
- Department of Emerging Infectious Diseases, Institute of Tropical Medicine, Nagasaki University, Nagasaki, Japan
Ebolavirus (EBOV) is an enveloped, non-segmented, negative-stranded RNA virus, which consists of five species: Zaire ebolavirus, Sudan ebolavirus, Tai Forest ebolavirus, Bundibugyo ebolavirus, and Reston ebolavirus. EBOV causes a lethal hemorrhagic fever in both humans and non-human primates. The EBOV RNA genome encodes seven viral proteins: NP, VP35, VP40, GP, VP30, VP24, and L. VP40 is a matrix protein and is essential for virus assembly and release from host cells. Expression of VP40 in mammalian cells is sufficient to generate extracellular virus-like particles, which resemble authentic virions. Tetherin/BST-2, which was identified as an effective cellular factor that prevents human immunodeficiency virus-1 release in the absence of viral accessory protein Vpu, has been reported to inhibit ZEBOV VP40-induced VLP release. Tetherin/BST-2 appears to inhibit virus release by physically tethering viral particles to the cell surface via its N-terminal transmembrane domain and C-terminal glycosylphosphatidylinositol anchor. Replication of ZEBOV is not inhibited by tetherin/BST-2 expression, although tetherin/BST-2 was expected to inhibit EBOV release as well as VLP release. Recently, it was reported that viral glycoprotein of EBOV, GP, antagonizes the antiviral effect of tetherin/BST-2. However, the mechanism by which GP antagonizes the antiviral activity of tetherin/BST-2 and whether GP of the other EBOV species function as antagonists of tetherin/BST-2 remain unclear.
Introduction
Ebolavirus (EBOV) is a member of the family Filoviridae in the order Mononegavirales (MNV), and causes a lethal hemorrhagic fever in both humans and non-human primates (Peters, 2005). Five species of EBOV have been defined to date on the basis of genetic divergence: Zaire ebolavirus (ZEBOV), Sudan ebolavirus (SEBOV), Tai Forest ebolavirus (TFEBOV), Reston ebolavirus (REBOV), and Bundibugyo ebolavirus (BEBOV). ZEBOV, SEBOV, TFEBOV, and BEBOV cause clinical symptoms in humans and non-human primates, while REBOV causes disease only in non-human primates, and not in humans. At present, there are no licensed vaccines or effective therapies for EBOV infection. Recently, tetherin/BST-2 was identified as a cellular factor that inhibits the release of a wide variety of enveloped viruses, including retroviruses, herpesviruses, and Lassa virus, and the production of virus-like particles (VLPs) of filoviruses and Nipa virus (Jouvenet et al., 2009; Kaletsky et al., 2009; Sakuma et al., 2009a; Radoshitzky et al., 2010). Tetherin/BST-2 may function as a host innate antiviral system against a wide variety of viruses, as tetherin/BST-2 is broadly induced by treatment with type I interferons (IFNs) in various cell types (Ishikawa et al., 1995; Blasius et al., 2006). This review will focus on EBOV replication and antiviral function of tetherin/BST-2.
Ebolavirus and Its Replication
Electron microscopic studies have indicated that EBOV is morphologically pleomorphic, with U-shaped, figure 6-shaped, or circular configurations, or as elongated filamentous forms of varying length (up to 14000 nm). The virions are usually 80 nm in diameter and 800–1000 nm in length and enveloped with a lipid bilayer (envelope) that is derived from the host cell, anchoring a glycoprotein that projects spikes 7–10 nm in length from its surface (Sanchez et al., 2007).
The genome is approximately 19 kb in length and encodes the viral proteins in the order NP–VP35–VP40–GP/sGP–VP30–VP24–L (Figure 1). The extragenic sequence at the 3′ end, which is called the leader, of EBOV is short, ranging from 50 to 70 bases in length, while the length of the 5′ end sequence, which is called the trailer, varies between species, ranging from 25 to 677 bases (25 bases for REBOV and 677 bases for ZEBOV). The extreme 3′ and 5′ end sequences are conserved and potentially form stem-loop structures (Geisbert and Jahrling, 1995; Sanchez et al., 2007). These sequences contain the encapsidation signals as well as the replication origin and transcription promoter.

Figure 1. Schematic representation of EBOV genome.
The NP and VP30 proteins are the major and minor viral nucleoproteins, respectively, which are phosphorylated, and interact strongly with the genomic RNA molecule to form the viral nucleocapsid along with VP35 and L (Mühlberger et al., 1999). The L and VP35 proteins form the viral polymerase complex, which transcribes and replicates the viral genome. The L protein has the RNA-dependent RNA polymerase activity of the complex, and possesses motifs linked to RNA binding, phosphodiester bonding, and ribonucleotide triphosphate binding. VP35 is thought to play an essential role as a cofactor that affects the mode of viral transcription and replication. VP35 also functions as an antagonist against the type I IFN signaling pathway (Basler et al., 2000).
The GP gene contains a translational stop codon in the middle, thus preventing synthesis of full-length glycoprotein. Twenty percent of the mRNA was shown to be edited, containing one additional non-template A in a stretch of seven consecutive A residues. The edited mRNA species encode full-length GP, whereas the primary gene product is a smaller secreted glycoprotein (sGP), which is produced in large amounts. The GP precursor (preGP), is synthesized as type I membrane protein in the endoplasmic reticulum (ER). preGP is cleaved by the cellular proprotein convertase furin into the N-terminal fragment GP1 and the C-terminal fragment GP2 which are linked by a disulfide bond forming the GP1, 2 complex in the trans-Golgi network (TGN; Volchkov et al., 1998). GP1 and GP2 function as a surface protein and a transmembrane protein, respectively. The trimeric spike of GP1, 2 is the only envelope glycoprotein of the virion and is assumed to be responsible for binding to cellular receptors and for fusion of the envelope with the cellular membrane in the course of viral entry into the host cell. GP has marked effects on viral pathogenesis and antigenicity. Recent report suggested that sGP can substitute for GP1 and present on virion as a structural protein, although sGP had been considered as a non-structural protein (Iwasa et al., 2011). The function of sGP is still unknown. sGP may contribute to disease progression, as it has been reported that large amounts of sGP are present in the blood of acutely infected patients (Volchkov et al., 1995; Sanchez et al., 1996).
The VP40 and VP24 proteins are viral matrix proteins and are associated with the virion envelope. VP40 is the most abundant protein in the virion and plays a key role in virus assembly and budding as viral matrix protein. Expression of VP40 in mammalian cells is sufficient to generate extracellular VLPs, which resemble authentic virions (Harty et al., 2000; Yasuda et al., 2003). Only small amounts of VP24 are present in the virion. VP24 has been reported to function as an antagonist of the type I IFN signaling pathway, along with VP35 (Reid et al., 2006).
Tetherin/BST-2
Tetherin/BST-2 (also known as CD317 or HM1.24) has been identified as an effective cellular factor that prevents human immunodeficiency virus (HIV)-1 release in the absence of the viral accessory protein Vpu (Neil et al., 2008; Van Damme et al., 2008). Tetherin/BST-2 consists of four domains, i.e., an N-terminal cytoplasmic tail (CT), a single transmembrane domain, an extracellular domain, and a putative C-terminal glycosylphosphatidylinositol (GPI) anchor, and therefore both ends of this molecule are associated with the plasma membrane (Figure 2A). This molecule is localized to lipid rafts at the cell surface and membranes of the TGN and recycling compartments (Kupzig et al., 2003; Blasius et al., 2006; Rollason et al., 2007; Evans et al., 2010). In addition, tetherin/BST-2 has two putative N-linked glycosylation sites in the extracellular domain and forms a homodimer by intermolecular disulfide bonds (Ohtomo et al., 1999; Kupzig et al., 2003; Figure 2A).
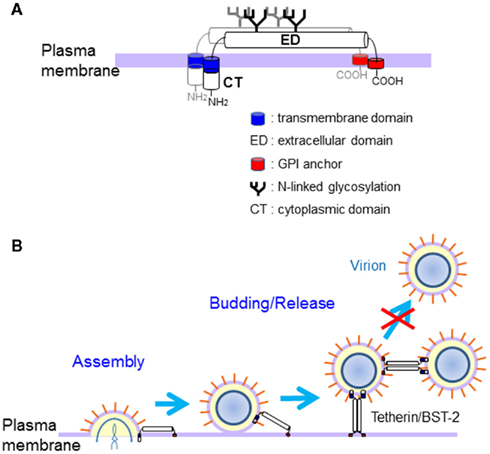
Figure 2. Structure and antiviral action of Tetherin/BST-2. (A) Structural representation of tetherin/BST-2. Tetherin/BST-2 has two putative N-linked glycosylation sites in the extracellular domain and forms a homodimer by intermolecular disulfide bonds. Both ends of tetherin/BST-2 are associated with the plasma membrane via the N-terminal transmembrane domain and C-terminal GPI anchor. (B) Proposed model for the antiviral mechanism of tetherin/BST-2. Tetherin/BST-2 appears to inhibit the release of progeny viruses by directly tethering virions to cells, briefly by anchoring one end of the molecule on the cell membrane and the other end on the viral envelope. Progeny virions could be also directly tethered to each other by tetherin/BST-2.
Previous studies showed that tetherin/BST-2 is constitutively expressed in terminally differentiated B cells, bone marrow stromal cells, and plasmacytoid dendritic cells, and is also broadly induced by treatment with type I IFN in various cell types (Ishikawa et al., 1995; Blasius et al., 2006). In fact, the upstream region of tetherin/BST-2 contains a tandem repeat of IL-6 response elements, STAT3 binding site, and the IFN response elements IRF-1/2 and ISGF3 (Ohtomo et al., 1999). Therefore, tetherin/BST-2 may be involved in antiviral host defense as a mechanism of innate immunity. However, the physiological function of tetherin/BST-2 is not yet clear.
Recent analysis for in vivo expression of tetherin/BST-2 showed that tetherin/BST-2 was expressed to varying degrees in most organs and a number of specialized cell types, including hepatocytes, pneumocytes, ducts of major salivary glands, pancreas and kidney, Paneth cells, epithelia, Leydig cells, plasma cells, bone marrow stromal cells, monocytes, and vascular endothelium, without IFN stimulation (Erikson et al., 2011). Therefore, IFN may only partially regulate tetherin/BST-2 in vivo.
Antiviral Activities of Tetherin/BST-2
We have shown that tetherin/BST-2 also efficiently inhibits the egress of VLPs of Marburgvirus and Lassa virus and retains VLPs on the cell surface (Sakuma et al., 2009a). Furthermore, tetherin/BST-2 has also been reported to inhibit the release of retroviruses other than HIV-1, Kaposi’s sarcoma-associated herpesvirus (KSHV), and the production of VLPs of EBOV and Nipa virus (Jouvenet et al., 2009; Kaletsky et al., 2009; Mansouri et al., 2009; Radoshitzky et al., 2010).
The N-linked glycosylation of tetherin/BST-2 is dispensable for the antiviral activity, while both N-terminal transmembrane domain and C-terminal GPI anchor are required for the activity (Neil et al., 2008; Andrew et al., 2009; Sakuma et al., 2009a). Interestingly, tetherin/BST-2 mutant with complete loss of dimerization activity (the cysteine mutant) still showed apparent inhibitory activity for the production of Lassa and Marburg VLPs, although its activity was reduced (Sakuma et al., 2009b). For HIV-1, the cysteine mutant showed much weaker antiviral activity (Andrew et al., 2009; Perez-Caballero et al., 2009). Nevertheless, these observations suggested that dimerization of tetherin/BST-2 is important but not essential for its antiviral activity. A recent study showed that tetherin/BST-2 inhibits HIV-1 release by directly tethering virions to cells, briefly by anchoring one end of the molecule on the cell membrane and the other end on the viral envelope (Perez-Caballero et al., 2009; Figure 2B). Progeny virions released from cells could also be directly tethered to each other by tetherin/BST-2. It is likely that tethering of virion by tetherin dimer is stronger than that by tetherin monomer because of stronger association with the membrane. Therefore, tetherin/BST-2 appears to inhibit release of a wide variety of enveloped viruses from host cells by a similar mechanism.
Counteraction of Tetherin/BST-2 Antiviral Action by GP
Human immunodeficiency virus-1 Vpu is known to antagonize the antiviral function of tetherin/BST-2 (Neil et al., 2008; Van Damme et al., 2008). Moreover, recent studies have shown that HIV-2 and simian immunodeficiency virus (SIV) Env, SIV Nef, KSHV K5, and EBOV GP also function as antagonists of tetherin/BST-2, suggesting that tetherin/BST-2 plays an important role in host defense against viral infection and these viruses have evolutionarily acquired viral-encoded antagonists to counteract the antiviral function of tetherin/BST-2 (Gupta et al., 2009b; Jia et al., 2009; Kaletsky et al., 2009; Le Tortorec and Neil, 2009; Mansouri et al., 2009; Zhang et al., 2009).
Kaletsky et al. (2009) demonstrated that EBOV GP interacts directly with tetherin/BST-2 and abrogates the inhibition of VP40-induced VLP release by tetherin/BST-2. In addition, analysis using infectious virus showed that the expression of tetherin/BST-2 had no effect on ZEBOV replication and spread (Radoshitzky et al., 2010).
One of the major functions of HIV-1 Vpu for counteraction of antiviral action of tetherin/BST-2 is downregulation of the surface expression of tetherin/BST-2. Several studies demonstrated that Vpu directs the degradation of human BST-2 via a β-TrCP-dependent mechanism. Vpu acts as an adapter molecule linking tetherin/BST-2 to the β-TrCP/SCF E3 ubiquitin ligase complex to induce the trafficking to late endosomes, lysosomes, and proteasomes, and subsequent lysosomal and/or proteasomal degradation of tetherin/BST-2. Vpu is thought to remove tetherin/BST-2 from the cell surface by these pathways (Douglas et al., 2009; Mitchell et al., 2009). Recent report suggests that β-TrCP-independent mechanism is also involved in the downregulation of cell surface expression of tetherin/BST-2 by Vpu, since mutations in the β-TrCP-binding motif of Vpu did not completely abrogate its antagonism of tetherin/BST-2. Schmidt et al. (2011) have reported that Vpu inhibited both the anterograde transport of newly synthesized tetherin/BST-2 and the recycling of tetherin/BST-2 to the cell surface and trapped trafficking tetherin/BST-2 molecules at the TGN. Vpu interacts with tetherin/BST-2 through species-specific determinants in their respective transmembrane domains (Gupta et al., 2009a; McNatt et al., 2009). HIV-1 Vpu specifically antagonizes the tetherin/BST-2s from human, chimpanzee, and gorilla, which are susceptible to HIV-1 infection, but not those from African green monkey, rhesus macaque, and mouse, which are not susceptible to this virus (McNatt et al., 2009; Kühl et al., 2011).
In contrast, Ebola GP counteracted tetherin/BST-2 from different primate species, including rhesus macaque and African green monkey (Kühl et al., 2011). Ebola GP does not seem to require a specific tetherin/BST-2 sequence for its activity (Lopez et al., 2010). It has been reported that tetherin/BST-2 interacts with the GP2 subunit of EBOV GP, although the antogonism of tetherin/BST-2 function by GP2 have not examined (Kühl et al., 2011). Vpu reduces cell surface expression of tetherin/BST-2, while Ebola GP appears to counteract tetherin/BST-2 without removing it from the cell surface, suggesting that both proteins employ different mechanisms to counteract tetherin/BST-2 (Lopez et al., 2010; Kühl et al., 2011). Ebola GP and tetherin/BST-2 colocalize in intracellular compartment, but not on the plasma membrane (Kühl et al., 2011). The sequestration of tetherin/BST-2 in the specific intracellular compartment may be one of the mechanisms of antagonism by Ebola GP. So far, the mechanism by which EBOV GP antagonizes tetherin/BST-2 remains unclear. Further investigations are required to understand the mechanism by which EBOV GP counteracts the antiviral function of tetherin/BST-2.
Future Perspectives
Tetherin/BST-2 inhibits the production of a wide variety of enveloped viruses. On the other hand, several viruses have evolved viral-encoded antagonists to counteract antiviral action of tetherin/BST-2. EBOV also appears to have evolved GP as an antagonist of tetherin/BST-2. However, it has been reported that high-level expression of tetherin/BST-2 inhibits ZEBOV production even in the presence of GP (Kühl et al., 2011). Furthermore, it may be possible to identify tetherin/BST-2 mutants that are not counteracted by EBOV GP.
Therefore, regulation of the progeny EBOV release may be possible by in vivo induction or exogenous expression of tetherin/BST-2. Tetherin/BST-2 has great potential for the development of novel antiviral therapeutic strategies against EBOV infection.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research in our laboratory was supported by grants from the Ministry of Health, Labor, and Welfare of Japan, the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Bio-oriented Technology Research Advancement Institution, and the Japan Society for the Promotion of Science (JSPS).
References
Andrew, A. J., Miyagi, E., Kao, S., and Strebel, K. (2009). The formation of cysteine-linked dimers of BST-2/tetherin is important for inhibition of HIV-1 virus release but not for sensitivity to Vpu. Retrovirology 6, 80.
Basler, C. F., Wang, X., Mühlberger, E., Volchkov, V., Paragas, J., Klenk, H. D., García-Sastre, A., and Palese, P. (2000). The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. U.S.A. 97, 12289–12294.
Blasius, A. L., Giurisato, E., Cella, M., Schreiber, R. D., Shaw, A. S., and Colonna, M. (2006). Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 177, 3260–3265.
Douglas, J. L., Viswanathan, K., McCarroll, M. N., Gustin, J. K., Früh, K., and Moses, A. V. (2009). Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a bTrCP-dependent mechanism. J. Virol. 83, 7931–7947.
Erikson, E., Adam, T., Schmidt, S., Lehmann-Koch, J., Over, B., Goffinet, C., Harter, C., Bekeredjian-Ding, I., Sertel, S., Lasitschka, F., and Keppler, O. T. (2011). In vivo expression profile of the antiviral restriction factor and tumor-targeting antigen CD317/BST-2/HM1.24/tetherin in humans. Proc. Natl. Acad. Sci. U.S.A. 108, 13688–13693.
Evans, D. T., Serra-Moreno, R., Singh, R. K., and Guatelli, J. C. (2010). BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol. 18, 388–396.
Geisbert, T. W., and Jahrling, P. B. (1995). Differentiation of filoviruses by electron microscopy. Virus Res. 39, 129–150.
Gupta, R. K., Hué, S., Schaller, T., Verschoor, E., Pillay, D., and Towers, G. J. (2009a). Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 5, e1000443. doi:10.1371/journal.ppat.1000443
Gupta, R. K., Mlcochova, P., Pelchen-Matthews, A., Petit, S. J., Mattiuzzo, G., Pillay, D., Takeuchi, Y., Marsh, M., and Towers, G. J. (2009b). Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST-2/CD317 by intracellular sequestration. Proc. Natl. Acad. Sci. U.S.A. 106, 20889–20894.
Harty, R. N., Brown, M. E., Wang, G., Huibregtse, J., and Hayes, F. P. (2000). A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl. Acad. Sci. U.S.A. 97, 13871–13876.
Ishikawa, J., Kaisho, T., Tomizawa, H., Lee, B. O., Kobune, Y., Inazawa, J., Oritani, K., Itoh, M., Ochi, T., Ishihara, K., and Hirano, T. (1995). Molecular cloning and chromosomal mapping of a bone marrow stromal cell surface gene, BST2, that may be involved in pre-B-cell growth. Genomics 26, 527–534.
Iwasa, A., Shimojima, M., and Kawaoka, Y. (2011). sGP serves as a structural protein in Ebola viru infection. J. Infect. Dis. 204, S897–S903.
Jia, B., Serra-Moreno, R., Neidermyer, W., Rahmberg, A., Mackey, J., Fofana, I. B., Johnson, W. E., Westmoreland, S., and Evans, D. T. (2009). Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 5, e1000429. doi:10.1371/journal.ppat.1000429
Jouvenet, N., Neil, S. J., Zhadina, M., Zang, T., Kratovac, Z., Lee, Y., McNatt, M., Hatziioannou, T., and Bieniasz, P. D. (2009). Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 83, 1837–1844.
Kaletsky, R. L., Francica, J. R., Agrawal-Gamse, C., and Bates, P. (2009). Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 106, 2886–2891.
Kühl, A., Banning, C., Marzi, A., Votteler, J., Steffen, I., Bertram, S., Glowacka, I., Konrad, A., Stürzl, M., Guo, J. T., Schubert, U., Feldmann, H., Behrens, G., Schindler, M., and Pöhlmann, S. (2011). The Ebola virus glycoprotein and HIV-1 Vpu employ different strategies to counteract the antiviral factor tetherin. J. Infect. Dis. 204, S850–S860.
Kupzig, S., Korolchuk, V., Rollason, R., Sugden, A., Wilde, A., and Banting, G. (2003). Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 4, 694–709.
Le Tortorec, A., and Neil, S. J. (2009). Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 83, 11966–11978.
Lopez, L. A., Yang, S. J., Hauser, H., Exline, C. M., Haworth, K. G., Oldenburg, J., and Cannon, P. M. (2010). Ebola virus glycoprotein counteracts BST-2/tetherin restriction in a sequence-independent manner that does not require tetherin surface removal. J. Virol. 84, 7243–7255.
Mansouri, M., Viswanathan, K., Douglas, J. L., Hines, J., Gustin, J., Moses, A. V., and Früh, K. (2009). Molecular mechanism of BST2/tetherin downregulation by K5/MIR2 of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 83, 9672–9681.
McNatt, M. W., Zang, T., Hatziioannou, T., Bartlett, M., Fofana, I. B., Johnson, W. E., Neil, S. J., and Bieniasz, P. D. (2009). Species-specific activity of HIV-1 Vpu and positive selection of tetherin transmembrane domain variants. PLoS Pathog. 5, e1000300. doi:10.1371/journal.ppat.1000300
Mitchell, R. S., Katsura, C., Skasko, M. A., Fitzpatrick, K., Lau, D., Ruiz, A., Stephens, E. B., Margottin-Goguet, F., Benarous, R., and Guatelli, J. C. (2009). Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 5, e1000450. doi:10.1371/journal.ppat.1000450
Mühlberger, E., Weik, M., Volchkov, V. E., Klenk, H. D., and Becker, S. (1999). Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J. Virol. 73, 2333–2342.
Neil, S. J. D., Zang, T., and Bieniasz, P. D. (2008). Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430.
Ohtomo, T., Sugamata, Y., Ozaki, Y., Ono, K., Yoshimura, Y., Kawai, S., Koishihara, Y., Ozaki, S., Kosaka, M., Hirano, T., and Tsuchiya, M. (1999). Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem. Biophys. Res. Commun. 258, 583–591.
Perez-Caballero, D., Zang, T., Ebrahimi, A., McNatt, M. W., Gregory, D. A., Johnson, M. C., and Bieniasz, P. D. (2009). Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139, 499–511.
Peters, C. J. (2005). Marburg and Ebola – arming ourselves against the deadly filoviruses. N. Engl. J. Med. 352, 2571–2573.
Radoshitzky, S. R., Dong, L., Chi, X., Clester, J. C., Retterer, C., Spurgers, K., Kuhn, J. H., Sandwick, S., Ruthel, G., Kota, K., Boltz, D., Warren, T., Kranzusch, P. J., Whelan, S. P., and Bavari, S. (2010). Infectious Lassa virus, but not filo viruses, is restricted by BST-2/tetherin. J. Virol. 84, 10569–10580.
Reid, S. P., Leung, L. W., Hartman, A. L., Martinez, O., Shaw, M. L., Carbonnelle, C., Volchkov, V. E., Nichol, S. T., and Basler, C. F. (2006). Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 80, 5156–5167.
Rollason, R., Korolchuk, V., Hamilton, C., Schu, P., and Banting, G. (2007). Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J. Cell Sci. 120, 3850–3858.
Sakuma, T., Noda, T., Urata, S., Kawaoka, Y., and Yasuda, J. (2009a). Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 83, 2382–2385.
Sakuma, T., Sakurai, A., and Yasuda, J. (2009b). Dimerization of tetherin is not essential for its antiviral activity against Lassa and Marburg viruses. PLoS ONE 4, e6934. doi:10.1371/journal.pone.0006934
Sanchez, A., Geisbert, T. W., and Feldmann, H. (2007). “Filoviridae: Marburg and Ebola viruses,” in Fields Virology, 5th Edn, Vol. 1, eds D. M. Knipe, and P. M. Howley (Philadelphia, PA: Lippincott Williams and Wilkins), 1409–1448.
Sanchez, A., Trappier, S. G., Mahy, B. W., Peters, C. J., and Nichol, S. T. (1996). The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. U.S.A. 93, 3602–3607.
Schmidt, S., Fritz, J. V., Bitzegeio, J., Fackler, O. T., and Keppler, O. T. (2011). HIV-1 Vpu blocks recycling and biosynthetic transport of the intrinsic immunity factor CD317/tetherin to overcome the virion release restriction. MBio 2, e00036-11.
Van Damme, N., Goff, D., Katsura, C., Jorgenson, R. L., Mitchell, R., Johnson, M. C., Stephens, E. B., and Guatelli, J. (2008). The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3, 245–252.
Volchkov, V. E., Becker, S., Volchkova, V. A., Ternovoj, V. A., Kotov, A. N., Netesov, S. V., and Klenk, H. D. (1995). GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 214, 421–430.
Volchkov, V. E., Feldmann, H., Volchkova, V. A., and Klenk, H. D. (1998). Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. U.S.A. 95, 5762–5767.
Yasuda, J., Nakao, M., Kawaoka, Y., and Shida, H. (2003). Nedd4 regulates egress of Ebola virus-like particles from host cells. J. Virol. 77, 9987–9992.
Keywords: ebolavirus, tetherin/BST-2, GP, antagonist, VLP
Citation: Yasuda J (2012) Ebolavirus replication and tetherin/BST-2. Front. Microbio. 3:111. doi: 10.3389/fmicb.2012.00111
Received: 15 December 2011; Accepted: 09 March 2012;
Published online: 02 April 2012.
Edited by:
Masako Nomaguchi, The University of Tokushima Graduate School, JapanReviewed by:
Takayuki Miyazawa, Kyoto University, JapanKenzo Tokunaga, National Institute of Infectious Diseases, Japan
Yoshinao Kubo, Graduate School of Biomedical Sciences, Nagasaki University, Japan
Copyright: © 2012 Yasuda. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jiro Yasuda, Department of Emerging Infectious Diseases, Institute of Tropical Medicine, Nagasaki University, 1-12-4 Sakamoto, Nagasaki 852 8523, Japan. e-mail:ai15YXN1ZGFAbmFnYXNha2ktdS5hYy5qcA==