing growth temperatures. (2) C. hydrogenoformans and T. carboxydivorans exhibit different types of cell wall structure and are members of widely divergent classes of the Phylum Firmicutes: the Clostridia and Negativicutes, respectively. (3) Despite their divergent lineages, their [Ni,Fe]-CODHs and the linked energy-converting hydrogenases (ECH) are very similar. The genomes of these bacteria were analyzed to determine whether the [Ni,Fe]-CODH–ECH gene cluster may have been subject to recent HGT.
The US Department of Energy’s Integrated Microbial Genome (IMG) database was searched using BLASTp to query the protein database (Altschul et al., 1990) for sequences corresponding to [Ni,Fe]-CODHs. [Ni,Fe]-CODHs are subdivided into two types: the type called CooS, which is more frequent in bacteria, and the type called Cdh, occurring almost exclusively in archaea. The ligands of the Ni,Fe-containing active site (Dobbek’s C cluster; Dobbek et al., 2001) are conserved, with few exceptions, in CooS- and Cdh-type CODHs (Lindahl, 2002). However, Cdh-type CODHs harbor two additional [Fe4S4] clusters (Gencic et al., 2010), and, on the whole, there is rather low homology between CooS- and Cdh-type CODHs. Therefore, the IMG database was searched using a CooS-type sequence (CooS-I from C. hydrogenoformans) and a Cdh-type sequence (Cdh-1 from the archaeon Archeoglobus fulgidus). Overlapping results and low scoring hits (Quality scores less than 200) were removed from the final [Ni,Fe]-CODH database (Table S3 in Supplemental Material). Many of the low scoring hits were often genes annotated as hydroxylamine reductases. This refined database was then used for further analysis.
The [Ni,Fe]-CODH sequences were aligned using the MUSCLE alignment program (Edgar, 2004a,b). This multiple sequence alignment was used to construct phylogenetic trees using Maximum Likelihood (ML) and Bayesian inference (BI). ML trees were constructed using the RAxML-HPC2 program (version 7.2.8; Stamatakis, 2006; Stamatakis et al., 2008) on the CIPRES servers (Miller et al., 2010). ML trees were constructed using the WAG + Γ + I model which was selected as the best-fit model for our database by the model selection tool implemented in Topali2 (Milne et al., 2009). Node support was assessed by 1,000 bootstrap replicates with the same model. BI trees were constructed using the BEAST program (Drummond and Rambaut, 2007) with the WAG + Γ + I model as was selected as the best-fit model by the Topali2 package. Searches were run with four chains of 7,500,000 generations, for which the first 750,000 were discarded as “burn-in.” Trees were sampled every 1,000 generations. Stabilization of chain parameters was determined using the program TRACER (Rambaut and Aj, 2007). A maximum credibility clade tree was annotated using the TreeAnnotator program (part of the BEAST package). Trees were drawn using the FigTree program1. Subtrees were drawn of each of the clades and are shown in (Figure A2 in Appendix).
The analysis of the genomic contexts was done using the information available at Gene Detail pages of the IMG database and, where necessary, applying tblastn with appropriate queries at the IMG website2. This information was used to identify whether the [Ni,Fe]-CODH gene was located (1) within an ACS gene cluster, (2) adjacent to an ECH gene cluster, (3) located at a distance less than 3 kb away from a gene encoding CooF, a ferredoxin-like FeS protein (Kerby et al., 1992) which carries out electron transfer from CooS to a variety of electron acceptors, or (4) not clustered with a cooF gene.
Thermosinus carboxydivorans was grown as previously described (Sokolova et al., 2004). DNA was extracted using previously described protocols (Wu et al., 2005). The genome of T. carboxydivorans Nor1 was sequenced at the Joint Genome Institute (JGI) using a combination of 3, 8, and 40 kb (fosmid) DNA libraries. In addition to Sanger sequencing, 454 pyrosequencing was done to a depth of 20× coverage. All general aspects of library construction and sequencing performed at the JGI can be found at http://www.jgi.doe.gov/.
Draft assemblies were based on 28,812 total reads. All three libraries provided 9.3× coverage of the genome. The Phred/Phrap/Consed software package3 was used for sequence assembly and quality assessment (Ewing and Green, 1998; Ewing et al., 1998; Gordon et al., 1998). After the shotgun stage, reads were assembled with parallel Phrap (High Performance Software, LLC). Possible mis-assemblies were corrected with Dupfinisher (Han and Chain, 2006) or transposon bombing of bridging clones (Epicentre Biotechnologies, Madison, WI, USA). Gaps between contigs were closed by editing in Consed, custom primer walk or PCR amplification (Roche Applied Science, Indianapolis, IN, USA).
A total of 5919 additional reactions were necessary to close gaps and to elevate the quality of the finished sequence. The completed genome sequences of T. carboxydivorans Nor1 contains 36,788 reads, achieving an average of 10-fold sequence coverage per base with an error rate less than 1 in 100,000. The draft genome sequence was deposited into GenBank (Accession Numbers AAWL01000001–AAWL01000049).
The nucleotide sequences for the [Ni,Fe]-CODH–ECH gene cluster were extracted (including genes of all hydrogenase subunits) from the C. hydrogenoformans and the T. carboxydivorans genomes. These sequences and the whole genome sequences for both organisms were used for further analysis. Additionally, the Escherichia coli K12 genome (GenBank Accession Number U00096.2) was used as a control.
G + C content was determined using GC-Profile program (Gao and Zhang, 2006). Tetranucleotide frequency was determined using the TETRA program (Teeling et al., 2004). Codon usage was determined from tables in the codon usage database4. Overall codon usage for the 10 genes of the [Ni,Fe]-CODH-ECH gene cluster was determined by averaging the codon frequency of each gene in the gene cluster.
The Kullback–Leibler (K–L) divergence metric (Kullback and Leibler, 1951) was used to determine whether the differences in G + C content, tetranucleotide frequency, and codon usage were significant or not. These three metrics and the K–L divergence were chosen based on a recent paper that suggests that codon usage and tetranucleotide frequency combined with K–L divergence are the most accurate metrics for determining HGT (Becq et al., 2010). K–L divergence was determined using the following formula:
where g is the a parameter (e.g., frequency of a particular tetranucleotide or frequency of usage of a particular codon) for the gene or operon and G is that same parameter for the whole genome. This calculation is repeated for all of the tetranucleotide frequencies and the resulting values are added together to determine the K–L divergence for tetranucleotide frequency. The same basic calculation is done for the frequency of usage for each codon to determine the K–L divergence for codon usage between a gene or operon and a genome.
BLAST searches of the IMG database revealed that a surprising number of organisms encode [Ni,Fe]-CODH genes. Of the 2887 extant bacterial and archaeal genomes, 185 genomes (>6%) encoded at least one [Ni,Fe]-CODH gene. Of these 185 genomes, 43% encoded more than one [Ni,Fe]-CODH (Listed in Table S2 in Supplemental Material). The highest number of [Ni,Fe]-CODHs detected in a single genome is five, found in the genome sequences of C. hydrogenoformans, the Mono Lake isolate delta Proteobacterium MLMS-1, and the methanogenic archaeon Methanosarcina acetivorans. Many of the species that encode multiple [Ni,Fe]-CODHs have been described as having the ability to utilize CO for both energy conservation and carbon acquisition, or energy conservation alone. Several of the gene clusters have genomic contexts that indicate probable primary functions such as metal reduction, oxidative stress responses, and cofactor reduction. In addition, the important human pathogens Clostridium difficile and Clostridium botulinum both possess two copies of [Ni,Fe]-CODHs, suggesting a role for [Ni,Fe]-CODHs in their physiology. The majority of [Ni,Fe]-CODHs found in our analysis occur in strains whose modes of CO utilization are not yet established, and therefore more CO-related physiological traits are likely to be discovered in the future.
A dataset of all of the [Ni,Fe]-CODHs from the IMG database was used to reconstruct the phylogeny of [Ni,Fe]-CODHs. Both ML and BI phylogenetic analyses were used. The phylogenetic trees (Figures 1A,B) constructed from both methods were congruent, therefore only the ML trees are shown in subsequent figures. These trees both detail six distinct, strongly supported clades (greater than 50% boostraps for ML trees, with most nodes supported at >86%, and Bayesian posterior support of greater than or equal to 0.99). While the branching pattern within clades differs slightly between ML and BI, the composition of all six clades in both ML and BI trees are identical (Table S3 in Supplemental Material). The distribution of sequences within these clades greatly expands our knowledge of the organisms that encode [Ni,Fe]-CODHs as well as the potential mechanisms for their evolution.
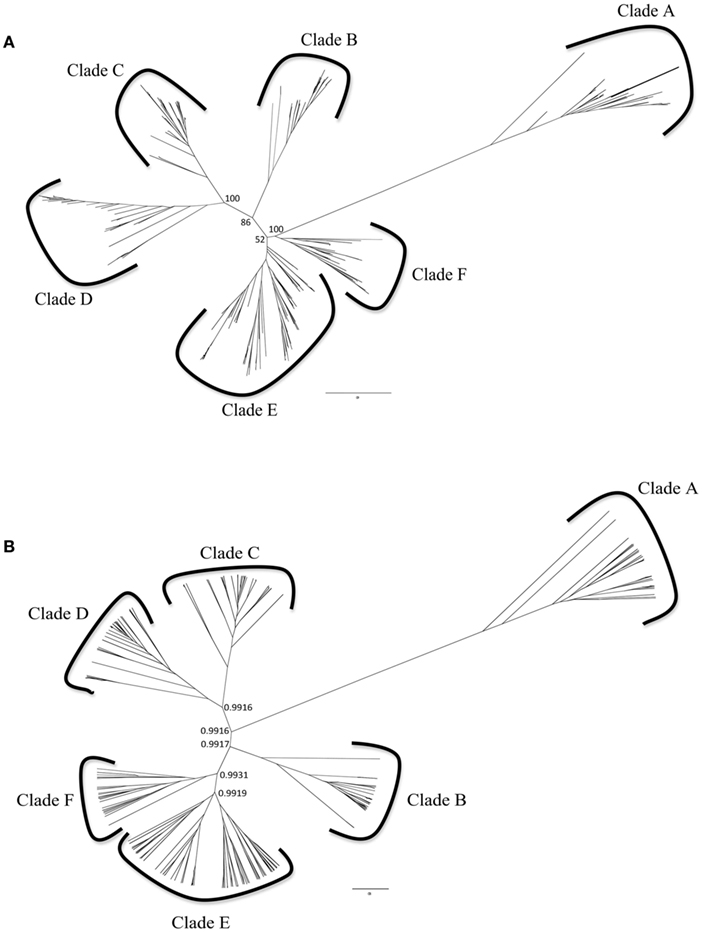
Figure 1. Unrooted phylogenetic trees of the [Ni,Fe]-CODH amino acid sequences encoded by IMG genomes were constructed from (A) Maximum Likelihood and (B) Bayesian Inference algorithms. Bootstrap values as a percentage of 1000 replicates or Bayesian posterior probabilities are shown for the deep branches defining the six [Ni,Fe]-CODH clades. The compositions of all six clades in both ML and BI trees are identical (see Figure A1 in Appendix and Table S3 in Supplemental Material).
Analysis of the genomic context was performed so that the [Ni,Fe]-CODH sequences were binned according to four categories: (1) within an acetyl-CoA synthase gene cluster, (2) adjacent to an ECH gene cluster, (3) adjacent to a cooF electron transfer protein, and (4) not adjacent to a cooF gene. These categories were plotted on the ML phylogenetic tree displayed in a circular format which was color coded according to the genomic context of the CODHs (Figure 2).
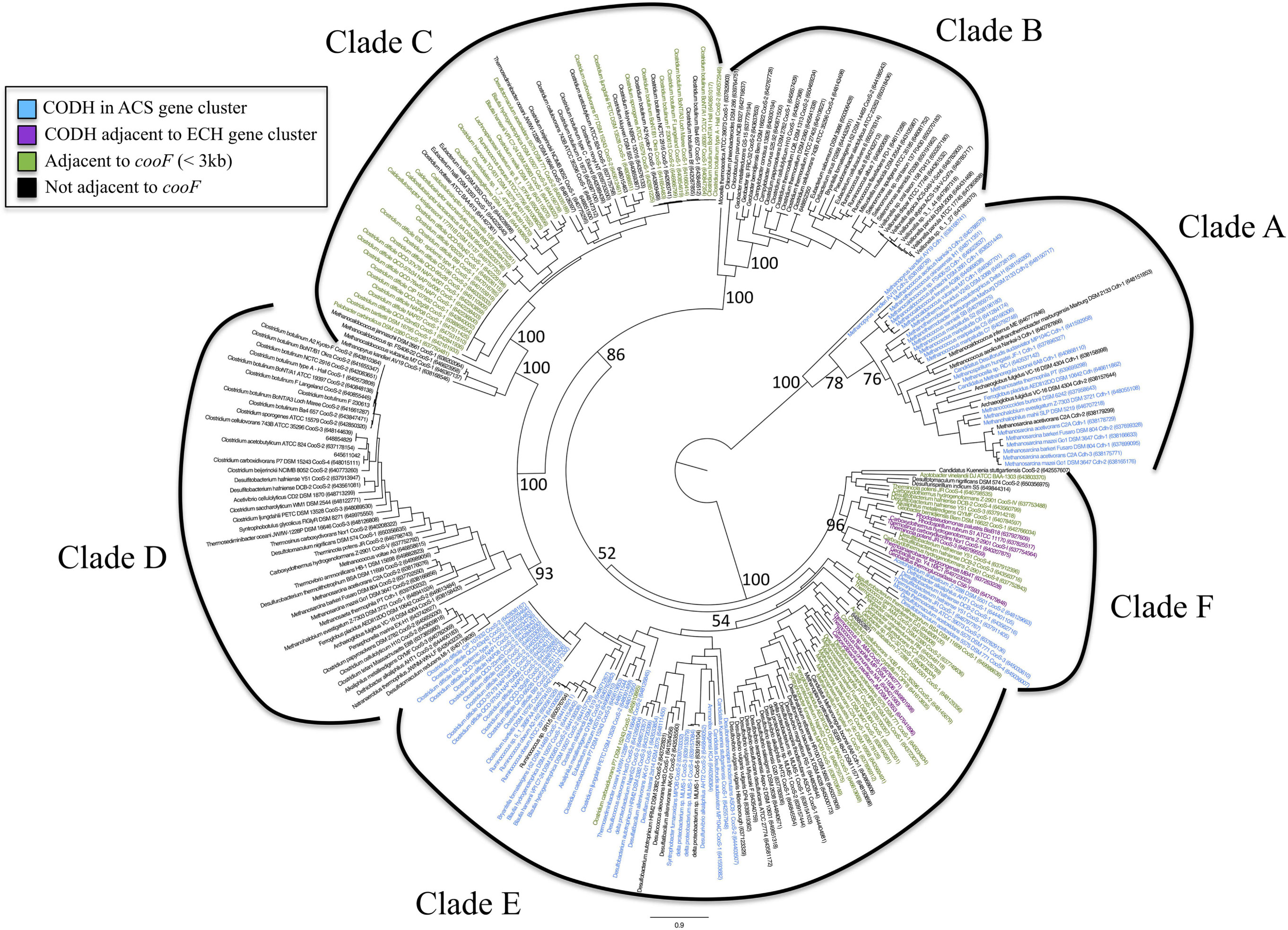
Figure 2. Maximum likelihood phylogeny from Figure 1A drawn as an unrooted circular tree. IMG Gene ID numbers are shown in parentheses after sequence names. The genomic context for each of the [Ni,Fe]-CODH sequences is shown by coloring of the tip labels. Blue indicates CODH occurrence within an ACS gene cluster. Purple indicates CODH adjacent to an energy-converting hydrogenase gene cluster. Green indicates that the CODH is adjacent to a cooF gene. Black indicates that the CODH is not linked to a cooF gene. Bootstrap values are shown as a percentage derived from 1000 replicates.
Clade A is composed of sequences corresponding to the Cdh-type CODHs, which occur almost exclusively in the Archaea and for the most part cluster in the genomes as part of the five-subunit acetyl-CoA decarbonylase/synthase (ACDS) complex (Terlesky et al., 1986; Grahame, 1991; Kocsis et al., 1999; Gencic et al., 2010). Only one bacterium, the deep subsurface pioneer species Candidatus Desulforudis audaxviator, has a typical archaeal Cdh-type ACDS complex (Chivian et al., 2008).
Clade B contains [Ni,Fe]-CODHs from an eclectic group of Bacteria, the majority comprising either gut microbiota or putative cellulose degraders. This clade includes species found in diverse enteric environments ranging from cow rumen isolates such as Ruminococcus spp. to constituents of the human gut microbiome such as Campylobacter spp. However, this clade also includes metal reducing Geobacter species, as well as two phototrophs, Chlorobium phaeobacteroides and Chlorobaculum parvum. These phototrophs and the second of two CooS sequences from the homoacetogen Moorella thermoacetica form the most deeply branching members of this clade. None of the sequences in Clade B is linked to any other known CO-related genes.
Clade C is composed primarily of clostridial species. Most of the strains within this clade are found as members of the human microbiome. A large proportion of the [Ni,Fe]-CODH sequences within this clade are one of two CooS homologs found in many strains of the human pathogens C. botulinum and C. difficile. A large number of these sequences are found adjacent to cooF genes, suggesting that these [Ni,Fe]-CODHs are able to use CooF to transfer electrons to other functional proteins. Thus it is possible that these [Ni,Fe]-CODHs are coupled to a broad range of functional processes including disease-related physiology. Further biochemical investigation is required to determine the function of the [Ni,Fe]-CODHs within this clade. The most deeply branching members of this clade are unlinked [Ni,Fe]-CODHs from hyperthermophilic methanogens including several Methanocaldococcus species and Methanopyrus kandleri. The organisms within this clade thus span an extraordinary phylogenetic range, from Bacteria through Euryarchaeota and likely participate in diverse pathways for CO utilization.
All of the [Ni,Fe]-CODH sequences within Clade D were found to be “lone” cooS genes, that is, not found in genomic context with known CO-metabolism related genes. The most deeply branching group of Clade D is a subclade composed of a few alkaliphilic bacteria. Another deeply branching group in Clade D is from Euryarchaeal species, either methanogens or Archaeoglobaceae. One of the subgroups of Clade D contains sequences from the second class of C. botulinum [Ni,Fe]-CODH sequences among other Clostridium spp. A third group of sequences within Clade D is composed of sequences similar to CooS-V from C. hydrogenoformans. The C. hydrogenoformans CooS-V was described in the genome analysis of C hydrogenoformans as being unique, since it is the only one of the five [Ni,Fe]-CODH genes in this genome without a putative function, and is not directly linked to other genes that could provide a contextual clue as to its function (Wu et al., 2005). The majority of Clade D [Ni,Fe]-CODH genes are encoded in genomes that also encode other [Ni,Fe]-CODH genes.
Clade E is the largest and most eclectic clade, composed of several subclades. Representatives of each of the four functional categories used in this study are found in Clade E. One of the Clade E subclades is dominated by clostridial and delta proteobacterial [Ni,Fe]-CODHs found within ACS gene clusters. This group of sequences includes the majority of the known bacterial CODH/ACS gene clusters. Another large subclade in Clade E contains sequences from sulfate reducing bacteria as well as methanogenic archaea. A noteable feature of one more subclade within Clade E is a group of [Ni,Fe]-CODHs from Thermococcus spp. that are linked to ECH in these Euryarchaeota. These thermococci have the ability to couple the anaerobic oxidation of CO to hydrogen production, via a conserved [Ni,Fe]-CODH-ECH gene cluster (Lim et al., 2010).
Clade F is numerically small, however members in all of the four functional categories are represented in this group of CODHs. The four C. hydrogenoformans [Ni,Fe]-CODH gene clusters in Clade F have been assigned different metabolic functions based on biochemical characterization as well as their genetic linkages to other functional genes (Wu et al., 2005). For example, the well-characterized [Ni,Fe]-CODH–ECH gene clusters originally described in R. rubrum and C. hydrogenoformans are assigned to this clade. In addition to these well-characterized [Ni,Fe]-CODH–ECH operons there are several other ECH-clustered CODHs within Clade F. Another subgroup within Clade F contains bacterial CODH/ACS gene clusters.
Clade F also contains many characterized [Ni,Fe]-CODHs with representatives from each of the four functional classes used in this study. Many of these have been characterized structurally and biochemically (Svetlitchnyi et al., 2001, 2004; Soboh et al., 2002; Volbeda and Fontecilla-Camps, 2005). These biochemical studies establish Clade F as encompassing the greatest known functional diversity of [Ni,Fe]-CODH gene clusters.
The genome of T. carboxydivorans encodes three [Ni,Fe]-CODH homologs. One of these homologs is highly similar to the hydrogenase-linked [Ni,Fe]-CODH from C. hydrogenoformans. A second [Ni,Fe]-CODH is highly similar to the CODH-V from C. hydrogenoformans. The third [Ni,Fe]-CODH homolog however is distinct from all known [Ni,Fe]-CODH sequences (Figure 3). Other known [Ni,Fe]-CODH sequences are 600–800 amino acid residues long. This divergent mini-CooS has only 482 amino acid residues and its short length does not result from frameshift truncation. Upon alignment with other [Ni,Fe]-CODH sequences, it is apparent that its small size can be accounted for by many deletions of varying size relative to standard size CooS genes (Figure 4), however the mini-CooS seems to retain all of the important domains of typical CODHs, suggesting that it may be functional (Figure 4). Dobbek et al. (2004) solved the prototype crystal structure for the CooS-II from C. hydrogenoformans which revealed three metal binding clusters (Clusters B, C, and D), each with separate metal coordinating domains with conserved Cys and His residues essential for CODH activity. Interestingly, the 11 Cys residues and the His residue coordinating these clusters are all conserved in the minimal CooS from T. carboxydivorans. Hydroxylamine reductases share some sequence similarity with CooS, but they all lack some of the conserved Cys residues that locate the metal clusters in CooS. The proximity in the genome of the minimal cooS gene to a cooF gene provides further evidence for its involvement in CO-metabolism.
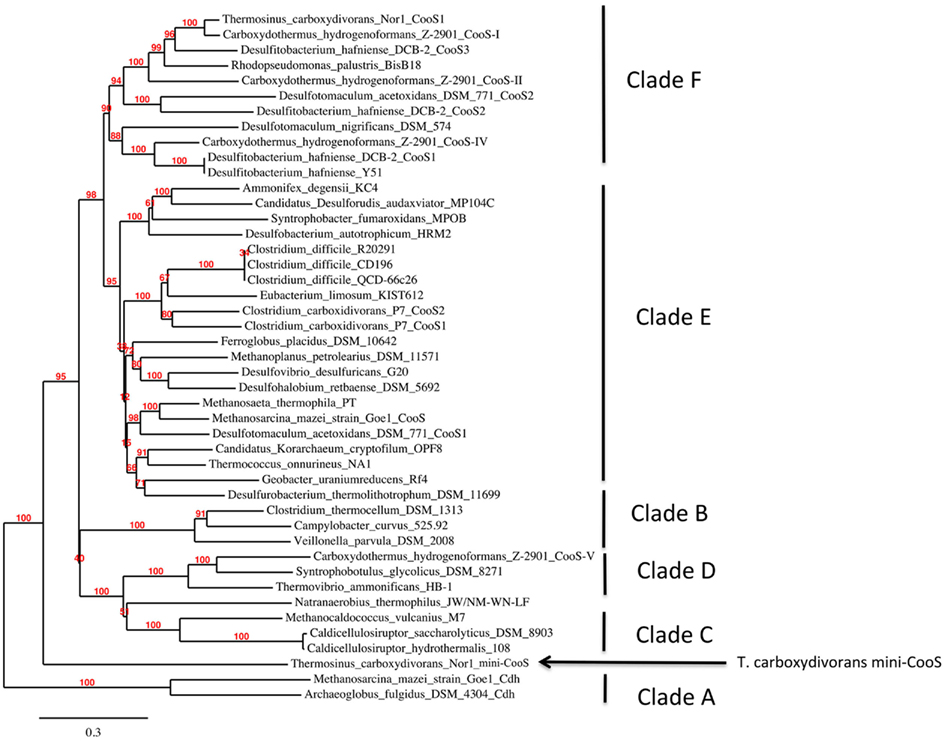
Figure 3. Phylogenetic position of the mini-CooS from Thermosinus carboxydivorans. A maximum likelihood tree was built from a MUSCLE alignment of representative sequences selected from all of the [Ni,Fe]-CODH clades defined by Figure 1 as well as the mini-CooS from T. carboxydivorans. The tree was bootstrapped with 1000 replicates,which are shown as percentage values at the nodes in the tree.

Figure 4. The mini-CooS from Thermosinus carboxydivorans contains conserved residues essential for CODH activity. An alignment of Carboxydothermus hydrogenoformans CooS-II, the Cdh-type CODH of Methanothermobacter thermautotrophicus (MTH1708), Thermosinus carboxydivorans mini-CooS, and Thermincola potens hydroxylamine reductase TherJR_2940 was constructed. Dobbek et al. (2001) defined three sites as being essential for CooS activity. Cluster C (purple), B (red), and D (green). All of these residues are present in the C. hydrogenoformans CooS-II, M. thermoautotrophicus Cdh, and T. carboxydivorans minimal CooS. Many of these residues are absent in the hydroxylamine reductase. The sequences were aligned with MAFFT v6.861b (http://mafft.cbrc.jp/alignment/server/index.html).
The genome sequence of T. carboxydivorans provides insights into the ability of microbes to utilize CO as a versatile electron donor as well as potential mechanisms for acquisition of [Ni,Fe]-CODH operons and the evolution of multi-[Ni,Fe]-CODH genotypes. As mentioned previously, the draft T. carboxydivorans genome sequence revealed three CODH gene clusters. The [Ni,Fe]-CODH-ECH gene cluster is highly similar in sequence and gene content to the homologs found in C. hydrogenoformans and R. rubrum as shown in Figure 5. In R. rubrum, this gene cluster has been shown to comprise two operons: a CODH operon, which contains a cooS gene and the gene for a nickel–iron, electron transfer protein (cooF), and the adjacent membrane-bound hydrogenase operon. The [Ni,Fe]-CODH-ECH gene clusters are composed of similar sets of genes in R. rubrum, C. hydrogenoformans, and T. carboxydivorans. In C. hydrogenoformans, the [Ni,Fe]-CODH–ECH gene cluster probably comprises two operons (there is an intergenic space approximately 100 nt long between the hypA gene and the cooF gene). In T. carboxydivorans, there is no intergenic space or putative promoter sequences between the ECH and CODH sections of the gene cluster, suggesting that it forms a single operon.
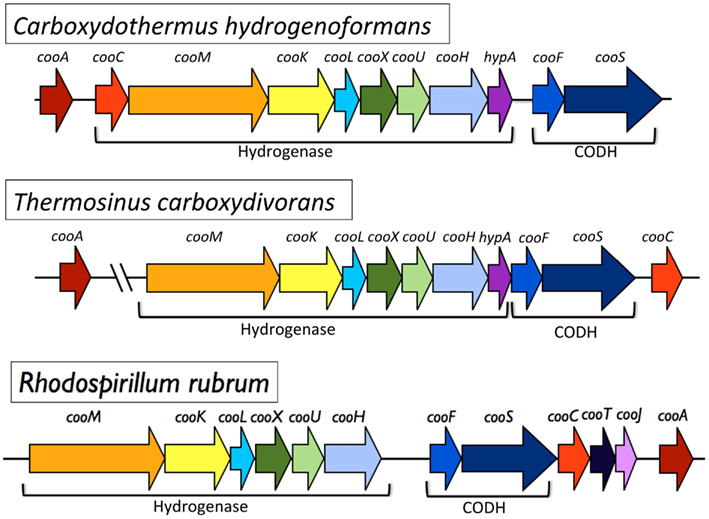
Figure 5. Gene topology for the hydrogenase – CODH gene clusters from C. hydrogenoformans, T. carboxydivorans, and R. rubrum. Homologous genes are colored similarly between operons. The C. hydrogenoformans and T. carboxydivorans gene clusters are identical except for displacement of the associated cooC gene and presence of intergenic space between hypA and the cooF in C. hydrogenoformans. Additionally the T. carboxydivorans cooA gene is located 254 kb away from the rest of the gene cluster. While the R. rubrum gene cluster has many of the same genes, there are notable differences in the genes encoding the hydrogenase accessory proteins (i.e., presence of cooT and cooJ in R. rubrum instead of hypA).
Nevertheless, the CODH and the clustered genes encoding the seven subunits of the membrane-bound hydrogenase from T. carboxydivorans are highly similar to the corresponding genes from C. hydrogenoformans, in both gene order and in nucleotide sequence. The 11.3 kb operon that comprises the [Ni,Fe]-CODH–ECH gene cluster in T. carboxydivorans is 84% identical on the nucleotide level to the homologous gene cluster in C. hydrogenoformans, an observation that prompted us to examine these gene clusters for quantifiable evidence of HGT.
Several in silico methods have been proposed as indicators of HGT. G + C content has been a well-established and simple method for predicting HGT (Lawrence and Ochman, 1998). The G + C content of the T. carboxydivorans genome is 51.6% whereas the G + C content of its 11.3-kb [Ni,Fe]-CODH–ECH operon is 43.4%, which is similar to the overall G + C content of C. hydrogenoformans genome (42.0%). The G + C% of the [Ni,Fe]-CODH-ECH gene cluster from C. hydrogenoformans (44%) is very similar to the overall G + C% of the C. hydrogenoformans genome (Wu et al., 2005).
Because G + C content alone is considered to be an inadequate criterion to detect HGT (Garcia-Vallve et al., 2000) we sought other criteria to distinguish the [Ni,Fe]-CODH–ECH gene cluster in T. carboxydivorans from the genome at large. In a recent paper, additional methods for determining HGT were evaluated (Becq et al., 2010). Tetranucleotide frequency and codon usage similarity quantified with K–L divergence were proposed as the most accurate metrics for predicting HGT. We compared the [Ni,Fe]-CODH–ECH gene clusters of T. carboxydivorans and C. hydrogenoformans with the full genome sequences of T. carboxydivorans, C. hydrogenoformans, with K–L analysis using the genome of E. coli K12 as a control to determine if these two CODH-I gene clusters are divergent from the genomes at large (Table 1).
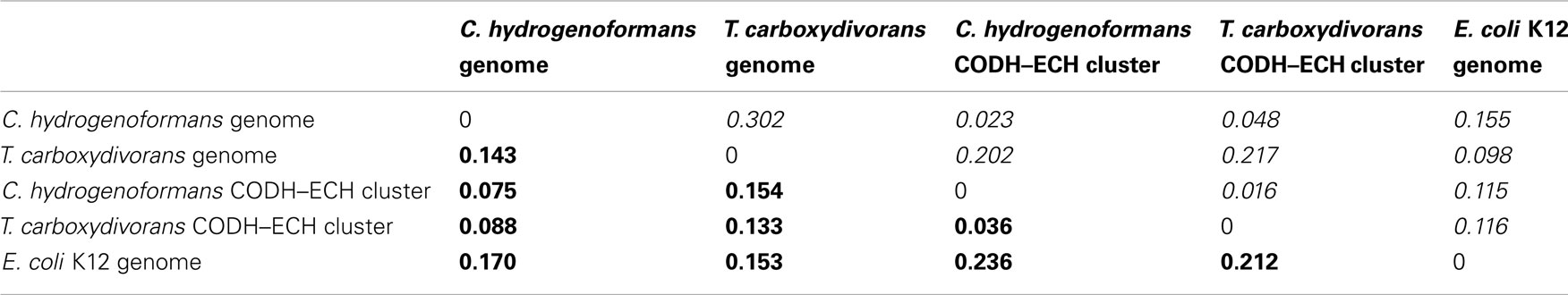
Table 1. Kullback–Leibler divergence comparing codon usage (bold) and tetranucleotide frequency (Italics) for various combinations of the C. hydrogenoforman s, T. carboxydivoran s, and E. coli K12 genomes and the C. hydrogenoforman s and T. carboxydivoran s CODH-ECH gene clusters.
The T. carboxydivorans [Ni,Fe]-CODH–ECH gene cluster was compared to the T. carboxydivorans genome using tetranucleotide frequency, resulting in a K–L divergence of 0.217. When codon usage was used as the metric, the K–L divergence was 0.133. The threshold values of 0.05 and 0.1 for K–L divergence of codon usage and tetranucleotide frequency respectively were used to determine similarity. By both metrics the T. carboxydivorans [Ni,Fe]-CODH–ECH gene cluster is significantly different from the T. carboxydivorans genome. As a control, the tetranucleotide frequency and codon usage of the genes encoding the T. carboxydivorans [Ni,Fe]-CODH–ECH gene cluster were compared to those of the E. coli K12 genome. K–L divergences for this comparison are 0.116 and 0.212 for tetranucleotide frequency and codon usage respectively. These results confirmed that the threshold values are appropriate for specifying similarity. The K–L divergence of the T. carboxydivorans [Ni,Fe]-CODH–ECH gene cluster vs. the T. carboxydivorans genome is thus greater than the K–L distance of the T. carboxydivorans [Ni,Fe]-CODH–ECH gene cluster vs. the E. coli K12 genome. Based on these metrics it appears that the [Ni,Fe]-CODH–ECH gene cluster from T. carboxydivorans is significantly divergent from the T. carboxydivorans genome at large.
The same analysis was performed to examine whether the C. hydrogenoformans [Ni,Fe]-CODH–ECH gene cluster is similar to rest of the C. hydrogenoformans genome. The K–L divergences comparing tetranucleotide frequency and codon usage for the C. hydrogenoformans [Ni,Fe]-CODH–ECH gene cluster against the C. hydrogenoformans genome are 0.023 and 0.075 respectively. Again E. coli K12 was used as a control and the K–L divergences for tetranucleotide frequency and codon usage are 0.155 and 0.236 respectively. The K–L divergences comparing the C. hydrogenoformans [Ni,Fe]-CODH–ECH gene cluster with the rest of the genome are well below the cutoff values and therefore suggest that the C. hydrogenoformans CODH-I gene cluster is equilibrated with the C. hydrogenoformans genome.
The K–L divergences for the comparison of tetranucleotide frequency and codon usage of the T. carboxydivorans [Ni,Fe]-CODH–ECH gene cluster against the C. hydrogenoformans genome are 0.048 and 0.088. These K–L divergences are below the cutoff values, indicating that the T. carboxydivorans [Ni,Fe]-CODH–ECH gene cluster is remarkably similar in composition to the C. hydrogenoformans genome.
The burgeoning release of microbial genomes has revealed that the ability to utilize CO is a surprisingly common trait shared by a diverse collection of bacteria and archaea. Phylogenetic analysis of [Ni,Fe]-CODHs revealed extensive diversity of [Ni,Fe]-CODH sequences in 6% of the released microbial genomes. Most of these species have primary physiologies not related to CO metabolism and have not been scored for CO utilization. Among [Ni,Fe]-CODH encoding species, more than 40% encode more than one CODHs. This previously unexplored diversity of sequences has the potential to provide insights into both the function and evolution of this important protein family. For the most part the multiple [Ni,Fe]-CODHs encoded by the same organism are not close relatives. This suggests that these multiple [Ni,Fe]-CODHs in one genome are not the result of recent gene duplications.
Our phylogenetic analysis confirms prior work which has shown that there is a clear distinction between the bacterial-type CODH/ACS and the archaeal-type CODH/ACDS. The clade composed of the archaeal-type CODH/ACDS is populated by archaea with one noteable exception. There is a sequence for an archaeal-like CODH/ACDS found in the genome of the deep subsurface bacterium Candidatus D. audaxviator, which was the sole detected inhabitant of subterranean effluent waters in a South African gold mine 2.8 km below the surface (Chivian et al., 2008). So far the CODH/ACDS from D. audaxviator is the only sequence of a CODH/ACDS found in a bacterial genome, supporting the notion that D. audaxviator acquired this CODH/ACDS via a HGT event from an archaeon.
Aside from D. audaxviator’s CODH/ACDS it is difficult to distinguish putative HGT events of [Ni,Fe]-CODHs from vertical descent solely from phylogenetic analysis. There are many examples of [Ni,Fe]-CODH clades that are composed of organisms that are not related to one another phylogenetically. For example, in clades C, D, and E, archaeal [Ni,Fe]-CODHs form deep branches of a clade composed primarily of [Ni,Fe]-CODHs from bacterial species, tempting us to infer that these sequences may be the result of ancient inter-domain HGTs. However, the disparity between the 16S rRNA phylogeny and the [Ni,Fe]-CODH phylogeny reconstructed here could result from functional distinctions between CODH lineages. We assume that the function of [Ni,Fe]-CODHs is determined by their associated proteins encoded within gene clusters, since this has been observed in many cases. Therefore, the CODH proteins from one clade may have evolved to associate with accessory proteins more often clustered with another clade member, resulting in [Ni,Fe]-CODHs with similar functions to have convergent sequence motifs, even if originating from different evolutionary starting points.
It is difficult at this stage to assess with accuracy whether the [Ni,Fe]-CODH phylogeny is due to divergence along functional lines as described above, due to the fact that only [Ni,Fe]-CODHs from clades A and F have been definitively characterized biochemically. In an effort to clearly elucidate the branching pattern seen in the global [Ni,Fe]-CODH tree shown in Figure 1, we have attempted to analyze the genomic context of each of the [Ni,Fe]-CODHs in the tree. The function of a [Ni,Fe]-CODH is often determined by the accessory proteins with which it associates. In many genomes, these accessory proteins are encoded in the same operon or adjacent to the [Ni,Fe]-CODH. The analysis of the genomic context and in turn the implied function of the [Ni,Fe]-CODH may help to distinguish vertical descent along function-determined lineages from HGT.
The functional tree (Figure 2) shows that on the whole there is co-occurrence of clades and genomic context. However there are exceptions where the genomic context is not congruent with clades. The best example of genomic context being congruent with phylogenetic lineage is in the case of the archaeal CODH/ACDS (Figure 2, Clade A). Aside from a few isolated cases, namely A. fulgidus and M. acetivorans, the remaining members are found within the context of the CODH/ACDS operon. It is most possible that in the cases of the exceptions, genomic context does not reflect functional cooperation. For the most part, however, Clade A is predominated by species from the domain Archaea and with [Ni,Fe]-CODHs with a conserved genomic context (ACDS clusters).
The bacterial side of the tree is not as straightforward. There are many cases in which genomic context does appear to be correlated with appearance in a common clade. Also, there are a few examples where similar functions are found in two different clades. The well-characterized physiology of hydrogenogenic carboxydotrophy is encoded by a [Ni,Fe]-CODH linked to an ECH. In this phylogenetic tree there are two places in which this gene cluster is found (Clade E and Clade F). While it may be difficult to explain this phenomenon in terms of vertical descent, it is clear that unlike the CODHs in Clade A the [Ni,Fe]-CODH–ECH gene cluster is not the result of simple vertical descent from an ancestral [Ni,Fe]-CODH–ECH.
Another example of correlation of the genomic context with the tree topology is provided by the absence of CooS/ACS gene clusters beyond clades E and F. This could indicate that CooS/ACS sequences have evolved by vertical inheritance and the presence of phylogenetically diverse hosts within these clades is not due to HGT but is a result of conservation of function. However, like the CODH–ECH gene cluster, the CODH/ACS gene cluster is present in three clades. Again this points to a more complex evolutionary history, which may indicate that this gene cluster has arisen more than once during the course of evolution or potentially a more complex mechanism involving HGT and genome rearrangements.
By overlaying known functional categories onto the phylogenetic tree we were able to clarify some of the phylogenetic history of [Ni,Fe]-CODHs. However much work needs to be done to further clarify the function of the many of the remaining [Ni,Fe]-CODHs. Since it appears that in some cases the clades fall along functional boundaries, the question of the role of HGT as a means of acquisition of [Ni,Fe]-CODHs could be more clearly addressed by examining unrelated species that possesses [Ni,Fe]-CODHs that are highly similar in sequence and function. One such example is found in the [Ni,Fe]-CODH–ECH of Clade F. C. hydrogenoformans and T. carboxydivorans are two thermophiles from the Phylum Firmicutes but are from two divergent classes, Clostridia and Negativicutes, respectively. While these organisms share a common physiology they represent widely separated phylogenetic lineages. They both possess [Ni,Fe]-CODH–ECH homologous gene clusters with high similarity to each other (84% nucleotide identity). For this reason we set out to investigate whether the [Ni,Fe]-CODH–ECH gene cluster could have been acquired by T. carboxydivorans via an HGT event. Analysis of G + C content, tetranucleotide frequency, and codon usage support the hypothesis that the CODH – ECH gene cluster was assimilated by T. carboxydivorans via HGT.
Based on the high similarity between the hydrogenase – CODH gene clusters of T. carboxydivorans and C. hydrogenoformans and on the fitness of the T. carboxydivorans [Ni,Fe]-CODH–ECH to the C. hydrogenoformans genome in terms of tetranucleotide frequency and codon usage patterns, it is tempting to propose that T. carboxydivorans obtained this operon by HGT from C. hydrogenoformans or a closely related species. However, closer examination of the data indicate that there may have been a recent transfer from a relative of C. hydrogenoformans or else a more ancient transfer from an ancestor of C. hydrogenoformans into the T. carboxydivorans lineage. First, the nucleotide identity of the gene clusters, although high (84%), is lower than the ANI value of 95–96% shown to delimit microbial species (Goris et al., 2007; Richter and Rossello-Mora, 2009; Tindall et al., 2010). Second, whereas in C. hydrogenoformans the cluster seems to comprise two operons, T. carboxydivorans appears likely to encode a single operon (Figure 5). Third, the order of genes in the gene clusters from T. carboxydivorans and C. hydrogenoformans has been switched by the relocation of cooC (Figure 5). The rearrangements suggest further evolution in T. carboxydivorans or C. hydrogenoformans following a horizontal transfer. The location of the regulator gene cooA apart from the [Ni,Fe]-CODH–ECH gene cluster in T. carboxydivorans also suggests that there is ongoing genome reordering of the CO-related genes in this isolate, or may be interpreted as the acquisition of cooA that occurred independently from the acquisition of the [Ni,Fe]-CODH–ECH gene cluster. One of the two cooA genes (cooA-1) in C. hydrogenoformans is directly upstream of its [Ni,Fe]-CODH–ECH gene cluster. The lone cooA homolog in the genome of T. carboxydivorans is a homolog of C hydrogenoformans cooA-1 and is separated by 252 kb from the hydrogenase – CODH gene cluster. Interestingly, the T. carboxydivorans cooA is flanked by transposes and phage-related genes, which are known to be mediators of genomic rearrangement and HGT (Ochman et al., 2000).
The T. carboxydivorans genome also contains the smallest gene predicted to contain all of the conserved CooS features. This putative CODH is 428 amino acid residues long compared with typical [Ni,Fe]-CODHs that range from approximately 600 to 800 residues long. This “mini-CooS” has been placed in a separate multiple alignment that gave rise to the ML analysis shown in Figure 3. Taking into account (i) the acquisition of the hydrogenase – [Ni,Fe]-CODH gene cluster via HGT, which we substantiate in this paper, (ii) the separate location of the cooA gene, and (iii) the fact that the G + C content of the gene encoding the unusual small cooS is about 10 mol% higher than that of the genome. Therefore, we speculate that the assemblage of the carboxydotrophic hydrogenogenic phenotype in T. carboxydivorans is an evolutionarily recent event, suggesting that rapid genome evolution is under way to accommodate selection for CO utilization.
Due to the large number of species that have been shown to encode multiple CODHs, it seems probable that multiple [Ni,Fe]-CODH operons increase the fitness of an organism in CO-rich milieus by providing divergent metabolic pathways through which CO can be metabolized. The consumption of CO by independent pathways providing energy conservation, carbon fixation, and nicotinamide coenzyme reduction are now well-established. On a broader scale, in microbial consortia, CO utilization has the additional communal benefit of dissipating a potentially toxic compound (Parshina et al., 2005; Techtmann et al., 2009). Our recent work revealed that adaptive regulation of expression of multiple [Ni,Fe]-CODH operons by dual cooA genes may partition CO between multiple competing pathways in response to varying needs for energy conservation and carbon acquisition across a wide range of CO concentrations (Techtmann et al., 2011).
In conclusion, we have developed a comprehensive phylogeny for [Ni,Fe]-CODHs which revealed the presence of six distinct clades. Comparison of the genomic contexts with CooS phylogeny suggests that several clades diverged based on function. These clades appear to be evolving via vertical descent. However there are a few CO-related metabolic functions that are spread throughout various clades on the tree either indicating different evolutionary events or HGT. It seems likely that both HGT and vertical transmission have driven the remarkable divergence of [Ni,Fe]-CODHs currently seen in both archaea and bacteria
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded by the US National Science Foundation through award numbers EAR 0747394 (Frank T. Robb), EAR 0747412 (Albert S. Colman), and MCB 0605301 (Frank T. Robb and Albert S. Colman). The work of Alexander V. Lebedinsky and Tatyana G. Sokolova has been supported by the Russian Foundation for Basic Research (project number 11-04-01723-a), federal targeted program for 2009–2013 “Scientific and Pedagogical Personnel of Innovative Russia” (GK number P646), and the program of the Presidium of the Russian Academy of Sciences “Molecular and Cell Biology.” The sequencing work conducted by the U.S. Department of Energy Joint Genome Institute is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We are indebted to our reviewers for helpful comments on phylogenetic methods. This is Contribution 11-230 from the Institute of Marine and Environmental Technology.
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Evolutionary_and_Genomic_Microbiology/10.3389/fmicb.2012.00132/abstract
Table S1. [Ni,Fe]-CODH sequences found in the IMG database as of October 1, 2011.
Table S2. Genomes encoding multiple [Ni,Fe]-CODH homologs.
Table S3. Composition of the six [Ni,Fe]-CODH clades of both the ML and BI phylogenetic trees.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Becq, J., Churlaud, C., and Deschavanne, P. (2010). A benchmark of parametric methods for horizontal transfers detection. PLoS ONE 5, e9989. doi:10.1371/journal.pone.0009989
Chivian, D., Brodie, E. L., Alm, E. J., Culley, D. E., Dehal, P. S., Desantis, T. Z., Gihring, T. M., Lapidus, A., Lin, L. H., Lowry, S. R., Moser, D. P., Richardson, P. M., Southam, G., Wanger, G., Pratt, L. M., Andersen, G. L., Hazen, T. C., Brockman, F. J., Arkin, A. P., and Onstott, T. C. (2008). Environmental genomics reveals a single-species ecosystem deep within Earth. Science 322, 275–278.
Dobbek, H., Gremer, L., Meyer, O., and Huber, R. (1999). Crystal structure and mechanism of CO dehydrogenase, a molybdo iron-sulfur flavoprotein containing S-selanylcysteine. Proc. Natl. Acad. Sci. U.S.A. 96, 8884–8889.
Dobbek, H., Svetlitchnyi, V., Gremer, L., Huber, R., and Meyer, O. (2001). Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni-4Fe-5S] cluster. Science 293, 1281–1285.
Dobbek, H., Svetlitchnyi, V., Liss, J., and Meyer, O. (2004). Carbon monoxide induced decomposition of the active site [Ni-4Fe-5S] cluster of CO dehydrogenase. J. Am. Chem. Soc. 126, 5382–5387.
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214. doi:10.1186/1471-2148-7-214
Edgar, R. C. (2004a). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113. doi:10.1186/1471-2105-5-113
Edgar, R. C. (2004b). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797.
Ewing, B., and Green, P. (1998). Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8, 186–194.
Ewing, B., Hillier, L., Wendl, M. C., and Green, P. (1998). Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8, 175–185.
Gao, F., and Zhang, C. T. (2006). GC-Profile: a web-based tool for visualizing and analyzing the variation of GC content in genomic sequences. Nucleic Acids Res. 34, W686–W691.
Garcia-Vallve, S., Romeu, A., and Palau, J. (2000). Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res. 10, 1719–1725.
Gencic, S., Duin, E. C., and Grahame, D. A. (2010). Tight coupling of partial reactions in the acetyl-CoA decarbonylase/synthase (ACDS) multienzyme complex from Methanosarcina thermophila: acetyl C-C bond fragmentation at the a cluster promoted by protein conformational changes. J. Biol. Chem. 285, 15450–15463.
Gnida, M., Ferner, R., Gremer, L., Meyer, O., and Meyer-Klaucke, W. (2003). A novel binuclear [CuSMo] cluster at the active site of carbon monoxide dehydrogenase: characterization by X-ray absorption spectroscopy. Biochemistry 42, 222–230.
Gonzalez, J. M., and Robb, F. T. (2000). Genetic analysis of Carboxydothermus hydrogenoformans carbon monoxide dehydrogenase genes cooF and cooS. FEMS Microbiol. Lett. 191, 243–247.
Gordon, D., Abajian, C., and Green, P. (1998). Consed: a graphical tool for sequence finishing. Genome Res. 8, 195–202.
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91.
Grahame, D. A. (1991). Catalysis of acetyl-CoA cleavage and tetrahydrosarcinapterin methylation by a carbon monoxide dehydrogenase-corrinoid enzyme complex. J. Biol. Chem. 266, 22227–22233.
Han, C. S., and Chain, P. (2006). “Finishing repeat regions automatically with Dupfinisher,” in Proceedings of the 2006 International Conference on Bioinformatics and Computational Biology, eds. H. R. Arabnia and H. Valafar (Las Vegas: CSREA Press), 141–146.
Hugendieck, I., and Meyer, O. (1992). The structural genes encoding CO dehydrogenase subunits (cox L, M and S) in Pseudomonas carboxydovorans OM5 reside on plasmid pHCG3 and are, with the exception of Streptomyces thermoautotrophicus, conserved in carboxydotrophic bacteria. Arch. Microbiol. 157, 301–304.
Kerby, R. L., Hong, S. S., Ensign, S. A., Coppoc, L. J., Ludden, P. W., and Roberts, G. P. (1992). Genetic and physiological characterization of the Rhodospirillum rubrum carbon monoxide dehydrogenase system. J. Bacteriol. 174, 5284–5294.
King, G. M. (2003). Uptake of carbon monoxide and hydrogen at environmentally relevant concentrations by mycobacteria. Appl. Environ. Microbiol. 69, 7266–7272.
King, G. M. (2006). Nitrate-dependent anaerobic carbon monoxide oxidation by aerobic CO-oxidizing bacteria. FEMS Microbiol. Ecol. 56, 1–7.
King, G. M., and Weber, C. F. (2007). Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat. Rev. Microbiol. 5, 107–118.
Kocsis, E., Kessel, M., Demoll, E., and Grahame, D. A. (1999). Structure of the Ni/Fe-S protein subcomponent of the acetyl-CoA decarbonylase/synthase complex from Methanosarcina thermophila at 26-A resolution. J. Struct. Biol. 128, 165–174.
Kullback, S., and Leibler, R. A. (1951). On information and sufficiency. Ann. Math. Stat. 22, 79–86.
Lawrence, J. G., and Ochman, H. (1998). Molecular archaeology of the Escherichia coli genome. Proc. Natl. Acad. Sci. U.S.A. 95, 9413–9417.
Lim, J. K., Kang, S. G., Lebedinsky, A. V., Lee, J. H., and Lee, H. S. (2010). Identification of a novel class of membrane-bound [NiFe]-hydrogenases in Thermococcus onnurineus NA1 by in silico analysis. Appl. Environ. Microbiol. 76, 6286–6289.
Lindahl, P. A. (2002). The Ni-containing carbon monoxide dehydrogenase family: light at the end of the tunnel? Biochemistry 41, 2097–2105.
Meyer, O., Frunzke, K., and Mörsdorf, G. (eds). (1993). Biochemistry of the Aerobic Utilization of Carbon Monoxide. Andover, MA: Intercept, Ltd.
Meyer, O., and Rajagopalan, K. V. (1984). Molybdopterin in carbon monoxide oxidase from carboxydotrophic bacteria. J. Bacteriol. 157, 643–648.
Meyer, O., and Schlegel, H. G. (1983). Biology of aerobic carbon monoxide-oxidizing bacteria. Annu. Rev. Microbiol. 37, 277–310.
Miller, M. A., Holder, M. T., Vos, R., Midford, P. E., Liebowitz, T., Chan, L., Hoover, P., and Warnow, T. (2010). The CIPRES Portals. CIPRES. Available at: http://www.phylo.org/sub_sections/portal
Milne, I., Lindner, D., Bayer, M., Husmeier, D., Mcguire, G., Marshall, D. F., and Wright, F. (2009). TOPALi v2: a rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics 25, 126–127.
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304.
Parshina, S. N., Kijlstra, S., Henstra, A. M., Sipma, J., Plugge, C. M., and Stams, A. J. (2005). Carbon monoxide conversion by thermophilic sulfate-reducing bacteria in pure culture and in co-culture with Carboxydothermus hydrogenoformans. Appl. Microbiol. Biotechnol. 68, 390–396.
Ragsdale, S. W. (2008). Enzymology of the wood-Ljungdahl pathway of acetogenesis. Ann. N. Y. Acad. Sci. 1125, 129–136.
Rambaut, A., and Aj, D. (2007). Tracer v1.4. Available at: http://beast.bio.ed.ac.uk/Tracer
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131.
Schubel, U., Kraut, M., Morsdorf, G., and Meyer, O. (1995). Molecular characterization of the gene cluster coxMSL encoding the molybdenum-containing carbon monoxide dehydrogenase of Oligotropha carboxidovorans. J. Bacteriol. 177, 2197–2203.
Seravalli, J., Kumar, M., Lu, W. P., and Ragsdale, S. W. (1995). Mechanism of CO oxidation by carbon monoxide dehydrogenase from Clostridium thermoaceticum and its inhibition by anions. Biochemistry 34, 7879–7888.
Seravalli, J., Kumar, M., Lu, W. P., and Ragsdale, S. W. (1997). Mechanism of carbon monoxide oxidation by the carbon monoxide dehydrogenase/acetyl-CoA synthase from Clostridium thermoaceticum: kinetic characterization of the intermediates. Biochemistry 36, 11241–11251.
Soboh, B., Linder, D., and Hedderich, R. (2002). Purification and catalytic properties of a CO-oxidizing:H2-evolving enzyme complex from Carboxydothermus hydrogenoformans. Eur. J. Biochem. 269, 5712–5721.
Sokolova, T. G., Gonzalez, J. M., Kostrikina, N. A., Chernyh, N. A., Slepova, T. V., Bonch-Osmolovskaya, E. A., and Robb, F. T. (2004). Thermosinus carboxydivorans gen. nov., sp. nov., a new anaerobic, thermophilic, carbon-monoxide-oxidizing, hydrogenogenic bacterium from a hot pool of Yellowstone National Park. Int. J. Syst. Evol. Microbiol. 54, 2353–2359.
Sokolova, T. G., Henstra, A. M., Sipma, J., Parshina, S. N., Stams, A. J., and Lebedinsky, A. V. (2009). Diversity and ecophysiological features of thermophilic carboxydotrophic anaerobes. FEMS Microbiol. Ecol. 68, 131–141.
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690.
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57, 758–771.
Svetlitchnyi, V., Dobbek, H., Meyer-Klaucke, W., Meins, T., Thiele, B., Romer, P., Huber, R., and Meyer, O. (2004). A functional Ni-Ni-[4Fe-4S] cluster in the monomeric acetyl-CoA synthase from Carboxydothermus hydrogenoformans. Proc. Natl. Acad. Sci. U.S.A. 101, 446–451.
Svetlitchnyi, V., Peschel, C., Acker, G., and Meyer, O. (2001). Two membrane-associated NiFeS-carbon monoxide dehydrogenases from the anaerobic carbon-monoxide-utilizing Eubacterium Carboxydothermus hydrogenoformans. J. Bacteriol. 183, 5134–5144.
Techtmann, S. M., Colman, A. S., Murphy, M. B., Schackwitz, W. S., Goodwin, L. A., and Robb, F. T. (2011). Regulation of multiple carbon monoxide consumption pathways in anaerobic bacteria. Front. Microbiol. 2:147. doi:10.3389/fmicb.2011.00147
Techtmann, S. M., Colman, A. S., and Robb, F. T. (2009). ‘That which does not kill us only makes us stronger’: the role of carbon monoxide in thermophilic microbial consortia. Environ. Microbiol. 11, 1027–1037.
Teeling, H., Waldmann, J., Lombardot, T., Bauer, M., and Glockner, F. O. (2004). TETRA: a web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinformatics 5, 163. doi:10.1186/1471-2105-5-163
Terlesky, K. C., Nelson, M. J., and Ferry, J. G. (1986). Isolation of an enzyme complex with carbon monoxide dehydrogenase activity containing corrinoid and nickel from acetate-grown Methanosarcina thermophila. J. Bacteriol. 168, 1053–1058.
Tindall, B. J., Rossello-Mora, R., Busse, H. J., Ludwig, W., and Kampfer, P. (2010). Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 60, 249–266.
Uffen, R. L. (1976). Anaerobic growth of a Rhodopseudomonas species in the dark with carbon monoxide as sole carbon and energy substrate. Proc. Natl. Acad. Sci. U.S.A. 73, 3298–3302.
Volbeda, A., and Fontecilla-Camps, J. C. (2005). Structural bases for the catalytic mechanism of Ni-containing carbon monoxide dehydrogenases. Dalton Trans. 21, 3443–3450.
Wu, M., Ren, Q., Durkin, A. S., Daugherty, S. C., Brinkac, L. M., Dodson, R. J., Madupu, R., Sullivan, S. A., Kolonay, J. F., Haft, D. H., Nelson, W. C., Tallon, L. J., Jones, K. M., Ulrich, L. E., Gonzalez, J. M., Zhulin, I. B., Robb, F. T., and Eisen, J. A. (2005). Life in hot carbon monoxide: the complete genome sequence of Carboxydothermus hydrogenoformans Z-2901. PLoS Genet. 1, e65. doi:10.1371/journal.pgen.0010065
Figure A1. Circular Bayesian Inference tree with sequence names shown (Same tree as Figure 1B). Posterior probabilities for deeply branching nodes are displayed.
Figure A2. Subtrees of maximum likelihood tree of [Ni,Fe]-CODHs (Figure 2) divided based on clades.
Keywords: carbon monoxide, thermophiles, hydrogenogens, carboxydotrophs, Thermosinus carboxydivorans, Carboxydothermus hydrogenoformans, carbon monoxide dehydrogenase
Citation: Techtmann SM, Lebedinsky AV, Colman AS, Sokolova TG, Woyke T, Goodwin L and Robb FT (2012) Evidence for horizontal gene transfer of anaerobic carbon monoxide dehydrogenases. Front. Microbio. 3:132. doi: 10.3389/fmicb.2012.00132
Received: 07 December 2011; Accepted: 20 March 2012;
Published online: 17 April 2012.
Edited by:
Martin G. Klotz, University of North Carolina at Charlotte, USAReviewed by:
Annette Summers Engel, University of Tennessee, USACopyright: © 2012 Techtmann, Lebedinsky, Colman, Sokolova, Woyke, Goodwin and Robb. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Frank T. Robb, Institute of Marine and Environmental Technology, 701 East Pratt Street, Baltimore, MD 21202, USA. e-mail:ZnJvYmJAc29tLnVtYXJ5bGFuZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.