
 Oleg Dmytrenko2
Oleg Dmytrenko2
- 1 School of Biology, Georgia Institute of Technology, Atlanta, GA, USA
- 2 Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA, USA
- 3 Department of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
Thioautotrophic endosymbionts in the Domain Bacteria mediate key sulfur transformations in marine reducing environments. However, the molecular pathways underlying symbiont metabolism and the extent to which these pathways are expressed in situ are poorly characterized for almost all symbioses. This is largely due to the difficulty of culturing symbionts apart from their hosts. Here, we use pyrosequencing of community RNA transcripts (i.e., the metatranscriptome) to characterize enzymes of dissimilatory sulfur metabolism in the model symbiosis between the coastal bivalve Solemya velum and its intracellular thioautotrophic symbionts. High-throughput sequencing of total RNA from the symbiont-containing gill of a single host individual generated 1.6 million sequence reads (500 Mbp). Of these, 43,735 matched Bacteria protein-coding genes in BLASTX searches of the NCBI database. The taxonomic identities of the matched genes indicated relatedness to diverse species of sulfur-oxidizing Gammaproteobacteria, including other thioautotrophic symbionts and the purple sulfur bacterium Allochromatium vinosum. Manual querying of these data identified 28 genes from diverse pathways of sulfur energy metabolism, including the dissimilatory sulfite reductase (Dsr) pathway for sulfur oxidation to sulfite, the APS pathway for sulfite oxidation, and the Sox pathway for thiosulfate oxidation. In total, reads matching sulfur energy metabolism genes represented 7% of the Bacteria mRNA pool. Together, these data highlight the dominance of thioautotrophy in the context of symbiont community metabolism, identify the likely pathways mediating sulfur oxidation, and illustrate the utility of metatranscriptome sequencing for characterizing community gene transcription of uncultured symbionts.
Introduction
Symbioses between thioautotrophic bacteria and invertebrates perform important steps in the marine sulfur cycle. In these associations, bacteria living intra- or extra-cellularly with a eukaryotic host oxidize reduced sulfur compounds as an energy source for autotrophic CO2 fixation, providing a substantial source of fixed carbon for the host (Cavanaugh et al., 2006; Dubilier et al., 2008). Such symbioses have been described in 7 host phyla, from ciliates to giant tubeworms (Stewart et al., 2005), and are routinely discovered in new species as marine environments become better explored (Losekann et al., 2008; Fujiwara et al., 2010; Rodrigues et al., 2010). In reducing habitats (e.g., anaerobic sediments, hydrothermal vents, hydrocarbon seeps), these symbioses may dominate the biomass, playing critical roles in local primary production and sulfur transformations (Sievert et al., 2007).
Despite their ubiquity and ecological importance, thioautotrophic symbioses are poorly described molecularly. Characterization of the genes and proteins mediating symbiont metabolism has been hindered by the inability (as of yet) to culture most symbionts outside their hosts. Genomic and metagenomic sequencing has helped reverse this trend, clarifying the genetic basis for diverse biochemical transformations in a handful of thioautotrophic symbioses (Woyke et al., 2006; Newton et al., 2007; Grzymski et al., 2008; Robidart et al., 2008). For example, a comparative genomics approach has been used to characterize the probable sulfur oxidation pathways in two endosymbionts of deep-sea vent clams (family Vesicomyidae; (Harada et al., 2009). However, DNA sequences do not reflect the actual metabolic activity of microorganisms. In contrast, methods that measure gene transcription (transcriptomics) and translation (proteomics) can provide more direct descriptions of symbiont function and metabolism, particularly if administered under in situ conditions (Markert et al., 2007; Harada et al., 2009; Wilmes and Bond, 2009).
Notably, technical advancements in RNA amplification and parallel pyrosequencing have made it possible to obtain hundreds of thousands of sequences from the microbial community RNA pool, i.e., the metatranscriptome. Such methods enable inferences of the relative metabolic activity of hundreds of diverse metabolic pathways using transcript abundance as a proxy for gene expression (Frias-Lopez et al., 2008; Stewart et al., 2011b). While metatranscriptomics has been increasingly used to study community metabolism in free-living microorganisms of the open ocean (Poretsky et al., 2009; Hewson et al., 2010; Stewart et al., 2011a), transcriptomic analysis of marine symbioses has been restricted to only a few taxa, and has focused primarily on host gene expression (Nyholm et al., 2008; Bettencourt et al., 2010). Here, we use pyrosequencing to analyze the in situ metatranscriptome of a model thiotrophic symbiont, the bacterial endosymbiont of the coastal bivalve Solemya velum, focusing specifically on transcripts encoding proteins of dissimilatory sulfur metabolism.
Studies of the protobranch S. velum and its endosymbiont were some of the first to describe the physiology and ecology of thioautotrophic symbioses (see review in Stewart and Cavanaugh, 2006). S. velum lives in shallow, sulfur-rich sediments along the Atlantic Coast (Florida to Canada), obtaining the bulk of its nutrition (>97% of host carbon) from a dense population of autotrophic bacteria living within specialized cells of the host gills (Cavanaugh, 1983; Krueger et al., 1992). Early molecular evidence (16S rRNA gene sequence) suggested that the symbiont population consists of a single, unique bacterial phylotype within the Gammaproteobacteria (Eisen et al., 1992). Sulfur-based autotrophy in the symbiont population was initially demonstrated through physiological and molecular studies showing (1) the activity and corresponding gene sequence for ribulose 1,5-bisphosphate carboxylase–oxygenase (RubisCO), the primary CO2-fixing enzyme of the Calvin cycle, in host gills (Cavanaugh, 1983; Schwedock et al., 2004), (2) an increase in symbiont CO2 fixation and oxygen consumption in response to sulfide and thiosulfate addition (Cavanaugh, 1983; Anderson et al., 1987), and (3) the activity of two enzymes in the APS pathway mediating sulfite oxidation, APS reductase, and ATP sulfurylase (Chen et al., 1987). While these data confirm that S. velum symbionts can oxidize reduced sulfur for energy, the full biochemical pathway through which this occurs, and the extent to which this pathway is expressed, remains uncharacterized.
Here, pyrosequencing of the symbiont-enriched RNA fraction from S. velum sampled from the environment revealed a strong representation by genes of dissimilatory sulfur metabolism in the total transcript pool. These results underscore the importance of thioautotrophy in the symbiosis, identify the likely pathways by which sulfur oxidation occurs, and illustrate the utility of metatranscriptome sequencing for characterizing symbiont community metabolism across thousands of genes.
Materials and Methods
Sample Collection
Individuals of S. velum were collected at low tide from Bluff Hill Cove, Point Judith Pond, Rhode Island (RI; 41.380°N, −71.502°W) on 5 December 2009. Bluff Hill Cove is connected to the ocean by a narrow inlet and bordered by extensive mud flats that support a stable and abundant population of S. velum. At the time of collection the average sediment temperature was 8°C. Individuals were dissected in the field upon collection, with symbiont-containing gill tissue placed immediately into RNAlater® and stored frozen until RNA extraction.
Symbiont RNA Extraction and Enrichment
Symbiont-containing gill tissue of a single host individual was lysed using a FastPrep FP120 tissue homogenizer (Savant) with 770–1180 μm glass beads. Total gill RNA (symbiont + host) was extracted with a chloroform–ethanol method using miRNeasy Mini Kit (Qiagen) according to the protocol for purification of total RNA, including small RNA, from animal cells. An aliquot of total RNA was then enriched for symbiont RNA using the MICROBEnrich™ kit (Ambion) according to the manufacturer’s instructions. The kit employs capture oligonucleotides and subtractive hybridization to selectively remove eukaryotic polyadenylated mRNAs, enriching for non-polyadenylated prokaryotic RNA. This kit is also designed to remove eukaryotic 18S and 28S rRNA. However, because capture oligonucleotides for rRNA removal are optimized for use with mammalian 18S/28S sequences (Ambion internal documentation), the removal efficiency of bivalve host rRNA was anticipated to be low.
RNA Amplification and CDNA Synthesis
Symbiont-enriched RNA was linearly amplified and converted to cDNA as described in Frias-Lopez et al. (2008), Shi et al. (2009). Briefly, using the MessageAmp™ II-Bacteria kit (Ambion), total RNA was polyadenylated, converted to double-stranded cDNA via reverse transcription, and then transcribed in vitro to produce large quantities (tens of micrograms) of single-stranded antisense RNA. Amplified RNA was then converted to double-stranded cDNA using the SuperScript® III First-Strand Synthesis System (Invitrogen) with priming via random hexamers for first-strand synthesis, and the SuperScript™ Double-Stranded cDNA synthesis kit (Invitrogen) for second-strand synthesis. cDNA was then purified with the QIAquick PCR purification kit (Qiagen), digested with BpmI to remove poly(A) tails, and used directly for pyrosequencing.
Sequencing
Purified cDNA was used for the generation of single-stranded DNA libraries and emulsion PCR via standard protocols (454 Life Sciences, Roche). Amplified library fragments were sequenced via a single full plate run on a Roche Genome Sequencer FLX instrument using Titanium series chemistry. Sequencing yields are shown in Table 1.

Table 1. Metatranscriptome read statistics.
Data Analysis
Duplicate sequence reads (reads with 100% nucleotide similarity and identical length), which may arise as artifacts of the pyrosequencing method, were identified as in Stewart et al. (2010) using the program CD-HIT (Li and Godzik, 2006) and removed from the dataset. Non-duplicate reads matching ribosomal RNA genes were identified by BLASTN searches against a custom database of prokaryotic and eukaryotic small and large subunit rRNA nucleotide sequences (5S, 16S, 18S, 23S, 28S rRNA, compiled from microbial genomes and sequences in the ARB SILVA databases, and including the published 16S rRNA gene sequence of the S. velum symbiont, http://www.arb-silva.de) and removed from the analysis. Protein-coding gene sequences were identified among the remaining non-duplicate, non-rRNA reads by BLASTX searches against the NCBI non-redundant protein database (NCBI-nr, as of May 2010). The top reference gene(s) matching each read above a bit score cutoff of 50 was used to designate each protein-coding read. For reads having multiple top matches with equal bit score, each matching reference gene was designated a top hit, with its representation scaled proportionate to the number of genes sharing an equal bit score. Read counts per gene were tabulated and normalized as a proportion of the total number of protein-coding reads (or protein-coding reads from the Domain Bacteria) in each sample. The taxonomic identity of protein-coding genes was inferred from the annotation affiliated with each gene, and tabulated according to the NCBI taxonomy using the program MEGAN (Huson et al., 2007).
Detailed analysis of all functional gene categories in the dataset is beyond the scope of this paper, and is the topic of a separate study (Dmytrenko et al., in preparation). Here, we focus only on transcripts matching genes of dissimilatory sulfur metabolism, as detected via BLASTX against all protein-coding genes in NCBI-nr. BLASTX results were manually queried to infer the relative abundance of genes encoding proteins of the primary pathways of dissimilatory sulfur metabolism, including: the dissimilatory sulfite reductase (DsrABCEFHMKLJOPNRS) pathway catalyzing sulfur oxidation to sulfite; the sulfur oxidation (SoxABCDEFGHRSVWXYZ) pathway responsible for thiosulfate oxidation; the APS pathway mediating the conversion of sulfite to sulfate via an APS intermediate, via the enzymes adenosine-5′-phosphosulfate (APS) reductase (AprAB), APS reductase membrane anchor (AprM), quinone-interacting membrane-bound oxidoreductase (QmoABC), and sulfate adenylyltransferase (SAT); and the sulfide oxidation step involving flavocytochrome c sulfide dehydrogenase (FccAB) and sulfide–quinone reductase (Sqr), which initiate electron flow from sulfide to the transport chain. When specified, sequence read counts per gene were normalized based on a best approximate gene length (estimated based on full-length open reading frames from sequenced genomes).
Sequence Data
All transcript sequences generated in this study have been deposited in the Community Cyberinfrastructure for Advanced Microbial Ecology Research and Analysis (CAMERA) Portal for access by the broader scientific community.
Results
Read Statistics
Pyrosequencing generated ~1.6 million sequence reads (~500 Mbp) from the symbiont-enriched gill metatranscriptome (Table 1). Reads matching ribosomal RNA represented 87% of the dataset. Of 284,553 ribosomal reads with top matches to the prokaryotic (Bacteria or Archaea) small subunit rRNA (16S rRNA) gene, 278,192 (98%) matched the published S. velum symbiont 16S rRNA sequence at 97% nucleotide identity or greater, with 233,904 (82%) matching at ≥ 99% identity. These results suggest a genetically homogenous microbial population within the S. velum gill. However, this pattern does not rule out the co-occurrence of distinct symbiont lineages (strains) within the same host or of non-symbiont microorganisms associated with the gill surface. Our dataset therefore reflects a “meta” transcriptome representing the transcriptional activity of the entire gill-associated microbial community.
Taxonomic Identities of Protein-Coding Reads
Removal of ribosomal RNA reads from the dataset left a total of 203,641 non-rRNA sequences, of which 70,427 (34.7%) matched protein-coding genes in the NCBI-nr database (above bit score 50). Of these, 62.1% matched genes from Bacteria and 37.3% matched genes from Eukarya, with the remainder matching taxonomically unclassified or Archaeal/viral genes (Table 1). The relative abundance of Bacteria reads suggests that the symbiont-enrichment protocol used here (see Methods), while not 100% efficient, likely did increase the proportion of symbiont mRNA in the total transcript pool. The proportion of total mRNA in the metatranscriptome could likely have been further enriched by implementing sample-specific methods to remove symbiont and host rRNA reads, e.g., subtractive hybridization with taxon-specific probes (Stewart et al., 2010).
The annotations of NCBI-nr genes can be used to infer the taxonomic relatedness of symbiont and host sequences to reference organisms. Approximately 80% of all protein-coding transcripts from Bacteria matched genes of Gammaproteobacteria as their closest sequence relative (BLASTX searches of NCBI-nr). Among the Gammaproteobacterial reads, nearly half matched species of the Order Chromatiales (Figure 1), with the freshwater purple sulfur bacterium Allochromatium vinosum DSM 180 constituting over 17% of the Bacteria protein-coding reads (>10% of the total protein-coding reads; Figures 1–3). Other highly represented Gammaproteobacteria included the haloalkaliphilic, sulfur-oxidizing species Thioalkalivibrio sp. HL-EbGR7 (Chromatiales) and the sulfur-oxidizing symbionts of marine invertebrates, notably Endoriftia persephone, the endosymbiont of the hydrothermal vent tubeworm Riftia pachyptila (Figure 2). The non-Bacteria protein-coding reads in the dataset matched eukaryotes from diverse phyla, suggesting a lack of close relatives to S. velum in the database (Figure 2).
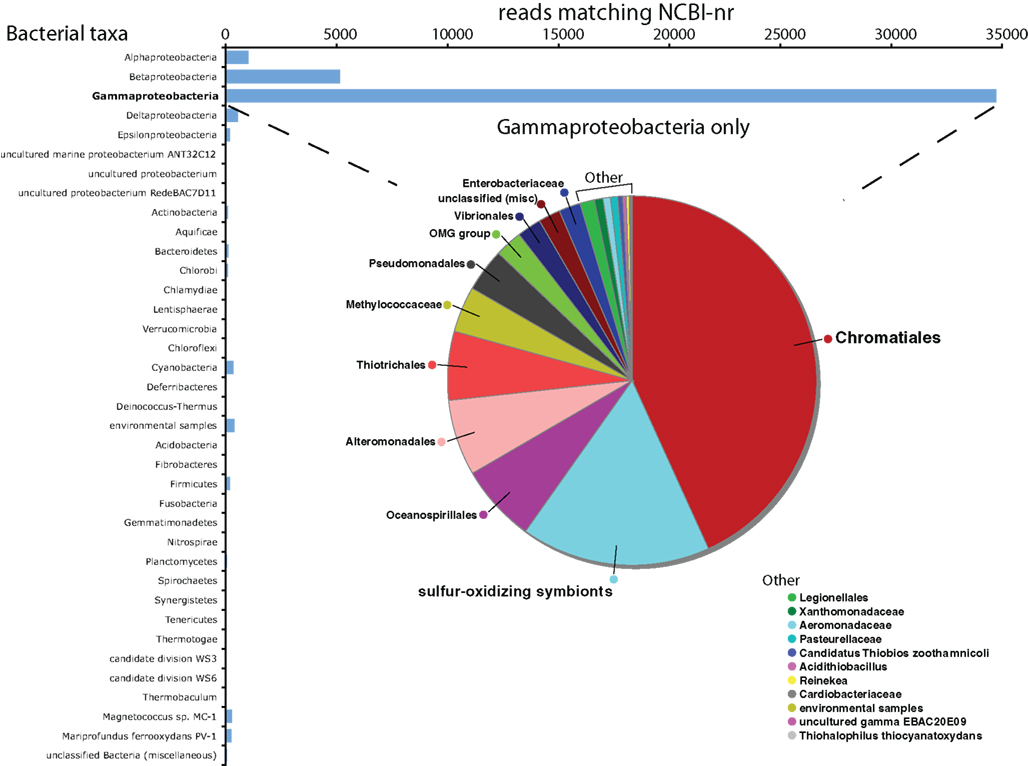
Figure 1. Taxonomic breakdown of all identified protein-coding transcripts assigned to the Domain Bacteria. Inset pie chart shows breakdown within the Gammaproteobacteria, which represented 79.4% of all protein-coding transcripts from the Domain Bacteria.
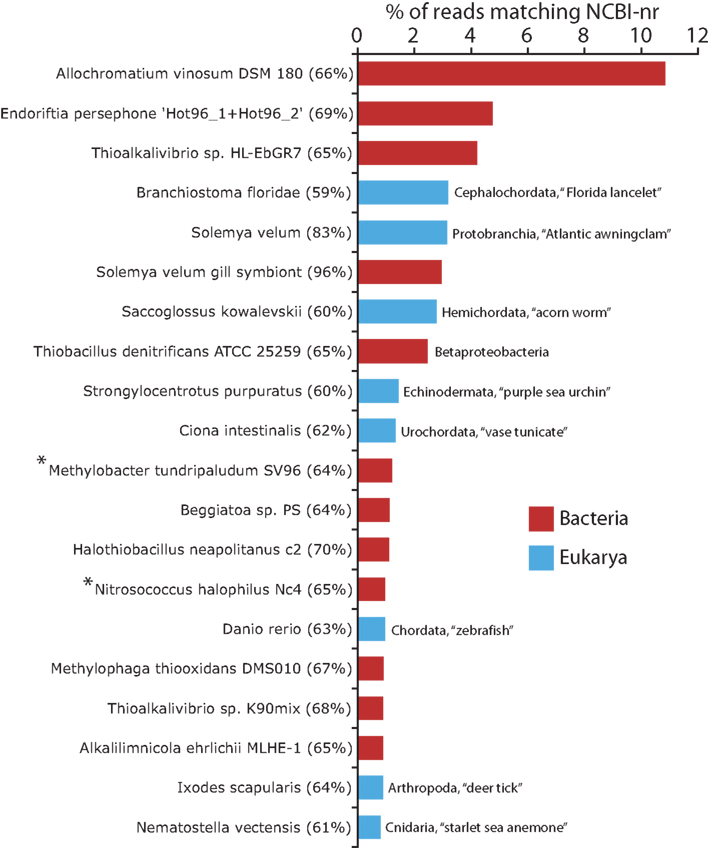
Figure 2. Top 20 taxa from the Domains Bacteria and Eukarya identified as best matches to protein-coding transcripts. All Bacteria taxa (red bars) belong to the Gammaproteobacteria and are putatively capable of chemolithotrophic growth on reduced sulfur compounds, unless otherwise marked (* = no evidence of sulfur-based chemolithotrophy). Eukarya (blue bars) are labeled according to phylum or subphylum designation. Numbers in parentheses are the amino acid identities averaged across all sequence reads matching a given taxon as a top BLASTX hit (above bit score 50). Identities reflect only the high-scoring segment pair (HSP) region for each BLAST result; gaps were not included in identity calculations.
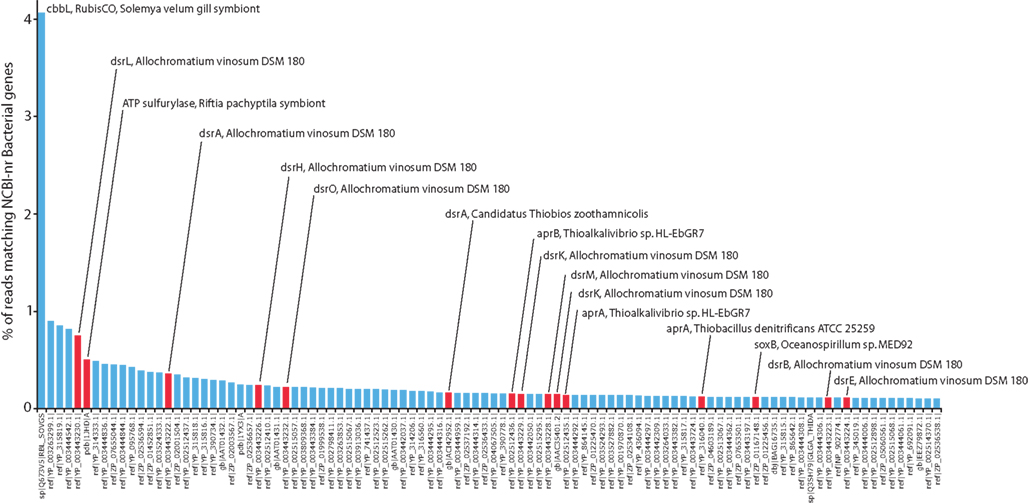
Figure 3. Top 100 most abundant reference genes (unique accession numbers) in the Domain Bacteria detected in the metatranscriptome, as a percentage of the total sequence reads matching Bacteria genes in the NCBI-nr database (accession numbers at bottom). Red bars indicate genes involved in sulfur-based energy metabolism.
Sulfur Oxidation Genes
BLASTX results revealed a dominant role for transcripts matching genes of dissimilatory sulfur oxidation pathways in the symbiont metatranscriptome. The putative functions of proteins encoded by these transcripts is inferred here based on the well characterized pathways from model sulfur-oxidizing bacteria, notably the Dsr pathway of the purple sulfur bacterium A. vinosum and the Sox pathway of the Alphaproteobacterium Paracoccus pantotrophus (for detailed descriptions of these pathways, see (Friedrich et al., 2001, 2005; Dahl et al., 2005; Frigaard and Dahl, 2009; Ghosh and Dam, 2009).
Our analysis revealed 2888 transcript sequences matching a total of 28 genes from the primary sulfur oxidation pathways (Figure 4). Of these, 65% (1886 reads) matched 14 genes encoding proteins of the Dsr pathway, including the dissimilatory sulfite reductase enzyme (DsrAB), the DsrAB-interacting flavoprotein DsrL, the transmembrane electron transport complex DsrKMJOP, and the putative sulfur relay molecules DsrEFH and DsrC, with the latter constituting the single most abundant sulfur gene in the transcriptome when read counts were corrected for variation in gene length (circles, Figure 4A).
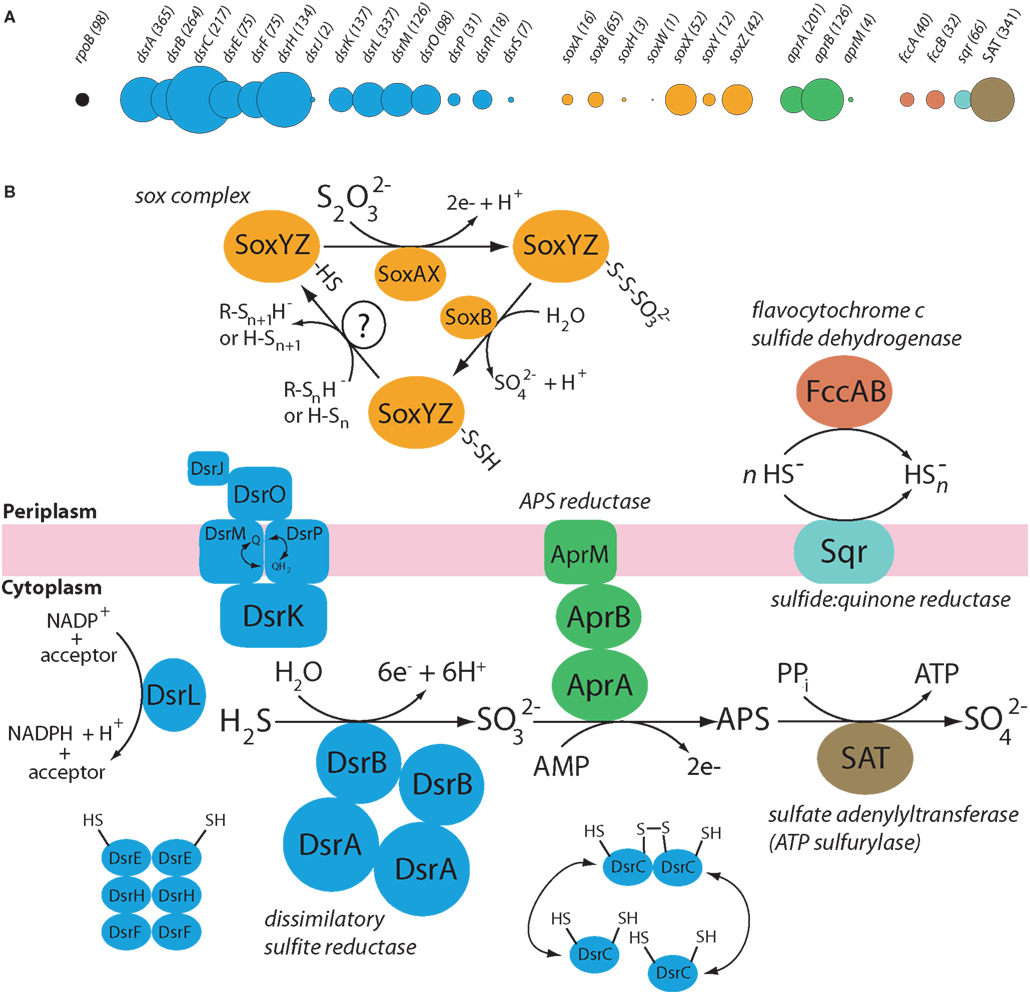
Figure 4. Relative abundance of transcripts encoding key enzymes of sulfur energy metabolism in the Solemya velum symbiont metatranscriptome. (A) Abundances of transcripts with top matches (BLASTX) to dissimilatory sulfur metabolism genes, shown relative to the abundance of transcripts matching RNA polymerase subunit B (rpoB), which typically occurs in a single copy per Bacteria genome. Numbers = read counts per gene. Circle area = read counts per gene normalized to the approximate gene length (in kilobases) inferred from published genomes. Sequences matching the same gene from different organisms have been pooled, in contrast to the data in Figure 3. (B) Putative metabolic function of corresponding enzymes, according to Dahl et al. (2005) and Frigaard and Dahl (2009). dsr = dissimilatory sulfite reductase gene cluster, sox = sulfur oxidation gene cluster; aprAB = adenosine-5’-phosphosulfate (APS) reductase; aprM, APS reductase membrane anchor; fccAB, flavocytochrome c sulfide dehydrogenase; SQR, sulfide–quinone reductase; SAT, sulfate adenylyltransferase.
Seven genes of the Sox system were detected, including five genes encoding the core proteins of the thiosulfate oxidation cycle: the SoxAX complex responsible for the fusion of thiosulfate to the carrier complex, the carrier complex SoxYZ, and SoxB, a putative thiol esterase mediating the hydrolytic release of sulfate from sulfur-bound SoxYZ (Figure 4B). Two additional sox genes, soxH, encoding a beta lactamase domain-containing protein, and soxW, encoding a periplasmic thioredoxin, were detected at low abundance. In P. pantotrophus, neither soxH or soxW appears necessary for autotrophic growth on thiosulfate (Rother et al., 2001; Bardischewsky et al., 2006).
Several additional sulfur energy metabolism genes were well represented in the transcriptome (Figure 4). These included four genes of the APS pathway for sulfite oxidation to sulfate: aprAB and aprM, encoding APS reductase (adenylylsulfate reductase) and the APS reductase membrane anchor, respectively, and SAT, encoding ATP sulfurylase (sulfate adenylyltransferase). Together, these four genes were represented by 672 sequences (23% of the reads matching sulfur genes). Finally, the analysis detected transcripts encoding two primary enzymes for sulfide oxidation: the membrane-bound sulfide–quinone reductase (Sqr), mediating electron transport to quinone, and flavocytochrome c sulfide dehydrogenase (FccAB), mediating transport to a c-type cytochrome (Dahl and Friedrich, 2008).
Discussion
This study provides the first molecular evidence that diverse genes of sulfur energy metabolism are present and actively transcribed in the S. velum symbiont population under in situ conditions. The metatranscriptome contained Bacteria transcripts encoding genes of several primary sulfur oxidation pathways, including the reverse dissimilatory sulfite reductase (Dsr) complex for sulfur oxidation to sulfite, the adenosine-5′-phosphate (APS) reductase pathway for sulfite oxidation, and the sulfur oxidation (Sox) pathway mediating thiosulfate oxidation. In total, the sulfur oxidation genes identified here (n = 28; Figure 4) represented 6.6% of the Bacteria protein-coding reads in the dataset (1 in 15 sequences). Several genes encoding enzymes for sulfide and thiosulfate oxidation, including the dsr genes and SAT (ATP sulfurylase), were among the top most abundant reference genes (i.e., unique accession numbers) detected in the study (Figure 3). Many of these were most closely related to homologs from known sulfur-oxidizing Gammaproteobacteria (Figures 1–3), in particular A. vinosum and Endoriftia persephone, the symbiont of the tubeworm Riftia pachyptila, consistent with the phylogenetic placement of the Solemya symbiont within this group (Eisen et al., 1992; Distel, 1998). Strikingly, a single reference gene, the previously characterized cbbL gene encoding the large subunit of the CO2 fixation enzyme RubisCO (ribulose 1,5-bisphosphate carboxylase–oxygenase) of the S. velum symbiont, accounted for over 4% of all protein-coding reads (Figure 3). The high abundance of transcripts for enzymes of sulfur compound oxidation and carbon fixation suggests that thioautotrophy dominated symbiont community metabolism at the time of collection in early winter.
Genes of the reverse Dsr pathway were among the most highly expressed in the symbiont metatranscriptome (Figures 3 and 4). In sulfate-reducing Bacteria and Archaea, dissimilatory sulfite reductase (DsrAB) mediates sulfite reduction to sulfide (Klein et al., 2001; Wagner et al., 2005). DsrAB homologs have been isolated from a wide variety of sulfur-oxidizing taxa (Loy et al., 2009), in which the enzymes are thought to operate in the oxidative direction in conjunction with a diverse set of accessory proteins, including the transmembrane complex DsrMKJOP, the sulfur shuttle molecules DsrEFH and DsrC, and the cytoplasmic flavoprotein DsrL (Dahl et al., 2005; Ghosh and Dam, 2009), all of which were detected in the Solemya symbiont transcriptome (Figure 4). It has been suggested that sulfide for oxidation by the DsrAB complex is released via the reduction of a polysulfur carrier molecule, as catalyzed by the NADH: (acceptor) oxidoreductase activity of DsrL (Dahl et al., 2005), which has been shown to be critical for sulfur oxidation in A. vinosum (Lubbe et al., 2006). In this species, the carrier molecule may be an organic perthiol involved with bringing stored elemental sulfur (sulfur globules) from the periplasm to the cytoplasm (Frigaard and Dahl, 2009). Stored sulfur globules in A. vinosum are produced during growth on sulfide by enzymes including FccAB and Sqr or via the activity of the Sox pathway during growth on thiosulfate (Frigaard and Dahl, 2009; Ghosh and Dam, 2009). Interestingly, the deposition of sulfur storage globules, while common in other thioautotrophic symbionts (e.g., Endoriftia persephone), has not been observed in Solemya, suggesting a potentially rapid turnover of elemental sulfur compounds by an active Dsr system. While the exact roles of Dsr enzymes remain to be confirmed in the Solemya symbiont, our data indicate a prominent role for sulfur oxidation to sulfite in this symbiosis.
Consistent with this finding, transcripts encoding the APS pathway for sulfite oxidation were abundant in the dataset. Use of the APS pathway by Solemya symbionts was established previously by enzyme assays showing the activity of APS reductase (AprAB) and ATP sulfurylase (SAT; Chen et al., 1987). Together, these enzymes catalyze the AMP-dependent oxidation of sulfite to APS and the subsequent ATP-generating conversion of APS to sulfate (Figure 4). Here, AprAB and SAT-encoding transcripts represented a combined 1.5% of all protein-coding Bacteria reads. Additionally, transcripts encoding the transmembrane anchor AprM were detected at low abundance. In other Bacteria taxa, electron transport from the cytoplasmic APS reductase to the membrane quinol/quinone pool is mediated via interactions with either AprM, which is more common in Gammaproteobacterial sulfur-oxidizers (e.g., A. vinosum), or the functionally associated membrane-bound redox complex QmoABC, common in green sulfur bacteria of the Order Chlorobiales (Meyer and Kuever, 2007). Here, detection of aprM but not qmoABC transcripts is consistent with the placement of the Solemya symbiont among the Gammaproteobacteria. Together, these data suggest a functional sulfite oxidation complex in this symbiosis.
The detection of transcripts matching genes of the Sox pathway also confirmed an active role for thiosulfate oxidation in S. velum symbionts. The Sox (sulfur oxidation) pathway mediates the oxidation of thiosulfate to sulfate in a variety of both non-photosynthetic and photosynthetic sulfur-oxidizing bacteria (Frigaard and Dahl, 2009; Ghosh and Dam, 2009; Sakurai et al., 2010). The pathway has been extensively studied in the Alphaproteobacterium Paracoccus pantotrophus, in which 15 genes soxRSVWXYZABCDEFGH comprise the sox operon (Friedrich et al., 2001, 2005; Rother et al., 2001). Of these, seven genes (soxABCDXYZ) encode the core periplasmic sox proteins, SoxB, SoxXA, SoxYZ, and SoxCD, with SoxYZ acting as the primary carrier molecule at every step in the pathway (Friedrich et al., 2001; Figure 4). Analysis of the S. velum symbiont metatranscriptome revealed transcripts matching two peripheral sox genes (soxHW) at low abundance and five of the seven core sox genes (soxABXYZ) at higher abundances (Figure 4). Transcripts encoding the sulfur dehydrogenase SoxCD were not detected in our analysis, consistent with the absence of these genes from the genomes of a variety of green and purple sulfur bacteria, including A. vinosum and the endosymbionts of deep-sea clams and tubeworms (Frigaard and Dahl, 2009; Harada et al., 2009). Alternatively, it is possible that soxC and soxD are present in the symbiont genome, but were not transcribed (above the level of detection) at the time of collection. In organisms lacking soxCD, the sulfur atom of the sulfane intermediate (SoxYZ-S-SH, Figure 4) is transferred to other acceptor substrates (RSnH− and −HSn− in Figure 4) and eventually into sulfur storage globules, or enters other sulfur oxidation pathways (Sauve et al., 2007; Frigaard and Dahl, 2009). The deposition of elemental sulfur globules has not yet been demonstrated for S. velum. However, the potential for globule storage under varying concentrations of thiosulfate or sulfide has not yet been exhaustively explored.
Implications
Our metatranscriptome data indicates the use of diverse pathways for both sulfide and thiosulfate oxidation by S. velum symbionts under environmental conditions, consistent with prior experiments showing that both substrates stimulate symbiont carbon fixation (Cavanaugh, 1983; Scott and Cavanaugh, 2007). The utilization of multiple sulfur oxidation pathways is not unusual among thioautotrophic bacteria and is likely related to substrate availability (Ghosh and Dam, 2009). For the endosymbionts of deep-sea clams, which share a complement of sulfur oxidation genes similar to that in S. velum symbionts (Kuwahara et al., 2007; Newton et al., 2007), it has been argued that the ability to oxidize both thiosulfate (Sox system) and sulfide (Dsr, Sqr/Fcc) is linked to the relative abundance of these compounds in the clam microhabitat (basaltic cracks and fissures in zones of diffuse-flow venting; Harada et al., 2009). In contrast, the endosymbiont of the tubeworm Riftia pachyptila putatively lacks the Sox pathway (Markert et al., 2007; Robidart et al., 2008), which may reflect an adaptation to a greater reliance on sulfide in the zones surrounding black smokers where this symbiosis occurs (Nelson and Fisher, 1995; Harada et al., 2009).
For S. velum, the relative abundance of reduced sulfur species available for symbiont oxidation has not been directly characterized. The host bivalve lives in coastal muds, positioning itself at the convergence of a Y-shaped burrow (Levinton, 1977) where it can access anoxic water from lower in the sediment. This water is likely enriched in sulfide but may also contain thiosulfate, perhaps resulting from chemical sulfide oxidation during burrow ventilation (Howarth et al., 1983; Elsgaard and Jorgensen, 1992). Alternatively, thiosulfate may also be available via sulfide oxidation by host mitochondria, as has been demonstrated in the Pacific congener S. reidi (Obrien and Vetter, 1990). Though both sulfide and thiosulfate can fuel symbiont autotrophy, sulfide has a much stronger effect (sevenfold) on carbon fixation rates in S. velum symbionts (Scott and Cavanaugh, 2007), suggesting that the symbiosis is adapted to use sulfide most efficiently.
Our transcriptome data, showing the relative abundance of transcripts in the Dsr and Sqr/Fcc pathways (Figure 4), potentially indicates a dominant role for sulfide metabolism in the S. velum symbiosis. However, transcript abundance may not be tightly coupled to protein abundance or enzyme activity (Taniguchi et al., 2010) and should be interpreted as a proxy for metabolic activity without supporting biochemical data. In other endosymbioses, namely those involving deep-sea hydrothermal vent clams, diverse sulfur pathways (Sox, Dsr, APS) have been shown to be constitutively expressed under variable environmental conditions (e.g., oxygen concentration; (Harada et al., 2009). However, compared to gene expression in the relatively stable environment of the deep sea, expression of S. velum symbiont genes, and the relative reliance on diverse reduced sulfur sources, may be more stochastic given the potential for diel and seasonal variation in the coastal environment.
Metatranscriptomics can now provide a comprehensive molecular framework for interpreting physiochemical measurements of metabolism in uncultured symbionts. Such analyses can be easily integrated into studies exploring the links between sulfur availability, environmental conditions, and symbiont gene expression. Our study confirms that genes of sulfur energy metabolism are a dominant component of the transcriptionally active symbiont gene pool in S. velum. Future experiments will help determine the extent to which genes of thioautotrophy co-vary with other metabolic pathways in the transcriptome (Dmytrenko et al., in preparation) and with the biochemical transformations of sulfur mediated by this model symbiosis.
Conflict of Interest Statement
This research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Rachel Barry for preparing cDNA samples for pyrosequencing. Generous support for this study came from the Gordon and Betty Moore Foundation (EFD) and the National Science Foundation (CMC, OCE-0453901).
References
Anderson, A. E., Childress, J. J., and Favuzzi, J. A. (1987). Net uptake of CO2 driven by sulfide and thiosulfate oxidation in the bacterial symbiont containing clam Solemya reidi. J. Exp. Biol. 133, 1–31.
Bardischewsky, F., Fischer, J., Holler, B., and Friedrich, C. G. (2006). SoxV transfers electrons to the periplasm of Paracoccus pantotrophus – an essential reaction for chemotrophic sulfur oxidation. Microbiology 152, 465–472.
Bettencourt, R., Pinheiro, M., Egas, C., Gomes, P., Afonso, M., Shank, T., and Santos, R. S. (2010). High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus. BMC Genomics 11, 559. doi: 10.1186/1471-2164-11-559
Cavanaugh, C. M. (1983). Symbiotic chemoautotrophic bacteria in marine invertebrates from sulfide rich habitats. Nature 302, 58–61.
Cavanaugh, C. M., McKiness, Z. P., Newton, I. L. G., and Stewart, F. J. (2006). “Marine chemosynthetic symbioses,” in The Prokaryotes, 3rd Edn, eds M. Dworkin, E. Rosenberg, K. H. Schleifer, and E. Stackebrandt (New York: Springer).
Chen, C., Rabourdin, B., and Hammen, C. S. (1987). The effect of hydrogen sulfide on the metabolism of Solemya velum and enzymes of sulfide oxidation in gill tissue. Comp. Biochem. Physiol. 88, 949–952.
Dahl, C., Engels, S., Pott-Sperling, A. S., Schulte, A., Sander, J., Lubbe, Y., Deuster, O., and Brune, D. C. (2005). Novel genes of the dsr gene cluster and evidence for close interaction of Dsr proteins during sulfur oxidation in the phototrophic sulfur bacterium Allochromatium vinosum. J. Bacteriol. 187, 1392–1404.
Distel, D. L. (1998). Evolution of chemoautotrophic endosymbioses in bivalves – Bivalve-bacteria chemosymbioses are phylogenetically diverse but morphologically similar. Bioscience 48, 277–286.
Dubilier, N., Bergin, C., and Lott, C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6, 725–740.
Eisen, J. A., Smith, S. W., and Cavanaugh, C. M. (1992). Phylogenetic relationships of chemoautotrophic bacterial symbionts of Solemya velum say (Mollusca, Bivalvia) determined by 16S ribosomal RNA gene sequence analysis. J. Bacteriol. 174, 3416–3421.
Elsgaard, L., and Jorgensen, B. B. (1992). Anoxic transformations of radiolabeled hydrogen sulfide in marine and fresh water sediments. Geochim. Cosmochim. Acta 56, 2425–2435.
Frias-Lopez, J., Shi, Y., Tyson, G. W., Coleman, M. L., Schuster, S. C., Chisholm, S. W., and DeLong, E. F. (2008). Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U.S.A. 105, 3805–3810.
Friedrich, C. G., Bardischewsky, F., Rother, D., Quentmeier, A., and Fischer, J. (2005). Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8, 253–259.
Friedrich, C. G., Rother, D., Bardischewsky, F., Quentmeier, A., and Fischer, J. (2001). Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl. Environ. Microbiol. 67, 2873–2882.
Frigaard, N. U., and Dahl, C. (2009). Sulfur metabolism in phototrophic sulfur bacteria. Adv. Microb. Physiol. 54, 103–200.
Fujiwara, Y., Kawato, M., Noda, C., Kinoshita, G., Yamanaka, T., Fujita, Y., Uematsu, K., and Miyazaki, J. I. (2010). Extracellular and mixotrophic symbiosis in the whale-fall mussel Adipicola pacifica: a trend in evolution from extra- to intracellular symbiosis. PLoS ONE 5, e11808. doi: 10.1371/journal.pone.0011808
Ghosh, W., and Dam, B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 33, 999–1043.
Grzymski, J. J., Murray, A. E., Campbell, B. J., Kaplarevic, M., Gao, G. R., Lee, C., Daniel, R., Ghadiri, A., Feldman, R. A., and Cary, S. C. (2008). Metagenome analysis of an extreme microbial symbiosis reveals eurythermal adaptation and metabolic flexibility. Proc. Natl. Acad. Sci. U.S.A. 105, 17516–17521.
Harada, M., Yoshida, T., Kuwahara, H., Shimamura, S., Takaki, Y., Kato, C., Miwa, T., Miyake, H., and Maruyama, T. (2009). Expression of genes for sulfur oxidation in the intracellular chemoautotrophic symbiont of the deep-sea bivalve Calyptogena okutanii. Extremophiles 13, 895–903.
Hewson, I., Poretsky, R. S., Tripp, H. J., Montoya, J. P., and Zehr, J. P. (2010). Spatial patterns and light-driven variation of microbial population gene expression in surface waters of the oligotrophic open ocean. Environ. Microbiol. 12, 1940–1956.
Howarth, R. W., Giblin, A., Gale, J., Peterson, B. J., and Luther, G. W. (1983). Reduced sulfur compounds in the pore waters of a New England salt marsh. Ecol. Bull. 35, 135–152.
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386.
Klein, M., Friedrich, M., Roger, A. J., Hugenholtz, P., Fishbain, S., Abicht, H., Blackall, L. L., Stahl, D. A., and Wagner, M. (2001). Multiple lateral transfers of dissimilatory sulfite reductase genes between major lineages of sulfate-reducing prokaryotes. J. Bacteriol. 183, 6028–6035.
Krueger, D. M., Gallager, S. M., and Cavanaugh, C. M. (1992). Suspension feeding on phytoplankton by Solemya velum, a symbiont-containing clam. Mar. Ecol. Prog. Ser. 86, 145–151.
Kuwahara, H., Yoshida, T., Takaki, Y., Shimamura, S., Nishi, S., Harada, M., Matsuyama, K., Takishita, K., Kawato, M., Uematsu, K., Fujiwara, Y., Sato, Y., Kato, C., Kitagawa, M., Kato, I., and Maruyama, T. (2007). Reduced genome of the thioautotrophic intracellular symbiont in a deep-sea clam, Calyptogena okutanii. Curr. Biol. 17, 881–886.
Levinton, J. (1977). “Ecology of shallow water – deposit-feeding communities, Quissett Harbor, Massachusetts,” in Ecology of Marine Benthos, ed. B. C. Coull (Columbia, SC: University of South Carolina Press).
Li, W. Z., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659.
Losekann, T., Robador, A., Niemann, H., Knittel, K., Boetius, A., and Dubilier, N. (2008). Endosymbioses between bacteria and deep-sea siboglinid tubeworms from an Arctic Cold Seep (Haakon Mosby Mud Volcano, Barents Sea). Environ. Microbiol. 10, 3237–3254.
Loy, A., Duller, S., Baranyi, C., Mussmann, M., Ott, J., Sharon, I., Beja, O., Le Paslier, D., Dahl, C., and Wagner, M. (2009). Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes. Environ. Microbiol. 11, 289–299.
Lubbe, Y. J., Youn, H. S., Timkovich, R., and Dahl, C. (2006). Siro(haem)amide in Allochromatium vinosum and relevance of DsrL and DsrN, a homolog of cobyrinic acid a,c-diamide synthase, for sulphur oxidation. FEMS Microbiol. Lett. 261, 194–202.
Markert, S., Arndt, C., Felbeck, H., Becher, D., Sievert, S. M., Hugler, M., Albrecht, D., Robidart, J., Bench, S., Feldman, R. A., Hecker, M., and Schweder, T. (2007). Physiological proteomics of the uncultured endosymbiont of Riftia pachyptila. Science 315, 247–250.
Nelson, D. C., and Fisher, C. R. (1995). “Chemoautotrophic and methanotrophic endosymbiotic bacteria at deep-sea vents and seeps,” in The Microbiology of Deep-Sea Hydrothermal Vents, ed. D. M. Karl (CRC Press).
Newton, I. L. G., Woyke, T., Auchtung, T. A., Dilly, G. F., Dutton, R. J., Fisher, M. C., Fontanez, K. M., Lau, E., Stewart, F. J., Richardson, P. M., Barry, K. W., Saunders, E., Detter, J. C., Wu, D., Eisen, J. A., and Cavanaugh, C. M. (2007). The Calyptogena magnifica chemoautotrophic symbiont genome. Science 315, 998–1000.
Nyholm, S. V., Robidart, J., and Girguis, P. R. (2008). Coupling metabolite flux to transcriptomics: insights into the molecular mechanisms underlying primary productivity by the hydrothermal vent tubeworm Ridgeia piscesae. Biol. Bull. 214, 255–265.
Obrien, J., and Vetter, R. D. (1990). Production of thiosulfate during sulfide oxidation by mitochondria of the symbiont-containing bivalve Solemya reidi. J. Exp. Biol. 149, 133–148.
Poretsky, R. S., Hewson, I., Sun, S. L., Allen, A. E., Zehr, J. P., and Moran, M. A. (2009). Comparative day/night metatranscriptomic analysis of microbial communities in the North Pacific subtropical gyre. Environ. Microbiol. 11, 1358–1375.
Robidart, J. C., Bench, S. R., Feldman, R. A., Novoradovsky, A., Podell, S. B., Gaasterland, T., Allen, E. E., and Felbeck, H. (2008). Metabolic versatility of the Riftia pachyptila endosymbiont revealed through metagenomics. Environ. Microbiol. 10, 727–737.
Rodrigues, C. F., Webster, G., Cunha, M. R., Duperron, S., and Weightman, A. J. (2010). Chemosynthetic bacteria found in bivalve species from mud volcanoes of the Gulf of Cadiz. FEMS Microbiol. Ecol. 73, 486–499.
Rother, D., Henrich, H. J., Quentmeier, A., Bardischewsky, F., and Friedrich, C. G. (2001). Novel genes of the sox gene cluster, mutagenesis of the flavoprotein SoxF, and evidence for a general sulfur-oxidizing system in Paracoccus pantotrophus GB17. J. Bacteriol. 183, 4499–4508.
Sakurai, H., Ogawa, T., Shiga, M., and Inoue, K. (2010). Inorganic sulfur oxidizing system in green sulfur bacteria. Photosyn. Res. 104, 163–176.
Sauve, V., Bruno, S., Berks, B. C., and Hemmings, A. M. (2007). The SoxYZ complex carries sulfur cycle intermediates on a peptide swinging arm. J. Biol. Chem. 282, 23194–23204.
Schwedock, J., Harmer, T. L., Scott, K. M., Hektor, H. J., Seitz, A. P., Fontana, M. C., Distel, D. L., and Cavanaugh, C. M. (2004). Characterization and expression of genes from the RubisCO gene cluster of the chemoautotrophic symbiont of Solemya velum: cbbLSQO. Arch. Microbiol. 182, 18–29.
Scott, K. M., and Cavanaugh, C. M. (2007). CO2 uptake and fixation by endosymbiotic chemoautotrophs from the bivalve Solemya velum. Appl. Environ. Microbiol. 73, 1174–1179.
Shi, Y. M., Tyson, G. W., and DeLong, E. F. (2009). Metatranscriptomics reveals unique microbial small RNAs in the ocean’s water column. Nature 459, 266–269.
Sievert, S. M., Kiene, R. P., and Schulz-Vogt, H. N. (2007). The sulfur cycle. Oceanography 20, 117–123.
Stewart, F. J., and Cavanaugh, C. M. (2006). Bacterial endosymbioses in Solemya (Mollusca: Bivalvia): model systems for studies of symbiont-host adaptation. Antonie Van Leeuwenhoek 90, 343–360.
Stewart, F. J., Newton, I. L. G., and Cavanaugh, C. M. (2005). Chemosynthetic endosymbioses: adaptations to oxic-anoxic interfaces. Trends Microbiol. 13, 439–448.
Stewart, F. J., Ottesen, E. A., and DeLong, E. F. (2010). Development and quantitative analyses of a universal rRNA-subtraction protocol for microbial metatranscriptomics. ISME J. 4, 896–907.
Stewart, F. J., Sharma, A. K., Bryant, J. A., Eppley, J. M., and Delong, E. F. (2011a). Community transcriptomics reveals universal patterns of protein sequence conservation in natural microbial communities. 12, R26.
Stewart, F. J., Ulloa, O., and DeLong, E. F. (2011b). Microbial metatranscriptomics in a permanent marine oxygen minimum zone. Environ. Microbiol. (in press). doi:10.1111/j.1462-2920.2010.02400x
Taniguchi, Y., Choi, P. J., Li, G. W., Chen, H. Y., Babu, M., Hearn, J., Emili, A., and Xie, X. S. (2010). Quantifying E. coli proteome and transcriptome with single molecule sensitivity in single cells. Science 329, 533–538.
Wagner, M., Loy, A., Klein, M., Lee, N., Ramsing, N. B., Stahl, D. A., and Friedrich, M. W. (2005). Functional marker genes for identification of sulfate-reducing prokaryotes. Environ. Microbiol. 397, 469–489.
Wilmes, P., and Bond, P. L. (2009). Microbial community proteomics: elucidating the catalysts and metabolic mechanisms that drive the Earth’s biogeochemical cycles. Curr. Opin. Microbiol. 12, 310–317.
Woyke, T., Teeling, H., Ivanova, N. N., Huntemann, M., Richter, M., Gloeckner, F. O., Boffelli, D., Anderson, I. J., Barry, K. W., Shapiro, H. J., Szeto, E., Kyrpides, N. C., Mussmann, M., Amann, R., Bergin, C., Ruehland, C., Rubin, E. M., and Dubilier, N. (2006). Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 443, 950–955.
Keywords: chemosynthesis, endosymbiosis, sulfide, thiosulfate, gene expression
Citation: Stewart FJ, Dmytrenko O, DeLong EF and Cavanaugh CM (2011) Metatranscriptomic analysis of sulfur oxidation genes in the endosymbiont of Solemya velum. Front. Microbio. 2:134. doi: 10.3389/fmicb.2011.00134
Received: 16 May 2011;
Paper pending published: 03 June 2011;
Accepted: 09 June 2011;
Published online: 20 June 2011.
Edited by:
Donald A. Bryant, The Pennsylvania State University, USAReviewed by:
Donald A. Bryant, The Pennsylvania State University, USAChristiane Dahl, Rheinische Friedrich-Wilhelms-Universität Bonn, Germany
Copyright: © 2011 Stewart, Dmytrenko, DeLong and Cavanaugh. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Frank J. Stewart, School of Biology, Georgia Institute of Technology, Ford ES&T Building, Room 1242, 311 Ferst Drive, Atlanta, GA 30332, USA. e-mail:ZnJhbmsuc3Rld2FydEBiaW9sb2d5LmdhdGVjaC5lZHU=