

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 29 March 2012
Sec. Microbiological Chemistry and Geomicrobiology
Volume 3 - 2012 | https://doi.org/10.3389/fmicb.2012.00112
This article is part of the Research Topic The microbial ferrous wheel: iron cycling in terrestrial, freshwater, and marine environments View all 17 articles
Microbial Fe(II) oxidation using NO3− as the terminal electron acceptor [nitrate-dependent Fe(II) oxidation, NDFO] has been studied for over 15 years. Although there are reports of autotrophic isolates and stable enrichments, many of the bacteria capable of NDFO are known organotrophic NO3−-reducers that require the presence of an organic, primary substrate, e.g., acetate, for significant amounts of Fe(II) oxidation. Although the thermodynamics of Fe(II) oxidation are favorable when coupled to either NO3− or NO2− reduction, the kinetics of abiotic Fe(II) oxidation by NO3− are relatively slow except under special conditions. NDFO is typically studied in batch cultures containing millimolar concentrations of Fe(II), NO3−, and the primary substrate. In such systems, NO2− is often observed to accumulate in culture media during Fe(II) oxidation. Compared to NO3−, abiotic reactions of biogenic NO2− and Fe(II) are relatively rapid. The kinetics and reaction pathways of Fe(II) oxidation by NO2− are strongly affected by medium composition and pH, reactant concentration, and the presence of Fe(II)-sorptive surfaces, e.g., Fe(III) oxyhydroxides and cellular surfaces. In batch cultures, the combination of abiotic and microbial Fe(II) oxidation can alter product distribution and, more importantly, results in the formation of intracellular precipitates and extracellular Fe(III) oxyhydroxide encrustations that apparently limit further cell growth and Fe(II) oxidation. Unless steps are taken to minimize or account for potential abiotic reactions, results of microbial NDFO studies can be obfuscated by artifacts of the chosen experimental conditions, the use of inappropriate analytical methods, and the resulting uncertainties about the relative importance of abiotic and microbial reactions. In this manuscript, abiotic reactions of NO3− and NO2− with aqueous Fe2+, chelated Fe(II), and solid-phase Fe(II) are reviewed along with factors that can influence overall NDFO reaction rates in microbial systems. In addition, the use of low substrate concentrations, continuous-flow systems, and experimental protocols that minimize experimental artifacts and reduce the potential for under- or overestimation of microbial NDFO rates are discussed.
To state the obvious, some geomicrobiological systems are easier to work with than others. In the same way that microbial, microaerophilic Fe(II) oxidation at circumneutral pH presents experimental challenges (Emerson and Floyd, 2005) microbial 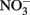 -dependent Fe(II) oxidation (NDFO) present a variety of complexities that can complicate experimental design and confuse interpretation of results. Fe(II) is usually initially provided as soluble Fe2+, but can also be introduced as sorbed Fe(II) or Fe(II)-bearing minerals of varying crystallinity and reactivity. The oxidized Fe(III) produced precipitates rapidly at circumneutral pH forming oxyhydroxides, thereby providing highly sorptive and reactive surfaces not initially present and frequently forming extensive cell coatings which can strongly effect microbial metabolism. Although usually not measured, the final product of
-dependent Fe(II) oxidation (NDFO) present a variety of complexities that can complicate experimental design and confuse interpretation of results. Fe(II) is usually initially provided as soluble Fe2+, but can also be introduced as sorbed Fe(II) or Fe(II)-bearing minerals of varying crystallinity and reactivity. The oxidized Fe(III) produced precipitates rapidly at circumneutral pH forming oxyhydroxides, thereby providing highly sorptive and reactive surfaces not initially present and frequently forming extensive cell coatings which can strongly effect microbial metabolism. Although usually not measured, the final product of 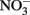 reduction, based on stoichiometry and lack of
reduction, based on stoichiometry and lack of 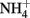 production, is typically assumed to be N2 or N2O. One of the reduction intermediates, i.e.,
production, is typically assumed to be N2 or N2O. One of the reduction intermediates, i.e., 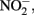 is more reactive with Fe(II) than
is more reactive with Fe(II) than 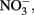 can accumulate in the medium, and result in abiotic reaction opportunities not present in abiotic controls.
can accumulate in the medium, and result in abiotic reaction opportunities not present in abiotic controls.
Rather than present new data, this manuscript will review potential abiotic reactions between either 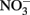 or
or 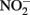 and homogeneous solutions of Fe2+ or heterogeneous systems containing solid-phase Fe(II). Numerous studies have been conducted for more than four decades (e.g., Chao and Kroontje, 1966), often under significantly different experimental conditions and resulting in varying reports of reactivity, reaction rates, and disparate distributions of products. This manuscript will describe previous abiotic studies in the context of microbial NDFO, attempt to provide deeper insights into the potential for simultaneous abiotic and biotic reactions of Fe and N, and assist in interpreting relative contributions of both reaction types in studies of microbial NDFO.
and homogeneous solutions of Fe2+ or heterogeneous systems containing solid-phase Fe(II). Numerous studies have been conducted for more than four decades (e.g., Chao and Kroontje, 1966), often under significantly different experimental conditions and resulting in varying reports of reactivity, reaction rates, and disparate distributions of products. This manuscript will describe previous abiotic studies in the context of microbial NDFO, attempt to provide deeper insights into the potential for simultaneous abiotic and biotic reactions of Fe and N, and assist in interpreting relative contributions of both reaction types in studies of microbial NDFO.
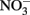
The oxidation of Fe2+ to goethite (α-FeOOH) by 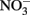 can be written such that either N2, N2O,
can be written such that either N2, N2O, 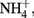 or
or 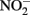 are produced as final products.
are produced as final products.
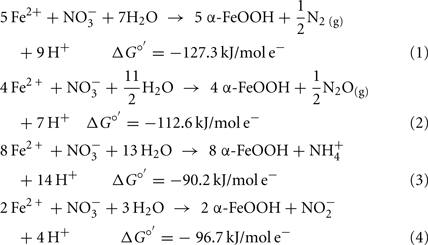
Calculations based on commonly used thermodynamic data (Stumm and Morgan, 1981) show that the standard free energy change at pH 7 for all reactions is favorable. Even though the thermodynamics are favorable, the kinetics of abiotic aqueous Fe2+ oxidation by 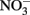 at circumneutral pH in the absence of catalytic ions or surfaces are relatively slow. With respect to catalysts for abiotic reactions, Cu2+ and other metal ions have been shown to increase reaction rates (Buresh and Moraghan, 1976; Ottley et al., 1997). In an oft-cited study, Buresh and Moraghan (1976) described the oxidation of aqueous Fe2+ by
at circumneutral pH in the absence of catalytic ions or surfaces are relatively slow. With respect to catalysts for abiotic reactions, Cu2+ and other metal ions have been shown to increase reaction rates (Buresh and Moraghan, 1976; Ottley et al., 1997). In an oft-cited study, Buresh and Moraghan (1976) described the oxidation of aqueous Fe2+ by 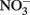 over a pH range of 6–10 in the presence of 0–160 μM Cu2+ as a catalyst. In the presence of 0.0 and 1.6 μM Cu2+,
over a pH range of 6–10 in the presence of 0–160 μM Cu2+ as a catalyst. In the presence of 0.0 and 1.6 μM Cu2+, 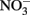 was stable over the 24-h experiment regardless of the pH. In the presence of Cu2+ at concentrations of 16 μM or greater,
was stable over the 24-h experiment regardless of the pH. In the presence of Cu2+ at concentrations of 16 μM or greater, 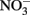 reduction occurred, and the extent of reduction increased with pH, becoming quite significant at pH 8 and above. When Cu-catalyzed
reduction occurred, and the extent of reduction increased with pH, becoming quite significant at pH 8 and above. When Cu-catalyzed 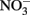 reduction occurred, Fe(OH)2 or other solid phases were presumed to be the reducing agents rather than soluble Fe2+ due to increased
reduction occurred, Fe(OH)2 or other solid phases were presumed to be the reducing agents rather than soluble Fe2+ due to increased 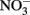 reduction at pH values >7 where extensive production of solid-phase Fe occurred. Ottley et al. (1997) further examined the reduction of
reduction at pH values >7 where extensive production of solid-phase Fe occurred. Ottley et al. (1997) further examined the reduction of 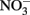 by Fe2+ in the presence of 7 μM to 7 mM Cu2+ and other trace metal ions at pH 7–8.5. Relatively rapid
by Fe2+ in the presence of 7 μM to 7 mM Cu2+ and other trace metal ions at pH 7–8.5. Relatively rapid 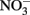 reduction rates in the presence of Cu2+ again increased at alkaline pH and the authors determined that solid-phase Cu produced during incubations was catalytically active rather than the soluble Cu2+.
reduction rates in the presence of Cu2+ again increased at alkaline pH and the authors determined that solid-phase Cu produced during incubations was catalytically active rather than the soluble Cu2+.
Solid-phase Fe(II), whether adsorbed or crystalline, has been shown to be a more effective reductant than aqueous Fe2+ for reactions involving both organic and inorganic compounds (Sung and Morgan, 1980; Tamura et al., 1980; Klausen et al., 1995; Cui and Eriksen, 1996; Kim and Picardal, 1999; Amonette et al., 2000; Williams et al., 2005; Neumann et al., 2009). The importance of solid-phase or adsorbed Fe(II) in the abiotic reduction of 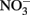 appears to be dependent upon the time scale of the studies, pH, and characteristics of the solid phase. Postma (1990) studied reduction of low-micromolar concentrations of
appears to be dependent upon the time scale of the studies, pH, and characteristics of the solid phase. Postma (1990) studied reduction of low-micromolar concentrations of 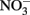 by Fe(II)-bearing silicate minerals at pH 2–7 over periods of 1000–2000 h. No aqueous Fe2+ was added and reduction relied on slow release of Fe2+ during silicate mineral dissolution. Maximal
by Fe(II)-bearing silicate minerals at pH 2–7 over periods of 1000–2000 h. No aqueous Fe2+ was added and reduction relied on slow release of Fe2+ during silicate mineral dissolution. Maximal 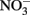 reduction rates were observed at pH 4 where small amounts of nitrite were also measured and reduction rates were thought to be dependent on precipitation of secondary Fe(III) minerals such as goethite. Overall
reduction rates were observed at pH 4 where small amounts of nitrite were also measured and reduction rates were thought to be dependent on precipitation of secondary Fe(III) minerals such as goethite. Overall 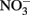 reduction rates were quite low, however, and reduction by Fe(II)-bearing silicate minerals was considered to be important only in groundwater systems with long residence times. Although the Postma study may have significance for microbial NDFO as an environmental process in suitable sediments, the acidic pH regime and low micromolar concentrations of
reduction rates were quite low, however, and reduction by Fe(II)-bearing silicate minerals was considered to be important only in groundwater systems with long residence times. Although the Postma study may have significance for microbial NDFO as an environmental process in suitable sediments, the acidic pH regime and low micromolar concentrations of 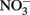 and Fe2+ used in his studies make it difficult to extrapolate his findings to laboratory studies of microbial NDFO.
and Fe2+ used in his studies make it difficult to extrapolate his findings to laboratory studies of microbial NDFO.
In the work of Ottley et al. (1997), weeklong incubations of 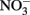 and initially aqueous Fe2+ in the absence of metal ion catalysts at pH 8 showed a 15%
and initially aqueous Fe2+ in the absence of metal ion catalysts at pH 8 showed a 15% 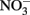 loss. Such losses would be important in microbial experiments where incubations are often conducted over similar or longer time scales. Since such losses of
loss. Such losses would be important in microbial experiments where incubations are often conducted over similar or longer time scales. Since such losses of 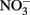 are typically not observed in microbiological experiments with aqueous Fe2+ (Straub et al., 1996; Weber et al., 2001; Blothe and Roden, 2009), it is not clear if trace amounts of O2 caused abiotic Fe2+ oxidation and subsequent
are typically not observed in microbiological experiments with aqueous Fe2+ (Straub et al., 1996; Weber et al., 2001; Blothe and Roden, 2009), it is not clear if trace amounts of O2 caused abiotic Fe2+ oxidation and subsequent 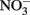 reduction by sorbed or solid-phase Fe(II). As also described by Ottley et al. addition of goethite to systems containing Fe2+ increased rates of
reduction by sorbed or solid-phase Fe(II). As also described by Ottley et al. addition of goethite to systems containing Fe2+ increased rates of 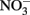 reduction. Since goethite is sometimes a product of microbial NDFO (described below), this shows the potential importance of solid phases and suggests that sorption of Fe2+ to some Fe(III) oxyhydroxides may increase abiotic
reduction. Since goethite is sometimes a product of microbial NDFO (described below), this shows the potential importance of solid phases and suggests that sorption of Fe2+ to some Fe(III) oxyhydroxides may increase abiotic 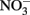 reduction rates.
reduction rates.
Petersen (1979) described the abiotic oxidation of 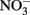 by Fe2+ over a pH range of 4–9. Reaction rates were very slow at pH ≤6 and maximal at pH 8. Petersen suggested that the reductant was a colloidal form of Fe(OH)2 which began to precipitate at pH 6. Although Petersen’s experiments were conducted at 70°C and should be carefully considered when working with hyperthermophiles, the applicability of his results to microbial experiments typically done at much lower temperatures is questionable.
by Fe2+ over a pH range of 4–9. Reaction rates were very slow at pH ≤6 and maximal at pH 8. Petersen suggested that the reductant was a colloidal form of Fe(OH)2 which began to precipitate at pH 6. Although Petersen’s experiments were conducted at 70°C and should be carefully considered when working with hyperthermophiles, the applicability of his results to microbial experiments typically done at much lower temperatures is questionable.
Select, crystalline Fe(II)-bearing minerals are also known to reduce 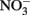 . Wüstite (FeO) has been shown to reduce
. Wüstite (FeO) has been shown to reduce 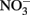 to
to 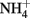 at temperature (3–41°C), pH (5.45–7.45), and concentration regimes appropriate to microbial studies (Rakshit et al., 2005). In addition, clear evidence of rapid abiotic reduction of
at temperature (3–41°C), pH (5.45–7.45), and concentration regimes appropriate to microbial studies (Rakshit et al., 2005). In addition, clear evidence of rapid abiotic reduction of 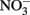 by green rust (GR) minerals has been provided by Hansen and coworkers (Hansen et al., 1994, 1996, 2001) and others (Choi and Batchelor, 2008). The GRs are highly reactive and consist of trioctahedral Fe(II)–Fe(III) hydroxide layers separated by hydrated anionic interlayers. The anions in the GR interlayer are variable, e.g., sulfate in
by green rust (GR) minerals has been provided by Hansen and coworkers (Hansen et al., 1994, 1996, 2001) and others (Choi and Batchelor, 2008). The GRs are highly reactive and consist of trioctahedral Fe(II)–Fe(III) hydroxide layers separated by hydrated anionic interlayers. The anions in the GR interlayer are variable, e.g., sulfate in 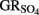 or chloride in GRCl. Both
or chloride in GRCl. Both 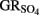 and GRCl have been shown to abiotically reduce
and GRCl have been shown to abiotically reduce 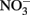 to
to 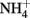 during oxidation of the GR to magnetite (Hansen et al., 1996, 2001). The kinetics of these reactions at circumneutral and slightly alkaline pH are sufficiently rapid to provide a competing pathway for microbial
during oxidation of the GR to magnetite (Hansen et al., 1996, 2001). The kinetics of these reactions at circumneutral and slightly alkaline pH are sufficiently rapid to provide a competing pathway for microbial 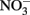 reduction when GR is present at sufficiently high concentrations.
reduction when GR is present at sufficiently high concentrations.
Considering all of the above studies, several generalities are possible. Firstly, Fe2+ and 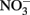 are generally stable at circumneutral pH over the time scale used in most microbial NDFO experiments. This is quite apparent in the killed-cell or uninoculated controls typically used in NDFO batch experiments (Straub et al., 1996; Weber et al., 2001, 2006a; Kappler et al., 2005). In addition, both
are generally stable at circumneutral pH over the time scale used in most microbial NDFO experiments. This is quite apparent in the killed-cell or uninoculated controls typically used in NDFO batch experiments (Straub et al., 1996; Weber et al., 2001, 2006a; Kappler et al., 2005). In addition, both 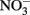 and Fe(II)-EDTA were stable in uninoculated and killed-cell controls in the limited number of experiments using Fe(II)-EDTA instead of Fe2+ (Kumaraswamy et al., 2006; Chakraborty et al., 2011). Although catalysis by Cu2+ and other trace metal ions may have importance in some natural, sedimentary environments, such catalyzed reactions will likely be unimportant in the culture media commonly used in laboratory NDFO experiments. Trace metal solutions used in anaerobic studies (Strąpoć et al., 2008; Wolfe et al., 2011) typically result in final Cu2+ concentrations more than one to two orders-of-magnitude less than the lowest effective catalytic concentration found in the above studies.
and Fe(II)-EDTA were stable in uninoculated and killed-cell controls in the limited number of experiments using Fe(II)-EDTA instead of Fe2+ (Kumaraswamy et al., 2006; Chakraborty et al., 2011). Although catalysis by Cu2+ and other trace metal ions may have importance in some natural, sedimentary environments, such catalyzed reactions will likely be unimportant in the culture media commonly used in laboratory NDFO experiments. Trace metal solutions used in anaerobic studies (Strąpoć et al., 2008; Wolfe et al., 2011) typically result in final Cu2+ concentrations more than one to two orders-of-magnitude less than the lowest effective catalytic concentration found in the above studies.
Solid-phase or adsorbed Fe(II) may reduce 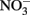 in some cases, but the effectiveness of the reductant is largely determined by the pH and the identity of the solid phase. The ability of the solid phase to function as a catalyst for
in some cases, but the effectiveness of the reductant is largely determined by the pH and the identity of the solid phase. The ability of the solid phase to function as a catalyst for 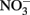 reduction may also be dependent on the simultaneous presence of Fe2+. In controls without supplemental Fe2+ using pasteurized, microbially reduced goethite, and other Fe(II)-bearing solid phases, concomitant losses of
reduction may also be dependent on the simultaneous presence of Fe2+. In controls without supplemental Fe2+ using pasteurized, microbially reduced goethite, and other Fe(II)-bearing solid phases, concomitant losses of 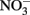 and Fe(II) were not observed (Weber et al., 2001; Chakraborty et al., 2011). GR minerals are effective reductants, especially at slightly alkaline pH, and their transient formation in experimental systems used in microbial NDFO experiments should always be considered. The usual method of GR synthesis involves either controlled oxidation of aqueous FeCl2 or FeSO4 (Tamaura et al., 1984b), the same compounds often used to study microbial Fe2+ oxidation, or an “induced hydrolysis” method involving reaction of Fe2+ with HFO in a pH 7, carbonate-buffered system (Hansen, 1989). In addition, GR-like minerals may also be transiently formed during conversion of other Fe(III) oxyhydroxides, e.g., lepidocrocite, to magnetite during incubation with Fe2+ (Tamaura et al., 1984a; Sørensen and Thorling, 1991). Since similar conditions needed for GR synthesis may exist during microbial NDFO studies, transient formation of minor GR-containing phases during microbial experiments must be taken into consideration. Typical uninoculated or killed-cell controls would not be suitable controls for a possible abiotic GR reaction since GR synthesis from aqueous Fe2+ requires Fe2+ oxidation.
and Fe(II) were not observed (Weber et al., 2001; Chakraborty et al., 2011). GR minerals are effective reductants, especially at slightly alkaline pH, and their transient formation in experimental systems used in microbial NDFO experiments should always be considered. The usual method of GR synthesis involves either controlled oxidation of aqueous FeCl2 or FeSO4 (Tamaura et al., 1984b), the same compounds often used to study microbial Fe2+ oxidation, or an “induced hydrolysis” method involving reaction of Fe2+ with HFO in a pH 7, carbonate-buffered system (Hansen, 1989). In addition, GR-like minerals may also be transiently formed during conversion of other Fe(III) oxyhydroxides, e.g., lepidocrocite, to magnetite during incubation with Fe2+ (Tamaura et al., 1984a; Sørensen and Thorling, 1991). Since similar conditions needed for GR synthesis may exist during microbial NDFO studies, transient formation of minor GR-containing phases during microbial experiments must be taken into consideration. Typical uninoculated or killed-cell controls would not be suitable controls for a possible abiotic GR reaction since GR synthesis from aqueous Fe2+ requires Fe2+ oxidation.
The Fe(III) mineral phases that form as products are often poorly crystalline and their mineralogy can be determined by factors such as pH, phosphate, or bicarbonate concentrations used in the culture media, rates of Fe(II) oxidation, presence of humic compounds or other ligands, and other factors. In cases where mineral identity has been characterized, end-products such as goethite, lepidocrocite, HFO, or Fe(III) phosphates are usually identified (Lack et al., 2002; Kappler et al., 2005; Senko et al., 2005; Miot et al., 2009; Larese-Casanova et al., 2010). Early work included one report of formation of GR and magnetite (Chaudhuri et al., 2001), but later work by the same authors using the same culture and media found formation of HFO with no evidence of GR formation (Lack et al., 2002). The authors attributed the disparate finding to differences in the rate of Fe(II) oxidation by variously treated cultures. Clear evidence of GR formation during Fe(II) oxidation by Acidovorax sp. strain BoFeN1 was recently reported (Pantke et al., 2012) and the relative contribution of enzymatic NDFO versus GR-catalyzed 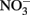 reduction cannot yet be determined in most cases. It should be noted that, although
reduction cannot yet be determined in most cases. It should be noted that, although 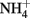 has been produced in some microbial NDFO studies that utilized sediments or sediment inoculum (Weber et al., 2006b; Coby et al., 2011) and with a pure culture of Geobacter metallireducens (Weber et al., 2006b), NDFO enrichment cultures and isolates generally reduce
has been produced in some microbial NDFO studies that utilized sediments or sediment inoculum (Weber et al., 2006b; Coby et al., 2011) and with a pure culture of Geobacter metallireducens (Weber et al., 2006b), NDFO enrichment cultures and isolates generally reduce 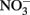 to N2 or N2O (Straub et al., 1996, 2004; Benz et al., 1998; Straub and Buchholz-Cleven, 1998; Weber et al., 2009). Since the products of abiotic oxidation of wüstite or GR by
to N2 or N2O (Straub et al., 1996, 2004; Benz et al., 1998; Straub and Buchholz-Cleven, 1998; Weber et al., 2009). Since the products of abiotic oxidation of wüstite or GR by 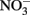 are magnetite and
are magnetite and 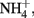 the absence of these products in most NDFO studies suggests that the abiotic reduction of
the absence of these products in most NDFO studies suggests that the abiotic reduction of 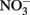 by transient amounts of GR minerals formed during Fe2+ oxidation may not be a significant contributor in many experiments.
by transient amounts of GR minerals formed during Fe2+ oxidation may not be a significant contributor in many experiments.
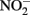
The thermodynamics of Fe2+ oxidation to goethite (α-FeOOH) coupled with the reduction of 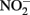 to either N2, N2O, or
to either N2, N2O, or 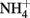 are also favorable.
are also favorable.
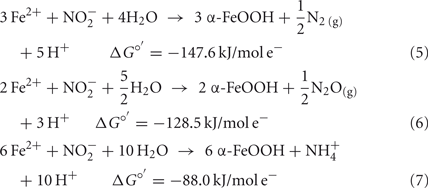
Compared to 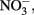 the kinetics of
the kinetics of 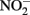 reduction by Fe(II) are generally more rapid in both homogeneous and heterogeneous reactions. In early work, Nelson and Bremner (1970) showed that only 4% of 3.6 mM
reduction by Fe(II) are generally more rapid in both homogeneous and heterogeneous reactions. In early work, Nelson and Bremner (1970) showed that only 4% of 3.6 mM 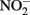 remained after 24 h incubation with 25 mM Fe2+ at pH 5. It is not clear, however, if air was excluded from their reaction system. Moraghan and Buresh (1977) conducted experiments with 14 mM Fe2+ under anoxic conditions and found complete removal of 1.8 mM
remained after 24 h incubation with 25 mM Fe2+ at pH 5. It is not clear, however, if air was excluded from their reaction system. Moraghan and Buresh (1977) conducted experiments with 14 mM Fe2+ under anoxic conditions and found complete removal of 1.8 mM 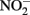 after 24 h at pH 8 with N2O as the primary product. At pH 6, only 12% of the
after 24 h at pH 8 with N2O as the primary product. At pH 6, only 12% of the 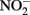 was reduced during the same period, although the presence of Cu2+ dramatically increased reaction rates. Since Fe(III) oxyhydroxide precipitates formed during their studies, the role of heterogeneous reactions could not be definitively ascertained. Van Cleemput and Baert (1983) examined reduction of
was reduced during the same period, although the presence of Cu2+ dramatically increased reaction rates. Since Fe(III) oxyhydroxide precipitates formed during their studies, the role of heterogeneous reactions could not be definitively ascertained. Van Cleemput and Baert (1983) examined reduction of 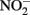 by aqueous Fe2+ under anoxic conditions over a pH range of 4–6. Rates of
by aqueous Fe2+ under anoxic conditions over a pH range of 4–6. Rates of 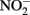 reduction were slowest at pH 6 and increased as the solution became more acidic. Fast rates of reaction between Fe2+ and
reduction were slowest at pH 6 and increased as the solution became more acidic. Fast rates of reaction between Fe2+ and 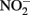 at acidic pH were also reported by Wullstein and Gilmour (1966).
at acidic pH were also reported by Wullstein and Gilmour (1966).
In addition to the presence of Cu2+ or low pH, abiotic  oxidation of Fe2+ is enhanced by the presence of solid phases. Van Cleemput and Baert (1983) reported that the addition of amorphous Fe(III) hydroxide (HFO) and, to a lesser extent, magnetite, greatly accelerated rates of reaction compared to systems containing Fe2+ alone. The effect of added HFO in heterogeneous systems was especially pronounced at pH 6 and 8 where rates of
oxidation of Fe2+ is enhanced by the presence of solid phases. Van Cleemput and Baert (1983) reported that the addition of amorphous Fe(III) hydroxide (HFO) and, to a lesser extent, magnetite, greatly accelerated rates of reaction compared to systems containing Fe2+ alone. The effect of added HFO in heterogeneous systems was especially pronounced at pH 6 and 8 where rates of 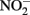 reduction were relatively slow absent the solid phase. Tai and Dempsey (2009) also showed that
reduction were relatively slow absent the solid phase. Tai and Dempsey (2009) also showed that 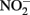 reduction rates at pH 6.8 were greatly enhanced in heterogeneous systems containing both HFO and Fe2+ compared to systems containing Fe2+ alone.
reduction rates at pH 6.8 were greatly enhanced in heterogeneous systems containing both HFO and Fe2+ compared to systems containing Fe2+ alone.
Sorption of Fe(II) to crystalline Fe(III) oxyhydroxides is also known to increase abiotic 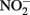 reduction rates. Sørensen and Thorling (1991) examined the effect of added lepidocrocite (γ-FeOOH) on the rates of abiotic Fe(II)-dependent
reduction rates. Sørensen and Thorling (1991) examined the effect of added lepidocrocite (γ-FeOOH) on the rates of abiotic Fe(II)-dependent 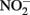 reduction over a pH range of 6–8.5 under anoxic conditions. Rates of reduction of
reduction over a pH range of 6–8.5 under anoxic conditions. Rates of reduction of 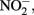 primarily to N2O, occurred much more rapidly in the presence of lepidocrocite than in its absence and increased progressively from pH 7.5 to 8.5. Fe2+ reacted with the solid phase to form magnetite (Fe3O4) and the authors suggest that a transient GR phase may have formed at pH 8 and above during magnetite formation. Sorption of Fe2+ to goethite was also found by Coby and Picardal (2005) to increase reduction of
primarily to N2O, occurred much more rapidly in the presence of lepidocrocite than in its absence and increased progressively from pH 7.5 to 8.5. Fe2+ reacted with the solid phase to form magnetite (Fe3O4) and the authors suggest that a transient GR phase may have formed at pH 8 and above during magnetite formation. Sorption of Fe2+ to goethite was also found by Coby and Picardal (2005) to increase reduction of 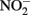 to N2O at pH 7 compared to systems lacking goethite.
to N2O at pH 7 compared to systems lacking goethite.
The ability of 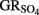 to reduce
to reduce 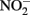 has been shown by Hansen et al. (1994) who proposed that
has been shown by Hansen et al. (1994) who proposed that 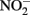 can be both reduced rapidly during GR formation and more slowly by Fe(II) in the GR lattice. In a recent report by Kampschreur et al. (2011), reduction of
can be both reduced rapidly during GR formation and more slowly by Fe(II) in the GR lattice. In a recent report by Kampschreur et al. (2011), reduction of 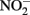 to NO and N2O by Fe2+ at pH 5.6–7.6 was attributed both to reaction with aqueous Fe2+ and the transient formation of GR-like compounds although a characterization was not done of the solid-phase formed during the reaction. With the exception of a recent report (Pantke et al., 2012) as described above, GR minerals are typically not found as final products during microbial NDFO exists in spite of conditions, i.e., presence of Fe2+ and freshly precipitated Fe(III) oxyhydroxides (Hansen, 1989), that may favor transient appearance during reaction progress.
to NO and N2O by Fe2+ at pH 5.6–7.6 was attributed both to reaction with aqueous Fe2+ and the transient formation of GR-like compounds although a characterization was not done of the solid-phase formed during the reaction. With the exception of a recent report (Pantke et al., 2012) as described above, GR minerals are typically not found as final products during microbial NDFO exists in spite of conditions, i.e., presence of Fe2+ and freshly precipitated Fe(III) oxyhydroxides (Hansen, 1989), that may favor transient appearance during reaction progress.
In addition to reactions with GR and Fe2+ sorbed to Fe(III) oxyhydroxides, other studies have shown abiotic reactions of 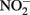 with various forms of solid-phase Fe(II). Weber et al. (2001) demonstrated that
with various forms of solid-phase Fe(II). Weber et al. (2001) demonstrated that 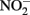 can abiotically oxidize Fe(II) in microbially reduced goethite and subsoils in addition to biogenic magnetite. In their experiments, neither biogenic nor chemically precipitated siderite (FeCO3) was abiotically oxidized by
can abiotically oxidize Fe(II) in microbially reduced goethite and subsoils in addition to biogenic magnetite. In their experiments, neither biogenic nor chemically precipitated siderite (FeCO3) was abiotically oxidized by 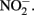 This contrasts with the subsequent experiments of Rakshit et al. (2008) who demonstrated abiotic reduction of
This contrasts with the subsequent experiments of Rakshit et al. (2008) who demonstrated abiotic reduction of 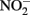 to N2O by chemically precipitated siderite at increasing rates as the pH was reduced from 7.9 to 5.5. In a separate study, Rakshit et al. (2005) also showed that wüstite (FeO) was able to reduce
to N2O by chemically precipitated siderite at increasing rates as the pH was reduced from 7.9 to 5.5. In a separate study, Rakshit et al. (2005) also showed that wüstite (FeO) was able to reduce 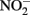 to
to 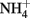 at rates significantly greater than corresponding
at rates significantly greater than corresponding 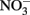 reduction rates.
reduction rates.
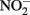 may also be reactive with Fe(II)-EDTA although results are equivocal. In 6-day, abiotic experiments with 2.5 mM
may also be reactive with Fe(II)-EDTA although results are equivocal. In 6-day, abiotic experiments with 2.5 mM 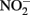 , 5 mM
, 5 mM 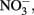 and 5 mM Fe(II)-EDTA at pH 7, no losses of any compound were observed (Chakraborty and Picardal, unpublished data). In the experiments of Kumaraswamy et al. (2006) at the same pH, however, 18% of 10 mM Fe(II)-EDTA was oxidized after 24 h. The greater reactivity of
and 5 mM Fe(II)-EDTA at pH 7, no losses of any compound were observed (Chakraborty and Picardal, unpublished data). In the experiments of Kumaraswamy et al. (2006) at the same pH, however, 18% of 10 mM Fe(II)-EDTA was oxidized after 24 h. The greater reactivity of 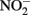 has also been highlighted in studies (Cooper et al., 2003; Coby and Picardal, 2005) which described abiotic reactions between
has also been highlighted in studies (Cooper et al., 2003; Coby and Picardal, 2005) which described abiotic reactions between 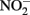 and Fe(II) sorbed to microbial cell surfaces. The products of this reaction were N2O and Fe(III) oxyhydroxide encrustations on cell surfaces that impeded subsequent transport of soluble substrates into cells.
and Fe(II) sorbed to microbial cell surfaces. The products of this reaction were N2O and Fe(III) oxyhydroxide encrustations on cell surfaces that impeded subsequent transport of soluble substrates into cells.
A unified and comprehensive understanding of abiotic reactions between 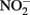 and Fe2+ is difficult to develop due to sometimes conflicting results in some of the above studies, likely due to the wide range of reactant concentrations (micromolar to tens of millimolar), different pH regimes, varying experimental conditions, incomplete characterization of aqueous:solid-phase Fe speciation, potential transient formation of reactive species and complexes, and often unspecified final reaction products. It is clear, however, that
and Fe2+ is difficult to develop due to sometimes conflicting results in some of the above studies, likely due to the wide range of reactant concentrations (micromolar to tens of millimolar), different pH regimes, varying experimental conditions, incomplete characterization of aqueous:solid-phase Fe speciation, potential transient formation of reactive species and complexes, and often unspecified final reaction products. It is clear, however, that 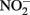 and soluble Fe2+ are reactive at acidic pHs and the Fe2+ sorption to amorphous and crystalline Fe(III) oxyhydroxides can increase rates of
and soluble Fe2+ are reactive at acidic pHs and the Fe2+ sorption to amorphous and crystalline Fe(III) oxyhydroxides can increase rates of 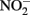 reduction. Sorption of Fe2+ to cell surfaces or transient formation of reactive GR minerals may also create artifacts under certain experimental conditions.
reduction. Sorption of Fe2+ to cell surfaces or transient formation of reactive GR minerals may also create artifacts under certain experimental conditions.
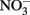 -Dependent Fe(II) Oxidation and Potential Abiotic Interactions
-Dependent Fe(II) Oxidation and Potential Abiotic InteractionsNitrate-dependent Fe(II) oxidation was first described by Straub et al. (1996) and is catalyzed by phylogenetically diverse bacteria in a variety of environments (Hafenbradl et al., 1996; Benz et al., 1998; Straub and Buchholz-Cleven, 1998; Straub et al., 2004; Kumaraswamy et al., 2006; Muehe et al., 2009; Weber et al., 2009). NDFO has been demonstrated in a variety of sediment enrichments, microbial consortia, and pure cultures, including both autotrophic and mixotrophic cultures. In addition to the autotrophic enrichment culture originally described by Straub et al. (1996), autotrophic growth has been reported for Pseudogulbenkiania sp. Strain 2002 (Weber et al., 2006a, 2009), Paracoccus ferrooxidans using organically complexed Fe(II) (Kumaraswamy et al., 2006), and a hyperthermophilic archaeum (Hafenbradl et al., 1996). Most pure cultures capable of NDFO, however, are organotrophic, 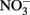 -reducing bacteria that oxidize Fe(II) in the presence of an organic cosubstrate such as acetate, either cometabolically or through a mixotrophic physiology. Examples of such bacteria include Paracoccus denitrificans (Muehe et al., 2009), various Acidovorax sp. (Straub et al., 2004; Kappler et al., 2005; Muehe et al., 2009; Byrne-Bailey et al., 2010; Chakraborty et al., 2011), Aquabacterium sp. Strain BrG2 (Straub et al., 2004), G. metallireducens (Finneran et al., 2002; Weber et al., 2006b), Azospira oryzae (“Dechlorosoma suillum”) (Lack et al., 2002), and other members of the α-, β-, γ-, and δ-subgroups of the Proteobacteria in addition to Gram-positive bacteria (Straub and Buchholz-Cleven, 1998; Straub et al., 2004; Kappler and Straub, 2005).
-reducing bacteria that oxidize Fe(II) in the presence of an organic cosubstrate such as acetate, either cometabolically or through a mixotrophic physiology. Examples of such bacteria include Paracoccus denitrificans (Muehe et al., 2009), various Acidovorax sp. (Straub et al., 2004; Kappler et al., 2005; Muehe et al., 2009; Byrne-Bailey et al., 2010; Chakraborty et al., 2011), Aquabacterium sp. Strain BrG2 (Straub et al., 2004), G. metallireducens (Finneran et al., 2002; Weber et al., 2006b), Azospira oryzae (“Dechlorosoma suillum”) (Lack et al., 2002), and other members of the α-, β-, γ-, and δ-subgroups of the Proteobacteria in addition to Gram-positive bacteria (Straub and Buchholz-Cleven, 1998; Straub et al., 2004; Kappler and Straub, 2005).
Fe(II) oxidation by mixotrophic organisms may be linked to energy conservation and growth or, in some cases, result from a summation of both enzymatic reactions and abiotic side reactions that occur fortuitously during organotrophic growth. Fe(II)-oxidation-enhanced growth has been clearly shown during mixotrophic growth by some Acidovorax sp. In experiments with two different Acidovorax isolates (Muehe et al., 2009; Chakraborty et al., 2011), increases in growth yield in batch cultures using 4–10 mM Fe2+, 8–10 mM 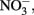 and 1.5–5 mM acetate were consistently greater in the presence of Fe2+ than in its absence. In the experiments of Chakraborty et al. growth typically ceased after several days, regardless of the presence of remaining substrate, likely as a result of Fe(III) oxyhydroxide coatings that develop on cells. When Fe(II)-EDTA was used instead of Fe2+ in their experiments, oxyhydroxide encrustations did not form and further additions of substrate could be utilized. In a continuous-flow system using 50–250 μM Fe2+, 100 μM
and 1.5–5 mM acetate were consistently greater in the presence of Fe2+ than in its absence. In the experiments of Chakraborty et al. growth typically ceased after several days, regardless of the presence of remaining substrate, likely as a result of Fe(III) oxyhydroxide coatings that develop on cells. When Fe(II)-EDTA was used instead of Fe2+ in their experiments, oxyhydroxide encrustations did not form and further additions of substrate could be utilized. In a continuous-flow system using 50–250 μM Fe2+, 100 μM 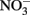 , and 20 μM acetate, they did not observe formation of cell encrustations and cell growth was sustained over a 14-day period rather than the 3–4 days observed using higher concentrations in batch cultures. Overall, it is clear that NDFO can enhance growth of some mixotrophic Fe(II) oxidizers and that the formation of cell encrustations in batch systems at millimolar substrate concentrations can limit growth and substrate utilization. These cell encrustations are commonly observed during NDFO in batch culture and their characterization has received detailed scrutiny (Kappler et al., 2005, 2010; Miot et al., 2009; Schädler et al., 2009).
, and 20 μM acetate, they did not observe formation of cell encrustations and cell growth was sustained over a 14-day period rather than the 3–4 days observed using higher concentrations in batch cultures. Overall, it is clear that NDFO can enhance growth of some mixotrophic Fe(II) oxidizers and that the formation of cell encrustations in batch systems at millimolar substrate concentrations can limit growth and substrate utilization. These cell encrustations are commonly observed during NDFO in batch culture and their characterization has received detailed scrutiny (Kappler et al., 2005, 2010; Miot et al., 2009; Schädler et al., 2009).
Although it is clear that NDFO is a biological process that can increase growth yields in some bacteria, the possibilities of abiotic reactions between Fe(II) and oxidized N species during the course of batch NDFO studies requires that experimenters consider both biotic and abiotic mechanisms when evaluating results. This is particularly important since typical abiotic controls in such experiments do not effectively mimic the changing aqueous and solid-phase chemical conditions that occur in live-culture replicates. Uninoculated or killed-cell controls in microbiological NDFO experiments typically consist of anoxic bottles containing culture medium components, ferrous iron (usually aqueous Fe2+) and 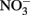 (Straub et al., 1996; Weber et al., 2001). Controls of this type have established that
(Straub et al., 1996; Weber et al., 2001). Controls of this type have established that 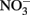 is usually unreactive with Fe2+ or the solid-phase Fe(II) initially added over the time period used in typical experiments.
is usually unreactive with Fe2+ or the solid-phase Fe(II) initially added over the time period used in typical experiments.
Such controls, however, do not contain the freshly precipitated Fe(III) oxyhydroxides that form in live-culture replicates. These materials likely include a spectrum of metastable oxyhydroxides of unknown mineralogy that are able to sorb Fe2+ from the aqueous phase and which are subject to continuing phase changes over the course of the experiment. The final phases formed will be a function of the solution chemistry, buffer choice, Fe2+ concentration, and Fe(II) oxidation rate. Although the predominant mineral phases present at the conclusion of the experiment have been characterized in a number of studies (Lack et al., 2002; Kappler et al., 2005; Miot et al., 2009), these identified phases represent a “snapshot” at a particular time and may also not capture highly reactive minor phases. Such reactive phases, especially in the presence of the 5- to 10-mM Fe2+ concentrations used in typical batch experiments, may lead to limited abiotic reactions with 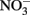 and, to a greater extent, with biogenic
and, to a greater extent, with biogenic 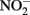 .
.
The accumulation of 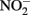 in live cultures can potentially be more problematic than
in live cultures can potentially be more problematic than 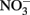 in producing abiotic artifacts due to greater possibility of reactions with Fe2+ sorbed to cellular materials or biogenic Fe(III) oxyhydroxides.The accumulation of 0.5–1.5 mM
in producing abiotic artifacts due to greater possibility of reactions with Fe2+ sorbed to cellular materials or biogenic Fe(III) oxyhydroxides.The accumulation of 0.5–1.5 mM 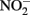 in NDFO batch cultures has been noted in several studies (Kappler et al., 2005; Larese-Casanova et al., 2010; Chakraborty et al., 2011). This
in NDFO batch cultures has been noted in several studies (Kappler et al., 2005; Larese-Casanova et al., 2010; Chakraborty et al., 2011). This 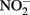 accumulation did not occur during organotrophic growth with acetate and often coincides with Fe2+ oxidation. In some studies, controls have been used to attempt to discount the contribution of abiotic Fe(II) oxidation by
accumulation did not occur during organotrophic growth with acetate and often coincides with Fe2+ oxidation. In some studies, controls have been used to attempt to discount the contribution of abiotic Fe(II) oxidation by 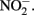 These controls contained
These controls contained 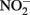 and either soluble Fe2+ (Kappler et al., 2005) or solid-phase Fe(II) (Weber et al., 2001), but not both. Since the abiotic studies described above have shown that
and either soluble Fe2+ (Kappler et al., 2005) or solid-phase Fe(II) (Weber et al., 2001), but not both. Since the abiotic studies described above have shown that 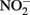 can react with Fe2+ sorbed to Fe-containing minerals, controls lacking both a Fe(III) oxyhydroxide and millimolar Fe2+ concentrations are not truly representative of conditions present in live cultures and may underestimate abiotic reaction kinetics. It is therefore currently difficult to accurately ascertain the relative contributions of abiotic and biotic reaction pathways in most studies where
can react with Fe2+ sorbed to Fe-containing minerals, controls lacking both a Fe(III) oxyhydroxide and millimolar Fe2+ concentrations are not truly representative of conditions present in live cultures and may underestimate abiotic reaction kinetics. It is therefore currently difficult to accurately ascertain the relative contributions of abiotic and biotic reaction pathways in most studies where 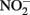 accumulates in batch culture media.
accumulates in batch culture media.
The reason why 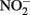 accumulates in mixotrophic, Fe(II)-oxidizing batch cultures but not in cultures growing organotrophically is also subject to speculation. One possibility is that electrons from Fe(II) enter the respiratory electron transport chain at a branch point where electron transport is possible to
accumulates in mixotrophic, Fe(II)-oxidizing batch cultures but not in cultures growing organotrophically is also subject to speculation. One possibility is that electrons from Fe(II) enter the respiratory electron transport chain at a branch point where electron transport is possible to 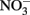 but not to
but not to 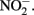 Alternately, Fe2+ could diffuse into the periplasm and sorb to a periplasmic
Alternately, Fe2+ could diffuse into the periplasm and sorb to a periplasmic 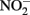 reductase. Since studies have shown that Fe2+ sorbed to cellular material can reduce
reductase. Since studies have shown that Fe2+ sorbed to cellular material can reduce 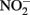 (Coby and Picardal, 2005), abiotic oxidation of the sorbed Fe2+ by biogenic
(Coby and Picardal, 2005), abiotic oxidation of the sorbed Fe2+ by biogenic 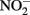 could result in accumulation of Fe(III)-mineral precipitates on the
could result in accumulation of Fe(III)-mineral precipitates on the 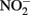 reductase and in the periplasm. Periplasmic accumulations of Fe(III) minerals have indeed been observed (Miot et al., 2009, 2011; Schädler et al., 2009). Although it is not clear if these periplasmic mineral accumulations are the result of abiotic or enzymatic Fe2+ oxidation, the recent observation (Miot et al., 2011) of mineralized protein globules in the periplasm and outer face of the plasma membrane supports the hypothesis that
reductase and in the periplasm. Periplasmic accumulations of Fe(III) minerals have indeed been observed (Miot et al., 2009, 2011; Schädler et al., 2009). Although it is not clear if these periplasmic mineral accumulations are the result of abiotic or enzymatic Fe2+ oxidation, the recent observation (Miot et al., 2011) of mineralized protein globules in the periplasm and outer face of the plasma membrane supports the hypothesis that 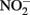 accumulation in Fe(II)-oxidizing cultures results from inhibition of the nitrite reductase by mineral deposition. Ultimately, very little is definitively known at the current time about mechanisms of microbial NDFO and the location of Fe(II) oxidation. The demonstration of
accumulation in Fe(II)-oxidizing cultures results from inhibition of the nitrite reductase by mineral deposition. Ultimately, very little is definitively known at the current time about mechanisms of microbial NDFO and the location of Fe(II) oxidation. The demonstration of 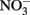 -dependent oxidation of solid-phase Fe(II) (Weber et al., 2001), however, strongly suggests that physiological oxidation takes place outside the cell since solid-phase Fe(II) is unlikely to enter cells.
-dependent oxidation of solid-phase Fe(II) (Weber et al., 2001), however, strongly suggests that physiological oxidation takes place outside the cell since solid-phase Fe(II) is unlikely to enter cells.
Although alternate mechanisms governing production of mineral cell encrustations during mixotrophic NDFO have been proposed (Kappler and Newman, 2004; Kappler et al., 2005; Schädler et al., 2009), the sorption of Fe2+ to cell surfaces in media containing 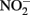 is sufficient for abiotic cell encrustation by Fe(III) oxyhydroxides (Coby and Picardal, 2005). When Fe(II)-EDTA was used in batch cultures instead of Fe2+ in the studies of Chakraborty et al. (2011) encrusted cells did not develop and utilization of additional amendments of soluble substrate was not blocked, even though >1 mM
is sufficient for abiotic cell encrustation by Fe(III) oxyhydroxides (Coby and Picardal, 2005). When Fe(II)-EDTA was used in batch cultures instead of Fe2+ in the studies of Chakraborty et al. (2011) encrusted cells did not develop and utilization of additional amendments of soluble substrate was not blocked, even though >1 mM 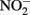 accumulated. In continuous-flow systems in which advective transport and low-micromolar Fe2+, acetate, and
accumulated. In continuous-flow systems in which advective transport and low-micromolar Fe2+, acetate, and 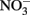 concentrations prevent
concentrations prevent 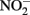 accumulation, they observed no significant cell encrustations. Lack of encrustations in continuous-flow systems may also be a result of advective removal of nanoscale Fe(III) oxide precipitates before they have an opportunity to aggregate and accumulate on cells.
accumulation, they observed no significant cell encrustations. Lack of encrustations in continuous-flow systems may also be a result of advective removal of nanoscale Fe(III) oxide precipitates before they have an opportunity to aggregate and accumulate on cells.
The use of such continuous-flow systems may be helpful in establishing whether Fe(II)-oxidation can be coupled to energy conservation and growth in other isolates. Since measurements of substrate conversion are problematic in a continuous-flow system operated at low micromolar substrate concentrations (described in Chakraborty et al., 2011), batch systems are still likely required to examine reaction stoichiometry and achieve a mass balance of reactants and products. In such cases, reducing concentrations of one or all substrates may lower abiotic reaction rates relative to microbial rates, reduce 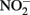 accumulation, and thereby minimize development of cell encrustations. Alternately, it may be possible to develop opposing-gradient culture systems based on those used to study microaerophilic Fe(II) oxidation (Emerson and Floyd, 2005). Additional studies are needed to determine if such approaches are useful alternatives to the use of 5–10 mM concentrations of Fe2+ and
accumulation, and thereby minimize development of cell encrustations. Alternately, it may be possible to develop opposing-gradient culture systems based on those used to study microaerophilic Fe(II) oxidation (Emerson and Floyd, 2005). Additional studies are needed to determine if such approaches are useful alternatives to the use of 5–10 mM concentrations of Fe2+ and 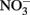 commonly used in batch NDFO systems.
commonly used in batch NDFO systems.
It is also important to consider abiotic reactions when measuring Fe species in NDFO experiments in which 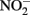 accumulates. Total Fe(II), i.e., sorbed and aqueous Fe2+, are commonly measured using 0.5 M HCl extraction. As described above,
accumulates. Total Fe(II), i.e., sorbed and aqueous Fe2+, are commonly measured using 0.5 M HCl extraction. As described above, 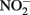 oxidizes Fe2+ relatively rapidly in acidic solution and a sequential extraction (Cooper et al., 2000; Weber et al., 2001) is necessary to avoid overestimating the extent of Fe(II) oxidation. A review of the literature reveals that this is not always done, raising doubts about Fe speciation data in such studies whenever
oxidizes Fe2+ relatively rapidly in acidic solution and a sequential extraction (Cooper et al., 2000; Weber et al., 2001) is necessary to avoid overestimating the extent of Fe(II) oxidation. A review of the literature reveals that this is not always done, raising doubts about Fe speciation data in such studies whenever 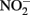 is present.
is present.
Although the focus of this paper has been on potential abiotic reactions between Fe(II) and 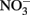 or
or 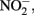 it should be emphasized that the microbial oxidation of Fe(II) can clearly be enzymatically catalyzed by microorganisms and conservation of energy from the reaction can, at least in some cases, be coupled to growth. Microaerobic Fe(II) oxidizers active at circumneutral pH must compete with the abiotic oxidation of Fe(II) by O2 and potential Fe(III) oxide encrustations by specializing in environments, e.g., at oxic:anoxic boundaries with low substrate concentrations, where abiotic reactions are not overly detrimental (Emerson, 2000). Likely to a lesser extent,
it should be emphasized that the microbial oxidation of Fe(II) can clearly be enzymatically catalyzed by microorganisms and conservation of energy from the reaction can, at least in some cases, be coupled to growth. Microaerobic Fe(II) oxidizers active at circumneutral pH must compete with the abiotic oxidation of Fe(II) by O2 and potential Fe(III) oxide encrustations by specializing in environments, e.g., at oxic:anoxic boundaries with low substrate concentrations, where abiotic reactions are not overly detrimental (Emerson, 2000). Likely to a lesser extent, 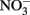 -dependent Fe(II) oxidizers must similarly exist in natural aquatic systems in a defined environmental milieu or substrate concentration regime where abiotic and biotic reactions can coexist, inhibitory accumulations of mineral encrustations do not form, and
-dependent Fe(II) oxidizers must similarly exist in natural aquatic systems in a defined environmental milieu or substrate concentration regime where abiotic and biotic reactions can coexist, inhibitory accumulations of mineral encrustations do not form, and 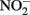 accumulations are minimized. These environments certainly differ from the “unnatural” conditions used in most batch studies. As in microaerobic systems, although possibly to a lesser extent, experimenters will need to deal with special challenges that arise from the potential abiotic reactions when designing experiments and interpreting experimental results.
accumulations are minimized. These environments certainly differ from the “unnatural” conditions used in most batch studies. As in microaerobic systems, although possibly to a lesser extent, experimenters will need to deal with special challenges that arise from the potential abiotic reactions when designing experiments and interpreting experimental results.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by National Science Foundation Biogeosciences Program Grant 0525069 to F. Picardal.
Amonette, J. E., Workman, D. J., Kennedy, D. W., Fruchter, J. S., and Gorby, Y. A. (2000). Dechlorination of carbon tetrachloride by Fe(II) associated with goethite. Environ. Sci. Technol. 34, 4606–4613.
Benz, M., Brune, A., and Schink, B. (1998). Anaerobic and aerobic oxidation of ferrous iron at neutral pH by chemoheterotrophic nitrate-reducing bacteria. Arch. Microbiol. 169, 159–165.
Blothe, M., and Roden, E. E. (2009). Composition and activity of an autotrophic Fe(II)-oxidizing, nitrate-reducing enrichment culture. Appl. Environ. Microbiol. 75, 6937–6940.
Buresh, R. J., and Moraghan, J. T. (1976). Chemical reduction of nitrate by ferrous iron. J. Environ. Qual. 5, 320–325.
Byrne-Bailey, K. G., Weber, K. A., Chair, A. H., Bose, S., Knox, T., Spanbauer, T. L., Chertkov, O., and Coates, J. D. (2010). Completed genome sequence of the anaerobic iron-oxidizing bacterium Acidovorax ebreus strain TPSY. J. Bacteriol. 192, 1475–1476.
Chakraborty, A., Roden, E. E., Schieber, J., and Picardal, F. (2011). Enhanced growth of Acidovorax sp. strain 2AN during nitrate-dependent Fe(II) oxidation in batch and continuous-flow systems. Appl. Environ. Microbiol. 77, 8548–8566.
Chao, T.-T., and Kroontje, W. (1966). Inorganic nitrogen transformations through the oxidation and reduction of iron. Soil Sci. Soc. Am. J. 30, 193–196.
Chaudhuri, S., Lack, J., and Coates, J. (2001). Biogenic magnetite formation through anaerobic biooxidation of Fe(II). Appl. Environ. Microbiol. 67, 2844–2848.
Choi, J., and Batchelor, B. (2008). Nitrate reduction by fluoride green rust modified with copper. Chemosphere 70, 1108–1116.
Coby, A. J., Picardal, F., Shelobolina, E., Xu, H. F., and Roden, E. E. (2011). Repeated anaerobic microbial redox cycling of iron. Appl. Environ. Microbiol. 77, 6036–6042.
Coby, A. J., and Picardal, F. W. (2005). Inhibition of NO3− and NO2− reduction by microbial Fe(III) reduction: evidence of a reaction between NO2− and cell surface-bound Fe2+. Appl. Environ. Microbiol. 71, 5267–5274.
Cooper, D. C., Picardal, F., Rivera, J., and Talbot, C. (2000). Zinc immobilization and magnetite formation via ferric oxide reduction by Shewanella putrefaciens 200. Environ. Sci. Technol. 34, 100–106.
Cooper, D. C., Picardal, F. W., Schimmelmann, A., and Coby, A. J. (2003). Chemical and biological interactions during nitrate and goethite reduction by Shewanella putrefaciens 200. Appl. Environ. Microbiol. 69, 3517–3525.
Cui, D., and Eriksen, T. E. (1996). Reduction of pertechnetate by ferrous iron in solution: influence of sorbed and precipitated Fe(II). Environ. Sci. Technol. 30, 2259–2262.
Emerson, D. (2000). “Microbial oxidation of Fe(II) and Mn(II) at circumneutral pH,” in Environmental Microbe-Metal Interactions, ed. D. R. Lovley (Washington, DC: ASM Press), 31–52.
Emerson, D., and Floyd, M. M. (2005). Enrichment and isolation of iron-oxidizing bacteria at neutral pH. Meth. Enzymol. 397, 112–123.
Finneran, K. T., Housewright, M. E., and Lovley, D. R. (2002). Multiple influences of nitrate on uranium solubility during bioremediation of uranium-contaminated subsurface sediments. Environ. Microbiol. 4, 510–516.
Hafenbradl, D., Keller, M., Dirmeier, R., Rachel, R., Roßnagel, P., Burggraf, S., Huber, H., and Stetter, K. O. (1996). Ferroglobus placidus gen. nov., sp. nov., a novel hyperthermophilic archaeum that oxidizes Fe2+ at neutral pH under anoxic conditions. Arch. Microbiol. 166, 308–314.
Hansen, H. C. B. (1989). Composition, stabilization, and light absorption of Fe(II)Fe(III) hydroxy-carbonate (‘green rust’). Clay Miner. 24, 663–669.
Hansen, H. C. B., Borggaard, O. K., and Sørensen, J. (1994). Evaluation of the free energy of formation of Fe(II)-Fe(III) hydroxide-sulphate (green rust) and its reduction of nitrite. Geochim. Cosmochim. Acta 58, 2599–2608.
Hansen, H. C. B., Guldberg, S., Erbs, M., and Bender Koch, C. (2001). Kinetics of nitrate reduction by green rusts-effects of interlayer anion and Fe(II):Fe(III) ratio. Appl. Clay Sci. 18, 81–91.
Hansen, H. C. B., Koch, C. B., Nancke-Krogh, H., Borggaard, O. K., and Sørensen, J. (1996). Abiotic nitrate reduction to ammonium: key role of green rust. Environ. Sci. Technol. 30, 2053–2056.
Kampschreur, M. J., Kleerebezem, R., De Vet, W. W. J. M., and Van Loosdrecht, M. C. M. (2011). Reduced iron induced nitric oxide and nitrous oxide emission. Water Res. 45, 5945–5952.
Kappler, A., Johnson, C. M., Crosby, H. A., Beard, B. L., and Newman, D. K. (2010). Evidence for equilibrium iron isotope fractionation by nitrate-reducing iron(II)-oxidizing bacteria. Geochim. Cosmochim. Acta 74, 2826–2842.
Kappler, A., and Newman, D. K. (2004). Formation of Fe(III)-minerals by Fe(II)-oxidizing photoautotrophic bacteria. Geochim. Cosmochim. Acta 68, 1217–1226.
Kappler, A., Schink, B., and Newman, D. K. (2005). Fe(III) mineral formation and cell encrustation by the nitrate-dependent Fe(II)-oxidizer strain BoFeN1. Geobiology 3, 235–245.
Kappler, A., and Straub, K. L. (2005). Geomicrobiological cycling of iron. Rev. Mineral. Geochem. 59, 85–108.
Kim, S., and Picardal, F. W. (1999). Enhanced anaerobic biodegradation of carbon tetrachloride in the presence of reduced iron oxides. Environ. Toxicol. Chem. 18, 2142–2150.
Klausen, J., Tröber, S. P., Haderlein, S. B., and Schwarzenbach, R. (1995). Reduction of substituted nitrobenzenes by Fe(II) in aqueous mineral solutions. Environ. Sci. Technol. 29, 2396–2404.
Kumaraswamy, R., Sjollema, K., Kuenen, G., Van Loosdrecht, M., and Muyzer, G. (2006). Nitrate-dependent [Fe(II)EDTA]2− oxidation by Paracoccus ferrooxidans sp nov., isolated from a denitrifying bioreactor. Syst. Appl. Microbiol. 29, 276–286.
Lack, J. G., Chaudhuri, S. K., Chakraborty, R., Achenbach, L. A., and Coates, J. D. (2002). Anaerobic biooxidation of Fe(II) by Dechlorosoma suillum. Microb. Ecol. 43, 424–431.
Larese-Casanova, P., Haderlein, S. B., and Kappler, A. (2010). Biomineralization of lepidocrocite and goethite by nitrate-reducing Fe(II)-oxidizing bacteria: effect of pH, bicarbonate, phosphate, and humic acids. Geochim. Cosmochim. Acta 74, 3721–3734.
Miot, J., Benzerara, K., Morin, G., Kappler, A., Bernard, S., Obst, M., Férard, C., Skouri-Panet, F., Guigner, J.-M., Posth, N., Galvez, M., Brown, G. E. Jr., and Guyot, F. (2009). Iron biomineralization by anaerobic neutrophilic iron-oxidizing bacteria. Geochim. Cosmochim. Acta 73, 696–711.
Miot, J., Maclellan, K., Benzerara, K., and Boisset, N. (2011). Preservation of protein globules and peptidoglycan in the mineralized cell wall of nitrate-reducing, iron(II)-oxidizing bacteria: a cryo-electron microscopy study. Geobiology 9, 459–470.
Moraghan, J. T., and Buresh, R. J. (1977). Chemical reduction of nitrite and nitrous oxide by ferrous iron. Soil Sci. Soc. Am. J. 41, 47–50.
Muehe, E. M., Gerhardt, S., Schink, B., and Kappler, A. (2009). Ecophysiology and the energetic benefit of mixotrophic Fe(II) oxidation by various strains of nitrate-reducing bacteria. FEMS Microbiol. Ecol. 70, 3–11.
Nelson, D. W., and Bremner, J. M. (1970). Role of soil minerals and metallic cations in nitrite decomposition and chemodenitrification in soils. Soil Biol. Biochem. 2, 1–8.
Neumann, A., Hofstetter, T. B., Skarpeli-Liati, M., and Schwarzenbach, R. P. (2009). Reduction of polychlorinated ethanes and carbon tetrachloride by structural Fe(II) in smectites. Environ. Sci. Technol. 43, 4082–4089.
Ottley, C. J., Davison, W., and Edmunds, W. M. (1997). Chemical catalysis of nitrate reduction by iron (II). Geochim. Cosmochim. Acta 61, 1819–1828.
Pantke, C., Obst, M., Benzerara, K., Morin, G., Ona-Nguema, G., Dippon, U., and Kappler, A. (2012). Green rust formation during Fe(II) oxidation by the nitrate-reducing Acidovorax sp. strain BoFeN1. Environ. Sci. Technol. 46, 1439–1446.
Postma, D. (1990). Kinetics of nitrate reduction by detrital Fe(II)-silicates. Geochim. Cosmochim. Acta 54, 903–908.
Rakshit, S., Matocha, C. J., and Coyne, M. S. (2008). Nitrite reduction by siderite. Soil Sci. Soc. Am. J. 72, 1070–1077.
Rakshit, S., Matocha, C. J., and Haszler, G. R. (2005). Nitrate reduction in the presence of wüstite. J. Environ. Qual. 34, 1286–1292.
Schädler, S., Burkhardt, C., Hegler, F., Straub, K. L., Miot, J., Benzerara, K., and Kappler, A. (2009). Formation of cell-iron-mineral aggregates by phototrophic and nitrate-reducing anaerobic Fe(II)-oxidizing bacteria. Geomicrobiol. J. 26, 93–103.
Senko, J. M., Dewers, T. A., and Krumholz, L. R. (2005). Effect of oxidation rate and Fe(II) state on microbial nitrate-dependent Fe(III) mineral formation. Appl. Environ. Microbiol. 71, 7172–7177.
Sørensen, J., and Thorling, L. (1991). Stimulation by lepidocrocite (γ-FeOOH) of Fe(II)-dependent nitrite reduction. Geochim. Cosmoschim. Acta 55, 1289–1294.
Straub, K., Schonhuber, W., Buchholz-Cleven, B., and Schink, B. (2004). Diversity of ferrous iron-oxidizing nitrate-reducing bacteria and their involvement in oxygen-independent iron cycling. Geomicrobiol. J. 21, 371–378.
Straub, K. L., Benz, M., Schink, B., and Widdel, F. (1996). Anaerobic, nitrate-dependent microbial oxidation of ferrous iron. Appl. Environ. Microbiol. 62, 1458–1460.
Straub, K. L., and Buchholz-Cleven, B. E. E. (1998). Enumeration and detection of anaerobic ferrous iron-oxidizing, nitrate-reducing bacteria from diverse European sediments. Appl. Environ. Microbiol. 64, 4846–4856.
Strąpoć, D., Picardal, F. W., Turich, C., Schaperdoth, I., Macalady, J. L., Lipp, J. S., Lin, Y.-S., Ertefai, T. F., Schubotz, F., Hinrichs, K.-U., Mastalerz, M., and Schimmelmann, A. (2008). Methane-producing microbial community in a coal bed of the Illinois Basin. Appl. Environ. Microbiol. 74, 2424–2432.
Sung, W., and Morgan, J. J. (1980). Kinetics and product of ferrous iron oxygenation in aqueous systems. Environ. Sci. Technol. 14, 561–568.
Tai, Y.-L., and Dempsey, B. A. (2009). Nitrite reduction with hydrous ferric oxide and Fe(II): stoichiometry, rate, and mechanism. Water Res. 43, 546–552.
Tamaura, Y., Saturno, M., Yamada, K., and Katsura, T. (1984a). The transformation of γ-FeO(OH) to Fe3O4 and green rust II in an aqueous solution. Bull. Chem. Soc. Jpn. 57, 2417–2421.
Tamaura, Y., Yoshida, T., and Katsura, T. (1984b). The synthesis of green rust II(FeIII1-FeII2) and its spontaneous transformation into Fe3O4. Bull. Chem. Soc. Jpn. 57, 2411–2416.
Tamura, H., Kawamura, S., and Hagayama, M. (1980). Acceleration of the oxidation of Fe2+ ions by Fe(III)-oxyhydroxides. Corros. Sci. 20, 963–971.
Van Cleemput, O., and Baert, L. (1983). Nitrite stability influenced by iron compounds. Soil Biol. Biochem. 15, 137–140.
Weber, K., Pollock, J., Cole, K., O’Conner, S., Achenbach, L., and Coates, J. (2006a). Anaerobic nitrate-dependent iron (II) bio-oxidation by a novel lithoautotrophic betaproteobacterium, strain 2002. Appl. Environ. Microbiol. 72, 686–694.
Weber, K. A., Urrutia, M. M., Churchill, P. F., Kukkadapu, R. K., and Roden, E. E. (2006b). Anaerobic redox cycling of iron by freshwater sediment microorganisms. Environ. Microbiol. 8, 100–113.
Weber, K. A., Hedrick, D. B., Peacock, A. D., Thrash, J. C., White, D. C., Achenbach, L. A., and Coates, J. D. (2009). Physiological and taxonomic description of the novel autotrophic, metal oxidizing bacterium, Pseudogulbenkiania sp strain 2002. Appl. Microbiol. Biotechnol. 83, 555–565.
Weber, K. A., Picardal, F. W., and Roden, E. E. (2001). Microbially catalyzed nitrate-dependent oxidation of biogenic solid-phase Fe(II) compounds. Environ. Sci. Technol. 35, 1644–1650.
Williams, A. G. B., Gregory, K. B., Parkin, G. F., and Scherer, M. M. (2005). Hexahydro-1,3,5-trinitro-1,3,5-triazine transformation by biologically reduced ferrihydrite: evolution of Fe mineralogy, surface area, and reaction rates. Environ. Sci. Technol. 39, 5183–5189.
Wolfe, R. S., Amy, C. R., and Stephen, W. R. (2011). “Techniques for cultivating methanogens,” in Methods in Enzymology, Vol. 494, eds A. Rosenzweig and S. Ragsdale (Salt Lake: Academic Press), 1–22.
Keywords: nitrate-dependent Fe(II) oxidation, nitrate reduction, anoxic Fe(II) oxidation, abiotic Fe(II) oxidation
Citation: Picardal F (2012) Abiotic and microbial interactions during anaerobic transformations of Fe(II) and NOx−. Front. Microbio. 3:112. doi: 10.3389/fmicb.2012.00112
Received: 22 November 2011; Accepted: 09 March 2012;
Published online: 29 March 2012.
Edited by:
Eric Roden, University of Wisconsin–Madison, USAReviewed by:
John Senko, The University of Akron, USACopyright: © 2012 Picardal. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Flynn Picardal, School of Public and Environmental Affairs, Indiana University, MSBII – Room 418, 702 North Walnut Grove Avenue, Bloomington, IN 47405-2204, USA. e-mail:cGljYXJkYWxAaW5kaWFuYS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.