

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol., 30 May 2011
Sec. Cellular and Infection Microbiology - closed section
volume 2 - 2011 | https://doi.org/10.3389/fmicb.2011.00093
This article is part of the Research TopicHuman health and disease in a microbial worldView all 13 articles
Members of the diverse bacterial phylum Bacteroidetes have colonized virtually all types of habitats on Earth. They are among the major members of the microbiota of animals, especially in the gastrointestinal tract, can act as pathogens and are frequently found in soils, oceans and freshwater. In these contrasting ecological niches, Bacteroidetes are increasingly regarded as specialists for the degradation of high molecular weight organic matter, i.e., proteins and carbohydrates. This review presents the current knowledge on the role and mechanisms of polysaccharide degradation by Bacteroidetes in their respective habitats. The recent sequencing of Bacteroidetes genomes confirms the presence of numerous carbohydrate-active enzymes covering a large spectrum of substrates from plant, algal, and animal origin. Comparative genomics reveal specific Polysaccharide Utilization Loci shared between distantly related members of the phylum, either in environmental or gut-associated species. Moreover, Bacteroidetes genomes appear to be highly plastic and frequently reorganized through genetic rearrangements, gene duplications and lateral gene transfers (LGT), a feature that could have driven their adaptation to distinct ecological niches. Evidence is accumulating that the nature of the diet shapes the composition of the intestinal microbiota. We address the potential links between gut and environmental bacteria through food consumption. LGT can provide gut bacteria with original sets of utensils to degrade otherwise refractory substrates found in the diet. A more complete understanding of the genetic gateways between food-associated environmental species and intestinal microbial communities sheds new light on the origin and evolution of Bacteroidetes as animals’ symbionts. It also raises the question as to how the consumption of increasingly hygienic and processed food deprives our microbiota from useful environmental genes and possibly affects our health.
The phylum Bacteroidetes is a very diverse bacterial phylum, the name of which changed several times over the past years. It is also known as the Cytophaga–Flexibacter–Bacteroides (CFB) group, an appellation that reflects the diversity of organisms found in this phylogenetic group (Woese, 1987; Woese et al., 1990). According to the Bergey’s Manual of Systematic Bacteriology (Bergey’s, 2011), the Bacteroidetes phylum comprises four classes: Bacteroidia, Flavobacteria, Sphingobacteria, and Cytophagia, representing around 7000 different species (NCBI, October 2010). The largest class is the Flavobacteria, grouping together around four times more species than the three others (Table 1). These bacteria are all Gram negative, cover a mixture of physiological types, from strictly anaerobic Bacteroides to strictly aerobic Flavobacteria. They are non-motile, flagellated, or move by gliding.

Table 1. Census of the phylogenetic divisions inside the Bacteroidetes phylum (NCBI, October 2010).
Members of the phylum Bacteroidetes have colonized many different ecological niches, including soil, ocean, freshwater, and the gastrointestinal tract (GIT) of animals, where they display various biological functions. In particular, they are well known degraders of polymeric organic matter. This review describes current knowledge on the role and mechanisms of polysaccharide degradation by Bacteroidetes in their respective habitats. We emphasize the features shared by members of the phylum that allow this functional -specialization in various environments. We address the links between these different microbial communities through food consumption, which raise the question of the evolution of gut microbes.
Microbes that live in and on humans (known as microbiota) can represent up to 100 trillion cells, 10 times more than the eukaryotic stem and somatic cells (Ley et al., 2006b; Turnbaugh et al., 2007). The large majority of these microbes reside in our GIT, and belong either to the Firmicutes or Bacteroidetes phyla (Marchesi, 2010). In fact, these two bacterial phyla account for >98% of the 16S rRNA sequences detected in the gut microbiota of mammals (Ley et al., 2006b). Bacteroidetes have colonized all the different parts of the GIT, despite the different conditions they have to face in terms of, e.g., pH, nutrients, and oxygen availability. Due to their easier accessibility, the mouth and colon microbiota have been the most studied. The large intestine is the most colonized compartment of the GIT, with bacterial densities reaching 1011–1012 cells/ml (Whitman et al., 1998). Members of the Bacteroidetes phylum are well known colonizers of the colon. They account for about 50% of the 16S rRNA sequences detected from healthy human mucosal tissues (Eckburg et al., 2005). Among this phylum, members of the genus Bacteroides are the most abundantly represented in the fecal microbiota (Moore and Holdeman, 1974; Sghir et al., 2000). Bacteroidetes have also been found in the normal microbiota of the oral cavity, either in the saliva or dental plaque (Keijser et al., 2008; Nasidze et al., 2009a,b). In the throat, Andersson et al. (2008) found that Bacteroidetes account for ∼20% of the reads using barcoding pyrosequencing, and identified Prevotella genus as the second most represented after Streptococcus. The same proportions were found in the distal esophagus (Pei et al., 2004). Due to its low pH, the stomach is a harsh ecological niche, quite different from the other GIT compartments. However, even if Helicobacter pylori represents a major part of the stomach microbiota, Bacteroidetes were still found to account for 10–20% of the bacteria (Bik et al., 2006), including members of the genera Prevotella, Capnocytophaga, Bergeyella, Porphyromonas, and Tannerella. In fact, Prevotella was the third most abundant genus in the stomach microbiota, after Helicobacter and Streptococcus.
Bacteroidetes are also encountered in the microbiota of other mammals, such as mice (Dubos et al., 1965; Savage et al., 1968), dogs (Middelbos et al., 2010), pigs (Leser et al., 2002), and ruminants (Tajima et al., 1999; Leng et al., 2010). They appear in the GIT of domesticated and wild birds, such as chickens (Zhu et al., 2002), turkeys (Scupham et al., 2008), goose (Lu et al., 2009), and ostrichs (Matsui et al., 2010). They have been demonstrated as dominant in the microbiota of echinoderms (Balakirev et al., 2008), millipedes (Knapp et al., 2010), and the last compartment of termite gut (Schmitt-Wagner et al., 2003).
The interaction between Bacteroidetes and their animal host is now known to be mutualism rather than commensalism since the fitness of both partners is increased (Backhed et al., 2005). Several studies have shown the implication of Bacteroidetes for the normal development of the GIT. For example, it has been shown that germ-free animals display an altered GIT compared to conventional ones, leading to changes in morphology and function (see for review, Abrams, 1983). Other contributions of Bacteroidetes to the health of their host include interactions with the immune system for the activation of T-cell mediated responses (Mazmanian et al., 2008; Wen et al., 2008), and limitation of the GIT colonization by potential pathogenic bacteria (Mazmanian, 2008). Gut Bacteroidetes generally produce butyrate, an end product of colonic fermentation which is thought to have antineoplastic properties and thus plays a role in maintaining a healthy gut (Kim and Milner, 2007). They are also involved in bile acid metabolism and transformation of toxic and/or mutagenic compounds (Smith et al., 2006).
However, their main and most studied biological function as symbionts is the degradation of biopolymers in the large intestine, and in particular polysaccharides. Carbohydrates represent the bulk of normal human and animal diets, thus forming a main source of nutrients for both the host and the microbiota. Mammals can easily absorb simple sugars (e.g., glucose, galactose) via active transport in the proximal small intestine (Ferraris, 2001) or degrade some disaccharides (e.g., lactose, maltose, sucrose) to monomers. They also possess enzymes which allow the degradation of starch to glucose. However, they are generally under-equipped for the degradation of other complex polysaccharides, which are resistant to the action of digestive enzymes and reach the colon relatively intact. The microbiota and especially Bacteroidetes are believed to complement eukaryotic genomes with degradation enzymes targeting resistant dietary polymers, such as plant cell wall compounds (e.g., cellulose, pectin, and xylan). Bacteroidetes also degrade host-derived carbohydrates, primarily coming from GIT secretions, such as N-glycans found in mucins or chondroitin sulfates (Salyers et al., 1977). With this respect, one well studied example is Bacteroides thetaiotaomicron, a prominent member of the human microbiota that has been shown to principally forage on host mucin O-glycans. This has an impact on their colonization and probably represents an important evolved component of their adaptation to the gut habitat (Martens et al., 2008). The bacteria-mediated fermentation of these food, and host-derived polysaccharides in the colon leads to the release of volatile, short-chain fatty acids (mainly acetate, propionate, and butyrate) that are reabsorbed by the host. Therefore these intestinal bacteria help the host to gain energy from otherwise refractory carbohydrate sources. In herbivorous animals such as ruminants, the diet consists largely of plant cell wall compounds resistant to the host’s digestive enzymes. Thus, assimilation of short-chain fatty acids produced by microbial fermentation of polysaccharides can support more than 50% of the total caloric supply (Carroll and Hungate, 1954). In omnivorous mammals, notably humans, this extra-source of energy accounts for 7–10% of the daily allowance (Smith and Bryant, 1979; Hooper et al., 2002). In rats, it has been shown that germ-free animals excrete 87% more calories within the feces than their normal counterparts, and need to ingest 30% more food to maintain their body weight (Wostmann et al., 1983). Hence, the presence of the intestinal microbiota is required for the optimal uptake of energy from the diet.
Members of the phylum Bacteroidetes have colonized virtually all types of environments encountered on Earth. This versatility is reflected by the diversity of sources from which cultivated strains have been isolated, such as soil, activated sludge, decaying plant material, compost, freshwater, and marine samples, algae, dairy products, and diseased animals (Bernardet and Nakagawa, 2006; Reichenbach, 2006). While the GIT microbiota is mainly composed of species from the Bacteroidia class, environmental Bacteroidetes belong primarily to the Flavobacteria, Cytophagia, and Sphingobacteria classes.
Over the past years, the diversity of environmental bacterial communities has been re-evaluated due to the use of culture-independent techniques. Bacteroidetes are increasingly recognized as an important compartment of the bacterioplankton in marine environments (Jooste and Hugo, 1999; Kirchman, 2002), especially in pelagic oceans. They are highly abundant as shown by fluorescent in situ hybridization (FISH) experiments using specific probes on seawater samples. They account for as much as half of the identified cells potentially identified and outnumber alternative species (Glöckner et al., 1999; Simon et al., 1999; Cottrell and Kirchman, 2000). Using 16S rRNA gene clone libraries from nine different sampling sites (including coastal and oceanic waters at 5 m-depth in temperate, tropical, and polar regions), Pommier et al. (2007) found that Bacteroidetes and Proteobacteria were dominant. Marine Bacteroidetes are also often demonstrated as living on marine inert or living surfaces, either in sediments (Llobet-Brossa et al., 1998; Julies et al., 2010), in biofilms (Edwards et al., 2010), in hydrothermal vents (Sievert et al., 2000), associated with corals (Frias-Lopez et al., 2002; Rohwer et al., 2002) or on the surface of macroalgae (Beleneva and Zhukova, 2006; Staufenberger et al., 2008; Salaun et al., 2010) and angiosperms (Crump and Koch, 2008).
Bacteroidetes also colonize freshwater environments to a significant extent. Using FISH, they were found in all the water samples collected in lakes from different locations (Austria, Switzerland, Germany, and Siberia), with a median abundance ranging from 2 to 12% which tends to increase with depth (Glöckner et al., 1999). Bacteroidetes accounted for 40–60% of the detectable bacteria in river samples from Spain (Simek et al., 2001). An integrative analysis of the available bacterial 16S rDNA sequences recovered from freshwater lakes and rivers over the past 15 years concluded that these environments host a specific planktonic bacterial community, different from the adjacent terrestrial habitats and sediments (Zwart et al., 2002). Bacteroidetes were amongst the major bacterial lineages in this community, together with Proteobacteria, Actinobacteria, and Verrucomicrobia.
Soil-associated bacterial communities also comprise members of the Bacteroidetes phylum. Using different techniques they have been detected in soil samples from various locations, including cultivated fields (Borneman et al., 1996; Martinez-Alonso et al., 2010), greenhouse soils (Kim et al., 2006) and unexploited areas (Buckley and Schmidt, 2003; Zhou et al., 2009). Recently, a study including soils from 88 different places revealed a positive correlation between the pH of the substrate and the relative abundance of Bacteroidetes (Lauber et al., 2009), ranging from 1.7% at low pH (<4) to 17% in basic soils (pH > 8). The surface of leaves, or phyllosphere, is another location of Bacteroidetes colonization in terrestrial environments. In a recent study, they accounted for 21.5% of the OTUs sequenced in samples from 56 tree species. Indeed, the most common group of bacteria found on the leaves was Sphingobacteriales which represented 21.3% of all sequences (Redford et al., 2010). Interestingly, Bacteroidetes tend to be more abundant on conifers (gymnosperms), reaching 70% of the sequences detected on the leaves, and less abundant on leaves from angiosperms.
Environmental Bacteroidetes are thought to be specialized in the degradation of complex organic matter in the biosphere, especially in the form of polysaccharides and proteins (Church, 2008). As a group, they are very versatile in the range of biopolymers they can use as carbon and energy source, e.g., plant, algal, or animal compounds. The description of new taxa often results from screening of environmental samples to discover original enzymatic activities with potential biotechnological applications (Barbeyron et al., 2001, 2008; Pankratov et al., 2006; Lee et al., 2010).
Several lines of evidence designate Bacteroidetes as crucial degraders of complex organic matter in the environment and not only in laboratory, many of them resulting from research on aquatic ecosystems (Kirchman, 2002). In the ocean, particulate and dissolved organic matter tends to aggregate to form particulate detritus (>0.5 mm) known as marine snow (Chin et al., 1998). These sinking particles are hotspots of organic carbon and thus play a key role in the export of matter from the euphotic surface to the sediment of the deep ocean (Azam and Malfatti, 2007). Marine snow hosts a considerable bacterial population, with densities reaching 106–108 cells per aggregate (Alldredge et al., 1986). Numerous studies characterizing these particles-associated populations have revealed that Bacteroidetes account for a large part (Rath et al., 1998; Ploug et al., 1999) and can even be the most abundant phylogenetic type detected (DeLong et al., 1993). Similar results have been found for estuarine particles, where Cytophaga spp. are amongst the dominant species in the bacterial assemblage (Crump et al., 1999). This enrichment of Bacteroidetes can be linked to their ability (i) to efficiently degrade complex and recalcitrant biopolymers sequestered in the particulate organic detritus and (ii) to colonize surfaces (Kirchman, 2002). Similarly, the immersion of insoluble cellulose for one month in the Irish Sea resulted in the colonization of the fibers by a specific bacterial biofilm, dominated by Bacteroidetes and Gammaproteobacteria (Edwards et al., 2010). In lakes, Glöckner et al. (1999) found that Bacteroidetes relative abundance in the water column increased with the depth of sampling. They proposed that this was due to the accumulation of slowly degradable, sinking macromolecules, a substrate that may have selected efficient degraders like Bacteroidetes.
Another evidence for consumption of high molecular weight (HMW) compounds by Bacteroidetes comes from several experiments where the bacterial community was followed after an increase of input of organic matter. Coastal and deep-sea environments typically receive seasonal pulses of organic matter after phytoplankton bloom events (Gooday, 2002; Kim et al., 2009). During such blooms of diatoms (Riemann et al., 2000) and dinoflagellates (Fandino et al., 2001), the population of Bacteroidetes increased significantly in a short time scale, concomitant with increasing activity of extracellular hydrolytic enzymes. Concordant with these results, a recent study of the communities of water masses from the North Atlantic Ocean revealed a specific enrichment of Flavobacteria in the space directly surrounding nanophytoplankton (Gomez-Pereira et al., 2010). In a freshwater lake, the virus-induced lysis of filamentous cyanobacteria led to the appearance of newly emerging bacterial species (van Hannen et al., 1999). The majority of these new microbial populations belonged to Cytophagales and Actinomycetes.
In alternative experiments, seasonal pulses were mimicked in laboratory conditions by an artificial increase of the organic carbon input. Gihring et al. (2009) simulated a phytodetritus deposition event by adding heat-killed Spirulina cyanobacteria on coastal sediments, resulting in an increase in the proportion of Bacteroidetes that was limited to the surface of the sediment (from 0 to 4 mm depth). This suggests a role of Bacteroidetes in the phytodetritus degradation in aerobic conditions (Gihring et al., 2009). The addition of exogenous protein to Californian coastal seawater mesocosms also triggered a change in the bacterial community, with an increase of the occurrence of several Bacteroidetes species (Pinhassi et al., 1999). Finally, by combining microautoradiography and FISH (MICRO–FISH), Cottrell and Kirchman (2000) compared the uptake of biopolymers (protein and chitin) and their constituent monomers (amino acids and N-acetylglucosamine, respectively) by natural bacterial assemblages from coastal waters of Delaware. This approach showed an over-representation of Bacteroidetes in the portion of bacteria consuming polymers, whereas Alphaproteobacteria dominated uptake of monomers (Cottrell and Kirchman, 2000).
Taken together, these results show that environmental Bacteroidetes specialize in the mineralization of HMW organic matter. Thus, this phylum represents a key compartment for carbon fluxes and budgets in ecosystems (Nagata, 2008).
As outlined above, Bacteroidetes are found in the normal bacterial community of many diverse habitats, where they play a beneficial role in the degradation of organic matter. On the contrary, some members of this phylum can have a strong pathogenic behavior toward different eukaryotic species. This is underlined by the number of Bacteroidetes strains isolated from clinical samples of human and animal origin (Hugo et al., 1999), including blood, urine, infected wounds, and feces. Some members of the Bacteroides genus, although belonging to the normal GIT microbiota, can cause opportunistic infections if the integrity of the intestinal mucosal barrier is broken (Smith et al., 2006). Indeed, the majority of species isolated from anaerobic infections falls into the Bacteroides genus, acting on different discrete sites such as peritoneal cavity, vagina, sinuses, skin, and heart (Wexler, 2007). These infections are usually polymicrobial, but B. fragilis and B. thetaiotaomicron are the most frequent species. Bacteroides are also associated with bacteremia, and B. fragilis is the most common blood isolate recovered from patients (Brook, 2010). Members of the genera Porphyromonas, Prevotella, and Tannerella are well known pathogens of the oral cavity, where they can notably cause periodontal disease and caries (Tanner et al., 1986; Shah and Collins, 1990; Gibson and Attardo Genco, 2006). The emergence of an infection seems to be linked to the assemblage of pathogens in bacterial consortia more than on the individual action of specific species (Jenkinson and Lamont, 2005).
The Flavobacteria class also contains opportunistic human pathogens, invading hosts with poor immune system (Bernardet and Nakagawa, 2006). One of them, Elizabethkingia meningoseptica, causes meningitis in infants (King, 1959; Ratner, 1984; Kim et al., 2005). Myroides has been reported in cases of surgery wound, urinary tract infections, septicemia, pneumonia, meningitis, fasciitis, ventriculitis, and nosocomial infections (Mammeri et al., 2002). The genera Empedobacter, Bergeyella, Weeksella, and Capnocytophaga also contain pathogenic species (Hugo et al., 2006a,b; Leadbetter, 2006).
Bacteroidetes infections can have devastating effects for farmed and wild fish (Austin and Austin, 2007), as reported a century ago and first observed in aquaria (Davis, 1922). Flavobacterium psychrophilum, F. columnare, F. branchiophilum, Tenacibaculum maritimum, and Chryseobacterium scophtalmum are some of the species with the most severe economic impact (Bernardet and Bowman, 2006). Members of the genera Riemerella, Ornithobacterium, and Coenonia can cause septicemia and respiratory tract infections in birds (Segers et al., 1993; Vandamme et al., 1994, 1999).
In addition, Bacteroidetes can affect various plants and algae. F. johnsoniae is incriminated as an opportunistic pathogen in the decay of fresh plants and vegetables called “soft rot” (Liao and Wells, 1986). Members of the genera Zobellia, Cellulophaga, and Kordia have algicidal activities (Skerratt et al., 2002; Sohn et al., 2004), and can therefore control blooms of microscopic algae. The Anaaki disease, severely damaging the red macroalga Porphyra yezoensis, is caused by Flavobacterium sp. LAD-1 (Sunairi et al., 1995). Uyenco (1977) isolated strains of Flavobacterium associated with “ice-ice disease” in decaying red alga Eucheuma striatum, characterized by whitening and hardening of the seaweed’s tissues. Similarly, a Cytophaga sp. promotes the ice–ice disease in stressed branches of the farmed Rhodophytes Kappaphycus alvarezii and Eucheuma denticulatum (Largo et al., 1995). Members of the Flavobacterium genus can also infect brown macroalga such as Undaria pinnatifida, where they are associated with “spot-rotting” disease and “shot hole disease” (Neill et al., 2008).
The pathogenicity of Bacteroidetes is at least partly due to the production of polymer-degrading enzymes targeting host cellular components. Animal pathogens produce a range of hydrolytic enzymes such as hyaluronidase, chondroitin sulfatase, heparinase, glucosidases, and N-acetylglucosaminidase (Rudek and Haque, 1976; Duerden, 1994). These enzymes damage the extracellular matrix of animals and thus are important both for tissue colonization and nutrients supply. In a study of the fish pathogen F. columnare, a chondroitin lyase was found to be related to the virulence (Suomalainen et al., 2006). Plant and algal pathogens are able to degrade cell wall compounds such as pectins and agars. Indeed, phytopathogenic strains of F. johnsoniae secrete much more pectate lyase than their non pathogenic counterparts (Liao and Wells, 1986). Bacteroidetes also secrete various proteases, and this has been documented as an important virulence factor. Proteases secreted by different pathogenic Bacteroides species can destroy human brush border digestive enzymes (Riepe et al., 1980). Greiner and Mayrand (1987) found that the pathogenic strains of Porphyromonas gingivalis had a higher collagenolytic activity than the non-virulent strains. In animal models, P. gingivalis mutants lacking the ability to produce various proteases lost their virulence (Loesche, 1993; Fletcher et al., 1995). Whatever their environment, it appears that the biological function of Bacteroidetes either as symbiont, environmental, or pathogen bacteria is connected with their propensity to degrade complex biopolymers, i.e., polysaccharides and proteins.
As a phylum, and especially due to their versatility in habitats, Bacteroidetes have access to an amazing diversity of carbon sources. Indeed, the chemical diversity of polysaccharides largely outnumbers the possibility for protein folds – it has been calculated that there are 1.05 × 1012 possible linear and branched forms of a single hexasaccharide (Laine, 1994). Moreover, these structural variations have been harnessed by living organisms to fulfill very different roles: e.g., structural, storage, specific signaling, -specific -recognition, host–pathogen interactions to name but a few (Carpita and Gibeaut, 1993; Graham et al., 2000; Stahl and Bishop, 2000). Consequently, carbohydrates account for around 75% of the biomass on Earth, a natural resource that was not lost on competing organisms that were developing their own strategies to utilize this chemical energy for their own survival.
But even more importantly several classes of polysaccharides are niche specific. While cell walls are a characteristic feature of all plants, they are not exclusive to plants, with most bacterial and algal cells as well as all fungal cells also being surrounded by extracellular, macromolecular barriers (extracellular matrix or ECM). The macromolecular composition, however, is characteristically different among the major evolutionary lineages of the living world, linking specific life style or nutritional habits to specifically encountered biopolymers. A vivid example is provided by the polysaccharides of the marine environment that are typically and to a large majority sulfated (carrageenans and fucans) or highly ionic (alginates) and unique to this particular habitat (Michel et al., 2010a,b; Popper et al., 2011). In contrast, the basic polysaccharide components of plant cell walls are cellulose and hemicellulose (pectins, xylans, mannans, xyloglucans, etc.), whereas fungal cell walls primarily consist of chitin (Niklas, 2004). In metazoa, the ECM will predominantly consist of chondroitin or dermatan, which are essentially made of sulfated polysaccharides referred to as glycosaminoglycans (GAGs; Sugahara and Kitagawa, 2002) that are interconnected by fibrillar proteins (collagens). Other sources of carbohydrates in animals are glycosylation sites, such as mucin that contain a high proportion of sialic acid in addition to GAGs (Raman et al., 2005).
Reflecting this chemical diversity of the substrate, glycosidases, the enzymes responsible for the breakdown of di-, oligo-, and polysaccharides, as well as glycoconjugates, are ubiquitous through all domains of life (Turnbaugh et al., 2010). Carbohydrate processing enzymes (CAZymes), including glycosidases and glycosyltransferases (the enzymes which transfer saccharides to other saccharide moieties, small molecules, lipids, or proteins), constitute between 1 and 3% of the genome of most organisms (Davies et al., 2005). Noteworthy, the genomes of Bacteroidetes species have revealed that they are champions with respect to the diversity and number of CAZymes they contain, reflecting the molecular strategies evolved by this microbial community to differentiate, capture, and degrade complex glycans. Consequently and as a result of this ability to degrade host and plant glycans, cultured (environmental or gut) species are often used to isolate specific enzymes for polysaccharide degradation (Berg et al., 1980; Tierny et al., 1994; Bernardet and Nakagawa, 2006; Reichenbach, 2006).
The first sequenced genome of a Bacteroidetes representative was published in 2002 for the human symbiont B. thetaiotaomicron (Xu et al., 2003). Since then, many sequencing projects have been conducted to increase the genomic knowledge on this phylum. To date, 33 Bacteroidetes genomes are complete, publicly available and published (Table 2). Many others are in a draft state or have not yet been published (total of 125 sequences censed on NCBI). During the last 10 years, sequencing efforts have indiscriminately concerned environmental, pathogen, and symbiotic/commensal species with the aim to better understand their biological -functions, including their capacity to interact with their habitats. This data allows comparing the different enzymatic capabilities of various genera, which sheds new light on the specialization of Bacteroidetes toward degradation of organic matter. A striking common feature revealed by this comparative genomic approach is the trend of Bacteroidetes genomes to encode many polymer-degrading enzymes, acting either on proteins or carbohydrates. The census of Carbohydrate-Active enzymes (CAZ Ymes) in the CAZY database1 (Cantarel et al., 2009) eases the comparison of the number of glycosylhydrolases (GH) and polysaccharide lyases (PL) in an increasing number of sequenced species. In each of the four classes of the Bacteroidetes phylum, there are examples of CAZYme-enriched species. In Bacteroidia, the proteomes of B. thetaiotaomicron, B. fragilis, and P. ruminicola comprise 272, 137, and 130 GH and PL respectively (Xu et al., 2003; Kuwahara et al., 2004), much more than other members of the gut microbiota, or outside the Bacteroidetes phylum, such as Clostridium perfringens (57 GH and PL) and Bifidobacterium longum (49 GH). The same is true for Flavobacteria [e.g., G. forsetii with 48 GH/PL (Bauer et al., 2006), Z. profunda with 120 GH (Qin et al., 2010), F. johnsoniae with 152 GH/PL (McBride et al., 2009)], Cytophagia [e.g., S. linguale with 151 GH/PL (Lail et al., 2010), D. fermentans with 98 GH/PL (Lang et al., 2009), and Sphingobacteria, e.g., C. pinensis with 184 and P. heparinus with 163 GH/PL, respectively (Han et al., 2009; Del Rio et al., 2010)]. In some cases, the prediction of CAZYme-encoding genes in newly sequenced organisms can even unveil unexpected catabolic capabilities toward specific substrates (McBride et al., 2009). Moreover, the predicted enzymatic battery of a bacterial species will help characterize its natural habitat (i.e., the available substrates) and its ecological function in organic matter recycling. Recently, this feature has been used for genome and habitat comparison, linking the number and occurrence of specific CAZyme-families to the environmental niche (Pope et al., 2010; Purushe et al., 2010). With this respect, the presence of a vast majority of exo-acting enzymes in the genome of B. thetaiotaomicron, suggests that the organism is able to use the saccharide decorations appended to the backbone of structural polysaccharides and glycoproteins (Xu et al., 2003). Another recent study elegantly demonstrates that this particularly evident expansion in exo-GHs of family GH92 enzymes (23 members) is indeed related to the α-mannosides present in the N-glycans of host and dietary glycoproteins (Zhu et al., 2010).
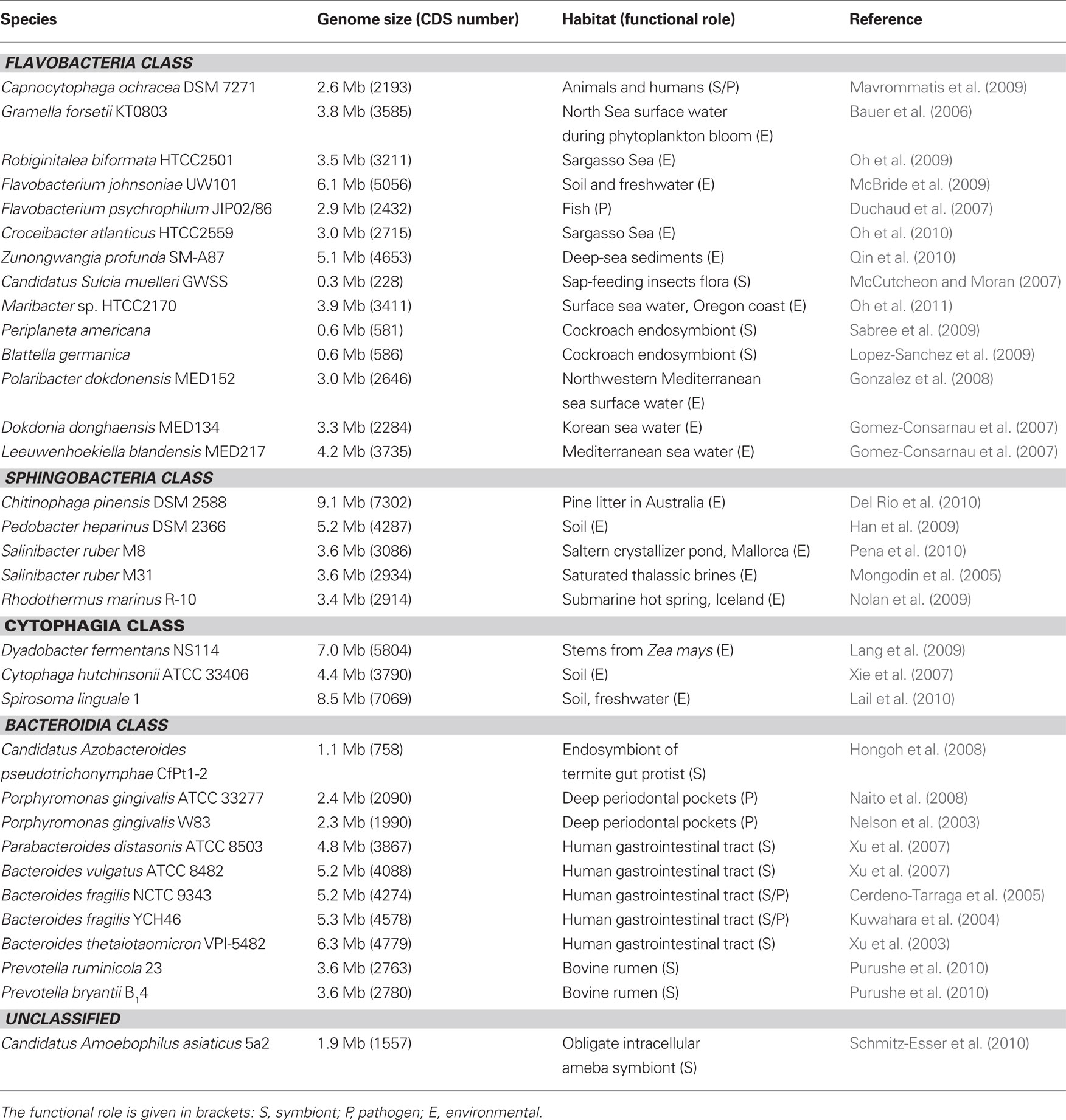
Table 2. List of Bacteroidetes with a fully sequenced and published genome, and their respective habitats.
However, one has to keep in mind that a rough analysis of the number of degradation enzymes in general is insufficient; one needs to go down to the enzymatic sub-family to infer a putative metabolism. Indeed, the CAZyme classification based on sequence -similarity1 has the consequence that gene families group together enzymes with widely different substrate or product specificities (Henrissat, 1991). Therefore, to derive knowledge useful for subsequent functional predictions, phylogenetic analyses defining subgroups that contain biochemically characterized representatives are needed to perform unambiguous assignments (Turnbaugh et al., 2010).
In spite of the general trend of Bacteroidetes to possess numerous degradation enzymes, there are several noteworthy exceptions. The fish pathogen F. psychrophilum genome harbors only 13 proteases, i.e., 4.5 proteases per megabase (Mb) and 3 GH per Mb (Duchaud et al., 2007). This is far less than its closely related soil-associated cousin F. johnsoniae, which possesses 20.5 proteases and 23 GH per Mb and has a genome more than twice as large. The reduced number of degradation enzymes in the fish pathogen can be explained by its dedication to the infection of animal tissues. For such a species, the relative restricted diversity of substrates (compared to a soil-associated species) would diminish the need of multiple families of hydrolases. A second, similar example is P. gingivalis that has a relatively lower number of hydrolases (only 24 GHs) compared to other members of the Bacteroidia class (Nelson et al., 2003). Again, the most plausible explanation is the high speciation of this species to dental plaque degradation that results in the restricted diversity of substrates utilized by this bacterium.
Some marine representatives of the Flavobacteria class seem to alternate between two life strategies depending on the abundance of carbon sources. This is notably the case of Polaribacter dokdonensis (strain MED152), Leeuwenhoekiella blandensis (strain MED217T), and Dokdonia donghaensis (strain MED134). On the one hand, commonly with other Bacteroidetes, they are very well equipped to attach to surfaces and depolymerize organic matter. For example, P. dokdonensis and D. donghaensis genomes encode many enzymes to degrade proteins (93 and 120 peptidases, respectively) and polysaccharides (30 and 22 GH, respectively; Gonzalez et al., 2008; Kirchman, 2008; Woyke et al., 2009). On the other hand, when polymeric substrates become scarce, these species switch to a free-living lifestyle adapted to carbon-poor environments. Genome analysis showed the presence of proteorhodopsin, a light-dependent H+ pump that can drive ATP synthesis (Beja et al., 2000). This protein allows the phototrophic production of sufficient energy to maintain the population growth when the concentration of organic carbon decreases (Gomez-Consarnau et al., 2007). Additionally, P. dokdonensis is enriched in enzymes involved in anaplerotic reactions, and assimilates CO2 faster in light conditions than in the dark (Gonzalez et al., 2008). Altogether, these results suggest that marine Bacteroidetes may cope as well with feast and famine, and complement the understanding of their role in carbon cycles (DeLong and Beja, 2010).
Beyond the prediction of numerous polysaccharide-degrading enzymes, two large paralogous families of proteins have been found in Bacteroidetes genomes, which likely participate in polysaccharide uptake. These include homologs of the outer membrane proteins SusC and SusD from B. thetaiotaomicron, which are involved in starch utilization. Seminal work of the Salyers’ group showed that this human gut symbiont degrades starch via a dedicated starch utilization system (Sus) with several proteins acting in coordination to sense, bind, and hydrolyze the substrate (Anderson and Salyers, 1989a,b; Shipman et al., 2000). The genes encoding these proteins cluster on the bacterial chromosome into typical polysaccharide utilization loci (PUL).
In B. thetaiotaomicron, the Sus locus comprises eight genes, susRABCDEFG (Martens et al., 2009). SusR is an inner membrane regulatory protein which activates the transcription of the other genes in the presence of maltose or starch (D’Elia and Salyers, 1996). SusC, SusD, SusE, and SusF are outer membrane proteins involved in the binding of the polysaccharide (Shipman et al., 2000). In fact, analyses of mutant strains have shown that SusC and SusD account together for 70% of the starch-binding capabilities of the wild type (Reeves et al., 1997). Surface-bound starch is hydrolyzed by the outer membrane α-amylase SusG, which acts endolytically and releases oligosaccharides larger than maltotriose (Shipman et al., 1999; Martens et al., 2009). These degradation products are then channeled to the periplasm through the TonB-dependent receptor, β-barrel-type SusC, where they are further cleaved by the neopullulanase SusA and α-glucosidase SusB. The atomic structures of several protein members have been resolved, namely the starch-binding SusD (Koropatkin et al., 2008), the α-glucosidase SusB (Kitamura et al., 2008), and the α-amylase SusG (Koropatkin and Smith, 2010). The Sus locus is organized into two transcriptional units under the control of SusR, one containing susA and the other containing susB to susG (D’Elia and Salyers, 1996; Reeves et al., 1997). This allows a co-regulation of the PUL (Anderson and Salyers, 1989b).
The characterization of this first starch-specific PUL was -followed by the discovery of numerous PULs in Bacteroidetes. susC-like and susD-like genes are strikingly frequent in Bacteroidetes genomes, often appearing in tandem and as the central units of substrate-specific PULs. B. thetaiotaomicron possesses 107 paralogs of susC, of which 101 are paired to a susD-like gene. 62 of these pairs are part of larger clusters, together with polysaccharide-degrading enzymes (Xu et al., 2003). In addition, some of these PULs comprise enzymes targeting glycan decorations, such as sulfatases or acetyl esterases. Thus, depending on the specificity of the predicted enzymes, one can infer the favorite substrate(s) of a given PUL. In total, PULs represent 18% of the genome of B. thetaiotaomicron (Martens et al., 2008). The closely related B. fragilis, B. vulgatus, and Parabacteroides distasonis also possess numerous PULs (Kuwahara et al., 2004; Xu et al., 2007). These PULs likely favor the success of Bacteroides spp. in the uptake of dietary and host-derived polysaccharides in the highly competitive gut habitat, and may explain their evolution as symbionts.
Interestingly, environmental species also harbor plenty of specific PULs. The annotation of the F. johnsoniae genome revealed 42 pairs of susC-like and susD-like genes, among which many were associated with CAZymes (McBride et al., 2009). The authors were notably able to predict PULs targeting starch (homologous the B. thetaiotaomicron Sus locus), chitin, and hemicelluloses. The genome of the marine Flavobacteria G. forsetii encodes 40 paralogs of SusC, and 14 clusters of susCD-like genes were detected (Bauer et al., 2006), often in the vicinity of CAZyme genes. This suggests that environmental Bacteroidetes as well as their gut-associated cousins use a unique and similar strategy to bind and degrade polymeric organic matter. Indeed, SusD homologs are only found in Bacteroidetes representatives. Thus, we speculate that the appearance of PULs including a susD-like gene in the ancestral Bacteroidetes could have at least partly driven the emergence of the phylum, and allowed its evolution as a group specialized in carbohydrate degradation.
The size of Bacteroidetes genomes varies considerably between species (Table 2). Among published sequencing projects, Chitinophaga pinensis has the largest genome (9.1 Mb) whereas Candidatus Sulcia muelleri has the smallest (0.3 Mb). This great discrepancy can be at least partly associated with the different ecological niches colonized and the biological functions played by Bacteroidetes (Figure 1). Obligate intracellular symbionts have a more reduced genome size, due to their peculiar lifestyle. These species have evolved through successive inactivation and loss of genes, affecting virtually every cellular process. The possible causes of this genome reduction are multiple, including the unusual stability and metabolic richness of the cytoplasmic compartment they inhabit (McCutcheon and Moran, 2010). Pathogenic Bacteroidetes, such as P. gingivalis and F. psychrophilum, have a small genome typically around 2 Mb. This can be linked to the dedication of their metabolic capabilities toward the infection of specific sites. Living in complex habitats and metabolizing a lot of different substrates, environmental, and intestinal species tend to have larger genomes (Figure 1), -correlating with their broader catabolic capabilities. This is also the case of the opportunistic pathogen B. fragilis, which is part of the -normal human gut microbiota but can cause infections at many other sites. To date, marine representatives harbor smaller genomes than their intestinal or terrestrial counterparts, but this will certainly progress as the number of complete genome sequences increases. For example, our group has recently annotated the genome of the marine Flavobacteria Zobellia galactanivorans. It comprises 5.5 Mb and encodes 4738 proteins, representing one of the largest genomes for a marine Bacteroidetes (Barbeyron et al., unpublished data). Interestingly, the proteorhodopsin-containing P. dokdonensis and D. donghaensis possess the smallest proteome among environmental species (2646 and 2284 proteins, respectively). Thus, the genome size fits the ecological niche. Big genomes increase the metabolic capacities, and hence broaden the spectrum of potential substrates for bacteria living in complex environments. In more stable habitats, bacteria tend to specialize toward specific functions and harbor smaller genomes. This raises the question of the nature of the ancestral Bacteroidetes genome. Indeed, the present variations in genome size could be due either to massive loss of genes from a large ancestral genome, or to successive acquisitions completing a small genome with genes representing a selective advantage.
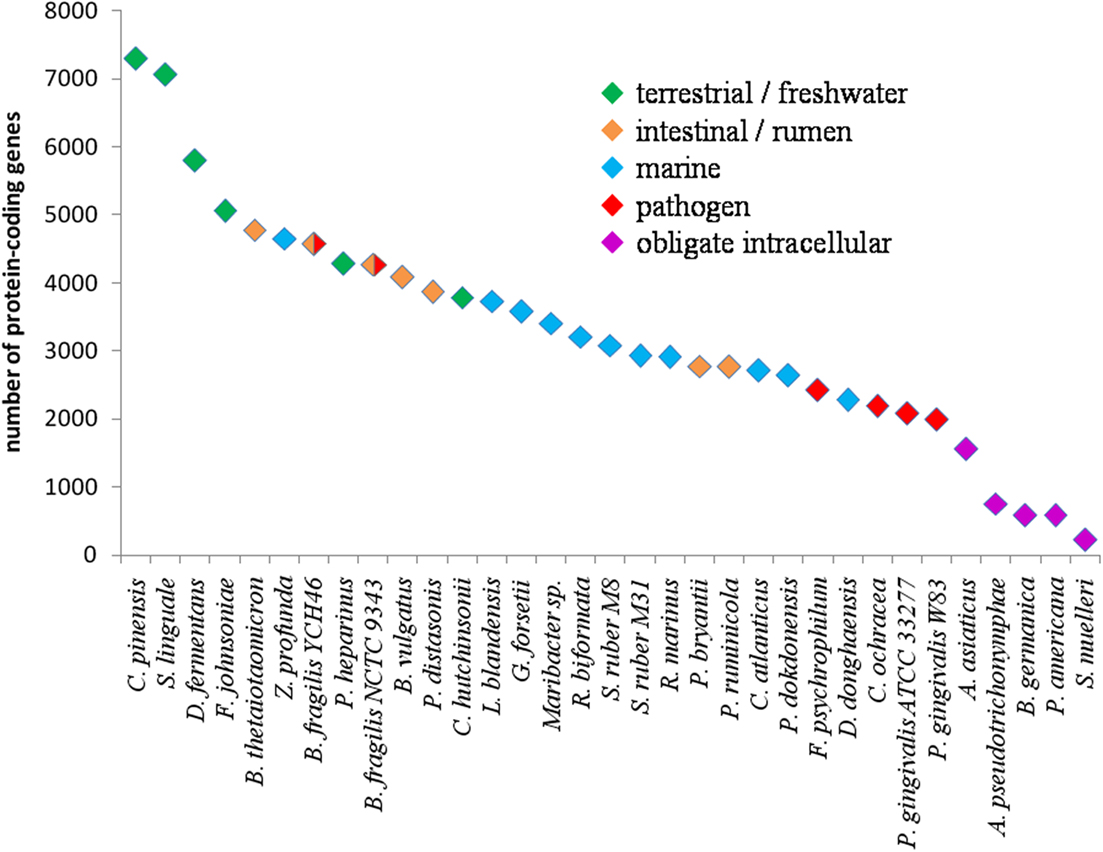
Figure 1. Schematic diagram representing the total number of coded proteins as a function of genome size of Bacteroidetes species. Only complete and published genomes of the Bacteroidetes phylum have been included (see also Table 2).
In this respect, the sequencing era unraveled the plasticity of Bacteroidetes genomes, which evolved, and probably still evolve, through dynamic processes. The outcome of this plasticity reflects in the rapid deterioration of the global synteny between evolutionary-related species living in the same environment, as revealed for gut and rumen Bacteroidetes (Xu et al., 2007; Purushe et al., 2010). Their evolution is driven by highly frequent genetic rearrangements, gene duplications, and lateral gene transfers (LGT) between species. Genome analysis of two B. fragilis strains revealed extensive DNA inversions affecting the expression patterns of several genes (Kuwahara et al., 2004; Cerdeno-Tarraga et al., 2005). These events control the antigenic composition of bacterial surface structures and likely help B. fragilis evading the immune system and colonize novel sites. Notably, the expression of 20 SusC-like proteins, most of them coupled with SusD homologs, is regulated through DNA inversions (Kuwahara et al., 2004). This may participate in the cell adaptation to degrade specific polysaccharides found at the infection sites. Bacteroidetes evolution is also characterized by frequent gene duplications and further divergence in sequence and function, leading to considerably expanded paralogous groups. Notably, GH and SusC/SusD-like proteins are amongst the largest paralogous families in several sequenced Bacteroidetes (Xu et al., 2003, 2007; Bauer et al., 2006). In addition to this intra-strain plasticity, Bacteroidetes genomes evolve through inter-species exchange of genetic material (Thomas and Nielsen, 2005). Using a phylogenetic approach, Xu et al. (2007) showed that around 5.5% of the genes in gut Bacteroidetes genomes were laterally acquired from non gut-associated bacteria, among which glycosyltransferases (GT) where significantly over-represented. These LGT events could partly explain the niche specialization of different species. The authors suggest that acquisition of new genes from outside the gut brought novel metabolic pathways to intestinal Bacteroidetes and broaden the spectrum of digestible substrates. Furthermore, it has been shown that the convergence of GT and GH repertoires in gut Bacteroidetes sharing the same habitat is largely due to massive LGT rather than gene duplications (Lozupone et al., 2008). Conjugative LGT events are also demonstrated to be responsible for antibiotic resistance spreading in natural communities of gut Bacteroides (Shoemaker et al., 2001). The exchange of genetic material is not necessarily restricted to closely related species and can overcome phylogenetic barriers. Indeed, glyceraldehyde-3-phosphate dehydrogenase genes have been horizontally transferred from a β-proteobacteria to a Bacteroidetes (Figge et al., 1999). Another example is the transfer of genes between an Archeae and the hyperhalophilic Sphingobacteria S. ruber (Mongodin et al., 2005).
Taken together, recent analyses of Bacteroidetes genome sequences have shown that: (i) there is a gradation in the size of the genomes correlated with the functional specialization; (ii) genomes can undergo massive reorganizations; (iii) highly frequent LGT events allow spreading of novel metabolic capabilities inside Bacteroidetes populations. As already mentioned, intestinal Bacteroidetes are specialized in the degradation of plant-derived polymers, a feature shared with environmental relatives. In the last part of this review, we will discuss the potential connections between these two communities that are not prima facie obviously interacting.
Several studies have shown that the diet strongly influences the intestinal microbiota. Early research focused on the comparison of fecal microbes retrieved from individuals with different nutritional habits. Benno et al. (1986) showed significant variations in the cultivable microbiota of rural Japanese and urban Canadians. They proposed that this discrepancy relates to the contrasted diet of the two populations. Similarly, in a rat model transplanted with human microbiota, the consumption of resistant starch changed the bacterial composition compared to a sucrose diet (Silvi et al., 1999). Yet, the use of cultivable bacterial counts is a limited method (Amann et al., 1995) and no statistical difference was found when comparing the fecal microbiota of strictly vegetarians and individuals consuming a general diet (Goldberg et al., 1977). The development of culture-independent techniques to assess bacterial abundance and diversity helped testing the influence of diet on the GIT microbiota. In a mouse model reproducing the human intestinal microbiome, the bacterial community composition and the representation of metabolic pathways was strongly dependant on the nature of the diet (Turnbaugh et al., 2009). The proportion of Bacteroidetes representatives decreased drastically when animals were switched from a chow containing low levels of fat and high level of plant polysaccharides to a Western diet (high fat, high sugar). Feeding on the diet rich in plant polysaccharides resulted in an enriched set of pathways including N-glycan, glycosaminoglycan, and starch degradation that are typical for Bacteroidetes (Turnbaugh et al., 2009). An independent study on a murine model showed that a high-fat diet was associated with a decrease in more than 30 lineages within the Bacteroidetes phylum, including in the Bacteroidaceae, Prevotellaceae, and Rikenellaceae families (Hildebrandt et al., 2009). Recent studies have investigated the diet impact on the human gut microbiota. The consumption of chemically modified resistant starch (RS4) instead of normal, digestible starch led to a shift in the bacterial community (Martinez et al., 2010). Even if results varied substantially between the 10 considered subjects, RS4 consumption was notably followed by enrichment in Bacteroidetes, among which Parabacteroides distasonis increased sevenfold. The observed changes were completely reversible within 1 week, demonstrating the high population dynamics (Martinez et al., 2010). In a recent comparison of the fecal microbiota of children from Burkina Faso, and Italy, De Filippo et al. (2010) showed a significant difference in the community composition. African children showed a higher proportion of Bacteroidetes (57 vs. 22%) and a lower proportion of Firmicutes (27 vs. 63%) than Europeans. The authors explained this difference by the higher dietary fiber content of the rural African food, mainly composed of cereals, legumes, and vegetables, which would favor the development of the polysaccharide-degrading Bacteroidetes. Interestingly, the genera Prevotella, Xylanibacter, Cytophaga, and Paludibacter were found exclusively in African microbiota. This is probably due to their increased fitness to grow on polysaccharides abundant in the Burkina Faso diet, such as xylan or cellulose (De Filippo et al., 2010). The control of gut microbiome composition by the diet quality likely denotes a selection of the population that optimally degrades the available substrates. In the case of Bacteroidetes, the selection criteria would primarily be based on the ability to digest complex polymers. Some species may have acquired specific catabolic pathways that others lack. This hypothesis has been recently tested in a mouse model. Germ-free mice were inoculated with two Bacteroides thetaiotaomicron and B. caccae strains of which the latter one can grow with inulin as carbon source. When the mice where fed with an inulin rich diet the ratio of the two species changed toward B. caccae. The ability to use inulin was associated with a GH32 absent in B. -thetaiotaomicron. This clearly showed that diet selects species composition in the animal intestine (Sonnenburg et al., 2010).
The question arises if different human populations with different diets contain specific food adaptations on the genetic level of their gut microbes. Recently, our group has shown that gut Bacteroidetes were able to get gene updates from environmental species to acquire novel functions (Hehemann et al., 2010; Rebuffet et al., 2011). Indeed, in the marine flavobacterium Zobellia galactanivorans, we have discovered and characterized the first porphyranases (Hehemann et al., 2010) as well as a 1,3-α-3,6-anhydro-L-galactosidase (Rebuffet et al., 2011). These enzymes are used by marine bacteria to degrade agarocolloïds, sulfated galactans only found in the cell walls of red algae, such as Porphyra or Gracilaria. When using these new sequences as lead sequences to probe publicly available databases, homologs were identified not only in other marine bacteria, but surprisingly also in the human gut isolate Bacteroides plebeius. The genome of B. -plebeius (DSM 17135) contains a porphyran/agar degradation locus, transferred from an ancestral marine Bacteroidetes (Hehemann et al., 2010; Rebuffet et al., 2011). This PUL was identified as a result of biochemical and structural characterization of the first two porphyranases belonging to the family GH16 (PorA and PorB; Hehemann et al., 2010) and the 1,3-α-3,6-anhydro-L-galactosidase AhgA belonging to the family GH117 (Rebuffet et al., 2011). The PUL contains sequences coding for two putative β-agarases (GH86), one β-agarase (GH16), two β-galactosidases (GH2), a sulfatase, a carbohydrate-binding module, and a susD-like gene associated with its TonB-dependant receptor (Figure 2). Altogether these enzymes form a complete system of detection and degradation for porphyran and agar, which provides B. plebeius with the set of utensils to use these polysaccharides as carbon source.
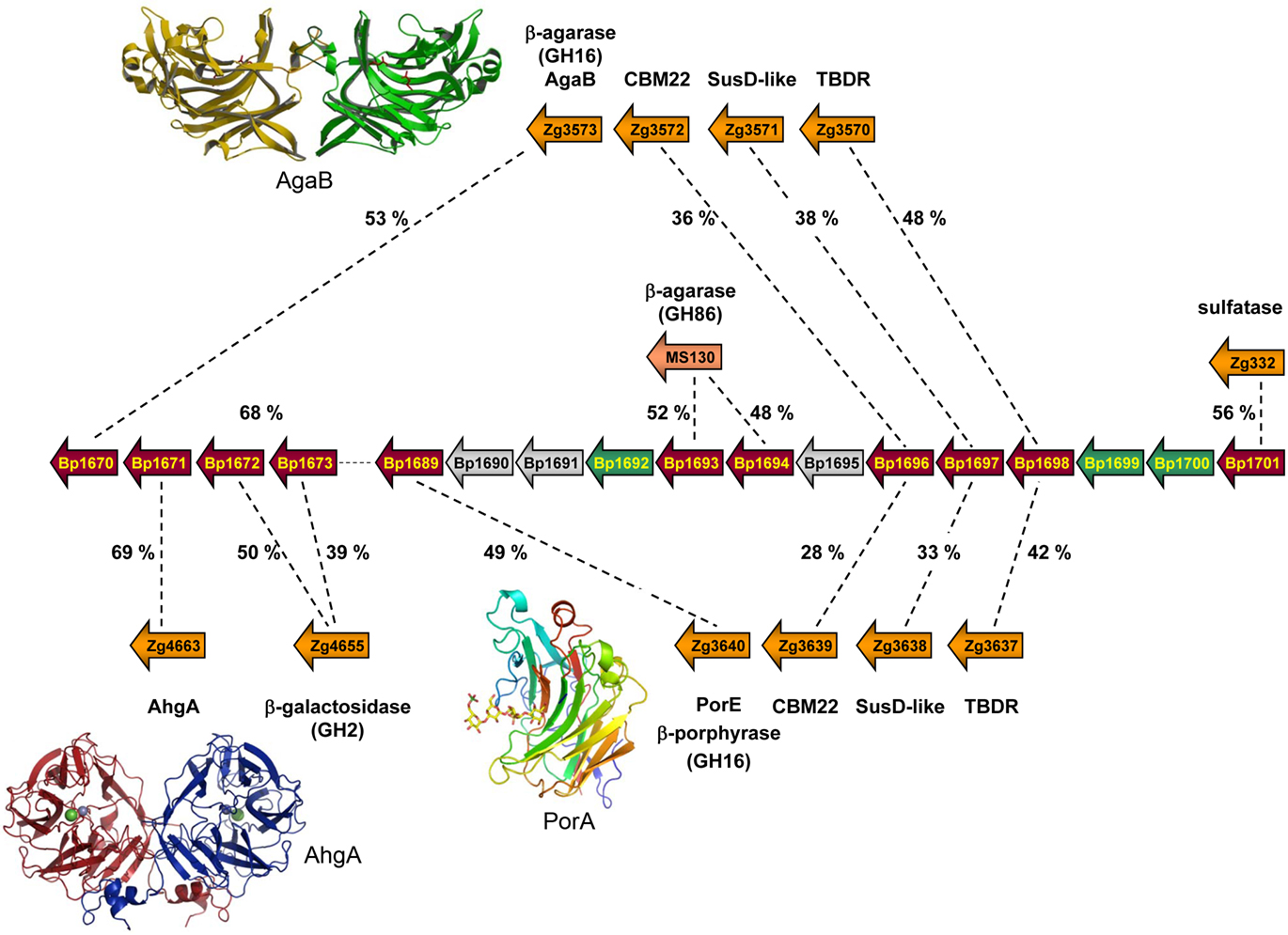
Figure 2. Schematic representation of a PUL region, present in the genome of Bacteroides plebeius and first identified as interesting because of the presence of a porphyranase gene (Bp1689; Hehemann et al., 2010). Besides the conserved Sus-like genes, the locus also contains carbohydrate-related genes which share highest identity with proteins used for red algal galactan degradation in two marine Bacteroides. Shown are the sequence identities between B. plebeius and one Microscilla sp. PRE1 protein, as well as with several Zobellia galactanivorans proteins. Six of these genes (Bp1670, Bp1671, Bp1689, Bp1693, Bp1694, and Bp1696) are conserved only with marine bacteria, and are absent in genomes of other gut Bacteroides. The crystal structures of marine homologous enzymes, coded by three of these genes (namely Bp1670, Bp1671, and Bp1689) have recently been determined and are illustrated as ribbon representations (PDB codes: AgaB – 1O4Z; AghA – 3P2N; and PorA – 3ILF). The 3D structures help determine the crucial residues for activity and substrate specificity that are all verified and present in the sequences of the B. plebeius proteins.
Metagenomic data revealed that porphyranases and 1,3-α-3,6-anhydro-L-galactosidases are absent in North American and Danish population but present in Spanish and Japanese populations with proportions of 10 and 38% respectively (Hehemann et al., 2010; Rebuffet et al., 2011). The biological rationale of “marine” enzymes in gut microbes could be linked to the high input of sea-derived products in the diet of these two populations. Indeed, the Japanese consume about 14.2 g seaweed per day and person (Fukuda et al., 2007), and the most popular seaweed is Nori (Porphyra spp.) used to make maki-sushi (Nisizawa et al., 1987). Similarly, Spain is the second largest consuming nation of seafood in the world (Manrique and Jensen, 2001). In both populations, contact between human-associated microbes and non-sterile seaweed or seafood could have created a favorable condition for a LGT from marine bacteria to human gut Bacteroidetes. Noteworthy, Nori is the only food that contains porphyran, which allowed associating the transfer of these genes to one special food source. This first evidence of a life style-associated adaptation of the genetic repertoire of the human gut microbiome could be detected due to the unique signature of seaweed degrading CAZymes. Therefore marine CAZymes may be particularly good probes to reveal such adaptations in the human gut microbiome.
These examples of LGT show that gut bacteria are able to acquire new functions via transfer of a complete degradation pathway from food-associated environmental bacteria. Interestingly, the Firmicutes Epulopiscium sp., which lives in the gut of grazer surgeonfish (Clements and Bullivant, 1991), also possesses a putative agar degradation locus acquired from marine bacteria (Rebuffet et al., 2011). One can therefore extrapolate that similar events occurred in omnivorous and herbivorous animals. During the course of evolution, the metabolic repertoire of their gut microbes could have been influenced by contacts with food-associated environmental bacteria. Thus, acquisition of selectively advantageous genes by successive LGT events could explain how gut symbionts acquired CAZymes involved in green plant polysaccharide degradation.
In conclusion, some crucial criteria seem to have particularly favored the adaptation of Bacteroidetes to such contrasting environments, rendering their distribution/dominance close to ubiquitous. Among these, we note the presence of specific Sus-like PULs that are dedicated to polysaccharide degradation (Xu et al., 2003; Flint et al., 2008) and their extreme genome flexibility (Xu et al., 2007) that assures the capacity to gain new functions by LGT from species living in different habitats. Consequently, the recently revealed LGT linking environmental and gut Bacteroidetes (Hehemann et al., 2010) raises the question as to how the ancestral members of the intestinal microbiota might have evolved from plant-degrading species, settled in the GIT of early herbivores, such as marine protists. Evidently, gut symbionts have co-evolved with their hosts (De Filippo et al., 2010), notably in response to the consumption of specific food, and this interdependence likely shaped and still shapes the lifestyle of human populations (Ley et al., 2006a). This “food connection” points toward the fact that recurrent contacts between environmental and gut microbes can have beneficial effects (Rook and Brunet, 2005). At the same time, it underlines the potential problem of our modern lifestyle and the consumption of hyper-hygienic, extensively processed food for human health, depriving us of the environmental reservoirs of microbial genes that allow adaptation by lateral transfer (De Filippo et al., 2010; Sonnenburg, 2010). On the other hand, global travel and trade are providing contact to new types of food and to diverse populations, perhaps giving access to new microbes harboring novel genes destined for integration into our microbiome.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abrams, G. D. (1983). “Impact of the intestinal microflora on intestinal structure and function,” in Human Intestinal Microflora in Health and Disease, ed. D. J. Hentges (New York, NY: Academic Press), 292–310.
Alldredge, A. L., Cole, J. J., and Caron, D. A. (1986). Production of heterotrophic bacteria inhabiting macroscopic organic aggregates (marine snow) from surface waters. Limnol. Oceanogr. 31, 68–78.
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169.
Anderson, K. L., and Salyers, A. A. (1989a). Biochemical evidence that starch breakdown by Bacteroides thetaiotaomicron involves outer membrane starch-binding sites and periplasmic starch-degrading enzymes. J. Bacteriol. 171, 3192–3198.
Anderson, K. L., and Salyers, A. A. (1989b). Genetic evidence that outer membrane binding of starch is required for starch utilization by Bacteroides thetaiotaomicron. J. Bacteriol. 171, 3199–3204.
Andersson, A. F., Lindberg, M., Jakobsson, H., Backhed, F., Nyren, P., and Engstrand, L. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3, e2836. doi: 10.1371/journal.pone.0002836
Austin, B., and Austin, D. A. (2007). Bacterial Fish Pathogens: Disease of Farmed and Wild Fish. Edinburgh: Springer Verlag.
Azam, F., and Malfatti, F. (2007). Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 5, 782–791.
Backhed, F., Ley, R. E., Sonnenburg, J. L., Peterson, D. A., and Gordon, J. I. (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915–1920.
Balakirev, E. S., Pavlyuchkov, V. A., and Ayala, F. J. (2008). DNA variation and symbiotic associations in phenotypically diverse sea urchin Strongylocentrotus intermedius. Proc. Natl. Acad. Sci. U.S.A. 105, 16218–16223.
Barbeyron, T., L’haridon, S., Corre, E., Kloareg, B., and Potin, P. (2001). Zobellia galactanovorans gen. nov., sp. nov., a marine species of Flavobacteriaceae isolated from a red alga, and classification of. Int. J. Syst. Evol. Microbiol. 51, 985–997.
Barbeyron, T., L’haridon, S., Michel, G., and Czjzek, M. (2008). Mariniflexile fucanivorans sp. nov., a marine member of the Flavobacteriaceae that degrades sulphated fucans from brown algae. Int. J. Syst. Evol. Microbiol. 58, 2107–2113.
Bauer, M., Kube, M., Teeling, H., Richter, M., Lombardot, T., Allers, E., Wurdemann, C. A., Quast, C., Kuhl, H., Knaust, F., Woebken, D., Bischof, K., Mussmann, M., Choudhuri, J. V., Meyer, F., Reinhardt, R., Amann, R. I., and Glockner, F. O. (2006). Whole genome analysis of the marine Bacteroidetes “Gramella forsetii ” reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 8, 2201–2213.
Beja, O., Aravind, L., Koonin, E. V., Suzuki, M. T., Hadd, A., Nguyen, L. P., Jovanovich, S. B., Gates, C. M., Feldman, R. A., Spudich, J. L., Spudich, E. N., and Delong, E. F. (2000). Bacterial rhodopsin: evidence for a new type of phototrophy in the sea. Science 289, 1902–1906.
Beleneva, I. A., and Zhukova, N. V. (2006). Bacterial communities of brown and red algae from Peter the Great Bay, the Sea of Japan. Mikrobiologiia 75, 410–419.
Benno, Y., Suzuki, K., Narisawa, K., Bruce, W. R., and Mitsuoka, T. (1986). Comparison of the fecal microflora in rural Japanese and urban Canadians. Microbiol. Immunol. 30, 521–532.
Berg, J. O., Lindqvist, L., and Nord, C. E. (1980). Purification of glycoside hydrolases from Bacteroides fragilis. Appl. Environ. Microbiol. 40, 40–47.
Bernardet, J.-F., and Nakagawa, Y. (2006). An introduction to the family of Flavobacteriaceae. Prokaryotes 7, 455–480.
Bik, E. M., Eckburg, P. B., Gill, S. R., Nelson, K. E., Purdom, E. A., Francois, F., Perez-Perez, G., Blaser, M. J., and Relman, D. A. (2006). Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. U.S.A. 103, 732–737.
Borneman, J., Skroch, P. W., O’sullivan, K. M., Palus, J. A., Rumjanek, N. G., Jansen, J. L., Nienhuis, J., and Triplett, E. W. (1996). Molecular microbial diversity of an agricultural soil in Wisconsin. Appl. Environ. Microbiol. 62, 1935–1943.
Buckley, D. H., and Schmidt, T. M. (2003). Diversity and dynamics of microbial communities in soils from agro-ecosystems. Environ. Microbiol. 5, 441–452.
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238.
Carpita, N. C., and Gibeaut, D. M. (1993). Structural models of primary cell walls in flowering plants: consistency of molecular structure with the physical properties of the walls during growth. Plant J. 3, 1–30.
Carroll, E. J., and Hungate, R. E. (1954). The magnitude of the microbial fermentation in the bovine rumen. Appl. Microbiol. 2, 205–214.
Cerdeno-Tarraga, A. M., Patrick, S., Crossman, L. C., Blakely, G., Abratt, V., Lennard, N., Poxton, I., Duerden, B., Harris, B., Quail, M. A., Barron, A., Clark, L., Corton, C., Doggett, J., Holden, M. T., Larke, N., Line, A., Lord, A., Norbertczak, H., Ormond, D., Price, C., Rabbinowitsch, E., Woodward, J., Barrell, B., and Parkhill, J. (2005). Extensive DNA inversions in the B. fragilis genome control variable gene expression. Science 307, 1463–1465.
Chin, W. C., Orellana, M. V., and Verdugo, P. (1998). Spontaneous assembly of marine dissolved organic matter into polymer gels. Nature 391, 568–572.
Church, M. J. (2008). “Resource control of bacterial dynamics in the sea,” in Microbial Ecology of the Oceans, 2nd Edn, ed. D. L. Kirchman (Hoboken, NJ: Wiley ’ Sons), 335–382.
Clements, K. D., and Bullivant, S. (1991). An unusual symbiont from the gut of surgeonfishes may be the largest known prokaryote. J. Bacteriol. 173, 5359–5362.
Cottrell, M. T., and Kirchman, D. L. (2000). Community composition of marine bacterioplankton determined by 16S rRNA gene clone libraries and fluorescence in situ hybridization. Appl. Environ. Microbiol. 66, 5116–5122.
Crump, B. C., Armbrust, E. V., and Baross, J. A. (1999). Phylogenetic analysis of particle-attached and free-living bacterial communities in the Columbia river, its estuary, and the adjacent coastal ocean. Appl. Environ. Microbiol. 65, 3192–3204.
Crump, B. C., and Koch, E. W. (2008). Attached bacterial populations shared by four species of aquatic angiosperms. Appl. Environ. Microbiol. 74, 5948–5957.
Davies, G. J., Gloster, T. M., and Henrissat, B. (2005). Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr. Opin. Struct. Biol. 15, 637–645.
Davis, H. S. (1922). “A new bacterial disease of fresh-water fishes,” in Bulletin of the U.S. Bureau of Fisheries, Washington, DC, 261–280.
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., Collini, S., Pieraccini, G., and Lionetti, P. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696.
D’Elia, J. N., and Salyers, A. A. (1996). Effect of regulatory protein levels on utilization of starch by Bacteroides thetaiotaomicron. J. Bacteriol. 178, 7180–7186.
Del Rio, T. G., Abt, B., Spring, S., Lapidus, A., Nolan, M., Tice, H., Copeland, A., Cheng, J.-F., Chen, F., Hugenholtz, P., Kyrpides, N. C., Klenk, H.-P., and Lucas, S. (2010). Complete genome sequence of Chitinophaga pinensis type strain (UQM 2034T). Stand. Genomic Sci. 2, 87–95.
DeLong, E. F., and Beja, O. (2010). The light-driven proton pump proteorhodopsin enhances bacterial survival during tough times. PLoS Biol. 8, e1000359. doi: 10.1371/journal.pbio.1000359
DeLong, E. F., Franks, D. G., and Alldredge, A. L. (1993). Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol. Oceanogr. 38, 924–934.
Dubos, R., Schaedler, R.W., Costello, R., and Hoet, P. (1965). Indigenous, normal, and autochthonous flora of the gastrointestinal tract. J. Exp. Med. 122, 67.
Duchaud, E., Boussaha, M., Loux, V., Bernardet, J. F., Michel, C., Kerouault, B., Mondot, S., Nicolas, P., Bossy, R., Caron, C., Bessieres, P., Gibrat, J. F., Claverol, S., Dumetz, F., Le Henaff, M., and Benmansour, A. (2007). Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat. Biotechnol. 25, 763–769.
Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., Gill, S. R., Nelson, K. E., and Relman, D. A. (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638.
Edwards, J. L., Smith, D. L., Connolly, J., Mcdonald, J. E., Cox, M. J., Joint, I., Edwards, C., and Mccarthy, A. J. (2010). Identification of carbohydrate metabolism genes in the metagenome of a marine biofilm community shown to be dominated by Gammaproteobacteria and Bacteroidetes. Genes 1, 371–384.
Fandino, L. B., Riemann, L., Steward, G. F., Long, R. A., and Azam, F. (2001). Variations in bacterial community structure during a dinoflagellate bloom analyzed by DGGE and 16S rDNA sequencing. Aquat. Microb. Ecol. 23, 119–130.
Ferraris, R. P. (2001). Dietary and developmental regulation of intestinal sugar transport. Biochem. J. 360, 265–276.
Figge, R. M., Schubert, M., Brinkmann, H., and Cerff, R. (1999). Glyceraldehyde-3-phosphate dehydrogenase gene diversity in eubacteria and eukaryotes: evidence for intra- and inter-kingdom gene transfer. Mol. Biol. Evol. 16, 429–440.
Fletcher, H. M., Schenkein, H. A., Morgan, R. M., Bailey, K. A., Berry, C. R., and Macrina, F. L. (1995). Virulence of a Porphyromonas gingivalis W83 mutant defective in the prtH gene. Infect. Immun. 63, 1521–1528.
Flint, H. J., Bayer, E. A., Rincon, M. T., Lamed, R., and White, B. A. (2008). Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat. Rev. Microbiol. 6, 121–131.
Frias-Lopez, J., Zerkle, A. L., Bonheyo, G. T., and Fouke, B. W. (2002). Partitioning of bacterial communities between seawater and healthy, black band diseased, and dead coral surfaces. Appl. Environ. Microbiol. 68, 2214–2228.
Fukuda, S., Saito, H., Nakaji, S., Yamada, M., Ebine, N., Tsushima, E., Oka, E., Kumeta, K., Tsukamoto, T., and Tokunaga, S. (2007). Pattern of dietary fiber intake among the Japanese general population. Eur. J. Clin. Nutr. 61, 99–103.
Gihring, T. M., Humphrys, M., Mills, H. J., Huettel, M., and Kostka, J. E. (2009). Identification of phytodetritus-degrading microbial communities in sublittoral Gulf of Mexico sands. Limnol. Oceanogr. 54, 1073–1083.
Glöckner, F. O., Fuchs, B. M., and Amann, R. (1999). Bacterioplankton compositions of lakes and oceans: a first comparison based on fluorescence in situ hybridization. Appl. Environ. Microbiol. 65, 3721–3726.
Goldberg, M. J., Smith, J. W., and Nichols, R. L. (1977). Comparison of the fecal microflora of seventh-day adventists with individuals consuming a general diet. Implications concerning colonic carcinoma. Ann. Surg. 186, 97–100.
Gomez-Consarnau, L., Gonzalez, J. M., Coll-Llado, M., Gourdon, P., Pascher, T., Neutze, R., Pedros-Alio, C., and Pinhassi, J. (2007). Light stimulates growth of proteorhodopsin--containing marine Flavobacteria. Nature 445, 210–213.
Gomez-Pereira, P. R., Fuchs, B. M., Alonso, C., Oliver, M. J., Van Beusekom, J. E., and Amann, R. (2010). Distinct flavobacterial communities in contrasting water masses of the north Atlantic Ocean. ISME J. 4, 472–487.
Gonzalez, J. M., Fernandez-Gomez, B., Fernandez-Guerra, A., Gomez-Consarnau, L., Sanchez, O., Coll-Llado, M., Del Campo, J., Escudero, L., Rodriguez-Martinez, R., Alonso-Saez, L., Latasa, M., Paulsen, I., Nedashkovskaya, O., Lekunberri, I., Pinhassi, J., and Pedros-Alio, C. (2008). Genome analysis of the proteorhodopsin-containing marine bacterium Polaribacter sp. MED152 (Flavobacteria). Proc. Natl. Acad. Sci. U.S.A. 105, 8724–8729.
Gooday, A. J. (2002). Biological responses to seasonally varying fluxes of organic matter to the ocean floor: a review. J. Oceanogr. 58, 305–332.
Graham, L. E., Cook, M. E., and Busse, J. S. (2000). The origin of plants: body plan changes contributing to a major evolutionary radiation. Proc. Natl. Acad. Sci. U.S.A. 97, 4535–4540.
Greiner, D., and Mayrand, D. (1987). Selected characteristics of pathogenic and nonpathogenic strains of Bacteroides gingivalis. J. Clin. Microbiol. 25, 738–740.
Han, C., Spring, S., Lapidus, A., Del Rio, T. G., Tice, H., Copeland, A., Cheng, J.-F., Lucas, S., Chen, F., Nolan, M., Bruce, D., Hugenholtz, P., Kyrpides, N. C., Klenk, H.-P., and Detter, J. C. (2009). Complete genome sequence of Pedobacter heparinus type strain (HIM 762-3T). Stand. Genomic Sci. 1, 54–62.
Hehemann, J. H., Correc, G., Barbeyron, T., Helbert, W., Czjzek, M., and Michel, G. (2010). Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464, 908–912.
Henrissat, B. (1991). A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 280, 309–316.
Hildebrandt, M. A., Hoffmann, C., Sherrill-Mix, S. A., Keilbaugh, S. A., Hamady, M., Chen, Y. Y., Knight, R., Ahima, R. S., Bushman, F., and Wu, G. D. (2009). High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137, 1716–1724, e1711–1712.
Hongoh, Y., Sharma, V. K., Prakash, T., Noda1, S., Toh, H., Taylor, T. D., Kudo1, T., Sakaki, Y., Toyoda, A., Hattori, M., and Ohkuma1, M. (2008). Genome of an endosymbiont coupling N2 fixation to cellulolysis within protist cells in termite gut. Science 322, 1108–1109.
Hooper, L. V., Midtvedt, T., and Gordon, J. I. (2002). How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 22, 283–307.
Hugo, C. J., Bruun, B., and Jooste, P. J. (2006a). The genera Bergeyella and Weeksella. Prokaryotes 7, 532–538.
Hugo, C. J., Bruun, B., and Jooste, P. J. (2006b). The genera Empedobacter and Myroides. Prokaryotes 7, 630–637.
Hugo, C. J., Jooste, P. J., Segers, P., Vancanneyt, M., and Kersters, K. (1999). A polyphasic taxonomic study of Chryseobacterium strains isolated from dairy sources. Syst. Appl. Microbiol. 22, 586–595.
Jenkinson, H. F., and Lamont, R. J. (2005). Oral microbial communities in sickness and in health. Trends Microbiol. 13, 589–595.
Jooste, P. J., and Hugo, C. J. (1999). The taxonomy, ecology and cultivation of bacterial genera belonging to the family Flavobacteriaceae. Int. J. Food Microbiol. 53, 81–94.
Julies, E. M., Fuchs, B. M., Arnosti, C., and Brüchert, V. (2010). Organic carbon degradation in anoxic organic-rich shelf sediments: biogeochemical rates and microbial abundance. Geomicrobiol. J. 27, 303–314.
Keijser, B. J., Zaura, E., Huse, S. M., Van Der Vossen, J. M., Schuren, F. H., Montijn, R. C., Ten Cate, J. M., and Crielaard, W. (2008). Pyrosequencing analysis of the oral microflora of healthy adults. J. Dent. Res. 87, 1016–1020.
Kim, B. Y., Weon, H. Y., Cousin, S., Yoo, S. H., Kwon, S. W., Go, S. J., and Stackebrandt, E. (2006). Flavobacterium daejeonense sp. nov. and Flavobacterium suncheonense sp. nov., isolated from greenhouse soils in Korea. Int. J. Syst. Evol. Microbiol. 56, 1645–1649.
Kim, H.-J., Miller, A. J., Mcgowan, J., and Carter, M. L. (2009). Coastal phytoplankton blooms in the Southern California Bight. Prog. Oceanogr. 82, 137–147.
Kim, K. K., Kim, M. K., Lim, J. H., Park, H. Y., and Lee, S. T. (2005). Transfer of Chryseobacterium meningosepticum and Chryseobacterium miricola to Elizabethkingia gen. nov. as Elizabethkingia meningoseptica comb. nov. and Elizabethkingia miricola comb. nov. Int. J. Syst. Evol. Microbiol. 55, 1287–1293.
Kim, Y. S., and Milner, J. A. (2007). Dietary modulation of colon cancer risk. J. Nutr. 137, 2576–2579.
King, E. O. (1959). Studies on a group of previously unclassified bacteria associated with meningitis in infants. Am. J. Clin. Pathol. 31, 241–247.
Kirchman, D. L. (2002). The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol. Ecol. 39, 91–100.
Kirchman, D. L. (2008). New light on an important microbe in the ocean. Proc. Natl. Acad. Sci. U.S.A. 105, 8487–8488.
Kitamura, M., Okuyama, M., Tanzawa, F., Mori, H., Kitago, Y., Watanabe, N., Kimura, A., Tanaka, I., and Yao, M. (2008). Structural and functional analysis of a glycoside hydrolase family 97 enzyme from Bacteroides thetaiotaomicron. J. Biol. Chem. 283, 36328–36337.
Knapp, B. A., Seeber, J., Rief, A., Meyer, E., and Insam, H. (2010). Bacterial community composition of the gut microbiota of Cylindroiulus fulviceps (diplopoda) as revealed by molecular fingerprinting and cloning. Folia Microbiol. (Praha) 55, 489–496.
Koropatkin, N. M., Martens, E. C., Gordon, J. I., and Smith, T. J. (2008). Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16, 1105–1115.
Koropatkin, N. M., and Smith, T. J. (2010). SusG: a unique cell-membrane-associated alpha-amylase from a prominent human gut symbiont targets complex starch molecules. Structure 18, 200–215.
Kuwahara, T., Yamashita, A., Hirakawa, H., Nakayama, H., Toh, H., Okada, N., Kuhara, S., Hattori, M., Hayashi, T., and Ohnishi, Y. (2004). Genomic analysis of Bacteroides fragilis reveals extensive DNA inversions regulating cell surface adaptation. Proc. Natl. Acad. Sci. U.S.A. 101, 14919–14924.
Lail, K., Sikorski, J., Saunders, E., Lapidus, A., Del Rio, T. G., Copeland, A., Tice, H., Cheng, J.-F., Lucas, S., Kyrpides, N. C., Klenk, H.-P., and Chen, F. (2010). Complete genome sequence of Spirosoma linguale type strain (1T). Stand. Genomic Sci. 2, 176–185.
Laine, R. A. (1994). A calculation of all possible oligosaccharide isomers both branched and linear yields 1.05 × 10(12) structures for a reducing hexasaccharide: the isomer barrier to development of single-method saccharide sequencing or synthesis systems. Glycobiology 4, 759–767.
Lang, E., Lapidus, A., Chertkov, O., Brettin, T., Detter, J. C., Han, C., Copeland, A., Del Rio, T. G., Nolan, M., Chen, F., Hugenholtz, P., Göker, M., Rohde, M., Kyrpides, N. C., and Klenk, H.-P. (2009). Complete genome sequence of Dyadobacter fermentans type strain (NS114T). Stand. Genomic Sci. 1, 133–140.
Largo, D. B., Fukami, K., and Nishijima, T. (1995). Occasional pathogenic bacteria promoting ice-ice disease in the carrageenan-producing red algae Kappaphycus alvarezii and Eucheuma denticulatum (Solieriaceae, Gigartinales, Rhodophyta). J. Appl. Phycol. 7, 545–554.
Lauber, C. L., Hamady, M., Knight, R., and Fierer, N. (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75, 5111–5120.
Lee, M., Woo, S. G., Park, J., and Yoo, S. A. (2010). Dyadobacter soli sp. nov., a starch-degrading bacterium isolated from farm soil. Int. J. Syst. Evol. Microbiol. 60, 2577–2582.
Leng, J., Xie, L., Zhu, R., Yang, S., Gou, X., Li, S., and Mao, H. (2010). Dominant bacterial communities in the rumen of Gayals (Bos frontalis ), Yaks (Bos grunniens ) and Yunnan Yellow Cattle (Bos taurs ) revealed by denaturing gradient gel electrophoresis. Mol. Biol. Rep.PMID: 21140219. [Epub ahead of print].
Leser, T. D., Amenuvor, J. Z., Jensen, T. K., Lindecrona, R. H., Boye, M., and Moller, K. (2002). Culture-independent analysis of gut bacteria: the pig gastrointestinal tract microbiota revisited. Appl. Environ. Microbiol. 68, 673–690.
Ley, R. E., Peterson, D. A., and Gordon, J. I. (2006a). Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848.
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006b). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023.
Liao, C. H., and Wells, J. M. (1986). Properties of Cytophaga johnsonae strains causing spoilage of fresh produce at food markets. Appl. Environ. Microbiol. 52, 1261–1265.
Llobet-Brossa, E., Rossello-Mora, R., and Amann, R. (1998). Microbial community composition of Wadden Sea sediments as revealed by fluorescence in situ hybridization. Appl. Environ. Microbiol. 64, 2691–2696.
Lopez-Sanchez, M. J., Neef, A., Peretó, J., Patiño-Navarrete, R., Pignatelli, M., Latorre, A., and Moya, A. (2009). Evolutionary convergence and nitrogen metabolism in Blattabacterium strain Bge, primary endosymbiont of the cockroach Blattella germanica. PLoS Genet. 5, e1000721.doi: 10.1371/journal.pgen.1000721
Lozupone, C. A., Hamady, M., Cantarel, B. L., Coutinho, P. M., Henrissat, B., Gordon, J. I., and Knight, R. (2008). The convergence of carbohydrate active gene repertoires in human gut microbes. Proc. Natl. Acad. Sci. U.S.A. 105, 15076–15081.
Lu, J., Santo Domingo, J. W., Hill, S., and Edge, T. A. (2009). Microbial diversity and host-specific sequences of Canada goose feces. Appl. Environ. Microbiol. 75, 5919–5926.
Mammeri, H., Bellais, S., and Nordmann, P. (2002). Chromosome-encoded beta-lactamases TUS-1 and MUS-1 from Myroides odoratus and Myroides odoratimimus (formerly Flavobacterium odoratum ), new members of the lineage of molecular subclass B1 metalloenzymes. Antimicrob. Agents Chemother. 46, 3561–3567.
Manrique, J., and Jensen, H. H. (2001). Spanish household demand for seafood. J. Agric. Econ. 52, 23–37.
Marchesi, J. R. (2010). Prokaryotic and eukaryotic diversity of the human gut. Adv. Appl. Microbiol. 72, 43–62.
Martens, E. C., Chiang, H. C., and Gordon, J. I. (2008). Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4, 447–457.
Martens, E. C., Koropatkin, N. M., Smith, T. J., and Gordon, J. I. (2009). Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J. Biol. Chem. 284, 24673–24677.
Martinez, I., Kim, J., Duffy, P. R., Schlegel, V. L., and Walter, J. (2010). Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS ONE 5, e15046. doi: 10.1371/journal.pone.0015046
Martinez-Alonso, M., Escolano, J., Montesinos, E., and Gaju, N. (2010). Diversity of the bacterial community in the surface soil of a pear orchard based on 16S rRNA gene analysis. Int. Microbiol. 13, 123–134.
Matsui, H., Kato, Y., Chikaraishi, T., Moritani, M., Ban-Tokuda, T., and Wakita, M. (2010). Microbial diversity in ostrich ceca as revealed by 16S ribosomal RNA gene clone library and detection of novel Fibrobacter species. Anaerobe 16, 83–93.
Mavrommatis, K., Gronow, S., Saunders, E., Land, M., Lapidus, A., Copeland, A., Del Rio, T. G., Nolan, M., Lucas, S., Chen, F., Tice, H., Cheng, J.-F., Bruce, D., Goodwin, L., Pitluck, S., Pati, A., Ivanova, N., Chen, A., Palaniappan, K., Chain, P., Hauser, L., Chang, Y.-J., Jeffries, C. D., Brettin, T., Detter, J. C., Han, C., Bristow, J., Göker, M., Rohde, M., Eisen, J. A., Markowitz, V., Kyrpides, N. C., Klenk, H.-P., and Hugenholtz, P. (2009). Complete genome sequence of Capnocytophaga ochracea type strain (VPI 2845T). Stand. Genomic Sci. 1, 101–109.
Mazmanian, S. K. (2008). Capsular polysaccharides of symbiotic bacteria modulate immune responses during experimental colitis. J. Pediatr. Gastroenterol. Nutr. 46(Suppl. 1), E11–E12.
Mazmanian, S. K., Round, J. L., and Kasper, D. L. (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625.
McBride, M. J., Xie, G., Martens, E. C., Lapidus, A., Henrissat, B., Rhodes, R. G., Goltsman, E., Wang, W., Xu, J., Hunnicutt, D. W., Staroscik, A. M., Hoover, T. R., Cheng, Y. Q., and Stein, J. L. (2009). Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl. Environ. Microbiol. 75, 6864–6875.
McCutcheon, J. P., and Moran, N. A. (2010). Functional convergence in reduced genomes of bacterial symbionts spanning 200 my of evolution. Genome Biol. Evol. 2, 708–718.
Michel, G., Tonon, T., Scornet, D., Cock, J. M., and Kloareg, B. (2010a). The cell wall polysaccharide metabolism of the brown alga Ectocarpus siliculosus. Insights into the evolution of extracellular matrix polysaccharides in Eukaryotes. New Phytol. 188, 82–97.
Michel, G., Tonon, T., Scornet, D., Cock, J. M., and Kloareg, B. (2010b). Central and storage carbon metabolism of the brown alga Ectocarpus siliculosus : insights into the origin and evolution of storage carbohydrates in Eukaryotes. New Phytol. 188, 67–81.
Middelbos, I. S., Vester Boler, B. M., Qu, A., White, B. A., Swanson, K. S., and Fahey, G. C. Jr. (2010). Phylogenetic characterization of fecal microbial communities of dogs fed diets with or without supplemental dietary fiber using 454 pyrosequencing. PLoS ONE 5, e9768. doi: 10.1371/journal.pone.0009768
Mongodin, E. F., Nelson, K. E., Daugherty, S., Deboy, R. T., Wister, J., Khouri, H., Weidman, J., Walsh, D. A., Papke, R. T., Sanchez Perez, G., Sharma, A. K., Nesbo, C. L., Macleod, D., Bapteste, E., Doolittle, W. F., Charlebois, R. L., Legault, B., and Rodriguez-Valera, F. (2005). The genome of Salinibacter ruber: convergence and gene exchange among hyperhalophilic bacteria and archaea. Proc. Natl. Acad. Sci. U.S.A. 102, 18147–18152.
Moore, W. E., and Holdeman, L. V. (1974). Special problems associated with the isolation and identification of intestinal bacteria in fecal flora studies. Am. J. Clin. Nutr. 27, 1450–1455.
Nagata, T. (2008). “Organic matter-bacteria interactions in seawater,” in Microbial Ecology in the Oceans, 2nd Edn, ed. D. L. Kirchman (Hoboken, NJ: Wiley & Sons), 207–242.
Naito, M., Hirakawa, H., Yamashita, A., Ohara, N., Shoji, M., Yukitake, H., Nakayama, K., Toh, H., Yoshimura, F., Kuhara, S., Hattori, M., Hayashi, T., and Nakayama, K. (2008). Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis. DNA Res. 15, 215–225.
Nasidze, I., Li, J., Quinque, D., Tang, K., and Stoneking, M. (2009a). Global diversity in the human salivary microbiome. Genome Res. 19, 636–643.
Nasidze, I., Quinque, D., Li, J., Li, M., Tang, K., and Stoneking, M. (2009b). Comparative analysis of human saliva microbiome diversity by barcoded pyrosequencing and cloning approaches. Anal. Biochem. 391, 64–68.
Neill, K., Heesch, S., and Nelson, W. (2008). Diseases, Pathogens and Parasites of Undaria pinnatifida. Wellington: Ministary of Agriculture and Foresty.
Nelson, K. E., Fleischmann, R. D., Deboy, R. T., Paulsen, I. T., Fouts, D. E., Eisen, J. A., Daugherty, S. C., Dodson, R. J., Durkin, A. S., Gwinn, M., Haft, D. H., Kolonay, J. F., Nelson, W. C., Mason, T., Tallon, L., Gray, J., Granger, D., Tettelin, H., Dong, H., Galvin, J. L., Duncan, M. J., Dewhirst, F. E., and Fraser, C. M. (2003). Complete genome sequence of the oral pathogenic Bacterium Porphyromonas gingivalis strain W83. J. Bacteriol. 185, 5591–5601.
Nisizawa, K., Noda, H., Kikuchi, R., and Watanabe, T. (1987). The main seaweed foods in Japan. Hydrobiologia 151/152, 5–29.
Nolan, M., Tindall, B. J., Pomrenke, H., Lapidus, A., Copeland, A., Glavina Del Rio, T., Lucas, S., Chen, F., Tice, H., Cheng, J. F., Saunders, E., Han, C., Bruce, D., Goodwin, L., Chain, P., Pitluck, S., Ovchinikova, G., Pati, A., Ivanova, N., Mavromatis, K., Chen, A., Palaniappan, K., Land, M., Hauser, L., Chang, Y. J., Jeffries, C. D., Brettin, T., Göker, M., Bristow, J., Eisen, J. A., Markowitz, V., Hugenholtz, P., Kyrpides, N. C., Klenk, H. P., and Detter, J. C. (2009). Complete genome sequence of Rhodothermus marinus type strain (R-10). Stand. Genomic Sci. 1, 283–290.
Oh, H. M., Giovannoni, S. J., Lee, K., Ferriera, S., Johnson, J., and Cho, J. C. (2009). Complete genome sequence of Robiginitalea biformata HTCC2501. J. Bacteriol. 191, 7144–7145.
Oh, H. M., Kang, I., Ferriera, S., Giovannoni, S. J., and Cho, J. C. (2010). Complete genome sequence of Croceibacter atlanticus HTCC2559(T). J. Bacteriol. 192, 4796–4797.
Oh, H. M., Kang, I., Yang, S. J., Jang, Y., Vergin, K. L., Giovannoni, S. J., and Cho, J. C. (2011). Complete genome sequence of strain HTCC2170, a novel member of the genus Maribacter in the family Flavobacteriaceae. J. Bacteriol. 193, 303–304.
Pankratov, T. A., Kulichevskaya, I. S., Liesack, W., and Dedysh, S. N. (2006). Isolation of aerobic, gliding, xylanolytic and laminarinolytic bacteria from acidic Sphagnum peatlands and emended description of Chitinophaga arvensicola Kampfer et al. 2006. Int. J. Syst. Evol. Microbiol. 56, 2761–2764.
Pei, Z., Bini, E. J., Yang, L., Zhou, M., Francois, F., and Blaser, M. J. (2004). Bacterial biota in the human distal esophagus. Proc. Natl. Acad. Sci. U.S.A. 101, 4250–4255.
Pena, A., Teeling, H., Huerta-Cepas, J., Santos, F., Yarza, P., Brito-Echeverría, J., Lucio, M., Schmitt-Kopplin, P., Meseguer, I., Schenowitz, C., Dossat, C., Barbe, V., Dopazo, J., Rosselló-Mora, R., Schüler, M., Glöckner, F. O., Amann, R., Gabaldón, T., and Antón, J. (2010). Fine-scale evolution: genomic, phenotypic and ecological differentiation in two coexisting Salinibacter ruber strains. ISME J. 4, 882–895.
Pinhassi, J., Azam, F., Hemphälä, J., Long, R. A., Martinez, J., Zweifel, U. L., and Hagström, A. (1999). Coupling between bacterioplankton species composition, population dynamics, and organic matter degradation. Aquat. Microb. Ecol. 17, 13–26.
Ploug, H., Grossart, H. P., Azam, F., and Jorgensen, B. B. (1999). Photosynthesis, respiration, and carbon turnover in sinking marine snow from surface waters of Southern California Bight: implications for the carbon cycle in the ocean. Mar. Ecol. Prog. Ser. 179, 1–11.
Pommier, T., Canback, B., Riemann, L., Bostrom, K. H., Simu, K., Lundberg, P., Tunlid, A., and Hagstrom, A. (2007). Global patterns of diversity and community structure in marine bacterioplankton. Mol. Ecol. 16, 867–880.
Pope, P. B., Denman, S. E., Jones, M., Tringe, S. G., Barry, K., Malfatti, S. A., Mchardy, A. C., Cheng, J. F., Hugenholtz, P., Mcsweeney, C. S., and Morrison, M. (2010). Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc. Natl. Acad. Sci. U.S.A. 107, 14793–14798.
Popper, Z. A., Michel, G., Hervé, C., Domozych, S. C., Willats, W. G. T., Tuohy, M. G., Kloareg, B., and Stengel, D. B. (2011). Evolution and diversity of plant cell walls: from algae to flowering plants. Annu. Rev. Plant Biol. 62, 567–590.
Purushe, J., Fouts, D. E., Morrison, M., White, B. A., Mackie, R. I., Coutinho, P. M., Henrissat, B., and Nelson, K. E. (2010). Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii : insights into their environmental niche. Microb. Ecol. 60, 721–729.
Qin, Q. L., Zhang, X. Y., Wang, X. M., Liu, G. M., Chen, X. L., Xie, B. B., Dang, H. Y., Zhou, B. C., Yu, J., and Zhang, Y. Z. (2010). The complete genome of Zunongwangia profunda SM-A87 reveals its adaptation to the deep-sea environment and ecological role in sedimentary organic nitrogen degradation. BMC Genomics 11, 247. doi: 10.1186/1471-2164-11-247
Raman, R., Sasisekharan, V., and Sasisekharan, R. (2005). Structural insights into biological roles of -protein-glycosaminoglycan interactions. Chem. Biol. 12, 267–277.
Rath, J., Wu, K. Y., Herndl, G. J., and Delong, E. F. (1998). High phylogenetic diversity in a marine-snow-associated bacterial assemblage. Aquat. Microb. Ecol. 14, 261–269.
Rebuffet, E., Groisillier, A., Thompson, A., Jeudy, A., Barbeyron, T., Czjzek, M., and Michel, G. (2011). Discovery and structural characterization of a novel glycosidase family of marine origin. Environ. Microbiol. 13, 1253–1270.
Redford, A. J., Bowers, R. M., Knight, R., Linhart, Y., and Fierer, N. (2010). The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ. Microbiol. 12, 2885–2893.
Reeves, A. R., Wang, G. R., and Salyers, A. A. (1997). Characterization of four outer membrane proteins that play a role in utilization of starch by Bacteroides thetaiotaomicron. J. Bacteriol. 179, 643–649.
Riemann, L., Steward, G. F., and Azam, F. (2000). Dynamics of bacterial community composition and activity during a mesocosm diatom bloom. Appl. Environ. Microbiol. 66, 578–587.
Riepe, S. P., Goldstein, J., and Alpers, D. H. (1980). Effect of secreted Bacteroides proteases on human intestinal brush border hydrolases. J. Clin. Invest. 66, 314–322.
Rohwer, F., Seguritan, V., Azam, F., and Knowlton, N. (2002). Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 243, 1–10.
Rudek, W., and Haque, R. U. (1976). Extracellular enzymes of the genus Bacteroides. J. Clin. Microbiol. 4, 458–460.
Sabree, Z. L., Kambhampati, S., and Moran, N. A. (2009). Nitrogen recycling and nutritional provisioning by Blattabacterium, the cockroach endosymbiont. Proc. Natl. Acad. Sci. U.S.A. 106, 19521–19526.
Salaun, S., Kervarec, N., Potin, P., Haras, D., Piotto, M., and La Barre, S. (2010). Whole-cell spectroscopy is a convenient tool to assist molecular identification of cultivatable marine bacteria and to investigate their adaptive metabolism. Talanta 80, 1758–1770.
Salyers, A. A., Vercellotti, J. R., West, S. E., and Wilkins, T. D. (1977). Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl. Environ. Microbiol. 33, 319–322.
Savage, D. C., Dubos, R., and Schaedler, R. W. (1968). The gastrointestinal epithelium and its autochthonous bacterial flora. J. Exp. Med. 127, 67–76.
Schmitt-Wagner, D., Friedrich, M. W., Wagner, B., and Brune, A. (2003). Phylogenetic diversity, abundance, and axial distribution of bacteria in the intestinal tract of two soil-feeding termites (Cubitermes spp.). Appl. Environ. Microbiol. 69, 6007–6017.
Schmitz-Esser, S., Tischler, P., Arnold, R., Montanaro, J., Wagner, M., Rattei, T., and Horn, M. (2010). The genome of the amoeba symbiont “Candidatus Amoebophilus asiaticus ” reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J. Bacteriol. 192, 1045–1057.
Scupham, A. J., Patton, T. G., Bent, E., and Bayles, D. O. (2008). Comparison of the cecal microbiota of domestic and wild turkeys. Microb. Ecol. 56, 322–331.
Segers, P., Mannheim, W., Vancanneyt, M., De Brandt, K., Hinz, K. H., Kersters, K., and Vandamme, P. (1993). Riemerella anatipestifer gene nov. comb. nov., the causative agent of septicemia anserum exsudativa, and its phylogenetic affiliation within the Flavobacterium-Cytophaga rRNA homology group. Int. J. Syst. Bacteriol. 43, 768–776.
Sghir, A., Gramet, G., Suau, A., Rochet, V., Pochart, P., and Dore, J. (2000). Quantification of bacterial groups within human fecal flora by oligonucleotide probe hybridization. Appl. Environ. Microbiol. 66, 2263–2266.
Shah, H. N., and Collins, D. M. (1990). Prevotella, a new genus to include Bacteroides melaninogenicus and related species formerly classified in the genus Bacteroides. Int. J. Syst. Bacteriol. 40, 205–208.
Shipman, J. A., Berleman, J. E., and Salyers, A. A. (2000). Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron. J. Bacteriol. 182, 5365–5372.
Shipman, J. A., Cho, K. H., Siegel, H. A., and Salyers, A. A. (1999). Physiological characterization of SusG, an outer membrane protein essential for starch utilization by Bacteroides thetaiotaomicron. J. Bacteriol. 181, 7206–7211.
Shoemaker, N. B., Vlamakis, H., Hayes, K., and Salyers, A. A. (2001). Evidence for extensive resistance gene transfer among Bacteroides spp. and among Bacteroides and other genera in the human colon. Appl. Environ. Microbiol. 67, 561–568.
Sievert, S. M., Kuever, J., and Muyzer, G. (2000). Identification of 16S ribosomal DNA-defined bacterial populations at a shallow submarine hydrothermal vent near Milos Island (Greece). Appl. Environ. Microbiol. 66, 3102–3109.
Silvi, S., Rumney, C. J., Cresci, A., and Rowland, I. R. (1999). Resistant starch modifies gut microflora and microbial metabolism in human flora-associated rats inoculated with faeces from Italian and UK donors. J. Appl. Microbiol. 86, 521–530.
Simek, K., Armengol, J., Comerma, M., Garcia, J. C., Kojecka, P., Nedoma, J., and Hejzlar, J. (2001). Changes in the epilimnetic bacterial community composition, production, and protist-induced mortality along the longitudinal axis of a highly eutrophic reservoir. Microb. Ecol. 42, 359–371.
Simon, M., Glockner, F. O., and Amann, R. (1999). Different community structure and temperature optima of heterotrophic picoplankton in various regions of the Southern Ocean. Aquat. Microb. Ecol. 18, 275–284.
Skerratt, J. H., Bowman, J. P., Hallegraeff, G., James, S., and Nichols, P. D. (2002). Algicidal bacteria associated with blooms of a toxic dinoflagellate in a temperate Australian estuary. Mar. Ecol. Prog. Ser. 244, 1–15.
Smith, C. J., and Bryant, M. P. (1979). Introduction to metabolic activities of intestinal bacteria. Am. J. Clin. Nutr. 32, 149–157.
Smith, C. J., Rocha, E. R., and Paster, B. J. (2006). The medically important Bacteroides spp. in health and disease. Prokaryotes 7, 381–427.
Sohn, J. H., Lee, J. H., Yi, H., Chun, J., Bae, K. S., Ahn, T. Y., and Kim, S. J. (2004). Kordia algicida gen. nov., sp. nov., an algicidal bacterium isolated from red tide. Int. J. Syst. Evol. Microbiol. 54, 675–680.
Sonnenburg, E. D., Zheng, H., Joglekar, P., Higginbottom, S. K., Firbank, S. J., Bolam, D. N., and Sonnenburg, J. L. (2010). Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell 141, 1241–1252.
Stahl, E. A., and Bishop, J. G. (2000). Plant-pathogen arms races at the molecular level. Curr. Opin. Plant Biol. 3, 299–304.
Staufenberger, T., Thiel, V., Wiese, J., and Imhoff, J. F. (2008). Phylogenetic analysis of bacteria associated with Laminaria saccharina. FEMS Microbiol. Ecol. 64, 65–77.
Sugahara, K., and Kitagawa, H. (2002). Heparin and heparan sulfate biosynthesis. IUBMB Life 54, 163–175.
Sunairi, M., Tsuchiya, H., Tsuchiya, T., Omura, Y., Koyanagi, Y., Ozawa, M., Iwabuchi, N., Murooka, H., and Nakajima, M. (1995). Isolation of a bacterium that causes anaaki disease of the red algae Porphyra yezoensis. J. Appl. Microbiol. 79, 225–229.
Suomalainen, L.-R., Tiirola, M., and Valtonen, E. T. (2006). Chondroitin AC lyase activity is related to virulence of fish pathogenic Flavobacterium columnare. J. Fish Dis. 29, 701–770.
Tajima, K., Aminov, R. I., Nagamine, T., Ogata, K., Nakamura, M., Matsui, H., and Benno, Y. (1999). Rumen bacterial diversity as determined by sequence analysis of 16S rDNA libraries. FEMS Microbiol. Ecol. 29, 159–169.
Tanner, A. C. R., Listgarten, M. A., Ebersole, J. L., and Strzempko, M. N. (1986). Bacteroides forsythus sp. nov., a slow-growing, Fusiform Bacteroides sp. from the human oral cavity. Int. J. Syst. Bacteriol. 36, 213–221.
Thomas, C. M., and Nielsen, K. M. (2005). Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 3, 711–721.
Tierny, Y., Bechet, M., Joncquiert, J. C., Dubourguier, H. C., and Guillaume, J. B. (1994). Molecular cloning and expression in Escherichia coli of genes encoding pectate lyase and pectin methylesterase activities from Bacteroides thetaiotaomicron. J. Appl. Bacteriol. 76, 592–602.
Turnbaugh, P. J., Henrissat, B., and Gordon, J. I. (2010). Viewing the human microbiome through three-dimensional glasses: integrating structural and functional studies to better define the properties of myriad carbohydrate-active enzymes. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 1261–1264.
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiome project. Nature 449, 804–810.
Turnbaugh, P. J., Ridaura, V. K., Faith, J. J., Rey, F. E., Knight, R., and Gordon, J. I. (2009). The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 1, 6ra14.
Uyenco, F. R. (1977). “Microbiological studies of diseased Eucheuma sp. and other seaweeds,” in National Seaweeds Symposium, Manilla, Philippines.
van Hannen, E. J., Zwart, G., Van Agterveld, M. P., Gons, H. J., Ebert, J., and Laanbroek, H. J. (1999). Changes in bacterial and eukaryotic community structure after mass lysis of filamentous cyanobacteria associated with viruses. Appl. Environ. Microbiol. 65, 795–801.
Vandamme, P., Segers, P., Vancanneyt, M., Van Hove, K., Mutters, R., Hommez, J., Dewhirst, F., Paster, B., Kersters, K., Falsen, E., Devriese, L. A., Bisgaard, M., Hinz, K. H., and Mannheim, W. (1994). Ornithobacterium -rhinotracheale gen. nov., sp. nov. isolated from the avian respiratory tract. Int. J. Syst. Bacteriol. 44, 24–37.
Vandamme, P., Vancanneyt, M., Segers, P., Ryll, M., Köhler, B., Ludwig, W., and Hinz, K. H. (1999). Coenonia anatina gen. nov., sp. nov., a novel bacterium associated with respiratory disease in ducks and geese. Int. J. Syst. Bacteriol. 49, 867–874.
Wen, L., Ley, R. E., Volchkov, P. Y., Stranges, P. B., Avanesyan, L., Stonebraker, A. C., Hu, C., Wong, F. S., Szot, G. L., Bluestone, J. A., Gordon, J. I., and Chervonsky, A. V. (2008). Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455, 1109–1113.
Wexler, H. M. (2007). Bacteroides: the good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 20, 593–621.
Whitman, W. B., Coleman, D. C., and Wiebe, W. J. (1998). Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U.S.A. 95, 6578–6583.
Woese, C. R., Yang, D., Mandelco, L., and Stetter, K. O. (1990). The flexibacter-flavobacter connection. Syst. Appl. Microbiol. 13, 161–165.
Wostmann, B. S., Larkin, C., Moriarty, A., and Bruckner-Kardoss, E. (1983). Dietary intake, energy metabolism, and excretory losses of adult male germfree Wistar rats. Lab. Anim. Sci. 33, 46–50.
Woyke, T., Xie, G., Copeland, A., Gonzalez, J. M., Han, C., Kiss, H., Saw, J. H., Senin, P., Yang, C., Chatterji, S., Cheng, J. F., Eisen, J. A., Sieracki, M. E., and Stepanauskas, R. (2009). Assembling the marine metagenome, one cell at a time. PLoS ONE 4, e5299. doi: 10.1371/journal.pone.0005299
Xie, G., Bruce, D. C., Challacombe, J. F., Chertkov, O., Detter, J. C., Gilna, P., Han, C. S., Lucas, S., Misra, M., Myers, G. L., Richardson, P., Tapia, R., Thayer, N., Thompson, L. S., Brettin, T. S., Henrissat, B., Wilson, D. B., and McBride, M. J. (2007). Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl. Environ. Microbiol. 73, 3536–3546.
Xu, J., Bjursell, M. K., Himrod, J., Deng, S., Carmichael, L. K., Chiang, H. C., Hooper, L. V., and Gordon, J. I. (2003). A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299, 2074–2076.
Xu, J., Mahowald, M. A., Ley, R. E., Lozupone, C. A., Hamady, M., Martens, E. C., Henrissat, B., Coutinho, P. M., Minx, P., Latreille, P., Cordum, H., Van Brunt, A., Kim, K., Fulton, R. S., Fulton, L. A., Clifton, S. W., Wilson, R. K., Knight, R. D., and Gordon, J. I. (2007). Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 5, e156. doi: 10.1371/journal.pone.0005299
Zhou, J. P., Huang, Y., and Mo, M. (2009). Phylogenetic analysis on the soil bacteria distributed in karst forest. Braz. J. Microbiol. 40, 827–837.
Zhu, X. Y., Zhong, T., Pandya, Y., and Joerger, R. D. (2002). 16S rRNA-based analysis of microbiota from the cecum of broiler chickens. Appl. Environ. Microbiol. 68, 124–137.
Zhu, Y., Suits, M. D., Thompson, A. J., Chavan, S., Dinev, Z., Dumon, C., Smith, N., Moremen, K. W., Xiang, Y., Siriwardena, A., Williams, S. J., Gilbert, H. J., and Davies, G. J. (2010). Mechanistic insights into a Ca2⩲-dependent family of alpha-mannosidases in a human gut symbiont. Nat. Chem. Biol. 6, 125–132.
Keywords: Bacteroidetes, adaptation to environmental niches, microbiota
Citation: Thomas F, Hehemann J-H, Rebuffet E, Czjzek M and Michel G (2011) Environmental and gut Bacteroidetes: the food connection. Front. Microbio. 2:93. doi: 10.3389/fmicb.2011.00093
Received: 12 January 2011; Accepted: 14 April 2011;
Published online: 30 May 2011.
Edited by:
Peter J. Turnbaugh, Harvard University, USAReviewed by:
Deborah Threadgill, North Carolina State University, USACopyright: © 2011 Thomas, Hehemann, Rebuffet, Czjzek and Michel. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Gurvan Michel, UMR 7139, CNRS/UPMC, Marine Plants and Biomolecules, Station Biologique de Roscoff, Place Georges Teissier, 29680 Roscoff, France. e-mail:Z3VydmFuQHNiLXJvc2NvZmYuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.