 Kalliopi Georgiades
Kalliopi Georgiades Didier Raoult*
Didier Raoult*
- Faculté de Médecine et de Pharmacie, Unité de Recherche sur les Maladies Infectieuses et Tropicales Emergentes, CNRS–IRD, UMR 6236, IFR48, Marseille, France
Actual definitions of bacterial species are limited due to the current criteria of definition and the use of restrictive genetic tools. The 16S ribosomal RNA sequence, for example, has been widely used as a marker for phylogenetic analyses; however, its use often leads to misleading species definitions. According to the first genetic studies, removing a certain number of genes from pathogenic bacteria removes their capacity to infect hosts. However, more recent studies have demonstrated that the specialization of bacteria in eukaryotic cells is associated with massive gene loss, especially for allopatric endosymbionts that have been isolated for a long time in an intracellular niche. Indeed, sympatric free-living bacteria often have bigger genomes and exhibit greater resistance and plasticity and constitute species complexes rather than true species. Specialists, such as pathogenic bacteria, escape these bacterial complexes and colonize a niche, thereby gaining a species name. Their specialization allows them to become allopatric, and their gene losses eventually favor reductive genome evolution. A pathogenic species is characterized by a gene repertoire that is defined not only by genes that are present but also by those that are lacking. It is likely that current bacterial pathogens will disappear soon and be replaced by new ones that will emerge from bacterial complexes that are already in contact with humans.
Current and Proposed Species Definitions
One hundred fifty years ago, Darwin’s evolutionary theory on the origin of species became the basis of modern biology. This theory has since been broadly adapted and discussed (Mayr, 1957). However, evolutionary genomic studies have shown that, quantitatively, natural selection is not the predominant force that shapes genome evolution (Koonin, 2009). Indeed, several studies of different species (Endo et al., 1996; Clark et al., 2003; Charlesworth and Eyre-Walker, 2006; Rubin et al., 2010) have found little evidence for natural selection. Therefore, it would be interesting to evaluate whether the elements gathered through the observation of visible species during the twentieth century play the same role in the bacterial world. The formidable explosion in the number of available sequenced genomes has facilitated the study of bacterial evolution. It would be useful to investigate if the evolutionary laws of eukaryotes can be applied to bacteria (Whitman, 2009).
The definition of bacterial species is extremely limited. Although the total biomass of the bacteria on earth is comparable to that of eukaryotes, only approximately 9,000 bacterial species have officially been described (Staley, 2006) compared to 1.5 million eukaryotic species (www.greenfacts.org/biodiversity/). This discrepancy is found because bacteria express few phenotypically identifiable characteristics compared to multicellular eukaryotes (Via, 2009). This shallow description of bacterial species is partly due to the criteria for the definition of bacterial species and the use of restrictive genetic tools (Rossello-Mora and Amann, 2001). For example, a single bacterial species may include strains with >70% DNA/DNA hybridization and a change in the melting temperature of the DNA hybrids of <5°C (Wayne, 1987; Rossello-Mora and Amann, 2001). Based on the same criteria, most primates would also be considered a single species (Sibley and Ahlquist, 1984).
The main characteristic of speciation in eukaryotes (geographic isolation or allopatry) is observed only in specialized bacteria. It is common for a currently recognized “bacterial species” to be, in fact, a complex of species with a large pan-genome (Tettelin et al., 2005; Via, 2009). This observation indicates that the species definition, which considers a species to be “a genomically coherent group of individual isolates sharing a high degree of similarity under standardized conditions” (Doolittle and Papke, 2006), recognizes the variety in non-microscopic species yet is not currently the basis for the definition of bacterial species.
We consider that a bacterial species complex is composed of “generalist” bacteria with sympatric lifestyles, a high level of horizontal gene transfers (HGT), a large pan-genome (Tettelin et al., 2005; Via, 2009), a large genome, and a significant number of ribosomal operons (Audic et al., 2007). The isolation of a specialist bacterium in a new niche or a stochastic population reduction will lead to the creation of a true species, which would be defined as an organism specialized in a given niche that presents an allopatric lifestyle, a smaller pan-genome and genome, and a reduced number of ribosomal operons (Figure 1; Merhej et al., 2009).
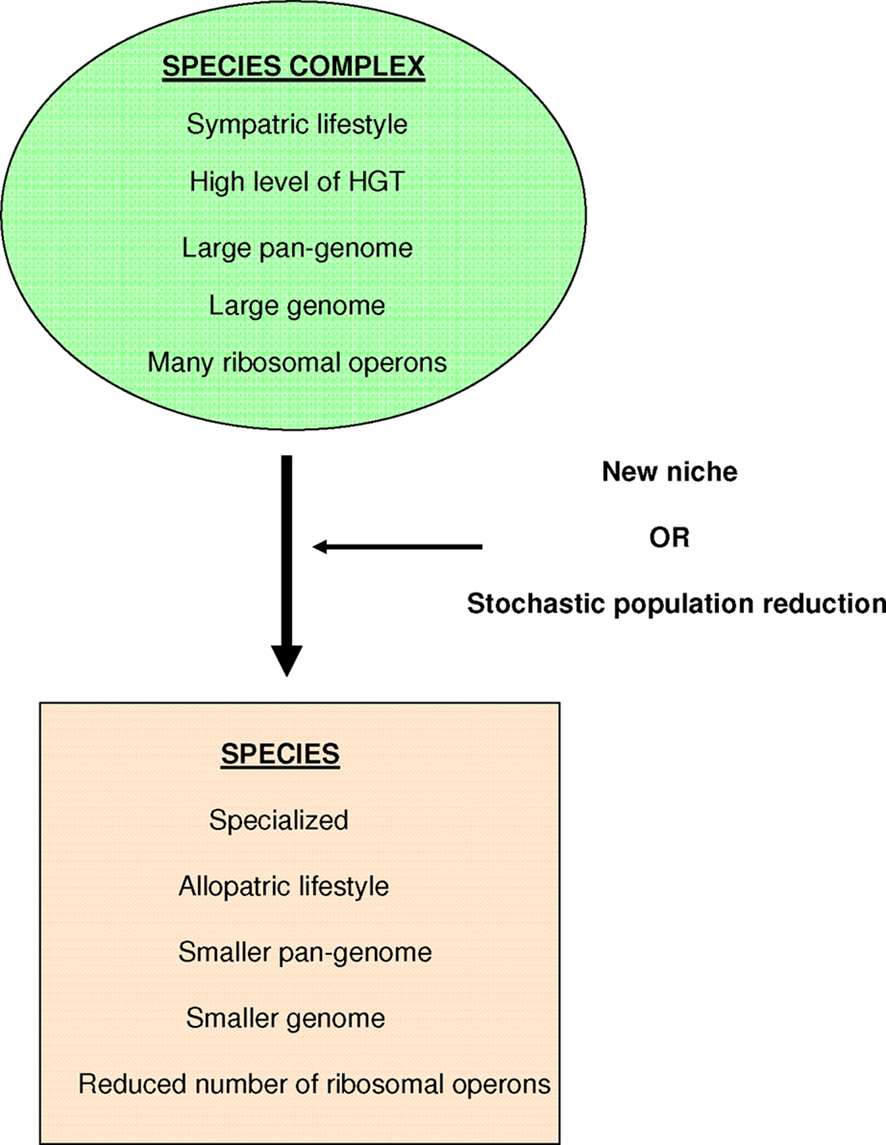
Figure 1. Our definition of bacterial species.
The History of Bacterial Taxonomy
Efforts to establish a system for bacterial classification date back to the seventeenth century, when microbes were regarded as a single species that could develop a variety of shapes (pleomorphism; Figure 2). At the end of the eighteenth century, attempts to create microbial classifications were based on morphological observations. It was not until the nineteenth century that pathogenic bacteria were classified separately as distinct species. Later, the isolation of organisms in pure cultures permitted the phenotypic description of bacteria (Logan, 1994; Rossello-Mora and Amann, 2001). In 1925, Edouard Chatton (Chatton, 1925, 1938) proposed a conceptual basis for taxa by recognizing two general patterns of cellular organization based on the existence of internal compartmentation: the prokaryotes (bacteria) and the eukaryotes (organisms with nucleated cells; Sapp, 2005). In the 1970s, Carl Woese used 16S ribosomal RNA (rRNA) analysis as a phylogenetic tool. Using the laborious molecular sequencing methods available in the 1970s, Woese achieved a comprehensive understanding of bacterial phylogeny (Woese et al., 1975). In doing so, he revealed two separate lineages among prokaryotes: the Archaea (Archaebacteria) and the Bacteria (Eubacteria). The name Archaea is derived from the Greek word “αρχαíα,” meaning “ancient things.” Woese and Fox (1977) recognized a series of methanogenic bacteria, presumed to have existed on earth for three to four billion years, as the most ancient group of bacteria ever detected. This classification of Eukaryotes, Archaea, and Bacteria was named “the three domain system” and replaced Chatton’s “two empire system” (Woese, 1994; Pace, 2006). The 16S rRNA sequencing method has been widely used as a marker for phylogenetic analyses in the description of most newly classified bacterial species (Rossello-Mora and Amann, 2001; Drancourt et al., 2004; Roux et al., 2004). However, the use of ribosomal DNA sequence identity often results in misleading species definitions and does not guarantee the accurate delineation of bacterial species (Fox et al., 1992; Rossello-Mora and Amann, 2001). More recently, our team has proposed a four domain classification, introducing large DNA viruses as the fourth domain of life (Boyer et al., 2010).

Figure 2. Pleomorphism of microbes. (A.1) Cocci, (A.2) diplococci, (A.3–5) streptococci, (A.6) staphylococci, (B.1) bacilli, (B.2) diplobacilli, (B.3) streptobacilli, and (B.4) coccobacilli.
There is a discrepant time-scale between mammalian and bacterial species. According to the molecular clock scale based on 16S rRNA, Actinobacteria appeared approximately three billion years ago and alpha-Proteobacteria approximately two billion years ago (Bromham and Penny, 2003). The first unicellular modern eukaryotes emerged between one and two billion years ago. Mitochondria originated from the proteobacteria Rickettsiales (Cavalier-Smith, 2002). The current definition of bacterial species sets a 16S rRNA divergence cut-off at 1.3% (Stackebrandt and Ebers, 2006). It has been proposed that a 16S rRNA divergence of 1–2% corresponds to approximately 50 million years of divergence (Ochman, et al., 1999; Ogata et al., 2001). Therefore, using this criterion, specialized bacteria within mammalian hosts, which diversified and specialized in a certain niche, are not defined as a species. The first human-specialized pathogenic bacteria, Mycobacterium tuberculosis, appeared only approximately 20,000 years ago (Wirth et al., 2008), much later than Homo sapiens, which emerged 250,000–400,000 years ago (Figure 3; The Smithsonian Institute, Human Origins Program, Feng et al., 1997). Therefore, species definitions cannot be based on the percentage divergence of rRNA because most bacteria having a divergence less than 1.3%, correspond to bacterial complexes rather than species (Doolittle and Papke, 2006).
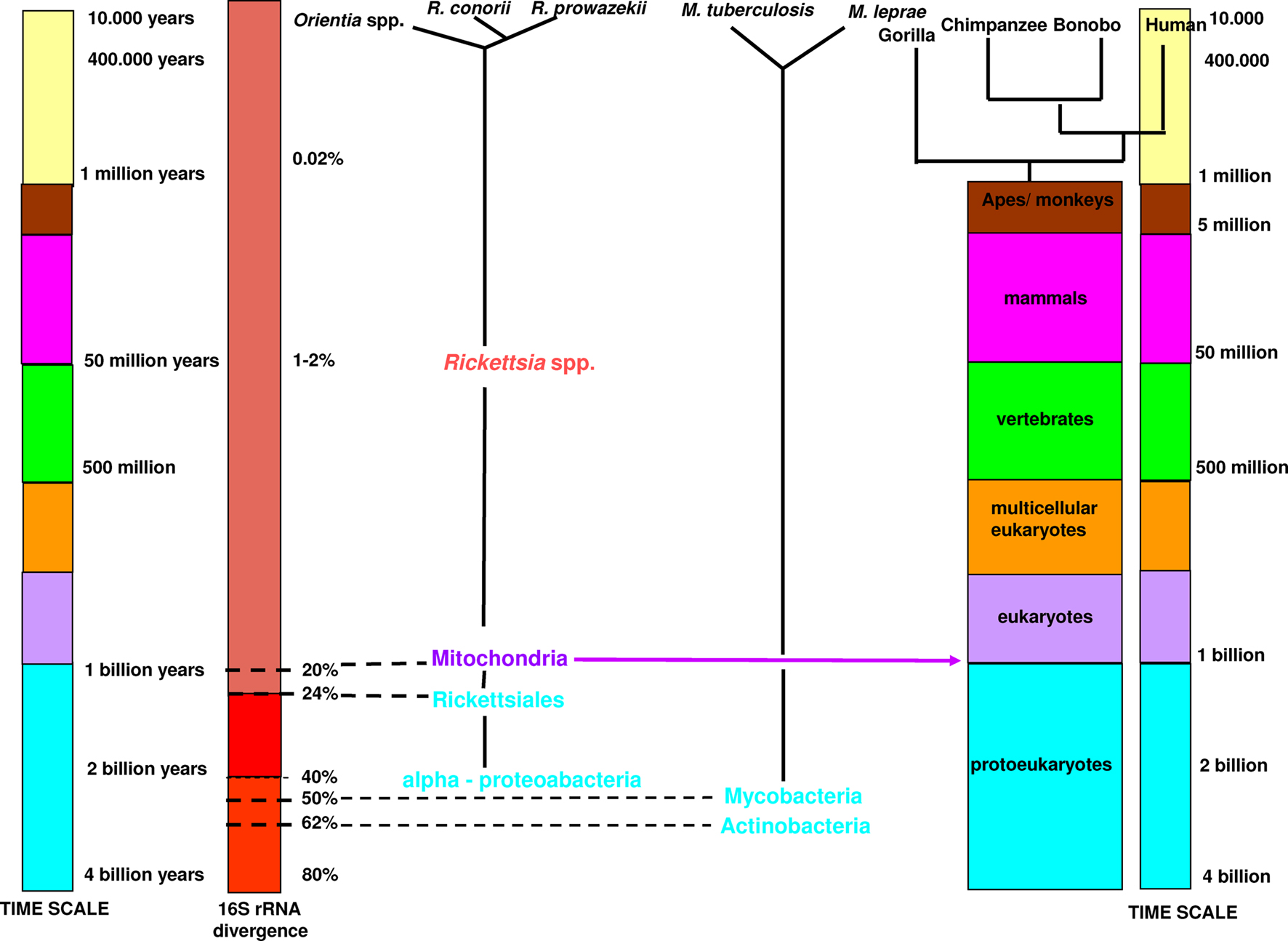
Figure 3. Time-scale comparison between animal and bacterial species.
During the early 1960s, the development of molecular biology techniques and the increasing knowledge of the properties of DNA suggested that bacteria might be classified by comparing their genomes (Rossello-Mora and Amann, 2001). Comparative genomics have shown that bacteria are capable of obtaining genes from distant species. Furthermore, the amounts, types, and sources of transferred genes vary, and gene transfer occurs in ways that are impossible to predict. Nevertheless, according to the core genome hypothesis proposed by Lan and Reeves (1996), despite such genomic fluidity, bacterial species can be rationally named and identified by considering their core genome (all genes present in at least 95% of the strains). However, the degree of gene transfer blurs the boundaries of bacterial groups to such an extent that systematic classification may not be possible (Doolittle and Papke, 2006).
The Imperfections of Species Definition
The allopatric speciation that characterizes eukaryotic species restricts their capacity to exchange genes (Via, 2009). Allopatric speciation is limited by genetic isolation, in which there is no possibility for gene exchange and, therefore, no possibility of a return to sympatry. Alternatively, sympatric speciation occurs between closely related species in which gene exchange is possible (Mayr, 1957; Ochman et al., 2005; Via, 2009; Moliner et al., 2010). Gene exchange in bacteria occurs through horizontal gene transfer mechanisms that can be mediated by viruses, selfish genes, transformation, and conjugation. The capacity for gene exchange depends on the natural environment of the bacteria (Ochman et al., 2005). Sympatric bacteria can easily exchange genes to such a degree that the species is at risk of destabilization (Yu et al., 1995; Halary et al., 2010). It is probable that sympatric species complexes are not yet fixed. However, certain species within a complex can become specialized. The best known examples are species that have a specialized role in mammalian diseases. Many “species,” such as Salmonella, are specialized in multiple animals (for example, chickens, mice, and humans), thus developing co-speciation (Zientz et al., 2001). While in the last century, the characterization of Salmonella as a species was based on essential phenotypic characteristics and geographic isolation, the application of DNA/DNA hybridization criterion has led to the suppression of all species of Salmonella that are pathogenic to animals and has grouped them as one unique species (Le Minor, 1988). The absence of a consistent standard in the field is evidenced by certain species, such as Bacillus anthracis and Shigella dysenteriae, not diverging greatly from their relatives but keeping their status as a species, in contrast to the various species of zoonotic Salmonella that are referenced as serotypes (Brenner et al., 2000).
Sympatric and Allopatric Species
Comparing the genomic content of bacteria to their lifestyle shows that their capacity to exchange genes depends on their ecosystem (Whitman, 2009). Reproductive isolation is a process that can only occur if geographical separation makes gene flow rare or impossible. This establishes allopatric speciation as the norm (Mayr, 1957). We recently proposed (Moliner et al., 2010; Raoult and Boyer, 2010) to systematically extend the notion of allopatry and sympatry, used by evolutionary scientists in the description of non-microscopic life forms, to the lifestyle of bacteria. Bacteria that live in aquatic communities (a sympatric environment) interact with many other bacteria belonging to divergent phyla, allowing them to exchange genes at an increased rate. These bacteria often have larger genomes, more genes, more ribosomal operons, better metabolic capacities, and greater resistance to physicochemical agents (Audic et al., 2007). Most bacteria that live in sympatric environments have conserved genomic and phenotypic plasticity and predominantly constitute species complexes rather than bona fide species. This characteristic is independent of intracellular life: bacteria that lead sympatric lifestyles within amebae have larger genomes than their relatives (Moliner et al., 2010). Therefore, genetic isolation, rather than parasitism (Zomorodipour and Andersson, 1999; Moran and Wernegreen, 2000; Moran, 2002), has a greater impact on genome reduction. Species complexes may have many different genomic repertoires and can therefore produce alternative phenotypes that are better adapted to their environment (Marco, 2008). According to Lawrence (2002), HGT is the main source of new metabolic abilities for microbial habitat colonization. Properties induced by HGT can serve as stable markers of bacterial species. Genes acquired from distant sources are likely to supply new traits that would serve to distance the recipient from its relatives (Ochman et al., 2005). Moreover, other major elements of microbial evolution, such as recombination, point mutations, and genome rearrangements, also have an effect in bacterial adaptation to new niches. These events, however, do not imply genetic isolation boundaries. Ecological separation accompanied by local isolation is enough to generate speciation, even when homologous recombination is not reduced by the ecological separation (Vetsigian and Goldenfeld, 2005). In mammals, bacteria that multiply in the digestive tube are sympatric and have a high gene exchange capacity. Ubiquitous bacteria are found in a pre-species situation which is consistent with Mayr’s hypothesis of the definition of species. In conclusion, sympatric bacteria frequently create new genomic repertoires that are the reservoir of future clonal specialists, and their specializations result in allopatry and a lack of gene acquisitions through HGT. This will result in a considerable genome reduction. This genomic size restriction is probably irreversible because a certain number of functions are lost (Merhej et al., 2009). Although it has been proposed that the genetic variation of allopatric species is partly influenced by mutator strains (Funchain et al., 2000), this is also the outcome of loss of functions due to genome reduction. The DNA replication and repair system is defective and, therefore, mutator strains introduce deleterious mutations that lead to further losses of functions.
Under these conditions, genome size is the outcome of the balance between gene gains and gene losses, resulting in an irreversible decay (Merhej et al., 2009).
General Genome Evolution of Specialists
Early genetic studies aimed at understanding what characterizes pathogenic bacteria have demonstrated that removing a certain number of genes from pathogens eliminates their capacity to infect hosts. The suppression of certain genes decreases their “fitness” in the ecosystem in which they evolved and became specialized. Each of the removed genes was therefore referred to as a “virulence factor” (Dobrindt et al., 2004). This reasoning is deeply biased by an anthropocentric perspective. Intuitively, we believe that bacteria that are dangerous to us are better armed than non-pathogenic bacteria, suggesting that the latter merely lack supplementary virulence factors (Lawrence, 1999; Ochman et al., 2005). However, recent studies based on comparative genomics have demonstrated that the specialization of bacteria for eukaryotic cells is associated with a massive gene loss (Nierman et al., 2004; Merhej et al., 2009) and the loss of identified “virulence factors” (Lescot et al., 2008). Specialization is also associated with a loss of translation regulation genes, which, apparently, constitutes a key step in the evolution toward a more intimate association with a specific host (Merhej et al., 2009). Indeed, in vitro specialization experiments have also led to dramatic genome reduction by Salmonella enterica (Nilsson et al., 2005). It is also possible that reducing the number of ribosomal operons constitutes a speciation factor. The DNA encoding rRNA genes of bacteria is typically organized in operons following the structure 16S-23S-5S (Klappenbach et al., 2000). Host-dependent bacteria have fewer copies of each rRNA gene than free-living bacteria and exhibit significantly more split or incomplete rRNA operons. Another speciation factor is the loss of a group of 100 genes by all obligate intracellular bacteria, which suggests a convergent evolution associated with stability and irreversible association with the host (Merhej et al., 2009). The most extreme form of genome decay is observed in allopatric host-associated bacteria, particularly endosymbionts that have been maintained in a static and undemanding intracellular niche (Andersson and Kurland, 1998; Moran and Wernegreen, 2000; Moran and Mira, 2001; Moran, 2002; Darby et al., 2007; Merhej et al., 2009).
Interestingly, the 12 deadliest bacteria that pose an epidemic risk to mankind have significantly smaller genomes than less dangerous species (p = 0.0009766; Table 1). Gene loss, associated with genetic isolation and multiplication in a new ecosystem, was the basis of vaccinations developed by Pasteur (Chamberland, 1883). While studying the agent of fowl cholera, he noticed that the causative bacterium, Pasteurella multocida, cultured many times in an axenic medium gradually lost its virulence in chickens without losing its ability to protect against later infections. Inoculation with the bacterium whose virulence was attenuated constituted the first vaccination resulting from a laboratory experiment (Figure 4; Chamberland, 1883; Walsh, 1913). Bacillus Calmette–Guérin vaccines are used to prevent tuberculosis, and they consist of mutant forms of the causative agent of bovine tuberculosis, M. bovis. Strains of the bacterium became attenuated between 1908 and 1921 through repeated culture passages under the same laboratory conditions. The attenuation of virulence was shown to coincide with the genomic loss of the RD1 region. Furthermore, any acquisition of DNA during laboratory propagation of M. bovis appears highly unlikely (Mostowy et al., 2003). Thus, multiple culture passage of a bacterium imitates specialization and allopatric isolation, which leads to speciation and gene loss rather than gene gain. Such changes render the bacterium unable to infect its former host. Consequently, selected mutants are more fit (“virulent”) in their new niche: the axenic medium.
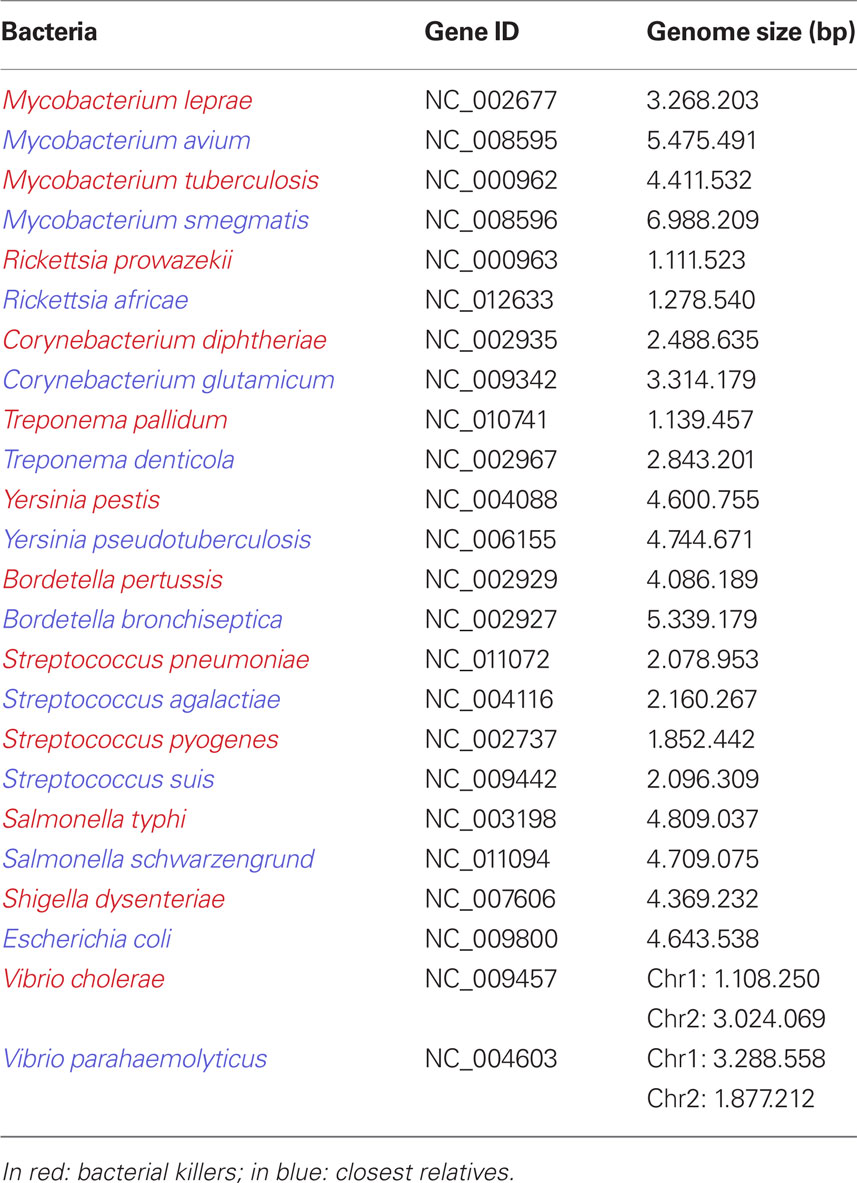
Table 1. Genome size of the 12 bigger bacterial epidemic killers of mankind, compared to their closest non-epidemic relatives.
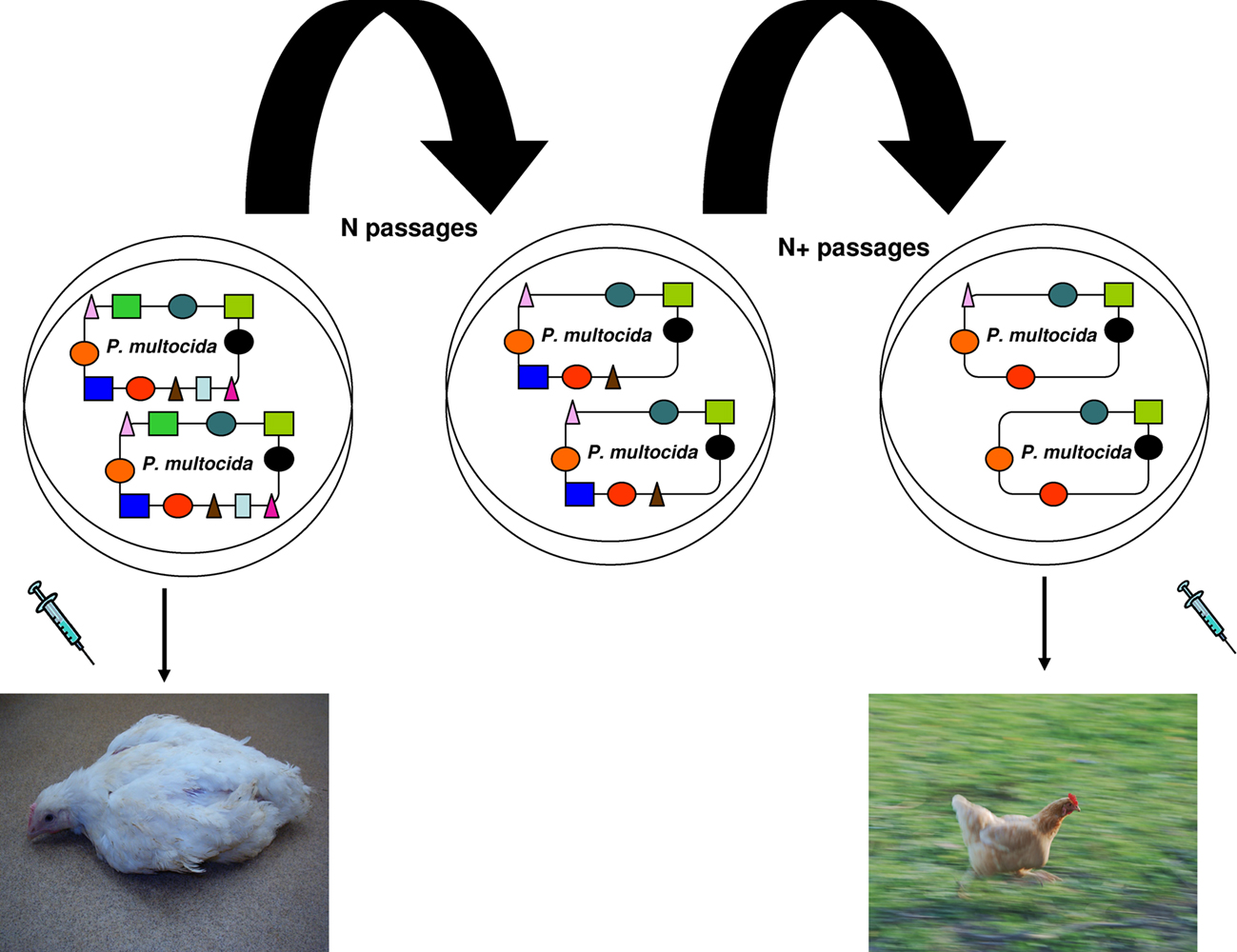
Figure 4. Vaccine principle established by Pasteur. P. multocida virulence is attenuated by multiple culture passages under the same laboratory conditions. The attenuation coincides with genomic loss. Injection of the attenuated strain protected chickens from future P. multocida infection.
Pathogenic Species
One of the best paradigms of reductive evolution is Rickettsia prowazekii, a specialized human pathogen (Merhej and Raoult, 2010). R. prowazekii is the most virulent species of the rickettsial genus. Surprisingly, 24% of its small genome is composed of pseudogenes and non-coding DNA (Andersson et al., 1998). Despite this fact, genome sequencing of rickettsial species has not found any identifiable virulence genes, only an area of genomic plasticity that appears to restore virulence in R. prowazekii and represents an example of adaptive mutation by this pathogen (Bechah et al., 2010). Intracellular motility has been considered a virulence factor in Shigella (Goldberg and Theriot, 1995) and Listeria monocytogenes (Tilney and Portnoy, 1989; Mounier et al., 1990). However, R. prowazekii does not possess this factor and is not mobile (Pollard, 2003; Kleba et al., 2010). Moreover, R. typhi, a closely related species that is less virulent than R. prowazekii, is mobile in the cytoplasm. Actin-based motility and cell-to-cell spread of R. rickettsii require the activities of two rickettsial proteins, Sca2 and RickA (Ogata et al., 2001), functioning together (Kleba et al., 2010), which suggests that these two proteins could be virulence factors for R. rickettsii. Yet, neither of these proteins is found in R. prowazekii, which lacks actin-based motility (Kleba et al., 2010; Merhej and Raoult, 2010), although the non-pathogenic R. montanensis possesses both of these genes (Balraj et al., 2008). This observation suggests that mobility is not a virulence factor per se but can be found in pathogens as part of a virulence repertoire. Motility is useful for R. typhi, R. rickettsii and some non-pathogenic Rickettsia but not for R. prowazekii. A comparison of R. prowazekii and R. conorii genomes found that R. prowazekii had few genes beyond that of R. conorii (Andersson et al., 1998; Ogata et al., 2001; Blanc et al., 2007; Gillespie et al., 2008; Ammerman et al., 2009). Many genes that code for proteins involved in amino acid biosynthesis are lost from the genome of R. prowazekii. Translation capacities are also decreased, and translation regulation factors are affected (Andersson and Kurland, 1998). Such losses could alter mechanisms that regulate invasion, replication, and transmission processes and induce a virulent phenotype. Interestingly, a recent study comparing R. africae and R. rickettsii demonstrated that the loss of essential genes, such as regulatory genes in R. rickettsii, was a possible factor involved in the development of pathogenicity (Fournier et al., 2009). In contrast, the retention of a relatively important repertoire of genes for recombination and repair processes could result from the need to offer protection from the host immune response or against the influx of mobile genetic elements (Moran, 2002). In the case of Rickettsia spp., the estimated rate of gene loss is highly variable: R. felis and R. bellii undergo fewer gene losses than other Rickettsia spp. (Blanc et al., 2007).
In the case of Mycobacteria, several studies performed by Cole and colleagues (Cole et al., 1998, 2001; Cole, 2002; Stinear et al., 2007, 2008; Demangel et al., 2009; Monot et al., 2009) have reported that human pathogens possess small, degraded genomes. The pathogenic bacterium M. leprae has the largest proportion of non-coding DNA of any sequenced bacterial genome. Reductive evolution in M. leprae is linked to the fact that it became an obligate pathogen during the last few million years (Cole et al., 2001). Only 49.5% of its genome contains protein-coding genes. Since diverging from the last common mycobacterial ancestor, the leprosy bacillus may have lost over 2,000 genes, including some that were involved in biosynthetic pathways. For example, its catabolism is limited, but all anabolic pathways seem to be intact (Ribeiro-Guimaraes and Pessolani, 2007; Demangel et al., 2009). Analyses of M. tuberculosis mutants have proven that a number of genes confer a hypervirulent phenotype in mice when they are deleted (Casali, 2003). These genes are found in different operons (mce1, CmaA2, and DosR). Mice inoculated with an M. tuberculosis mutant succumbed to infection earlier than mice infected with the parental strain (Casali, 2003). Another pathogenic Mycobacterium that presents reductive evolution is the Buruli ulcer-causing agent M. ulcerans (Demangel et al., 2009). Compared with its neighbor, M. marinum, M. ulcerans became specialized with the loss of many genes, including putative virulence factors and immunogens, such as the phenolic glycolipids genes, which are considered virulence factors for M. marinum and other mycobacterial pathogens (Demangel et al., 2009).
An outstanding example of a bacterial pathogen is S. dysenteriae, a clone from the Escherichia coli complex that was classified as a genus because of its role in human dysentery (Niyogi, 2005). Shigella spp. differ from E. coli by their poor phenotypic traits, such as extracellular immobility and an inability to ferment lactose and many sugars (Figure 5; Karaolis et al., 1994; Pupo et al., 2000). Shigella spp. and E. coli have always been considered closely related and, at times, were even placed in the same species (Pupo et al., 2000). Nevertheless, Shigella spp. are human pathogens, whereas E. coli strains (with the exception of some pathogenic clones) are mainly commensals of the human intestine, with a much larger genome repertoire (Maurelli et al., 1998). Similar to S. dysenteriae, the pathogenic enteroinvasive E. coli lacks lysine decarboxylase (LDC) activity, which may be connected with its virulence. In a study by Maurelli et al. (1998), the induction of LDC expression (achieved by introducing the cadA gene) by a transformed S. flexneri strain attenuated the virulence of this organism. The most plausible scenario is that Shigella evolved from the E. coli complex through a plasmid containing critical genes. Massive gene deletions followed, thereby increasing its virulence (Figure 5; Maurelli et al., 1998). Therefore, S. dysenteriae, like other pathogenic bacteria, has no more virulence genes than neighboring non-pathogenic bacteria.
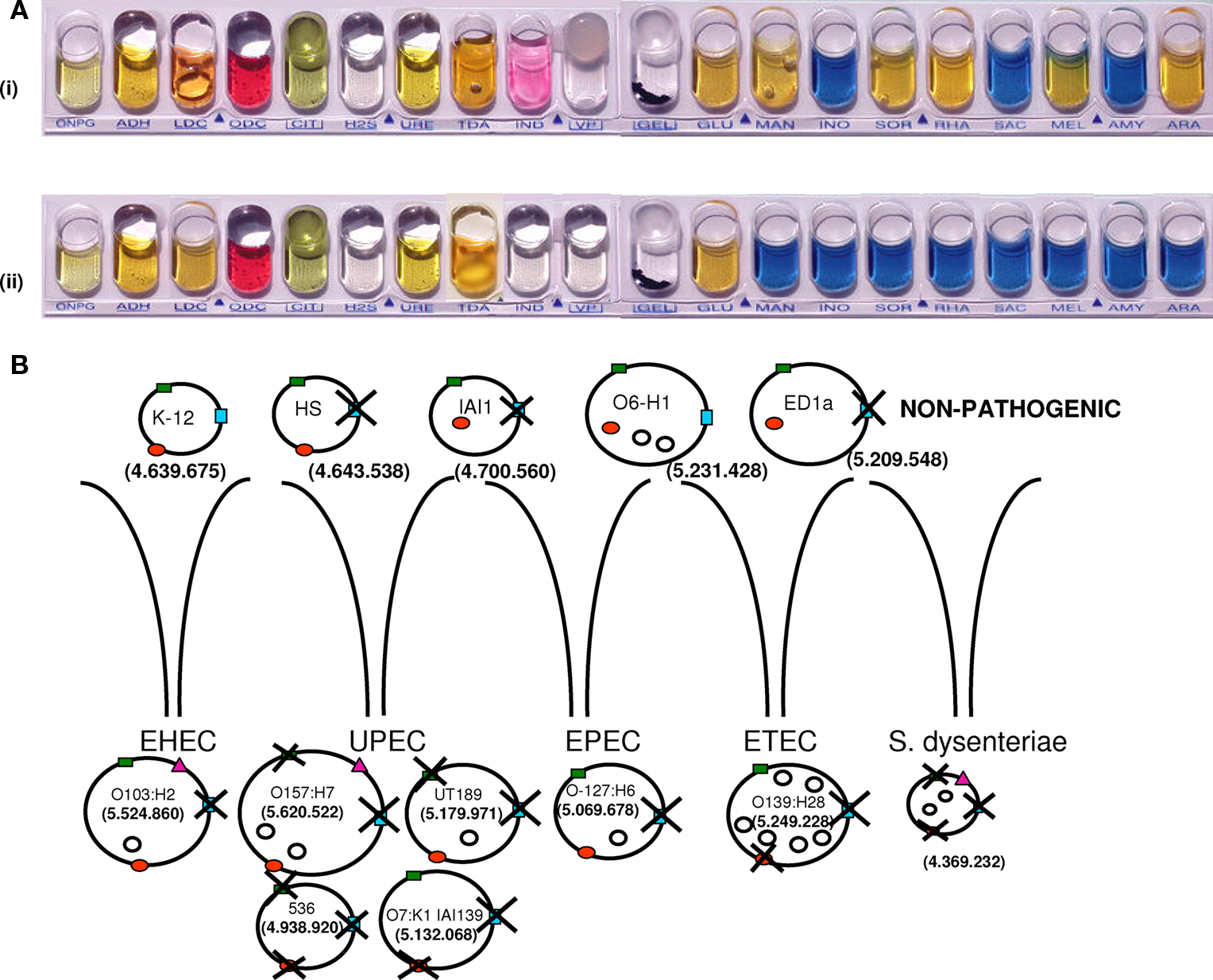
Figure 5. Shigella dysenteriae and Escherichia coli strains. (A) The metabolic capacity of S. dysenteriae (ii) is limited compared to E. coli (i). (B) Sequence deletions in non-pathogenic E. coli strains (K-12; O6-H1; ED1a, and HS, IAI1), pathogenic E. coli strains (O103:H2, O157:H7, UT189, 536, O7:K1, O-127:H6, and O1:139:H28), and S. dysenteriae. Pathogenic strains of E. coli are divided into four groups: enterohemorrhagic (EHEC), uropathogenic (UPEC), enteropathogenic (EPEC), and enterotoxigenic (ETEC). Examples of three different genomic regions around melA (green rectangle), cadA (red circle), and yjcV (blue rectangle) genes are shown. Deletions occurred around these genes in most pathogenic strains, but they remain in non-pathogenic strains. Most pathogenic strains have plasmids, represented by empty circles. Genome size is reported in base pairs. The pink triangle represents Shiga toxin, which is found in S. dysenteriae, E. coli O103:H2, and E. coli O157:H7 (cadA: gene coding for lysine decarboxylase; melA: gene coding for melA protein, alpha-galactosidase activity; and yjcV: gene coding for D-allose transport system permease protein).
Large-scale gene loss and inactivation also occurred during the evolution of two other host-restricted species, Bordetella pertussis and Bordetella parapertussis, leading to the modification of metabolic pathways and regulatory networks (Parkhill et al., 2003). These modifications have resulted in novel virulence characteristics, making infection of the host more effective (Parkhill et al., 2003; Viola et al., 2008).
In conclusion, a pathogenic species is not characterized by its virulence factors but by a considerable genome reduction resulting from its extreme specialization in a given, stable environment. Pathogenic bacteria have a pathogenic gene repertoire in which gene absence may be just as important as gene presence. Modifying any part of the repertoire may influence fitness in a given ecosystem.
Scenarios for Bacterial Speciation and Pathogenicity
Many currently defined bacterial species are actually species complexes with a large pan-genome (Tettelin et al., 2005). The pan-genome is the set of all genes present in the genomes of members of a group of organisms; it can shed light on bacterial biology and lifestyles and has implications in the definition of a species (Tettelin et al., 2008; Lapierre and Gogarten, 2009). The pan-genome is the sum of three groups of open reading frames (ORFs) found in an organism: (i) the extended core genome made up of ORFs that are found in nearly all genomes; (ii) the accessory pool, comprising fast-evolving genes present in only a few genomes, which was probably created de novo; and (iii) the character genes that comprise proteins encoded in only a portion of the genomes (Tettelin et al., 2005, 2008; Bentley, 2009; Lapierre and Gogarten, 2009). Comparative genome analyses have shown that nearly all genes have been exchanged or recombined at some point (Garcia and Olmos, 2007). Up to 30% of the genome-to-genome variation within a species is the result of HGT and gene loss. In some species, the pan-genome is considered open, meaning of unlimited size (Doolittle and Bapteste, 2007; Bentley, 2009). High proportions of genes are ORFans, resulting from de novo creation, fusion, or degradation, and do not belong to any phylogenetic tree (Raoult, 2009). We propose that a bacterial species should be defined as a species with a limited pan-genome.
The intervals of speciation in bacteria may be due to either minor or major changes that correspond to punctuated evolutions, in accordance with the SJ Gould theory, which proposes that most evolution is marked by long periods of evolutionary stability, separated by major stochastic changes (Eldredge and Gould, 1972). Most species experience few evolutionary changes during the major part of their history. When evolution occurs, it is localized in rare, rapid events of branching speciation. Punctuated equilibrium is commonly contrasted against the theory of phyletic gradualism, developed by Darwin (1859), that proposes that evolution occurs uniformly through steady and gradual transformations of entire lineages (Eldredge and Gould, 1972). In bacteria, we can find major elements associated with critical changes, such as the acquisition of genes through HGT, plasmids and virulence islands (Figure 6), or massive deletions (Nierman et al., 2004; Nilsson et al., 2005), such as the initial genome reduction in endosymbionts that occurs by rapid deletion of large blocks of DNA (Merhej et al., 2009). This spontaneous evolutionary tendency toward increased diversity is what McShea and Brandon (2010) call the zero-force evolutionary law. If an evolutionary system is in a zero-force state, it will experience an increase in divergence over time. If, however, the zero-force state is not observed, then the tendency to diversify will not increase. This tendency acts regardless of whether or not there is natural selection. Recently, it was demonstrated that a large expansion of insertion elements in host-adapted bacteria, such as Burkholderia mallei, may play an essential role during the genome reduction process because they seem to mediate genomic deletions and rearrangements (Song et al., 2010). During specialization, bacteria lose hundreds of genes and some ribosomal operons, which limits their translation and translation regulation factors (Whitman, 2009). Moreover, adaptation to a more stable niche is associated with reduced regulation.
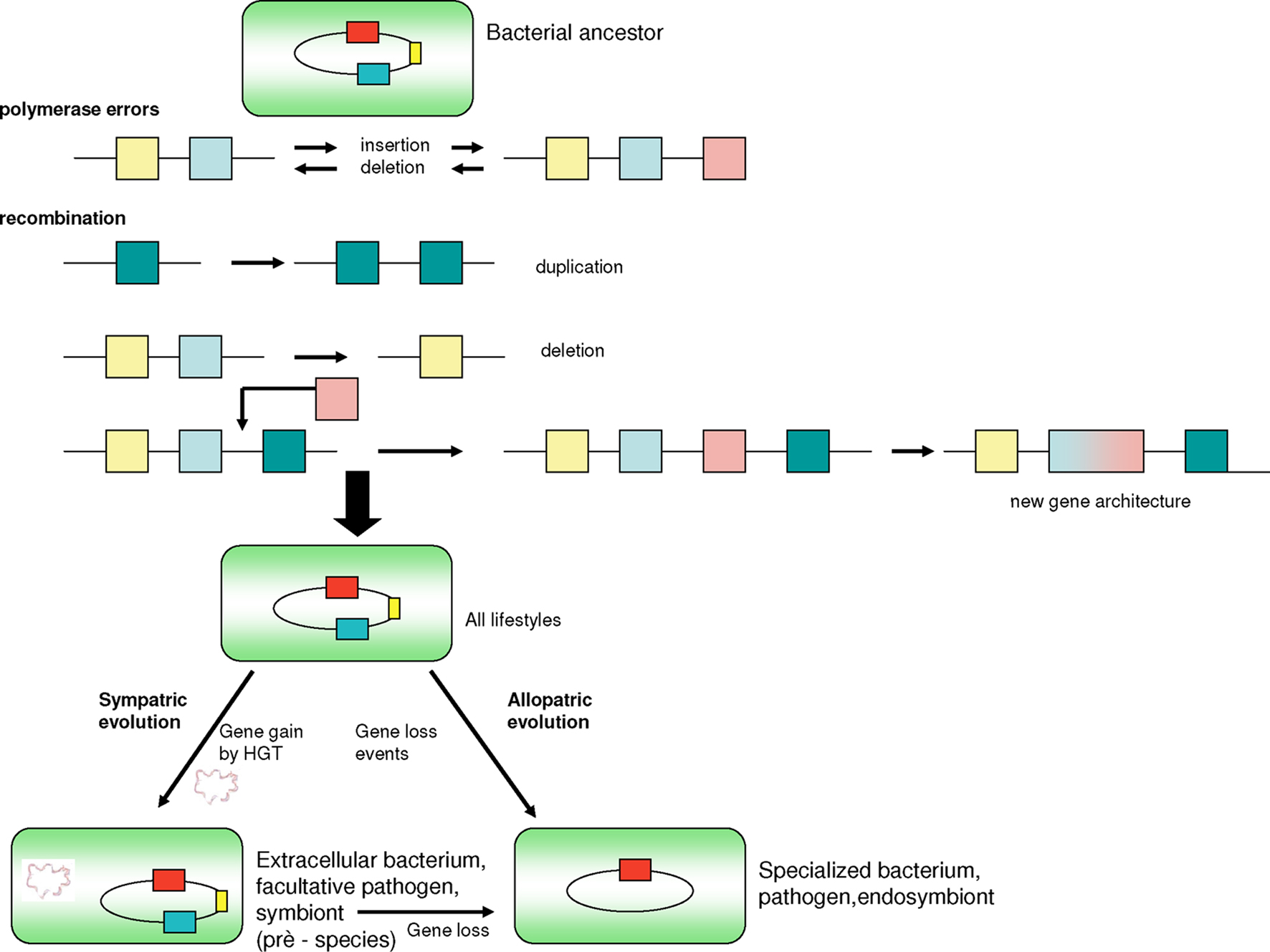
Figure 6. Genetic events leading to speciation of pathogenic bacteria. Polymerase errors can introduce new genes and/or delete existing ones. Recombination events cause duplication, deletion, or fusion of genes and yield new gene architecture. Bacteria may gain genes through horizontal gene transfers, thereby gaining fitness and the ability to better adapt to new environments. Finally, gene loss resulting from deletion events and a significant restriction of the number of ribosomal operons occurs once bacteria become specialized in a host organism whose metabolic substrates can be used by bacteria. Gene gain is subsequently decreased.
In summary, specialists regularly escape from bacterial complexes (Feil, 2010) to colonize a niche and, therefore, earn a species name. Because of their specialization, they will then become allopatric and attempt to maintain a balance between gene gain and gene loss but will favor gene loss, leading to stochastic genome reduction.
Factors Driving Speciation
The evolution of pathogenic bacteria could intuitively be best understood through the Red Queen theory. This model explains evolution as a balance of biotic competition between enemies and is characterized by the Red Queen’s statement to Alice in Lewis Carroll’s novel Through the Looking Glass: “It takes all the running you can do to keep in the same place” (Van Vallen, 1973). Pathogenic bacteria appear to be better armed to fight against humans. The Red Queen theory suggests that arming oneself is an essential element of evolution (Benton, 2009). However, given that pathogenic bacteria seem to be less regulated (bacteria are crazy) than well-armed (bacteria are smart; Merhej et al., 2009), the Court Jester theory, recalling the capricious behavior of the licensed fool of Medieval times (Benton, 2009), may be more descriptive of the evolution of pathogenic bacteria than the Red Queen theory. The Court Jester theory postulates that species diversity depends on fluctuations in climate, landscape, and food supply. According to this model, evolution, speciation, and extinction occur in response to unpredictable changes in the physical environment (Benton, 2009).
One of the initial sources of the description of bacterial pathogens is the analogy with the acquisition of resistance factors, as is compatible with the Red Queen theory (Fontaneto et al., 2007). There is a selective pressure from antibiotics that has nothing to do with that of specialization (Werner et al., 2004; Furuya and Lowy, 2006). Antibiotic resistance may result from the acquisition of resistance genes through HGT (Fontaneto et al., 2007). This arsenal of antibiotic resistance genes has been called the “resistome” (Fajardo et al., 2008). Therefore, acquiring resistance is an effective evolutionary strategy in specific environments. However, it is clear that resistance gained through HGT is more common in unspecialized environmental bacteria, such as Pseudomonas aeruginosa and Acinetobacter baumannii, than bacterial pathogens (Fournier et al., 2006; Maragakis and Perl, 2008). In large bacterial communities, such as the human gut, 10–100 trillion microorganisms live together (Turnbaugh et al., 2007). Bacteria associated with human mucosal tissues, in which genomic exchange rates are elevated, such as the gut (Streptococcus agalactiae; Lefebure and Stanhope, 2007), are capable of developing resistance intermediate to that of free-living bacteria (particularly aquatic bacteria; Audic et al., 2007) and the pathogenic bacteria specialized in humans (Figure 7). This phenomenon emphasizes the isolation of pathogenic human-specialized bacteria and their low capacity to exchange genes (Figure 8). Some of these bacteria spend a period of their lives in sympatric environments, but the proportion of time living in “communities” is smaller than with non-specialized bacteria, as they multiply intracellularly. This explains their limited gene exchange capacity.
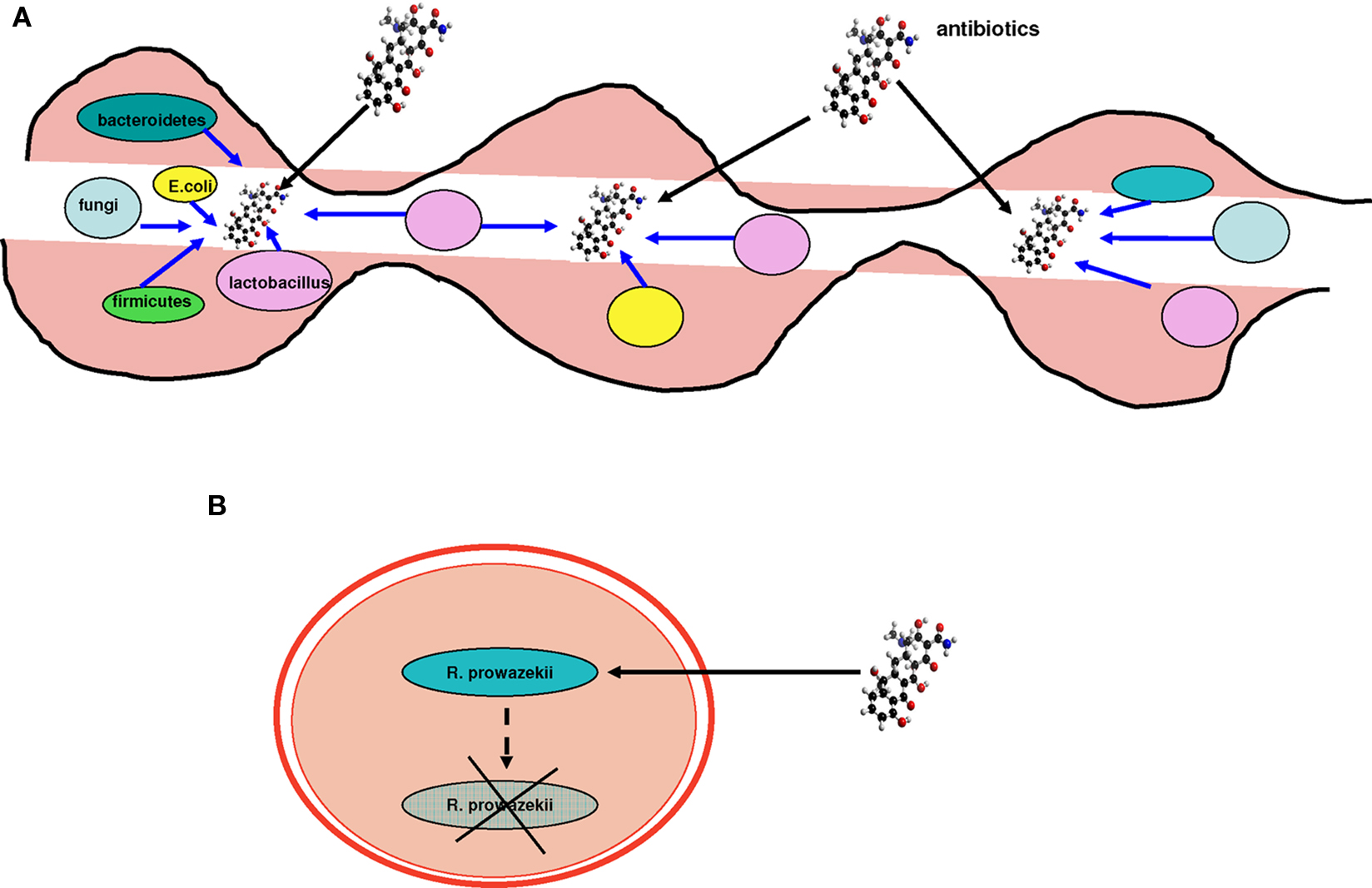
Figure 7. The resistome. Sympatric species have a higher resistance capacity compared to allopatric species, which are more sensitive to the action of antibiotics. (A) Gut microbes unaffected by antibiotics possess resistance genes, probably acquired through HGT. (B) Specialized species are more sensitive and may not resist some antibiotics. Their resistome is limited because of their isolation and their inability to exchange genes. Blue arrows represent resistance genes.
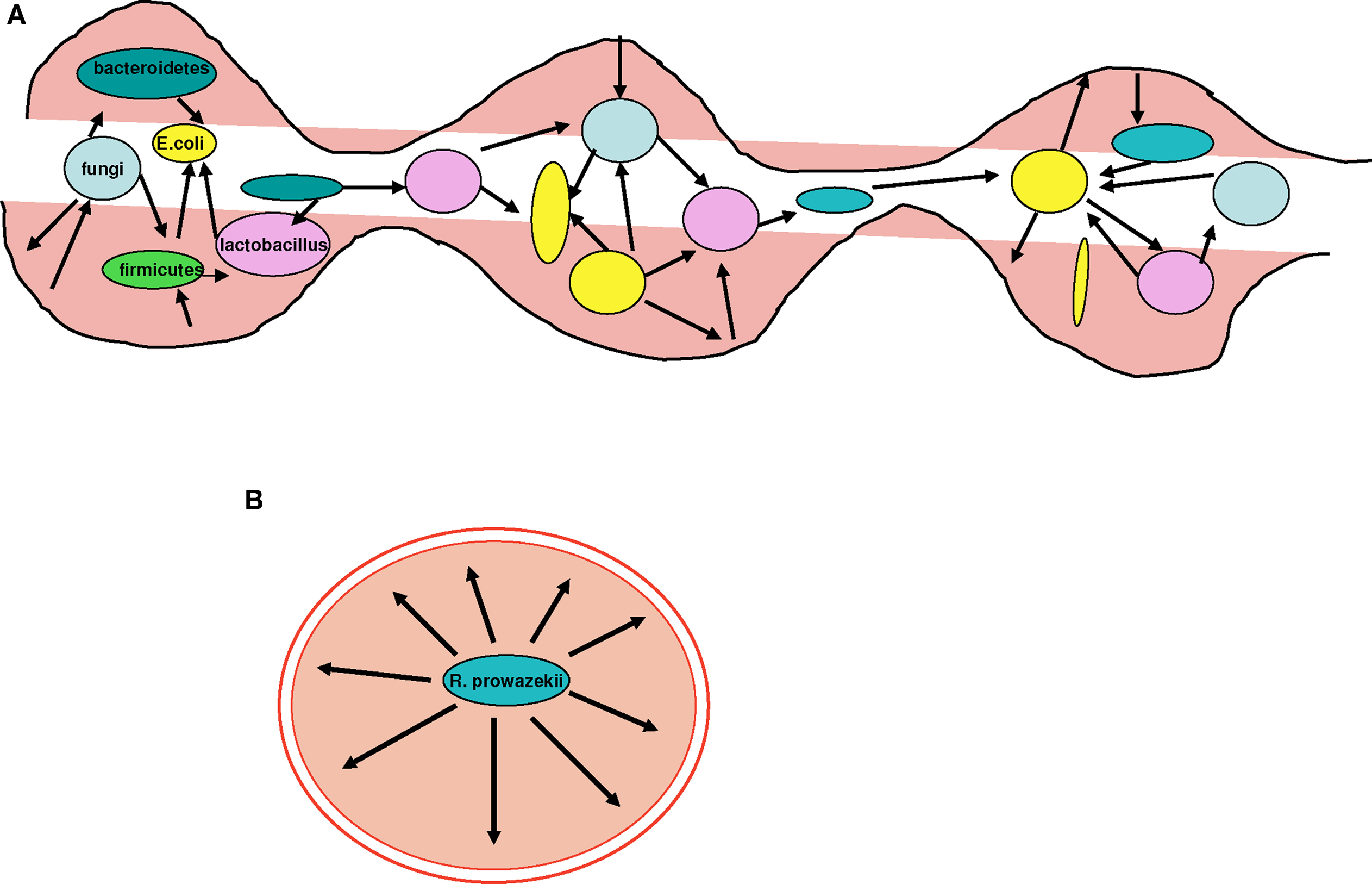
Figure 8. Examples of sympatric and allopatric bacteria. (A) The gut microbiota comprise millions of bacterial species, such as Bacteroidetes, Firmicutes, Lactobacillus, and Escherichia, and fungi. Each of these sympatric species exchanges genes at an intermediate rate, placing them between free-living non-specialized bacteria and isolated obligate intracellular parasites. Therefore, their resistance to antibiotics is also intermediate compared to free-living and pathogenic bacteria whose sensitivity to antibiotics is increased. (B) R. prowazekii inhabits the endothelium in humans. R. prowazekii is an allopatric bacterium and is therefore a specialized isolated pathogen that suffers from ongoing genome reduction. In contrast, it no longer gains any genes through HGT.
The vision of the world deduced from Darwin’s tree of life cannot be reconciled with our current bacterial knowledge (Bapteste et al., 2009). Bacteria represent a complex network of genes acquired from different sources, including viruses, eukaryotes, and Archaea. Gene exchange has allowed the emergence of potent bacteria, generating specialized species that will continue their evolution alone, outside of the species complex. Under these conditions, species complexes constitute melting pots from which specialized species regularly arise (Figure 9; Cohan, 2002; Doolittle and Papke, 2006; Staley, 2006). Species are chimeras composed of mosaics of genes with different origins whose associations are relevant in the beginning of the speciation process (Fraser et al., 2000). From this perspective, the evolution of species more closely resembles a rhizome from which species with genomic repertoires from various origins emerge, eventually allowing the multiplication of the species under favorable environmental conditions (Raoult, 2009). On a phylogenomic tree derived from the whole genome content (absent and present genes) of different E. coli strains and S. dysenteriae, pathogenic and non-pathogenic species form two distinct clusters (Figure 10). This corresponds to Adanson’s (1763)idea for plant phenotypic classification. He arranged plant species by equally considering all of the characteristics of organisms (morphological, structural, and biochemical). None of the characteristics received greater consideration than others (Adanson, 1763). This Adansonian or numerical classification was later used by Sneath for bacterial taxonomy (Sneath and Johnson, 1973). We conclude that the genomic repertoire defines the species and includes both present and absent genes.
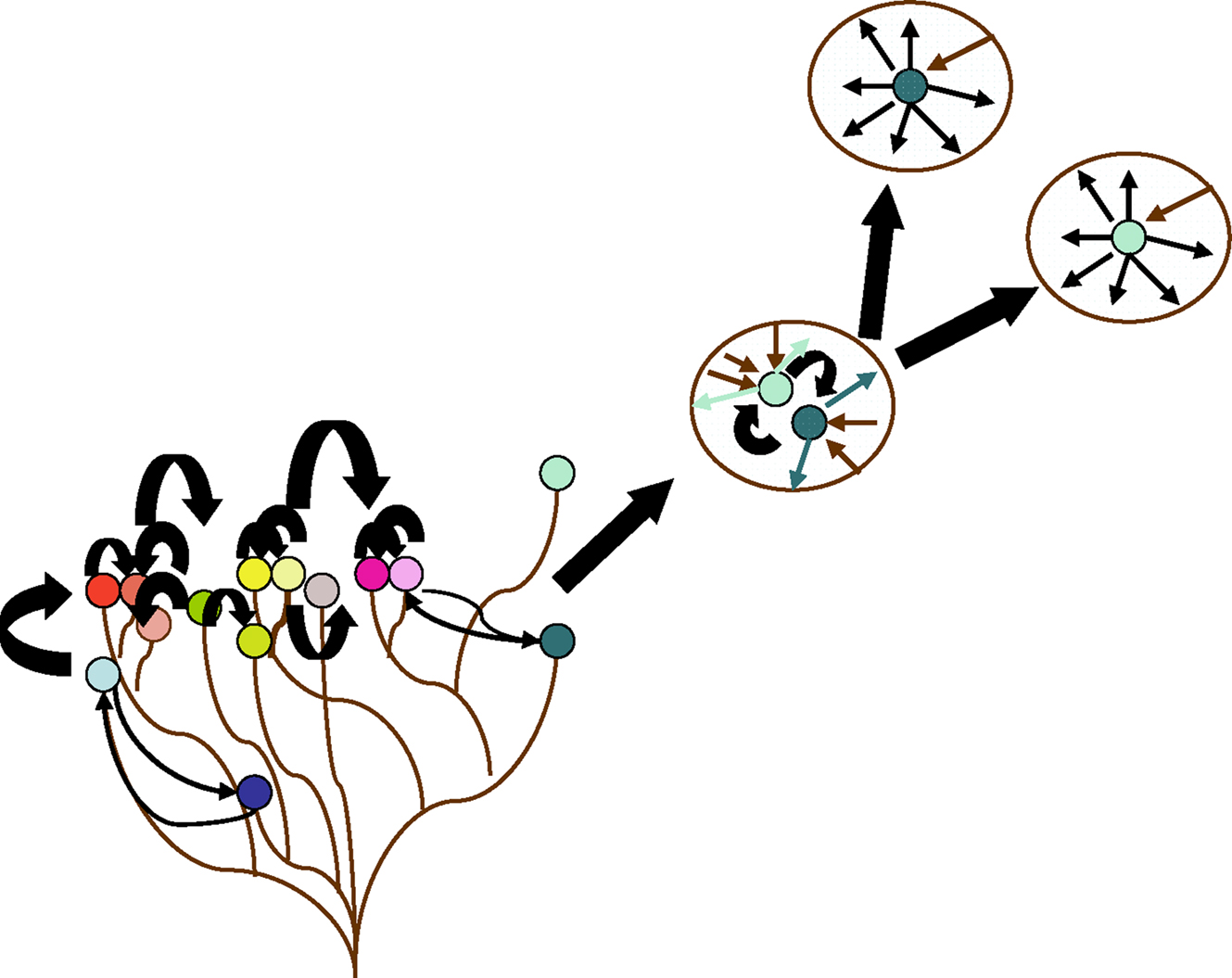
Figure 9. Species complexes and the rise of specialists. Non-specialized bacteria form complexes and engage in high rates of gene exchange. Some specialists may emerge from a complex and progressively adapt a balance between gene gain and gene loss (favoring gene loss). The more specialized a species is, the more important genome reduction becomes. This limits its capacity to obtain new characteristics.
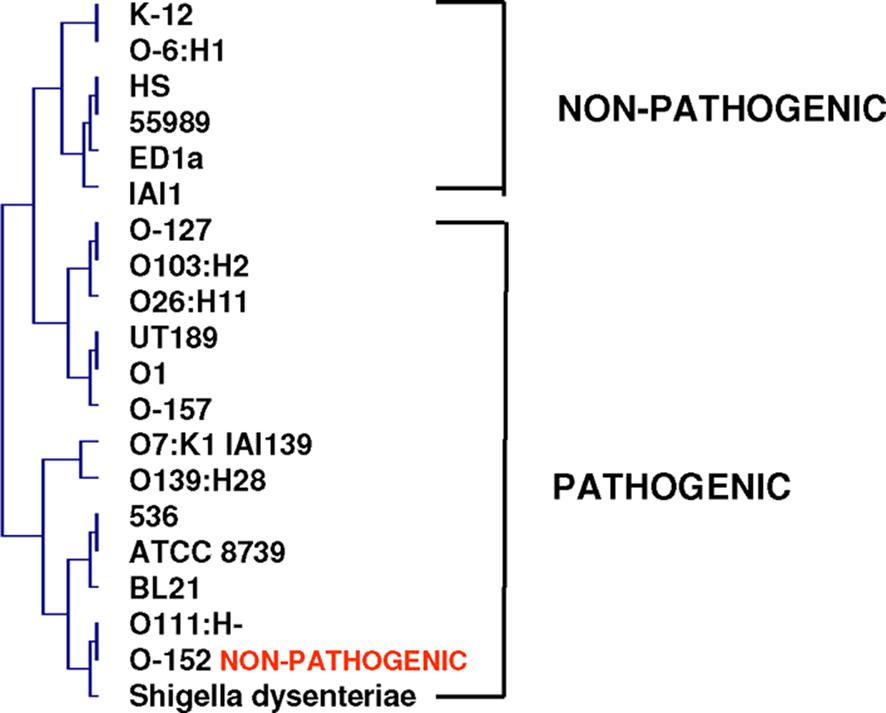
Figure 10. Phylogenomic tree of E. coli strains and S. dysenteriae based on their gene repertoires. Pathogenic and non-pathogenic species form two separate groups.
Where do we Go from there?
Bacterial speciation is accompanied by massive gene losses that can lead to a total loss of autonomy, such as in the cases of Carsonella (Nakabachi et al., 2006), Hodgkin Buchnera (Wixon, 2001), and mitochondria (Nakabachi et al., 2006). Speciation can be defined as a restricted capacity for a bacterium to obtain new characteristics, resulting from its isolation in a given ecosystem. This is associated with a limited capacity to adapt to ecological changes. Therefore, any significant change in ecosystem or any modifications of the niche may result in the disappearance of the bacterium (Wolfe et al., 2007). In contrast to the situation predicted by the Red Queen theory, most human pathogens of the nineteenth century, such as those that cause plague, typhus, leprosy, typhoid, and diphtheria, are found in a state of considerable decline, resulting from their low capacity to adapt. Therefore, commonly known bacterial pathogens are probably on their way to extinction, as has occurred for most bacterial species. However, they will likely be replaced by new pathogens specialized for human pathogenesis for as long as human beings are present. These species will emerge from bacterial complexes that are already in contact with humans, arising from human commensals (gut and other mucosal sites), animals (zoonotic agents), or the environment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We would like to thank Pierre Pontarotti for constructive remarks and reviews.
References
Adanson, M. (1763). Familles Naturelles des Plantes, Vol. 2. Paris: Chez Vincent, Imprimeur-Libraire de Mgr de Comte de Provence.
Ammerman, N. C., Gillespie, J. J., Neuwald, A. S., Sobral, B. W., and Azad, A. F. (2009). A typhus group-specific protease defies reductive evolution in Rickettsia. J. Bacteriol. 191, 7609–7613.
Andersson, S. G., Zomorodipour, A., Andersson, J. O., Sicheritz-Ponten, T., Alsmark, U. C. M., Podowski, R. M., Naslund, A. K., Eriksson, A.-S., Winkler, H. H., and Kurland, G. C. (1998). The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396, 133–140.
Andersson, S. G. E., and Kurland, C. G. (1998). Reductive evolution of resident genomes. Trends Microbiol. 6, 263–268.
Audic, S., Robert, C., Campagna, B., Parinello, H., Claverie, J. M., Raoult, D., and Drancourt, M. (2007). Genome analysis of Minibacterium massiliensis highlights the convergent evolution of water-living bacteria. PLoS Genet. 3, e138. doi: 10.1371/journal.pgen.0030138
Balraj, P., Karkouri, K. E., Vestris, G., Espinosa, L., Raoult, D., and Renesto, P. (2008). RickA expression is not sufficient to promote actin-based motility of Rickettsia raoultii. PLoS ONE 3, e2582. doi: 10.1371/journal.pone.0002582
Bapteste, E., O’Malley., M. A., Beiko, R. G., Ereshefsky, M., Gogarten, J. P., Franklin-Hall, L., Lapointe, F. J., Dupré, J., Dagan, T., Boucher, Y., and Martin, W. (2009). Prokaryotic evolution and the tree of life are two different things. Biol. Direct 3, 34.
Bechah, Y., Karkouri, K. E., Mediannikov, O., Leroy, Q., Pelletier, N., Robert, C. Medigue, C., Mege, J. L., and Raoult, D. (2010). Genomic, proteomic, and transcriptomic analysis of virulent and avirulent Rickettsia prowazekii reveals its adaptive mutation capabilities. Genome Res. 20, 655–663.
Benton, M. J. (2009). The red queen and the court jester: species diversity and the role of biotic and abiotic factors through time. Science 323, 728–732.
Blanc, G., Ogata, H., Robert, C., Audic, S., Suhre, K., Vestris, G., Claverie, J. M., and Raoult, D. (2007). Reductive genome evolution from the mother of Rickettsia. PLoS Genet. 3, e14. doi: 10.1371/journal.pgen.0030014
Boyer, M., Madoui, M. A., Gimenez, G., La Scola, B., and Raoult, D. (2010). Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4th domain of life including giant viruses. PLoS ONE 5, e15530. doi: 10.1371/journal.pone.0015530
Brenner, F. W., Villar, R. G., Angulo, F. J., Tauxe, R., and Swaminathan, B. (2000). Salmonella nomenclature – guest commentary. J. Clin. Microbiol. 38, 2465–2467.
Casali, N. (2003). Hypervirulent Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 26, 15918–15923.
Cavalier-Smith, T. (2002). The phagotrophic origin of eukaryotes and phylogenetic classification of protozoa. Int. J. Syst. Evol. Microbiol. 52, 297–354.
Charlesworth, J., and Eyre-Walker, A. (2006). The rate of adaptive evolution in enteric bacteria. Mol. Biol. Evol. 23, 1348–1356.
Chatton, E. (1925). Pansporella perplexa. Réflexions sur la biologie et la phylogénie de protozoaires. 10e serie. Ann. Sci. Nat. Zool. 7, 1–84.
Clark, A. G., Glanowski, S., Nielsen, R., Thomas, P. D., Kejariwal, A., Todd, M. A., Tanenbaum, D. M., Civello, D., Lu, F., Murphy, B., Ferriera, S., Wang, G., Zheng, X., White, T. J., Sninsky, J. J., Adams, M. D., and Cargill, M. (2003). Inferring nonneutral evolution from human–chimp–mouse orthologous gene trios. Science 302, 1960–1963.
Cole, S. T. (2002). Comparative and functional genomics of the Mycobacterium tuberculosis complex. Microbiology 148, 2919–2928.
Cole, S. T., Brosch,R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S. V., Eiglmeier, K., Gas, S., Barry III, C. E., Tekaia, F., Badcock, K., Basham, D., Brown, D., Chillingworth, T., Connor, R., Davies, R., Devlin, K., Feltwell, T., Gentles, S., Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Krogh, A., McLean, J., Moule, S., Murphy, L., Oliver, K., Osborne, J., Quail, M. A., Rajandream, M.-A., Rogers, J., Rutter, S., Seeger, K., Skelton, J., Squares, R., Squares, S., Sulston, J. E., Taylor, K., Whitehead, S., and Barrell, B. G. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544.
Cole, S. T., Eiglmeier, K., Parkhill, J., James, K. D., Thomson, N. R., Wheeler, P. R., Honoré, N., Garnier, T., Churcher, C., Harris, D., Mungall, K., Basham, D., Brown, D., Chillingworth, T., Connor, R., Davies, R. M., Devlin, K., Duthoy, S., Feltwell, T., Fraser, A., Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Lacroix, C., Maclean, J., Moule, S., Murphy, L., Oliver, K., Quail, M. A., Rajandream, M.-A., Rutherford, K. M., Rutter, S., Seeger, K., Simon, S., Simmonds, M., Skelton, J., Squares, R., Squares, S., Stevens, K., Taylor, K., Whitehead, S., Woodward J. R., and Barrell, B. G. (2001). Massive gene decay in the leprosy bacillus. Nature 409, 1007–1011.
Darby, A. C., Cho, N. H., Fuxelius, H. H., Westberg, J., and Andersson, S. G. E. (2007). Intracellular pathogens go extreme: genome evolution in the Rickettsiales. Trends Genet. 23, 511–520.
Demangel, C., Stinear, T. P., and Cole, S. T. (2009). Buruli ulcer: reductive evolution enhances pathogenicity of Mycobacterium ulcerans. Nat. Rev. Microbiol. 7, 50–60.
Dobrindt, U., Hochhut, B., Hentschel U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424.
Doolittle, W. F., and Bapteste, E. (2007). Pattern pluralism and the tree of life hypothesis. Proc. Natl. Acad. Sci. U.S.A. 104, 2043–2049.
Doolittle, W. F., and Papke, R. T. (2006). Genomics and the bacterial species problem. Genome Biol. 7, 116–123.
Drancourt, M., Berger, P., and Raoult, D. (2004). Systematic 16S rRNA gene sequencing of atypical clinical isolates identified 27 new bacterial species associated with humans. J. Clin. Microbiol. 42, 2197–2202.
Eldredge, N., and Gould, S. J. (1972). “Punctuated equilibria: an alternative to phyletic gradualism,” in Models in Paleobiology, ed. T. J. M. Schopf (San Francisco: Freeman, Cooper and Company), 82–115.
Endo, T., Ikeo, K., and Gojobori, T. (1996). Large scale search for genes on which positive selection may operate. Mol. Biol. Evol. 13, 685–690.
Fajardo, A., Martinez-Martin, N., Mercadillo, M., Galan, J. C., Ghysels, B., Matthijs, S., Cornelis, P., Wiehlmann, L., Tummler, B., Baquero, F., and Martinez, J. L. (2008). The neglected intrinsic resistome of bacterial pathogens. PLoS ONE 3, e1619. doi: 10.1371/journal.pone.0001619
Feil, J. E. (2010). “Linkage, selection, and the clonal complex,” in Bacterial Population Genetics in Infectious Disease, eds D. A. Robinson, D. Falush, and J. E. Feil (Hoboken, NJ: John Wiley and Sons, Inc.), 19–35.
Feng, D. F., Cho, G., and Doolittle, R. F. (1997). Determining divergence times with a protein clock: update and reevaluation. Proc. Natl. Acad. Sci. U.S.A. 94, 13028–13033.
Fontaneto, D., Herniou, E. A., Boschetti, C., Caprioli, M., Melone, G., Ricci, C., and Barraclough, T. G. (2007). Independently evolving species in asexual bdelloid rotifers. PLoS Biol. 5, 914–921. e87. doi: 10.1371/journal.pbio.0050087
Fournier, P. E., Karkouri, K. E., Leroy, Q., Robert, C., Giumelli, B., Renesto, P., Socolovschi, C., Parola, P., Audic, S., and Raoult, D. (2009). Analysis of the Rickettsia africae genome reveals that virulence acquisition in Rickettsia species may be explained by genome reduction. BMC Genomics 10, 166–180. doi: 10.1186/1471-2164-10-166
Fournier, P. E., Vallenet, D., Barbe, V., Audic, S., Ogata, H., Poirel, L., Richet, H., Robert,C., Mangenot, S., Abergel, C., Nordmann, P., Weissenbach, J., Raoult, D., and Claverie, J.-M. (2006). Comparative genomics in multidrug resistance in Acinetobacter baumannii. PLoS Genet. 2, e7. doi: 10.1371/journal.pgen.0020007
Fox, G. E., Wisotzkey, J. D., and Jurtshuk, P. (1992). How close is close – 16S ribosomal-RNA sequence identity may not be sufficient to guarantee species identity. Int. J. Syst. Bacteriol. 42, 166–170.
Fraser, C. M., Eisen, J. A., and Salzberg, S. L. (2000). Microbial genome sequencing. Nature 406, 799–803.
Funchain, P., Yeung, A., Steward, J. L., Lin, R., Slupska, M. M., and Miller, L. H. (2000). The consequences of growth of a mutator strain of Escherichia coli as measured by loss of function among multiple gene targets and loss of fitness. Genetics 154, 959–970.
Furuya, E. Y., and Lowy, F. D. (2006). Antimicrobial-resistant bacteria in the community setting. Nat. Rev. Microbiol. 4, 36–45.
Garcia, A. T., and Olmos, J. S. (2007). Quantification by fluorescent in situ hybridization of bacteria associated with Litopenaeus vannamei larvae in Mexican shrimp hatchery. Aquaculture 262, 211–218.
Gillespie, J. J., William, K., and Shukla, M. (2008). Rickettsia phylogenomics: unwinding the intricacies of obligate intracellular life. PLoS ONE 3, e2018. doi: 10.1371/journal.pone.0002018
Goldberg, M. B., and Theriot, J. A. (1995). Shigella flexneri surface protein Icsa is sufficient to direct actin-based motility. Proc. Natl. Acad. Sci. U.S.A. 92, 6572–6576.
Halary, S., Leigh, J. W., Cheaib, B., Lopez, P., and Bapteste, E. (2010). Network analyses structure genetic diversity in independent genetic words. Proc. Natl. Acad. Sci. U.S.A. 107, 127–132.
Karaolis, D. K. R., Lan, R. T., and Reeves, P. R. (1994). Sequence variation in Shigella sonnei (Sonnei), a pathogenic clone of Escherichia coli, over 4 continents and 41 years. J. Clin. Microbiol. 32, 796–802.
Klappenbach, J. A., Dunbar, J. M., and Schmidt, T. M. (2000). rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 66, 1328–1333.
Kleba, B., Clark, T. R., Lutter, E. L., Ellison, D. W., and Hackstadt, T. (2010). Disruption of the Rickettsia rickettsii Sca2 autotransporter inhibits actin based motility. Infect. Immun. 78, 2240–2247.
Koonin, E. V. (2009). Darwinian evolution in the light of genomics. Nucleic Acids Res. 37, 1011–1034.
Lan, R., and Reeves, P. R. (1996). Gene transfer is a major factor in bacterial evolution. Mol. Biol. Evol. 13, 47–55.
Lapierre, P., and Gogarten, J. P. (2009). Estimating the size of the bacterial pan-genome. Trends Genet. 25, 107–110.
Lawrence, J. (2002). Gene transfer in bacteria: speciation without species? Theor. Popul. Biol. 61, 449–460.
Lawrence, J. G. (1999). Gene transfer, speciation and the evolution of bacterial genomes. Curr. Opin. Microbiol. 2, 519–523.
Lefebure, T., and Stanhope, M. J. (2007). Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 8, R71.
Lescot, M., Audic, S., Robert, C., Nguyen, T. T., Blanc, G., Cutler, S. J., Wincker, P., Couloux, A., Claverie, J.-M., Raoult, D., and Drancourt, M. (2008). The genome of Borrelia recurrentis, the agent of deadly louse-borne relapsing fever, is a degraded subset of tick-born Borrelia duttonii. PLoS Genet. 4, e1000185. doi: 10.1371/journal.pgen.1000185
Maragakis, L. L., and Perl, T. M. (2008). Acinetobacter baumannii: Epidemiology antimicrobial resistance, and treatment options. Clin. Infect. Dis. 46, 1254–1263.
Maurelli, A. T., Fernandez, R. E., Bloch, C. A., Rode, C. K., and Fasano, A. (1998). Black holes and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 95, 3943–3948.
Mayr, E. (1957). The Species Problem. Washington, DC: American Association for the Advancement of Science.
McShea, D., and Brandon, R. (2010). Biology’s First Law: The Tendency of Diversity and Complexity to Increase in Evolutionary Systems. Chicago: The University of Chicago Press.
Merhej, V., and Raoult, D. (2010). Rickettsial evolution in the light of comparative genomics. Biol. Rev. doi: 10.1111/j.1469-185X.2010.00151.x. [Epub ahead of print].
Merhej, V., Royer-Carenzi, M., Pontarotti, P., and Raoult, D. (2009). Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct 4, 13.
Moliner, C., Fournier, P. E., and Raoult, D. (2010). Genome analysis of microorganisms living in amoeba reveals a melting pot of evolution. FEMS Microbiol. Rev. 34, 281–294.
Monot, M., Honoré, N., Garnier, T., Zidane, N., Sherafi, D., Paniz-Mondolfi, A., Matsuoka, M., Taylor, G. M., Donoghue, H. D., Bouwman, A., Mays, S., Watson, C., Lockwood, D., Khamesipour, A., Dowlati, Y., Jianping, S., Rea, T. H., Vera-Cabrera, L., Stefani, M. M., Banu, S., Macdonald, M., Sapkota, B. R., Spencer, J. S., Thomas, J., Harshman, K., Singh, P., Busso, P., Gattiker, A., Rougemont, J., Brennan, P. J., and Cole, S. T. (2009). Comparative genomic and phylogeographic analysis of Mycobacterium leprae. Nat. Genet. 41, 1282–1289.
Moran, N. A. (2002). Microbial minimalism: genome reduction in bacterial pathogens. Cell 108, 583–586.
Moran, N. A., and Mira, A. (2001). The process of genome shrinkage in the obligate symbiont Buchnera aphidicola. Genome Biol. 2, research0054.1–research0054.12.
Moran, N. A., and Wernegreen, J. J. (2000). Lifestyle evolution in symbiotic bacteria: insights from genomics. Trends Ecol. Evol. 15, 321–326.
Mostowy, S., Tsolaki, A. G., Small, P. M., and Behr, M. A. (2003). The in vitro evolution of BCG vaccines. Vaccine 21, 4270–4274.
Mounier, J., Ryter, A., Coquis-Rondon, M., and Sansonetti, P. J. (1990). Intracellular and cell-to cell spread of Listeria monocytogenes involves interaction with F-actin in the enterocyte like cell line Caco-2. Infect. Immun. 58, 1048–1058.
Nakabachi, A., Yamashita, A., Toh, H., Ishikawa, H., Dunbar, H. E., Moran, N. A., and Hattori, M. (2006). The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science 314, 267.
Nierman, W. C., DeShazer, D., Kim, H. S., Tettelin, H., Nelson, K. E., Feldblyum, T., Ulrich, R. L., Ronning, C. M., Brinkac, L. M., Daugherty, S. C., Davidsen, T. D., Deboy, R. T., Dimitrov, G., Dodson, R. J., Durkin, A. S., Gwinn, M. L., Haft, D. H., Khouri, H., Kolonay, J. F., Madupu, R., Mohammoud, Y., Nelson, W. C., Radune, D., Romero, C. M., Sarria, S., Selengut, J., Shamblin, C., Sullivan, S. A., White, O., Yu, Y., Zafar, N., Zhou, L., and Fraser, C. M. (2004). Structural flexibility in the Burkholderia mallei genome. Proc. Natl. Acad. Sci. U.S.A. 101, 14246–14251.
Nilsson, A. I., Koskiniemi, S., Eriksson, S., Kugelberg, E., Hinton, J. C. D., and Andersson, D. I. (2005). Bacterial genome size reduction by experimental evolution. Proc. Natl. Acad. Sci. U.S.A. 102, 11112–11116.
Ochman, H., Elwyn, S., and Moran, N. A. (1999). Calibrating bacterial evolution. Proc. Natl. Acad. Sci. U.S.A. 96, 12638–12643.
Ochman, H., Lerat, E., and Daubin, V. (2005). Examining bacterial species under the specter of gene transfer and exchange. Proc. Natl. Acad. Sci. U.S.A. 102, 6595–6599.
Ogata, H., Renesto-Audiffren, P., Fournier, P.-E., Barbe, V., Samson, D., Roux, V., Cossart, P., Weissenbach, J., Claverie, J.-M., and Raoult, D. (2001). Mechanisms of evolution in Rickettsia conorii and Rickettsia prowazekii. Science 293, 2093–2098.
Parkhill, J., Sebaihia, M., Preston, A., Murphy, L. D., Thomson, N., Harris, D. E., Holden, M. T., Churcher, C. M., Bentley, S. D., Mungall, K. L., Cerdeno-Tarraga, A. M., Temple, L., James, K., Harris, B., Quail, M. A., Achtman, M., Atkin, R., Baker, S., Basham, D., Bason, N., Cherevach, I., Chillingworth, T., Collins, M., Cronin, A., Davis, P., Doggett, J., Feltwell, T., Goble, A., Hamlin, N., Hauser, H., Holroyd, S., Jagels, K., Leathze, S., Moule, S., Norberczak, H., O’Neil, S., Ormond, D., Price, C., Rabbinowitsch, E., Rutter, S., Sanders, M., Saunders, D., Seeger, K., Sharp, S., Simmonds, M., Skelton, J., Squares, R., Squares, S., Stevens, K., Unwin, L., Whitehead, S., Barrell, B. G., and Maskell, D. J. (2003). Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat. Genet. 35, 32–40.
Pollard, T. D. (2003). The cytoskeleton, cellular motility and the reductionist agenda. Nature 422, 741–745.
Pupo, G. M., Lan, R. T., and Reeves, P. R. (2000). Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc. Natl. Acad. Sci. U.S.A. 97, 10567–10572.
Raoult, D., and Boyer, M. (2010). Amoebae as genitors and reservoirs of giant viruses. Intervirology 53, 321–329.
Ribeiro-Guimaraes, M. L., and Pessolani, M. C. V. (2007). Comparative genomics of mycobacterial proteases. Microb. Pathog. 43, 173–178.
Rossello-Mora, R., and Amann, R. (2001). The species concept for prokaryotes. FEMS Microbiol. Rev. 25, 39–67.
Roux, V., Drancourt, M., Stein, A., Riegel, P., Raoult, D., and La Scola, B. (2004). Corynebacterium species isolated from bone and joint infection identified by 16 rRNA gene sequence analysis. J. Clin. Microbiol. 42, 2231–2233.
Rubin, C. J., Zody, M. C., Eriksson, J., Meadows, J. R., Sherwood, E., Webster, M. T., Jiang, L., Ingman, M., Sharpe, T., Ka, S., Hallbook, F., Besnier, F., Carlborg, O., Bed’hom, B., Tixier-Boichard, M., Jensen, P., Siegel, P., Lindblad-Toh, K., and Andersson, L. (2010). Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464, 587–591.
Sapp, J. (2005). The prokaryote–eukaryote dichotomy: meanings and mythology. Microbiol. Mol. Biol. Rev. 69, 292–305.
Sibley, C. G., and Ahlquist, J. E. (1984). The phylogeny of the hominid primates as indicated by DNA/DNA hybridization. J. Mol. Evol. 20, 1–15.
Sneath, P. H. A., and Johnson, R. (1973). Numerical taxonomy of Haemophilus and related bacteria. Int. J. Syst. Bacteriol. 23, 405–418.
Song, H., Hwang, J., Yi, H., Ulrich, R. L., Yu, Y., Nierman, W. C., and Kim, H. S. (2010). The early stage of bacterial genome-reductive evolution in the host. PLoS Pathog. 6, e1000922. doi:10.1371/journal.ppat.1000922
Stackebrandt, E., and Ebers, J. (2006). Taxonomic parameters revisited: tarnished gold standards. Microbiol. Today 33, 152–155.
Staley, J. T. (2006). The bacterial species dilemma and the genomic-phylogenetic species concept. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 361, 1899–1909.
Stinear, T. P., Seemann, T., Harrison, P. F., Jenkin, J. A., Davies, J. K., Johnson, P. D. R., Abdellah, Z., Arrowsmith, C., Chillingworth, T., Churcher, C., Clarke, K., Cronin, A., Davis, P., Goodhead, I., Holroyd, N., Jagels, K., Lord, A., Moule, S., Mungall, K., Norbertczak, H., Quail, M. A., Rabbinowitsch, E., Walker, D., White, B., Whitehead, S., Small, L. C. P., Brosch, R., Ramakrishnan, L., Fischbach, M. A., Parkhill, J., and Cole, S. T. (2008). Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 18, 729–741.
Stinear, T. P., Seemann, T., Pido, S., Frigui, W., Reysset, G., Garnier, T., Meurice, G., Simon, D., Bouchier, C., Ma, L., Tichit, M., Porter, L. J., Ryan, J., Johnson, P. D. R., Davies, J. K., Jenkin, G. A., Small, P. L. C., Jones, L. M., Tekaia, F., Laval, F., Daffé, M., Parkhill, J., and Cole, S. T. (2007). Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Genome Res. 17, 192–200.
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Wasd, N. L., Angiuoli, S. V., Crabtree, J., Jones, A. L., Durkin, A. S., Deboy, R. T., Davidsen, T. M., Mora, M., Scarselli, M., Margarit y Ros, I., Peterson, J. D., Hauser, C. R., Sundaram, J. P., Nelson, W. C., Madupu, R., Brinkac, L. M., Dodson, R. J., Rosovitz, M. J., Sullivan, S. A., Daugherty, S. C., Haft, D. H., Selengut, J., Gwinn, M. L., Zhou, L., Zafar, N., Khouri, H., Radune, D., Dimitrov, G., Watkins,K., O’Connor, K. J., Smith, S., Utlerback, T. R., White, O., Rubens, C. E., Grandi, G., Madoff, L. C., Kasper, D. L., Telford, J. L., Wessels, M. R., Rappuoli, R., and Fraser, C. M. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial pan-genome. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955.
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477.
Tilney, L. G., and Portnoy, D. A. (1989). Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109, 1597–1608.
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiome project. Nature 449, 804–810.
Vetsigian, K., and Goldenfeld, N. (2005). Global divergence of microbial genome sequences mediated by propagating fronts. Proc. Natl. Acad. Sci. U.S.A. 102, 7332–7337.
Via, S. (2009). Natural selection in action during speciation. Proc. Natl. Acad. Sci. U.S.A. 106, 9939–9946.
Viola, R. E., Yerman, L., Fowler, J. M., Arvidson, C. G., and Brubaker, R. R. (2008). A missense mutation causes aspartate deficiency in Yersinia pestis. Microbiology 154, 1271–1280.
Wayne, L. G. (1987). Report of the Ad Hoc Committee on reconciliation of approaches to bacterial systematics. Int. J. Syst. Bacteriol. 37, 463–464.
Werner, C. A., Monnet, L. D., and Harbarth, S. (2004). Antibiotic selection pressure and resistance in Streptococcus pneumoniae and Streptococcus pyogenes. Emerg. Infect. Dis. 10, 514–517.
Whitman, W. B. (2009). The modern concept of the prokaryote. J. Bacteriol. doi: 10.1128/JB.00962–968. [Epub ahead of print].
Wirth, T., Hildebrand,F., Allix-Béguec, C., Wolbeling, F., Kubica, T., Kremer, K., van Soolingen, D., Rusche-Gerdes, S., Locht, C., Brisse, S., Meyer, A., Supply, P., and Niemann, S. (2008). Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 4, e1000160. doi: 10.1371/journal.ppat.1000160
Wixon, J. (2001). Reductive evolution in bacteria: Buchnera sp., Rickettsia prowazekii and Mycobacterium leprae. Comp. Funct. Genomics 2, 44–48.
Woese, C. R. (1994). There must be a prokaryote somewhere – microbiology search for itself. Microbiol. Rev. 58, 1–9.
Woese, C. R., and Fox, G. E. (1977). Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc. Natl. Acad. Sci. U.S.A. 74, 5088–5090.
Woese, C. R., Fox, G. E., Zablen, L., Uchida, T., Bonen, L., Pechman, Lewis, K. B. J., and Stahl, D. (1975). Conservation of primary structure in 16S ribosomal-RNA. Nature 254, 83–86.
Wolfe, N. D., Dunavan, C. P., and Diamond, J. (2007). Origins of major human infectious diseases. Nature 447, 279–283.
Yu, H., Schurr, M. J., and Deretic, V. (1995). Functional equivalence of Escherichia coli sigma (E) and Pseudomonas aeruginosa Algu – Escherichia coli Rpoe restores mucoidy and reduces sensitivity to reactive oxygen intermediates in Algu mutants of Pseudomonas aeruginosa. J. Bacteriol. 177, 3259–3268.
Zientz, E., Silva, F. J., and Gross, R. (2001). Genome independence in insect-bacterium symbioses. Genome Biol. 2, 1032.1–1032.6.
Keywords: speciation, evolution, pathogens, gene loss, allopatry, sympatry
Citation: Georgiades K and Raoult D (2011) Defining pathogenic bacterial species in the genomic era. Front. Microbio. 1:151. doi: 10.3389/fmicb.2010.00151
Received: 06 October 2010;
Accepted: 31 December 2010;
Published online: 17 January 2011.
Edited by:
Martin G. Klotz, University of Louisville, USAReviewed by:
Barbara Methé, The J. Craig Venter Institute, USAChristopher J. Marx, Harvard University, USA
Awdhesh Kalia, University of Louisville, USA
Copyright: © 2011 Georgiades and Raoult. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Didier Raoult, Faculté de Médecine et de Pharmacie, Unité de Recherche sur les Maladies Infectieuses et Tropicales Emergentes, CNRS–IRD, UMR 6236, IFR48, 27, Boulevard Jean Moulin, 13385 Marseille Cedex 5, France. e-mail:ZGlkaWVyLnJhb3VsdEBnbWFpbC5jb20=