 Caroline Deshayes1
Caroline Deshayes1 Md. Nasir Arafath
Md. Nasir Arafath Emilie Roger
Emilie Roger
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. Technol., 25 January 2022
Sec. Nano-Based Drug Delivery
Volume 3 - 2021 | https://doi.org/10.3389/fmedt.2021.778645
Antimicrobial peptides (AMPs) have a great potential to face the global expansion of antimicrobial resistance (AMR) associated to the development of multidrug-resistant (MDR) pathogens. AMPs are usually composed of 10–50 amino acids with a broad structural diversity and present a range of antimicrobial activities. Unfortunately, even if the oral route is the most convenient one, currently approved therapeutic AMPs are mostly administrated by the intravenous route. Thus, the development of novel drug delivery systems (DDSs) represents a promising opportunity to protect AMPs from chemical and enzymatic degradation through the gastrointestinal tract and to increase intestinal permeability leading to high bioavailability. In this review, the classification and properties as well as mechanisms of the AMPs used in infectiology are first described. Then, the different pharmaceutical forms existing in the market for oral administration are presented. Finally, the formulation technologies, including microparticle- and nanoparticle-based DDSs, used to improve the oral bioavailability of AMPs are reviewed.
Almost 100 years after the discovery of antibiotics, an increasing number of multidrug-resistant (MDR) pathogens are alarming. The overuse and misuse of antibiotics have led to antimicrobial resistance (AMR), which is one of the most important public health threats worldwide (1–4). AMR occurs when bacteria, viruses, or fungi develop resistance to antibiotics, antiviral, or antifungal drugs, respectively (5, 6). At least 700,000 people die annually worldwide due to drug-resistant diseases. Worst-case projections estimate that AMR could cause 10 million deaths each year by 2050, leading to a global economic output of 100 trillion USD (7). AMR is a complex issue of global concern with potentially dramatic health and economic consequences. In 2015, WHO launched the Global Action Plan on AMR and the Global AMR and Use Surveillance System (GLASS) (8, 9). In this context, antimicrobial peptides (AMPs) provide a great potential as an alternative strategy to fight against MDR microorganisms (10–12). AMPs are synthetic or naturally conserved molecules found in organisms ranging from prokaryotes to humans and are part of the body's first line of defense against pathogens (bacteria, viruses, fungi, and parasites). Most of them are positively charged with hydrophobic residues with a broad spectrum of antimicrobial activities. For decades, AMPs have shown a growing interest as potential therapeutic agents. Several hundreds of AMPs are under preclinical and clinical development, nevertheless, only a few AMPs are currently approved (13). They are used for the treatment of infectious diseases like bacterial skin infections, Clostridium difficile pseudomembranous colitis, HIV infection, or Candida infections. Thus, AMPs are a promising class of molecules active against bacteria (14), viruses (15), or fungi (16). Furthermore, they could be effective against MDR pathogens and have a potent activity against intracellular bacteria and also against the biofilms that are involved in antibiotic resistance (12, 17–20).
Now, for several reasons, the AMPs that are used as therapeutic agents are mainly administrated by the intravenous route rather than by the oral route. Indeed, the gastrointestinal environment affects the drug stability, solubility, and permeability across the mucosal barriers. In addition, due to their nature, AMPs have a low bioavailability and solubility, are easily degraded by proteases, and potentially present toxicity and immunogenicity, which limit their use as antimicrobial agents. However, the oral route has several advantages: easy to use, non-invasiveness, and convenience for self-administration (21). To minimize the drawbacks of AMPs and the oral route, the development of new drug delivery systems (DDSs) is required (10, 22, 23). This review describes (i) the properties and activities of the AMPs that are used against microbial pathogens, (ii) their indications for oral administration, and (iii) the micro- and nano-DDSs to improve oral bioavailability for local and systemic drug delivery.
The history of AMPs began in the early 1920s with the discovery by Alexander Fleming of a bacteriolytic substance in the tissues and secretions of human and animals, and in some vegetable tissues (24, 25). The name “lysozyme” has been applied to this enzyme, and the peptidic nature of this salivary antiseptic was subsequently established. Since this discovery nearly a century ago, the interest in AMPs has continued to grow, as evidenced by the number of articles published on this topic with an average of 15,000 articles per year in the last decade. Due to the pleiotropic functions not only killing microbes but also controlling host physiological functions such as inflammation, angiogenesis, and wound healing, alternative terms for AMPs have also appeared like “host defense peptides,” “alarmins,” and even “defensins.” AMPs are synthesized by virtually all living organisms, from bacteria to humans via plants. AMPs represent the first line of defense against invading pathogens, being a key part of the innate immune system. AMPs are either ribosomally synthesized oligopeptides or non-ribosomally synthesized peptides. In the latter case, peptides are assembled by multimodular enzymes designated as non-ribosomal peptide synthetases (NRPS). Several regularly updated databases such as APD3 (26), the Collection of AMPs (CAMP) (27), the Database of Antimicrobial Activity and Structure of Peptides (DBAASP) (28), or the Data Repository of AMPs (DRAMP) (29) provide information on sequences, structures, activities, or clinical status of thousands of AMPs identified so far.
Antimicrobial peptides are usually 10–50 amino acids long and lower than 10 kDa, and they contain a composition rich in cationic and hydrophobic amino acids. However, AMPs lack any consensus amino acid sequence and present a broad structural variety and range of antimicrobial activities. The diversity of AMPs causes difficulty in their classification. AMPs can be classified in numerous ways: biological sources, peptide properties, structure, or activity.
The biological sources of AMPs are wide. Indeed, natural peptides have been identified in all kingdoms of life, from bacteria, fungi, plants to animals (30, 31). Animal AMPs can be further classified into insect AMPs, amphibian AMPs, fish AMPs, reptile AMPs, and mammal AMPs (32–34) (Figure 1).
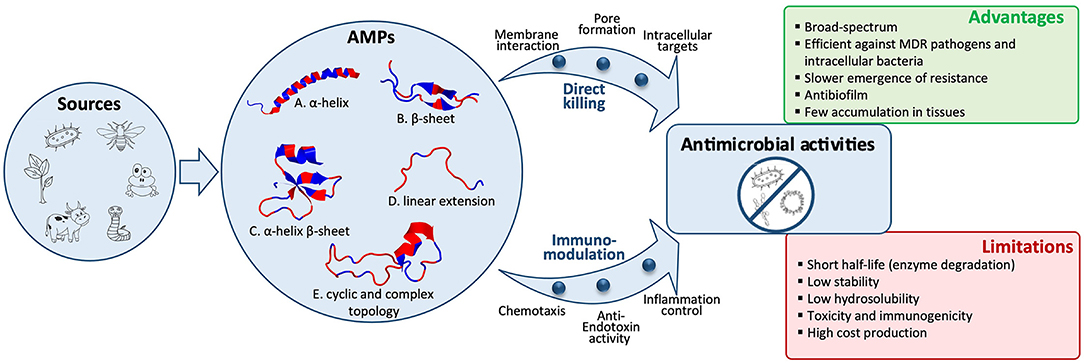
Figure 1. Structural diversity of antimicrobial peptides (AMPs) and their activities against bacteria, viruses, or fungi. A wide variety of biological sources, including microbes, insects, amphibians, reptiles, mammals, or plants, produce AMPs, which are classified into five structural classes. Representative examples of these five classes are shown as a cartoon representation and colored by hydrophobicity [sourced from the RCSB Protein Data Bank (https://www.rcsb.org/)]: (A) α-helical structure of human LL-37 (PDB entry: 2K6O); (B) β-sheet structure of bovine lactoferricin (PDB entry: 1LFC); (C) α-helix and β-sheet structure of human beta-defensin-1 (PDB entry: 1IJV); (D) Linear extension structure of bovine indolicidin (PDB entry: 1G89); (E) Cyclic structure of Bacillus subtilis Subtilisin A (PDB entry: 1PXQ). Direct pathogen killing and immunomodulatory activities of AMPs lead to antibacterial, antiviral, and antifungal activities. AMPs' advantages and limitations to treat infectious diseases are listed.
Antimicrobial peptides can be classified based on peptide properties such as charge, amino acid composition, hydrophobicity, and length. AMPs are oligopeptides containing a varying number of amino acids (usually 10–50 amino acids, ranging in size from 2 to 10 kDa). They can be sorted by size: short (10–24 aa), medium (25–50 aa), and long (50–100 aa). Based on net charge, there can be cationic, neutral, and anionic peptides although most of them are cationic and display a net positive charge ranging from +2 to +13 and may contain a specific cationic domain (35). The cationic nature can be attributed to the presence of lysine and arginine (and sometimes histidine) residues, which allow them to interact with negatively charged bacterial cell membranes, causing the direct destabilization of the surface of membranes with pore formation and subsequent cell lysis. Based on amino acid composition, AMPs can be predominantly rich in specific amino acids such as proline (e.g., apidaecin and pyrrhocoricin), tryptophan, arginine, glycine, histidine (e.g., Histatin-5), or rare modified amino acids (e.g., nisin) (30, 36). Based on hydrophobicity, hydrophobic, amphipathic, and hydrophilic peptides exist. Hydrophobicity governs the extent to which AMPs will be able to be partitioned into the membrane lipid bilayer. It is required for membrane permeabilization, whereas amphipathicity determines whether AMPs can be inserted into the bacterial cell membranes to form hydrophobic channels or pores.
Antimicrobial peptides are classically divided into four categories based on their structures, including linear α-helical peptides, β-sheet peptides, both α-helix and β-sheet peptides, and a linear extension structure. In addition to these four categories, a fifth referred to as the topologically complex AMPs, including various post-translational modifications (PTMs), has recently been added (37) (Figure 1).
The AMPs that adopt a linear α-helix structure represent the largest and best-studied group. They are predominantly found in the extracellular matrix of insects and amphibians like magainin from Xenopus laevis although some other well-known examples are produced by mammals like the human peptide LL-37, a member of cathelicidins (35, 37). Some of these AMPs are unstructured in solution and undergo conformational changes upon interactions with target membranes. Amidation at the C-terminus has been shown to increase its antimicrobial activity by stabilizing α-helical conformation and by eliminating the negative charge of the carboxyl group, therefore enhancing peptide binding to negatively charged target membranes (38). AMPs in the β-family are characterized by at least a pair of two β-strands in the structure. Almost all of these AMPs contain cysteine residues forming one or more disulfide bonds, which stabilize the structure (39). These peptides are therefore more structured in solution and do not undergo major structural changes in a membrane environment. The β-sheet peptides include, for example, bovine lactoferricin or human defensins. Some AMPs adopt a structure with both α-helix and β-sheet elements, such as the cis defensins superfamily (40). The fourth category represents AMPs with a linear extension structure, which do not fold into a particular 3D structure. They often contain a high proportion of certain amino acids such as arginine, tryptophan, or proline like indolicidin (37). A recent fifth group of AMPs has been proposed to gather AMPs with cyclic and complex topologies (37). These peptides do not adopt a linear structure unlike AMPs belonging to the first four classes. In this group, two types of cyclic AMPs are found: “head to tail” and “head to side chain” cyclic topologies. To stabilize their structure, most of the cyclic AMPs contain disulfide bonds or thioether bridges. Plant cyclotides represent a family of backbone-cyclic AMPs with three stabilizing disulfide bonds. Lasso peptides like bacterial microcin are part of head to side chain AMPs and consist of a macrolactam ring formed between the N-terminal α-amino group and an aspartate or glutamate side chain and a linear C-terminal peptide tail (41).
Antimicrobial peptides exhibit several mechanisms of action for an interaction with bacteria and other microorganisms such as viruses and fungi. In general, AMPs kill microorganisms by disturbing membrane integrity or by interacting with the synthesis of intracellular components such as DNA, RNA, and proteins. They can also exert a broad range of immunomodulatory activities (Figure 1).
Membrane interaction is a key factor for the direct activity of AMPs. Many AMPs cause disruption of the physical activity of the microbial membrane and/or translocation across the membrane into the cytoplasm of bacteria to act on an intracellular target. Some of them are presented in Table 1. AMPs kill bacteria by disturbing membrane integrity through membrane lysis (i.e., colistin, bacitracin, daptomycin, and polymyxin B), membrane poration (i.e., gramicidin D and tyrothricin), or the inhibition of cell wall synthesis (i.e., teicoplanin and vancomycin) (Table 1). Gram-negative and Gram-positive bacteria have molecules at the surface that confer a negative charge, allowing an electrostatic interaction with cationic peptides. Thus, teichoic acids in the cell wall of Gram-positive bacteria and lipopolysaccharides in the outer membrane of Gram-negative bacteria provide additional electronegative charge to the bacterial surface. Several models have been proposed to explain the interaction of AMPs with the bacterial membrane. Thus, three possible pathways to disrupt the inner membrane have been described: (i) in the barrel-stave model, peptides can perpendicularly insert into the membrane, promoting peptide–peptide interactions thanks to the AMP's amphipathic structure and resulting in the formation of a peptide-lined transmembrane pore, (ii) concerning the toroidal-pore model, the insertion of the peptides is induced by a curvature in the lipid layer, and a pore is generated by both the peptide and the phospholipid head group, and (iii) in the carpet-model, the peptide is absorbed onto the membrane, covering the entire surface leading to the loss of the membrane integrity and the formation of micelles (Figure 1). Moreover, peptides can translocate into the cytoplasm and directly inhibit the cell wall and protein synthesis, bacterial cell division, or DNA replication by interacting with specific proteins involved in the biological process (18, 19, 48, 50).
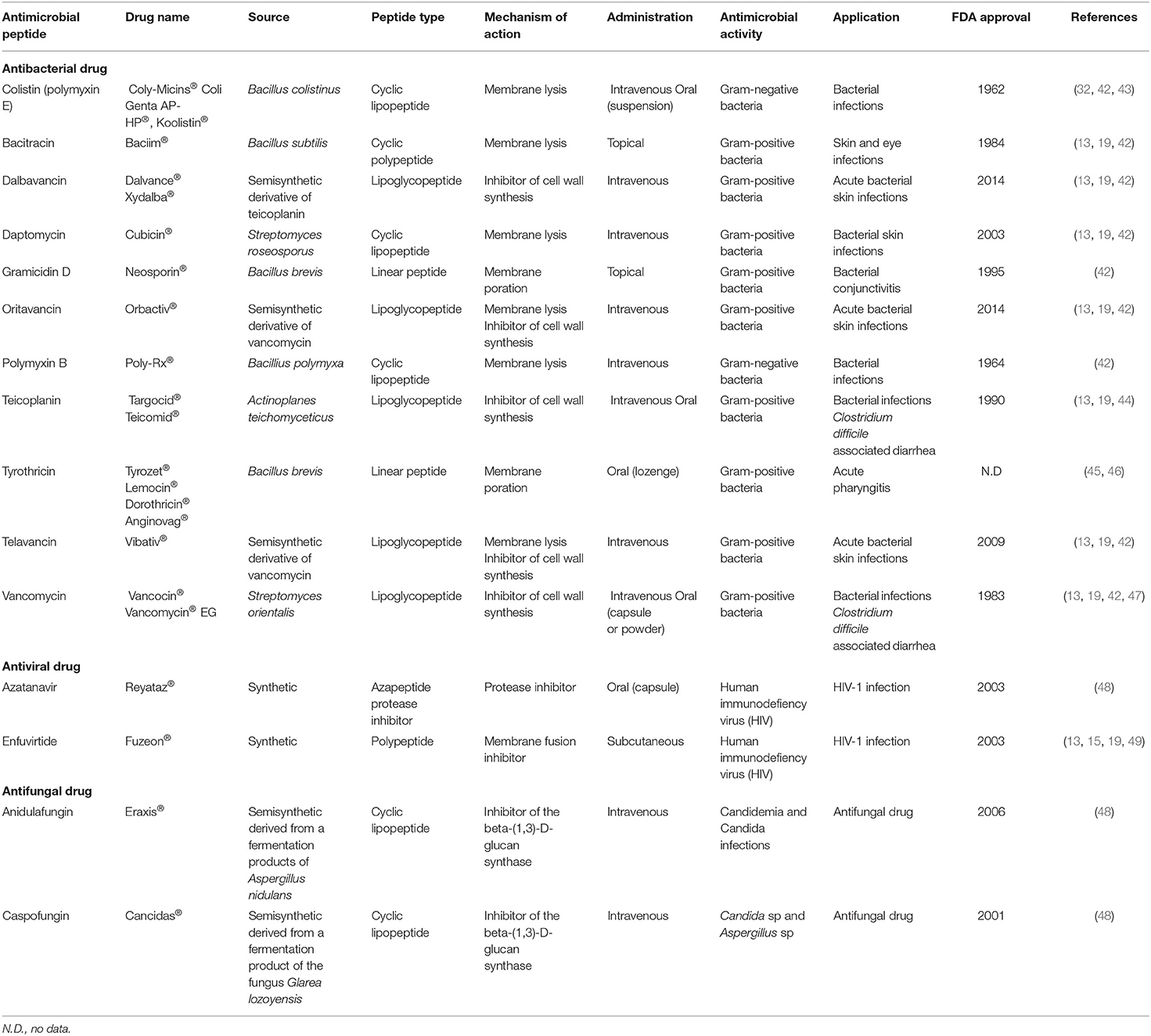
Table 1. The antimicrobial peptide (AMP) drugs approved by the Food and Drug Administration (FDA).
Some AMPs may present their activities against viruses. Antiviral peptides (AVPs) can cause membrane instability by integrating into viral envelopes. Both enveloped RNA and DNA viruses can be targeted by AVPs. AVPs can (i) interact with different glycoaminoglycans present on the cell surface competing with the virus for cellular binding sites, (ii) block the viral entry into the cell, (iii) suppress the cell fusion by interfering with the activity of ATPase protein, (iv) suppress viral gene expression, or (v) interfere with the assembly process of the viruses (13, 18, 19). As examples among the AVPs approved by the FDA, the polypeptide enfuvirtide that is a membrane fusion inhibitor block virus (HIV-1) from entering the host cells and azatanavir, an inhibitor of HIV-1 proteases, preventing the maturation of the proteins needed to assemble the viral capsid, can be cited (Table 1).
Some AMPs pass through the fungal membrane by pore formation or act on beta-glucan or chitin synthesis and others interact with the membrane and cause cell lysis of fungi. They can lead to fungi death by (i) the inhibition of DNA, RNA, or protein synthesis, (ii) induction of apoptosis, (iii) permeabilization of membrane, and (iv) inhibition of cell wall synthesis and enzyme activity (18). Thus, the cyclic lipopeptides (anidulafungin and caspofungin) that are used as antifungal drugs are the inhibitors of the beta-(1,3)-D-glucan synthase (Table 1).
In addition to a broad spectrum of antimicrobial activities, AMPs have anti-inflammatory and immunomodulatory properties. They are described as the effective modulators of inflammation and neutralizers of toxins. AMPs can indirectly promote pathogen clearance of the host by stimulating chemotaxis (by recruiting/activating immunocytes), immune cell differentiation, and the initiation of adaptive immunity, while also preventing harmful inflammation and sepsis by the inhibition of cytokine release and direct scavenging of bacterial endotoxins (20, 50). Indeed, some AMPs modulate host immunity by influencing Toll-like receptor (TLR) recognition of microbial products (i.e., the neutralization of bacterial products such as lipopolysaccharide and lipoteichoic acid to suppress inflammation) and nucleic acids released upon tissue damage to promote auto-inflammation (51) (Figure 1). AMPs having immunomodulatory activities belong to the two major families: defensins and cathelicidins. Human defensins exhibit chemotactic properties and are produced by multiple cell types (i.e., neutrophils, macrophages, lymphocytes, and intestinal epithelial cells). The human cathelicidin membrane disrupting peptide LL-37 acts as a chemoattractant for monocytes, neutrophils, mast cells, and T cells (11, 49, 51). In addition, the immunomodulatory properties of several marine-derived AMPs have been demonstrated. Thus, defensins from oyster or mytilus are the AMPs acting as host defense peptides that disrupt the membrane of microbial pathogens and play a major role in immunomodulation by acting on the innate and adaptive immune response (29). Among the approved AMPs that are used as antibacterial agents, the glycopeptide vancomycin exhibits pharmacological activity against Gram-positive bacteria and immunomodulatory activity affecting tumor necrosis factor-alpha (TNF-α) pathways. Thus, Arbabanel et al. showed that oral vancomycin can be used as an effective treatment for concomitant primary sclerosing cholangitis and inflammatory bowel disease in pediatric. Indeed, oral vancomycin-mediated disease resolution is associated with elevated peripheral TGF-β levels without alterations in Th1 or Th2 cytokine production patterns and increased regulatory T-cell levels (52).
Antimicrobial peptides have a broad spectrum of antimicrobial activities (antibacterial, antiviral, and antifungal) and are a promising class of drugs to face the development of MDR pathogens. They have advantages over conventional antibiotics or antifungals, which include slower emergence of resistance, antibiofilm activity, and an ability to modulate the host immune response. AMPs are less immunogenic than recombinant proteins and antibodies. In addition, they are in general considered to have a safety profile because their metabolites are natural amino acids and there are having short half-life, few peptides accumulate in tissues (10, 13). However, despite the beneficial properties of AMPs, they present some limitations such as: (i) short half-life because of a rapid degradation by proteolytic enzymes, both in the bloodstream and in the gastrointestinal system; (ii) plasma protein binding, which leads to their inactivation; (iii) low metabolic stability and low oral bioavailability; (iv) rapid excretion through the kidneys and liver; (v) high toxicity (i.e., nephrotoxicity) and immunogenicity; (vi) a poor correlation between in vitro antimicrobial activity and their efficacy in vivo; and (vii) high costs of production (22, 42, 50) (Figure 1). For these reasons, their use for in vivo applications has not been fully satisfactory and to date, only a few AMPs are approved by the FDA for clinical use (Table 1). Most of the commercialized AMPs (except colistin and polymyxin B) are used for treating Gram-positive bacterial infections, and the development of AMPs to treat Gram-negative bacterial infections is needed. Moreover, concerning the route of administration, therapeutic peptides are mostly restricted to topical administration (skin and eye infections) or to parenteral administration, while the oral route (p.o) is mostly convenient for medication adherence. Indeed, p.o provides treatment acceptability for patients and facilitates the administration with non-invasive and ambulatory treatments; it is also the main route for local treatment of the gastrointestinal tract. Accordingly, it constitutes the first investigated administration route during the pharmaceutical development of a new drug. Unfortunately, this oral administration route is most of the time not suitable for the emerging therapeutic peptides, and significant efforts to knock down the locks of peptide administration by p.o are conducted. Indeed, after oral administration, peptides are exposed to the aggressive biological environment (pH, enzymes, and gut microbiota) and a complex structure (mucus, epithelial barrier, and hepatic first pass) of the gastrointestinal tract before they have local action or reach the systemic circulation (Figure 2). Generally, gastrointestinal degradation and low permeability lead to oral peptide bioavailability <1–2% (53).
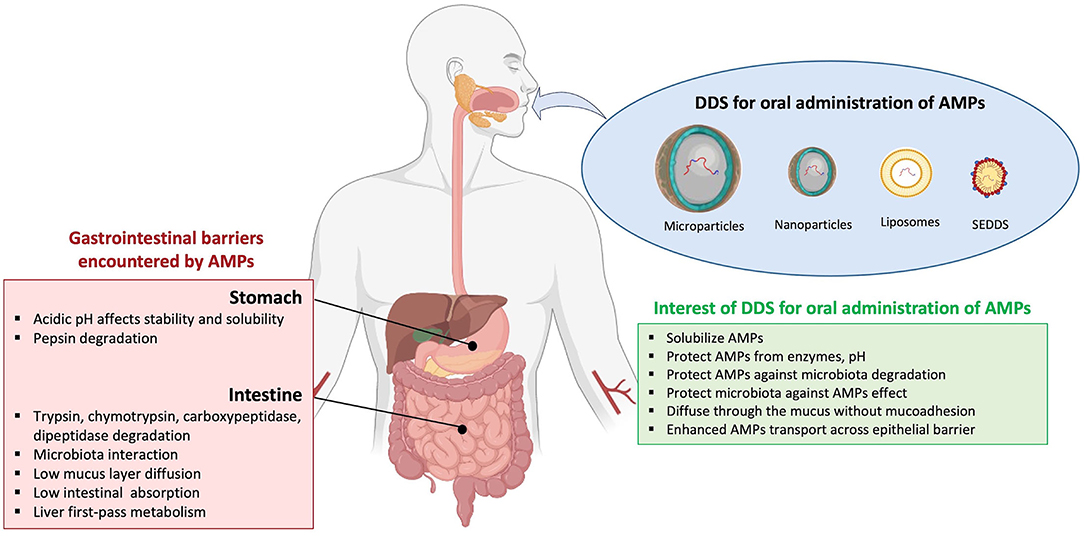
Figure 2. Barriers of AMP absorption and interest of drug delivery systems by oral route. Current drug delivery systems (DDS), including microparticles, nanoparticles, liposomes, and self-emulsifying drug delivery systems (SEDDS), are assessed for oral antimicrobial peptides (AMPs) administration. The encapsulation of AMPs in DDS presents advantages to avoid gastrointestinal barriers.
Some peptides are already used or currently used in clinical trials to treat local diseases of the gastrointestinal tract, including colon infection disease such as amoebiosis and C. difficile or Enterococcus infections. First, some AMPs present an interest after oral administration without pharmaceutical forms. For example, Stiefel et al. (54) administered ramoplanin, a glycolipodepsipeptide, in drinking water (at 100 μg/ml of water) in female mice and demonstrated that it could potentially be used to reduce cross-transmission of vancomycin-resistant Enterococcus. Nevertheless, this work demonstrated some limitations notably due to the administration of this AMP in drinking water that, for example, may not reproduce the pharmacokinetic of human dosing.
LFF571, a new investigational thiopeptide, was synthesized to treat C. difficile infections. The aqueous solubility of this analog was enhanced in comparison with 4-aminothiazolyl analogs of the antibiotic natural product GE2270 A. Pharmacokinetic studies of the infected hamster were performed, and LFF571 solution (prepared with PEG-400 or Cremophor®EL, two solubilizing agents) was administrated at a dosage of 20 mg.kg−1 (55). A very low bioavailability was observed (as expecting to treat local C. difficile infection) and no detectable levels of C. difficile toxins A and B were measured in cecal content (animals were successfully treated). A randomized clinical study (phase II exploratory study in USA and Canada) was performed to compare LFF571 and vancomycin safety and efficacy (56). Moreover, a study was conducted to evaluate the safety, tolerability, and pharmacokinetic of LFF571 in healthy human volunteers (57). In both studies, LFF571 was well-tolerated in patients and in comparison with vancomycin administration, and the frequencies of serious adverse events were similar. Unfortunately, this phase II study was discontinued in 2019.
Moreover, to date, a few small peptides are on the market: peptides with good stability and high potency. These peptides are administrated with conventional forms [composed of active pharmaceutical ingredient (AMPs) and excipient(s)]. They include solid dosage forms such as tablets, capsules, powders, granules, lozenges, and liquid dosage forms such as solutions, emulsions, suspensions, and syrups. For example, vancomycin is administered by the oral route for both local and systemic delivery (Table 1). For local delivery, vancomycin is formulated in 125- and 250-mg capsules or in powder for oral solution to treat the infection of the intestinal mucosa (pseudomembranous colitis induced by C. difficile). Indeed, after oral administration, vancomycin is not usually absorbed into the blood and is excreted almost exclusively in the feces. However, absorption is reported in patients with inflammatory disorders of the intestinal mucosa (47). Nisin containing pectin/hydroxypropyl methyl cellulose (HPMC) compression-coated tablets was formulated for the treatment of local colon diseases such as irritable bowel syndrome, inflammatory bowel disease, and ulcerative colitis (58). Authors propose to coat a tablet with pectin and HPMC (two hydrophilic polymers, which swell to form a hydrogel layer upon contact with aqueous media) to form an enzymatically controlled delivery system. Adding pectin limits the hydration and swelling of HPMC that allow to maintain tablet integrity and thus nisin stability. Furthermore, in vivo studies with pectin/HPMC-coated tablet formulation conducted in healthy volunteers have demonstrated an interesting interplay between the tablet position in the gut, the hydration of the matrix, and the subsequent release pattern. Thus, HPMC maintains tablet integrity until the colon (59). For surotomycin (CB-315), peptides were proposed to treat diarrhea with severe C. difficile infection, tablet, and age-appropriate oral solid formulation that can be dispersed or dissolved. During clinical trials phase I, an insignificant absorption of surotomycin was observed and the primary route of elimination was in the feces following oral administration (60). Throat lozenge pharmaceutical forms were also developed to administer tyrothricin by the oral route in the treatment of patients with acute pharyngitis. Finally, liquid forms prepared from powders were used. Indeed, teicoplanin, to treat diarrhea with severe C. difficile infection, is presented as powder for oral solution at the dosages of 100, 200, and 400 mg. During pharmacokinetic studies, when teicoplanin is administered by the oral route at a 250- or 500-mg single dosage to healthy subjects, it was demonstrated that teicoplanin was not detected in the serum or urine but only recovered in feces (about 45% of the administered dosage) as unchanged medicinal product (44). Colistin sulfate was administered orally as a suspension via a nasogastric tube in the gastrointestinal tract for selective digestive tract decontamination in intensive care units (43).
Despite limited oral absorption of peptides, some of them are still administered orally and indicated for systemic bacterial or viral infections. For example, capsules (200 mg) of a HIV protease inhibitor, atazanavir (ATV), are on the market. ATV has a low oral bioavailability, which can be enhanced clinically by a co-administration with ritonavir and food (Table 2). To avoid food and ritonavir co-administration uses, amorphous solid dispersion system of ATV was also prepared with sodium lauryl sulfate as a carrier and polyoxylglycerides (Gelucire® 50/13) as an absorption enhancer (Table 2). ATV solid dispersion showed a 4.7-fold increase in bioavailability compared with ATV alone (61).
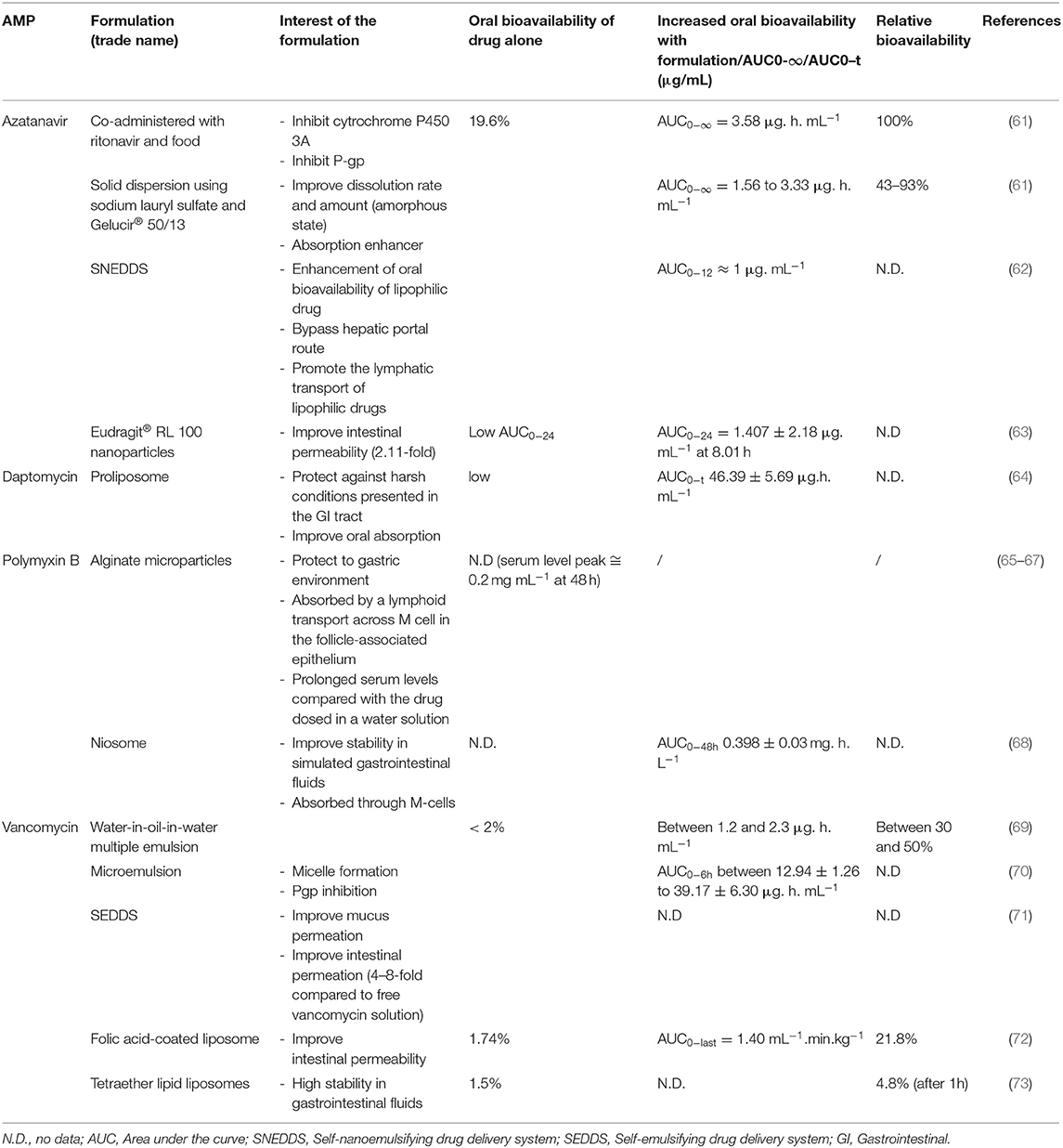
Table 2. Oral AMP formulations to improve bioavailability.
Currently, vancomycin was administered by the oral route only for local C. difficile treatment. Indeed, after oral administration, during pharmacokinetic studies in human adults and during multiple dosing of vancomycin hydrochloride capsule at 250 mg every 8 h for seven dosages, no blood concentration was detected (74). So, to extend the use of vancomycin, pharmaceutical forms have been studied to improve its oral bioavailability. For example, multiple emulsion (water-in-oil-in-water) was proposed to administer vancomycin (as the model drug). The emulsion incorporating C18 unsaturated fatty acid oil (linoleic acid or linolenic acid) has improved the bioavailability more than 40% after emulsion administration into the rat colon loop (at a dosage of 5 mg.kg–1 of vancomycin) (69) (Table 2). On the other hand, for emulsion incorporating C18 unsaturated fatty acid oil (oleic acid) or docosahexaenoic acid, enteral bioavailability was lower (more than 25%) (69). Similarly, the formulation of a solution with a co-administration of the surfactant as an absorption promoter [PEG-8 caprylic/capric glycerides (Labrasol®) and D-α-tocopheryl polyethylene glycol 1,000 succinate (TPGS)] was studied (70). Vancomycin solutions were administered at a dosage of 20 mg.kg–1 into the rat ileum. Formulation containing 50% of Labrasol® and 12.5% of TPGS increased the AUC0−6h value of the vancomycin about 2.4 times in comparison with the formulation with 50% of Labrasol® without TPGS (70).
Thus, the excipients that are used in pharmaceutical forms have demonstrated their importance to improve the absorption of AMPs. In addition, the stability, biocompatibility, safety, and efficacy of AMPs can be further improved through novel formulation strategies and design of new DDSs.
After oral ingestion peptides can be easily affected by an biological environment; pepsin, protease (trypsin and chymotrypsin), acidic, or neutral pH, temperature resulting in the loss of their bioactivity in the gastrointestinal tract. Moreover, after oral administration, to obtain systemic delivery, besides potential gastrointestinal degradation, peptides have also to be transport across mucosal, epithelial, and endothelial barriers (75). Broadly, different approaches to update the inconvenience of conventional dosage forms were proposed to protect and/or to improve intestinal absorption of peptides. In this context, a number of approaches such as chemical modification of the peptide structure, a co-administration of absorption enhancer and/or protease inhibitor and/or mucolytic agents, or peptide encapsulation in well-adapted DDSs are proposed (76, 77).
Drug delivery systems exhibit different chemical or physical properties; they can be a protected drug, modified drug pharmacokinetic, controlled drug release, and improved therapeutic efficacy with less side effects of drugs (78). Thus, these systems emerge as pioneering and promising forms to enhance therapeutic effectiveness. DDSs are particulate pharmaceutical forms such as microparticles and nanoparticles (including liposomes, polymer- and lipid-based nanoparticles, and micelles) that allow drug encapsulation (Figure 2).
The loading of AMPs into DDS could protect peptides from enzymatic degradation after oral administration for local treatment of the gastrointestinal tract. For example, the loading of nisin into the DDS is interesting due to its loss of bioactivity after an interaction with food (inactivation by enzymatic degradation, inactivity at alkaline pH). To demonstrate the interest of encapsulation, different nanoparticles as well as microparticle system were developed (79–85), but even if encapsulation improves the stability of nisin at alkaline pH or in the presence of enzymes, no pharmaceutical indications were described after oral administration. Thus, these different works demonstrated the interest of DDS to protect the peptide from gastrointestinal media. Similarly, the encapsulation of microcin J25 (the peptide with a bactericidal activity against a range of pathogenic enteric bacteria such as Escherichia coli and Salmonella) into the liposome coated with whey protein and pectin has protected significantly the peptide during in vitro digestion study (86). Finally, the efficacy of orally administered encapsulated cryptdin-2 onto chitosan nanoparticles (more than 105 nm particle size) was also demonstrated against Salmonella Typhimurium infection in mice (87). The property of chitosan can modulate the intestinal behavior of nanoparticles, which increase stability and protect cryptdin-2 against gastrointestinal conditions.
To enhance the absorption of AMPs following oral administration, microparticulate and more specifically, nanoparticulate systems introduced advanced features. Indeed, due to their size, surface charge, and/or targeting moieties on the surface, nanoparticles are expected to diffuse through the mucus layer and to transport peptides across the intestinal barrier to reach blood circulation by different pathways (mainly transcellular pathways) (88). As a consequence, a lot of nanoparticulate delivery systems are described in the literature to improve the oral bioavailability of AMPs (Table 2; Figure 2).
Self-emulsifying DDS (SEDDS) and self-nanoemulsifying DDS (SNEDDS) are defined as the isotropic mixtures of oil, surfactants, and co-solvents. They present advantages for oral drug delivery like the protection against enzymatic degradation, reduced first pass metabolism, exhibiting mucus permeating properties, and enhanced absorption (89). Accordingly, daptomycin was incorporated into SEDDS that was prepared with tricaprylin (Dermofeel®MCT) and mono-diglyceride of medium chain fatty acids (mainly caprylic and capric) (Capmul® MCM) as oil and PEG-40 hydrogenated castor oil (Cremophor® 40) as the surfactant. Daptomycin was studied for the treatment of complicated skin infections, bacteremia, and right-side endocarditis caused by multiresistant Gram-positive bacteria. In vitro mucus permeation study demonstrated that SEDDS formulation improved daptomycin permeation by a factor 2 in comparison to pure daptomycin and protected daptomycin against enzymatic degradation (90). Similarly, SEDDS containing vancomycin, 25% of glycerol monocaprylate (Capmul® 808G), 37.5% of PEG-40 (Cremophor® R40), 13.6% of diethylene glycol monoethyl ether (Transcutol® HP), and 26.5% of dimethyl sulfoxide (DMSO) was developed. SEDDS formulation improved intestinal in vitro mucosa permeating properties, nearly 45% of vancomycin permeated the mucus in SEDDS formulation within 4 h, whereas <5% permeated with free vancomycin (Table 2). Ex vivo permeation across porcine small intestinal mucosa confirmed these results (30 vs. 5% with SEDDS formulation and free vancomycin, respectively) (71). SNEDDS formulation of ATV was also developed. Optimized formulation was prepared with glyceryl monolinoleate (Maisin™35-1) as oil and diethylene glycol monoethyl ether (Transcutol®P) as the surfactant. After oral administration into rats at a dosage of 7.2 mg.kg–1 of ATV, the area under the curve (AUC) of ATV was improved 2.57-fold with SNEDDS compared to pure ATV (administered in the form of 0.3% carboxymethyl cellulose suspension) (62) (Table 2).
Microparticles (microspheres or microcapsules) are defined as particles with size larger than 1 μm. They are promising encapsulation systems for protecting peptides from degradation, enhancing peptide stability, and providing an increased surface to volume ratio for peptide release and gastrointestinal absorption (91). In this way, oral polymeric microparticulate as a carrier of polymyxin B was developed. Polymyxin B, which has a potent bactericidal activity against a broad range of Gram-negative bacteria (e.g., Pseudomonas aeruginosa), is not absorbed orally. After encapsulation into crosslinked alginate/chitosan microparticles, the biological activity of polymyxin B was conserved due to microparticle stability in a gastric environment (Table 2) (65–67). Moreover, these microparticles demonstrated their ability to target the gut-associated lymphoid tissue (GALT) by Peyer's patch uptake, but more experiments have to be performed to demonstrate an eventual improvement of polymyxin B absorption (67).
Liposomes (vesicles in which an aqueous volume is entirely surrounded by a bilayer phospholipid membrane) are the first nanoparticulate delivery systems that reached the market (92). Conventional formulations of liposomes were instable in the gastrointestinal environment, but the modification of their composition by incorporating a specific phospholipid (e.g., DSPC, DPPC, and tetraether lipids) (92, 93) or by polymer coating (e.g., chitosan and PEG) (93, 94) enhanced their stability. Recent research on oral delivery systems has shown that liposomes can be employed to improve the bioavailability of encapsulated materials by protecting them against the chemical or enzymatic degradation environment and by improving their intestinal absorption (95). Uhl et al. (73) prepared a vancomycin-loaded liposome containing glycerylcaldityltetraether lipid (TEL). For in vivo studies, different vancomycin formulations were administered by gavage in Wistar rats. The obtained bioavailability 1 h after oral administration demonstrated that a 3-fold increase of vancomycin was observed by using TEL-liposome. Anderson et al. (72) formulated another vancomycin-loaded liposomes coated with folic acid to target intestinal epithelial cells expressing folic acid receptors. After the oral administration of vancomycin formulations (61.75 mg.kg−1) in Sprague–Dawley rats, the absolute bioavailability was 1.74, 6.7, and 21.8% from vancomycin solution, uncoated liposome, and folic acid-coated liposome, respectively. Thus, the folic acid and the liposomal formulation increase by a 12.5-fold the vancomycin bioavailability compared to vancomycin solution. In the same way, Arregui et al. (64) prepared a proliposome formulation [defined as dry, free-flowing particles coated with phospholipids, which can immediately form a liposomal suspension when in contact with water (96)] containing diacetyl phosphate and stearylamine to increase drug loading and to enhance the oral absorption of daptomycin. Pharmacokinetic studies in rats demonstrated that, after the oral administration of daptomycin at a dosage of 40 mg.kg–1, a greater AUC0−t (46.39 ± 5.69 μg/ml/h), and higher Cmax (8.35 ± 0.64 μg/ml) were observed with proliposomal compared to free drug (AUC0−t and Cmax were less than the limit of quantification) (Table 2).
Nanoparticles (including nanospheres and nanocapsules) are lipid- and polymer-based nanocarriers. These nanoparticle approaches have some advantages for oral administration by protecting the encapsulated drug from enzymatic degradation, facilitating mucus diffusion, and membrane permeation (45). All these systems have already demonstrated their ability to improve peptide bioavailability (97–99).
Concerning AMPs, niosomes were developed. Niosomes (lipid-based nanoparticles) are similar to liposomes, but a bilayer is formed by a non-ionic surfactant and stabilized by the addition of cholesterol. These particles show a high stability in the gastric environment and a high permeability across the intestine (100). Polymyxin B niosomes were prepared using sorbitan monostearate (Span® 60) and cholesterol. Vesicles were stable in simulated gastric and intestinal fluids, and about 86.2 and 78.5% of polymyxin B were retained, respectively (Table 2). Pharmacokinetic studies (at 2.0 mg.kg–1 dosage of polymyxin B in rats) demonstrated that polymyxin B niosome administrated orally present pharmacokinetic parameters (AUC0−48, t1/2) similar to polymyxin B sulfate administrated intravenously (68).
Polymeric nanoparticles provide also the possibility to enhance oral absorption. Eudragit® RL 100 [a copolymer of poly (ethylacrylate and methyl-metacrylate)] nanoparticles were prepared by nanoprecipitation to encapsulate ATV. During in vivo pharmacokinetic studies in rats (at 7.2 mg.kg−1 dosage of ATV), the values of Cmax and AUC0−24 increased to 1.1- and 2.91-fold, with pure drug and optimized nanoparticle formulation, respectively. These results demonstrate the considerable performance of the nanoparticulate DDS in enhancing the bioavailability of ATV (63).
To date, there have been numerous studies investigating the interest of AMPs. Indeed, they display an antibiotic alternative and have already demonstrated their great potential to treat infectious diseases, including those caused by MDR strains. To be approved as therapeutic agents, AMPs must overcome their disadvantages, which limit their administration. Thus, in terms of future perspectives, one of the biggest challenges will be the development of pharmaceutical forms of the AMPs for all routes of administration and especially, oral administration, the main convenient administration route. Nevertheless, formulated with conventional forms, AMPs are always subjected to their limitations (enzymatic degradation, low bioavailability, rapid metabolism, opsonization, etc.). By this way, nano- and micro-DDSs emerge to play a critical role in determining the success of current and future products. Indeed, nanotechnology is an emerging field that offers a unique potential in comparison with conventional forms, including, for example, the protection against biological environment, the transport through barrier and cells, the improvement of bioavailability, or release modification. In this review, we have described the significance and high potential of AMPs to treat both systemic and local gastrointestinal infectious diseases as well as the oral AMP delivery systems available as innovative formulations. These new systems have to overcome the lack of specific regulatory guidelines notably for their characterizations (the absence of harmonized standard protocols), the complexity of their process of formulation and scale-up, and the uncertainties of their toxicity. They also require additional in vivo pharmacokinetic studies. In future, the encapsulation of AMPs into the DDS and their use in synergy with conventional antimicrobial drugs could be promising to obtain more effective oral treatments especially to treat MDR pathogen infections.
ER conceived the original idea. MA initiated the literature research and contributed Figure 2. CD, VA-M, and ER organized the literature research and wrote the final version. All authors contributed to this article and approved the submitted version.
MA was funded by NANOMED EMJMD supported by the European Union and the Erasmus+ Program by the European Union in the Framework Agreement Number 2016–2057/001-001 EMJMD, No. 574439-EPP-1-FR-EPPKA1-JMD-MOB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Marine Galloux for providing drawings for the design of Figure 1.
1. Baekkeskov E, Rubin O, Munkholm L, Zaman W. Antimicrobial Resistance as a Global Health Crisis. Oxford Research Encyclopedia of Politics. (2020). Retrieved from: https://oxfordre.com/politics/view/10.1093/acrefore/9780190228637.001.0001/acrefore-9780190228637-e-1626
2. Dadgostar P. Antimicrobial resistance: implications and costs. Infect Drug Resist. (2019) 12:3903–10. doi: 10.2147/IDR.S234610
3. Romandini A, Pani A, Schenardi PA, Pattarino GAC, De Giacomo C, Scaglione F. Antibiotic resistance in pediatric infections: global emerging threats, predicting the near future. Antibiotics (Basel). (2021) 10:393. doi: 10.3390/antibiotics10040393
4. Toner E, Adalja A, Gronvall GK, Cicero A, Inglesby TV. Antimicrobial resistance is a global health emergency. Health Secur. (2015) 13:153–5. doi: 10.1089/hs.2014.0088
5. Gupta AK, Renaud HJ, Quinlan EM, Shear NH, Piguet V. The growing problem of antifungal resistance in onychomycosis and other superficial mycoses. Am J Clin Dermatol. (2021) 22:149–57. doi: 10.1007/s40257-020-00580-6
6. Mason S, Devincenzo JP, Toovey S, Wu JZ, Whitley RJ. Comparison of antiviral resistance across acute and chronic viral infections. Antiviral Res. (2018) 158:103–12. doi: 10.1016/j.antiviral.2018.07.020
7. O'Neill J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. The Review on antimicrobial resistance (2016).
9. WHO. Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report 2021. Geneva: WHO (2021).
10. Magana M, Pushpanathan M, Santos AL, Leanse L, Fernandez M, Ioannidis A, et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect Dis. (2020) 20:e216–30. doi: 10.1016/S1473-3099(20)30327-3
11. Mahlapuu M, Bjorn C, Ekblom J. Antimicrobial peptides as therapeutic agents: opportunities and challenges. Crit Rev Biotechnol. (2020) 40:978–92. doi: 10.1080/07388551.2020.1796576
12. Mwangi J, Hao X, Lai R, Zhang Z-Y. Antimicrobial peptides: new hope in the war against multidrug resistance. Zool Res. (2019) 40:488. doi: 10.24272/j.issn.2095-8137.2019.062
13. Divyashree M, Mani MK, Reddy D, Kumavath R, Ghosh P, Azevedo V, et al. Clinical applications of antimicrobial peptides (AMPs): where do we stand now? Protein Pept Lett. (2020) 27:120–34. doi: 10.2174/0929866526666190925152957
14. Lewies A, Du Plessis LH, Wentzel JF. Antimicrobial peptides: the Achilles' heel of antibiotic resistance? Probiotics Antimicrob Proteins. (2019) 11:370–81. doi: 10.1007/s12602-018-9465-0
15. Vilas Boas LCP, Campos ML, Berlanda RLA, de Carvalho Neves N, Franco OL. Antiviral peptides as promising therapeutic drugs. Cell Mol Life Sci. (2019) 76:3525–42. doi: 10.1007/s00018-019-03138-w
16. Buda De Cesare G, Cristy SA, Garsin DA, Lorenz MC. Antimicrobial peptides: a new Frontier in antifungal therapy. mBio. (2020) 11. doi: 10.1128/mBio.02123-20
17. Buccini DF, Cardoso MH, Franco OL. Antimicrobial Peptides and Cell-Penetrating Peptides for Treating Intracellular Bacterial Infections. Front Cell Infect Microbiol. (2020) 10:612931. doi: 10.3389/fcimb.2020.612931
18. Di Somma A, Moretta A, Cane C, Cirillo A, Duilio A. Antimicrobial and antibiofilm peptides. Biomolecules. (2020) 10:652. doi: 10.3390/biom10040652
19. Lei J, Sun L, Huang S, Zhu C, Li P, He J, et al. The antimicrobial peptides and their potential clinical applications. Am J Transl Res. (2019) 11:3919–31.
20. Pfalzgraff A, Brandenburg K, Weindl G. Antimicrobial peptides and their therapeutic potential for bacterial skin infections and wounds. Front Pharmacol. (2018) 9:281. doi: 10.3389/fphar.2018.00281
21. Hua S. Advances in oral drug delivery for regional targeting in the gastrointestinal tract - influence of physiological, pathophysiological and pharmaceutical factors. Front Pharmacol. (2020) 11:e00524. doi: 10.3389/fphar.2020.00524
22. Martin-Serrano A, Gomez R, Ortega P, de la Mata FJ. Nanosystems as vehicles for the delivery of antimicrobial peptides (AMPs). Pharmaceutics. (2019) 11:448. doi: 10.3390/pharmaceutics11090448
23. Teixeira MC, Carbone C, Sousa MC, Espina M, Garcia ML, Sanchez-Lopez E, et al. Nanomedicines for the delivery of antimicrobial peptides (AMPs). Nanomaterials (Basel, Switzerland). (2020) 10:560. doi: 10.3390/nano10030560
24. Fleming A, Allison VD. Observations on a bacteriolytic substance (“lysozyme”) found in secretions and tissues. Br J Exp Pathol. (1922) 3:252–60.
25. Fleming A, Wright AE. On a remarkable bacteriolytic element found in tissues and secretions. Proc R Soc Lond Series B Cont Papers Biol Char. (1922) 93:306–17. doi: 10.1098/rspb.1922.0023
26. Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. (2016) 44:D1087–93. doi: 10.1093/nar/gkv1278
27. Waghu FH, Idicula-Thomas S. Collection of antimicrobial peptides database and its derivatives: applications and beyond. Protein Sci. (2020) 29:36–42. doi: 10.1002/pro.3714
28. Pirtskhalava M, Amstrong AA, Grigolava M, Chubinidze M, Alimbarashvili E, Vishnepolsky B, et al. v3: database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. (2021) 49:D288–97. doi: 10.1093/nar/gkaa991
29. Kang X, Dong F, Shi C, Liu S, Sun J, Chen J, et al. DRAMP 20, an updated data repository of antimicrobial peptides. Sci Data. (2019) 6:148. doi: 10.1038/s41597-019-0154-y
30. Huan Y, Kong Q, Mou H, Yi H. Antimicrobial peptides: classification, design, application and research progress in multiple fields. Front Microbiol. (2020) 11:e582779. doi: 10.3389/fmicb.2020.582779
31. Ageitos JM, Sanchez-Perez A, Calo-Mata P, Villa TG. Antimicrobial peptides (AMPs): ancient compounds that represent novel weapons in the fight against bacteria. Biochem Pharmacol. (2017) 133:117–38. doi: 10.1016/j.bcp.2016.09.018
32. Wu Q, Patočka J, Kuča K. Insect antimicrobial peptides, a mini review. Toxins. (2018) 10:461. doi: 10.3390/toxins10110461
33. Patocka J, Nepovimova E, Klimova B, Wu Q, Kuca K. Antimicrobial peptides: amphibian host defense peptides. Curr Med Chem. (2019) 26:5924–46. doi: 10.2174/0929867325666180713125314
34. Masso-Silva JA, Diamond G. Antimicrobial peptides from fish. Pharmaceuticals (Basel). (2014) 7:265–310. doi: 10.3390/ph7030265
35. Kumar P, Kizhakkedathu JN, Straus SK. Antimicrobial peptides: diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules. (2018) 8:4. doi: 10.3390/biom8010004
36. Mishra AK, Choi J, Moon E, Baek KH. Tryptophan-rich and proline-rich antimicrobial peptides. Molecules. (2018) 23:815. doi: 10.3390/molecules23040815
37. Koehbach J, Craik DJ. The vast structural diversity of antimicrobial peptides. Trends Pharmacol Sci. (2019) 40:517–28. doi: 10.1016/j.tips.2019.04.012
38. Mura M, Wang J, Zhou Y, Pinna M, Zvelindovsky AV, Dennison SR, et al. The effect of amidation on the behaviour of antimicrobial peptides. Eur Biophys J. (2016) 45:195–207. doi: 10.1007/s00249-015-1094-x
39. Nguyen LT, Haney EF, Vogel HJ. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. (2011) 29:464–72. doi: 10.1016/j.tibtech.2011.05.001
40. Shafee TM, Lay FT, Phan TK, Anderson MA, Hulett MD. Convergent evolution of defensin sequence, structure and function. Cell Mol Life Sci. (2017) 74:663–82. doi: 10.1007/s00018-016-2344-5
41. Hegemann JD, Zimmermann M, Xie X, Marahiel MA. Lasso peptides: an intriguing class of bacterial natural products. Acc Chem Res. (2015) 48:1909–19. doi: 10.1021/acs.accounts.5b00156
42. Carratalá JV, Serna N, Villaverde A, Vázquez E, Ferrer-Miralles N. Nanostructured antimicrobial peptides: the last push towards clinics. Biotechnol Adv. (2020) 44:107603. doi: 10.1016/j.biotechadv.2020.107603
43. Halaby T, Naiemi N, Al Kluytmans J, Van Der Palen J, Vandenbroucke-Grauls CMJE. Emergence of colistin resistance in Enterobacteriaceae after the introduction of selective digestive tract decontamination in an intensive care unit. Antimicrobial Agents Chemother. (2013) 57:3224–9. doi: 10.1128/AAC.02634-12
44. European Medicines Agency (EMA). Targocid Article-30 Referral - Annex III Summary of Product Characteristics, Labelling and Package Leaflet. Amsterdam: European Medicines Agency (EMA) (2014). p. 19–50.
45. Aguirre TAS, Teijeiro-Osorio D, Rosa M, Coulter IS, Alonso MJ, Brayden DJ. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv Drug Deliv Rev. (2016) 106:223–41. doi: 10.1016/j.addr.2016.02.004
46. European Medicines Agency E. Data Submission on Authorised Medicines (Article 57). (2021). Available online at: https://www.ema.europa.eu/en/human-regulatory/post-authorisation/data-medicines-iso-idmp-standards/data-submission-authorised-medicines-article-57 (accessed July 12, 2021).
47. European Medicines Agency (EMA). Annex III Amendments to Relevant Sections of the Summary of Vancomycin Product Characteristics and Package Leaflets. Amsterdam: European Medicines Agency (EMA) (2001). p. 45–8.
48. Chen CH, Lu TK. Development and challenges of antimicrobial peptides for therapeutic applications. Antibiotics (Basel). (2020) 9:24. doi: 10.3390/antibiotics9010024
49. Browne K, Chakraborty S, Chen R, Willcox MDP, Black DS, Walsh WR, et al. new era of antibiotics: the clinical potential of antimicrobial peptides. Int J Mol Sci. (2020) 21:1–23. doi: 10.3390/ijms21197047
50. Mahlapuu M, Hakansson J, Ringstad L, Bjorn C. Antimicrobial peptides: an emerging category of therapeutic agents. Front Cell Infect Microbiol. (2016) 6:194. doi: 10.3389/fcimb.2016.00194
51. Zhang LJ, Gallo RL. Antimicrobial peptides. Curr Biol. (2016) 26:R14–9. doi: 10.1016/j.cub.2015.11.017
52. Abarbanel DN, Seki SM, Davies Y, Marlen N, Benavides JA, Cox K, et al. Immunomodulatory effect of vancomycin on Treg in pediatric inflammatory bowel disease and primary sclerosing cholangitis. J Clin Immunol. (2013) 33:397–406. doi: 10.1007/s10875-012-9801-1
53. Han Y, Gao Z, Chen L, Kang L, Huang W, Jin M, et al. Multifunctional oral delivery systems for enhanced bioavailability of therapeutic peptides/proteins. Acta Pharmaceutica Sinica B. (2019) 9:902–22. doi: 10.1016/j.apsb.2019.01.004
54. Stiefel U, Pultz NJ, Helfand MS, Donskey CJ. Efficacy of oral ramoplanin for inhibition of intestinal colonization by vancomycin-resistant Enterococci in mice. Antimicrobial Agents Chemother. (2004) 48:2144–8. doi: 10.1128/AAC.48.6.2144-2148.2004
55. Lamarche MJ, Leeds JA, Amaral A, Brewer JT, Bushell SM, Deng G, et al. Discovery of LFF571: an investigational agent for Clostridium difficile infection. J Med Chem. (2012) 55:2376–87. doi: 10.1021/jm201685h
56. Mullane K, Lee C, Bressler A, Buitrago M, Weiss K, Dabovic K, et al. Multicenter, randomized clinical trial to compare the safety and efficacy of lff571 and vancomycin for Clostridium difficile infections. Antimicrobial Agents Chemother. (2015) 59:1435–40. doi: 10.1128/AAC.04251-14
57. Ting LSL, Praestgaard J, Grunenberg N, Yang JC, Leeds JA, Pertel P, et al. first-in-human, randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose study to assess the safety and tolerability of LFF571 in healthy volunteers. Antimicrobial Agents Chemother. (2012) 56:5946–51. doi: 10.1128/AAC.00867-12
58. Ugurlu T, Turkoglu M, Gurer US, Akarsu BG. Colonic delivery of compression coated nisin tablets using pectin/HPMC polymer mixture. Eur J Pharm Biopharm. (2007) 67:202–10. doi: 10.1016/j.ejpb.2007.01.016
59. Hodges LA, Connolly SM, Band J, O'Mahony B, Ugurlu T, Turkoglu M, et al. Scintigraphic evaluation of colon targeting pectin-HPMC tablets in healthy volunteers. Int J Pharm. (2009) 370:144–50. doi: 10.1016/j.ijpharm.2008.12.002
60. Chandorkar G, Zhan Q, Donovan J, Rege S, Patino H. Pharmacokinetics of surotomycin from phase 1 single and multiple ascending dose studies in healthy volunteers. BMC Pharmacol Toxicol. (2017) 18:1–8. doi: 10.1186/s40360-017-0123-z
61. Fukushima K, Terasaka S, Haraya K, Kodera S, Seki Y, Wada A, et al. Pharmaceutical approach to HIV protease inhibitor atazanavir for bioavailability enhancement based on solid dispersion system. Biol. Pharm. Bull. (2007) 30:733–8. doi: 10.1248/bpb.30.733
62. Singh G, Pai RS. Optimized self-nanoemulsifying drug delivery system of atazanavir with enhanced oral bioavailability: in vitro/in vivo characterization. Expert Opin Drug Deliv. (2014) 11:1023–32. doi: 10.1517/17425247.2014.913566
63. Singh G, Pai RS. Atazanavir-loaded eudragit RL 100 nanoparticles to improve oral bioavailability: optimization and in vitro/in vivo appraisal. Drug Deliv. (2016) 23:532–9. doi: 10.3109/10717544.2014.930760
64. Arregui JR, Kovvasu SP, Betageri GV. Daptomycin proliposomes for oral delivery: formulation, characterization, and in vivo pharmacokinetics. AAPS Pharm Sci Tech. (2018) 19:1802–9. doi: 10.1208/s12249-018-0989-0
65. Coppi G, Bondi M, Coppi A, Rossi T, Sergi S, Iannuccelli V. Toxicity and gut associated lymphoid tissue translocation of polymyxin B orally administered by alginate/chitosan microparticles in rats. J Pharm Pharmacol. (2010) 60:21–6. doi: 10.1211/jpp.60.1.0003
66. Coppi G, Sala N, Bondi M, Sergi S, Iannuccelli V. Ex-vivo evaluation of alginate microparticles for Polymyxin B oral administration. J Drug Target. (2006) 14:599–606. doi: 10.1080/10611860600864182
67. Coppi G, Iannuccelli V, Sala N, Bondi M. Alginate microparticles for Polymyxin B Peyer's patches uptake: microparticles for antibiotic oral administration. J Microencapsul. (2004) 21:829–39. doi: 10.1080/02652040400015437
68. Chauhan MK, Bhatt N. Bioavailability enhancement of polymyxin B with novel drug delivery: development and optimization using quality-by-design approach. J Pharm Sci. (2019) 108:1521–8. doi: 10.1016/j.xphs.2018.11.032
69. Kajita M, Morishita M, Takayama K, Chiba Y, Tokiwa S, Nagai T. Enhanced enteral bioavailability of vancomycin using water-in-oil-in-water multiple emulsion incorporating highly purified unsaturated fatty acid. J Pharm Sci. (2000) 89:1243–52. doi: 10.1002/1520-6017(200010)89:10<1243::AID-JPS2>3.0.CO;2-C
70. Prasad Y, Puthli SP, Eaimtrakarn S, Ishida M, Yoshikawa Y, Shibata N, et al. Enhanced intestinal absorption of vancomycin with Labrasol and D-α-tocopheryl PEG 1000 succinate in rats. Int J Pharm. (2003) 250:181–90. doi: 10.1016/S0378-5173(02)00544-6
71. Zaichik S, Steinbring C, Caliskan C, Bernkop-Schnürch A. Development and in vitro evaluation of a self-emulsifying drug delivery system (SEDDS) for oral vancomycin administration. Int J Pharm. (2019) 554:125–33. doi: 10.1016/j.ijpharm.2018.11.010
72. Anderson KE, Eliot LA, Stevenson BR, Rogers JA. Formulation and evaluation of a folic acid receptor-targeted oral vancomycin liposomal dosage form. Pharm Res. (2001) 18:316–22. doi: 10.1023/A:1011002913601
73. Uhl P, Pantze S, Storck P, Parmentier J, Witzigmann D, Hofhaus G, et al. Oral delivery of vancomycin by tetraether lipid liposomes. Eur J Pharm Sci. (2017) 108:111–8. doi: 10.1016/j.ejps.2017.07.013
74. U.S. Food and Drug Administration. Firvanq™ (Vancomycin Hydrochloride), for Oral Solution. Silver Spring, MD: U.S. Food and Drug Administration (2018).
75. Malhaire H, Gimel JC, Roger E, Benoît JP, Lagarce F. How to design the surface of peptide-loaded nanoparticles for efficient oral bioavailability? Adv Drug Deliv Rev. (2016) 106:320–36. doi: 10.1016/j.addr.2016.03.011
76. Brayden DJ, Hill TA, Fairlie DP, Maher S, Mrsny RJ. Systemic delivery of peptides by the oral route: formulation and medicinal chemistry approaches. Adv Drug Deliv Rev. (2020) 157:2–36. doi: 10.1016/j.addr.2020.05.007
77. Al-Hilal TA, Alam F, Byun Y. Oral drug delivery systems using chemical conjugates or physical complexes. Adv Drug Deliv Rev. (2013) 65:845–64. doi: 10.1016/j.addr.2012.11.002
78. Drucker DJ. Advances in oral peptide therapeutics. Nat Rev Drug Discov. (2020) 19:277–89. doi: 10.1038/s41573-019-0053-0
79. Ji S, Lu J, Liu Z, Srivastava D, Song A, Liu Y, et al. Dynamic encapsulation of hydrophilic nisin in hydrophobic poly (lactic acid) particles with controlled morphology by a single emulsion process. J Colloid Interface Sci. (2014) 423:85–93. doi: 10.1016/j.jcis.2014.02.025
80. Yamakami K, Tsumori H, Sakurai Y, Shimizu Y, Nagatoshi K, Sonomoto K. Sustainable inhibition efficacy of liposome-encapsulated nisin on insoluble glucan-biofilm synthesis by Streptococcus mutans. Pharm Biol. (2013) 51:267–70. doi: 10.3109/13880209.2012.717227
81. Yamakami K, Tsumori H, Shimizu Y, Sakurai Y, Nagatoshi K, Sonomoto K. Cationic lipid content in liposome-encapsulated nisin improves sustainable bactericidal activity against Streptococcus mutans. Open Dent J. (2016) 10:360–6. doi: 10.2174/1874210616021001360
82. Khaksar R, Hosseini SM, Hosseini H, Shojaee-Aliabadi S, Mohammadifar MA, Mortazavian AM., et al. Nisin-loaded alginate-high methoxy pectin microparticles: preparation and physicochemical characterization. Int J Food Sci Technol. (2014) 49:2076–82. doi: 10.1111/ijfs.12516
83. Prombutara P, Kulwatthanasal Y, Supaka N, Sramala I, Chareonpornwattana S. Production of nisin-loaded solid lipid nanoparticles for sustained antimicrobial activity. Food Control. (2012) 24:184–90. doi: 10.1016/j.foodcont.2011.09.025
84. Goudarzi F, Asadi A, Afsharpour M, Jamadi RH. In vitro characterization and evaluation of the cytotoxicity effects of nisin and nisin-loaded PLA-PEG-PLA nanoparticles on gastrointestinal (AGS and KYSE-30), hepatic (HepG2) and blood (K562) cancer cell lines. AAPS Pharm Sci Tech. (2018) 19:1554–66. doi: 10.1208/s12249-018-0969-4
85. Zohri M, Alavidjeh MS, Haririan I, Ardestani MS, Ebrahimi SES, Sani HT, et al. Comparative study between the antibacterial effect of nisin and nisin-loaded chitosan/alginate nanoparticles on the growth of Staphylococcus aureus in raw and pasteurized milk samples. Probiotics Antimicrob Proteins. (2010) 2:258–66. doi: 10.1007/s12602-010-9047-2
86. Gomaa AI, Martinent C, Hammami R, Fliss I, Subirade M. Dual coating of liposomes as encapsulating matrix of antimicrobial peptides: development and characterization. Front Chem. (2017) 5:e00103. doi: 10.3389/fchem.2017.00103
87. Rishi P, Bhogal A, Arora S, Pandey SK, Verma I, Kaur IP. Improved oral therapeutic potential of nanoencapsulated cryptdin formulation against Salmonella infection. Eur J Pharm Sci. (2015) 72:27–33. doi: 10.1016/j.ejps.2015.02.014
88. Roger E, Lagarce F, Garcion E, Benoit J-P. Biopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral delivery. Nanomedicine. (2010) 5:287–306. doi: 10.2217/nnm.09.110
89. Mahmood A, Bernkop-Schnürch A, SEDDS. A game changing approach for the oral administration of hydrophilic macromolecular drugs. Adv Drug Deliv Rev. (2019) 142:91–101. doi: 10.1016/j.addr.2018.07.001
90. Zupančič O, Partenhauser A, Lam HT, Rohrer J, Bernkop-Schnürch A. Development and in vitro characterisation of an oral self-emulsifying delivery system for daptomycin. Eur J Pharm Sci. (2016) 81:129–36. doi: 10.1016/j.ejps.2015.10.005
91. Singh MN, Hemant KSY, Ram M, Shivakumar HG. Microencapsulation: a promising technique for controlled drug delivery. Res Pharm Sci. (2010) 5:65–77.
92. Barenholz Y. Doxil® - The first FDA-approved nano-drug: lessons learned. J Control Release. (2012) 160:117–34. doi: 10.1016/j.jconrel.2012.03.020
93. Kokkona M, Kallinteri P, Fatouros D, Antimisiaris SG. Stability of SUV liposomes in the presence of cholate salts and pancreatic lipases: effect of lipid composition. Eur J Pharm Sci. (2000) 9:245–52. doi: 10.1016/S0928-0987(99)00064-0
94. He H, Lu Y, Qi J, Zhu Q, Chen Z, Wu W. Adapting liposomes for oral drug delivery. Acta Pharmaceutica Sinica B. (2019) 9:36–48. doi: 10.1016/j.apsb.2018.06.005
95. Nguyen TX, Huang L, Gauthier M, Yang G, Wang Q. Recent advances in liposome surface modification for oral drug delivery. Nanomedicine. (2016) 11:1169–85. doi: 10.2217/nnm.16.9
96. Nekkanti V, Venkatesan N, Betageri G. Proliposomes for oral delivery: progress and challenges. Curr Pharm Biotechnol. (2015) 16:303–12. doi: 10.2174/1389201016666150118134256
97. Choonara BF, Choonara YE, Kumar P, Bijukumar D, du Toit LC, Pillay V, et al. review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol Adv. (2014) 32:1269–82. doi: 10.1016/j.biotechadv.2014.07.006
98. Lakkireddy HR, Urmann M, Besenius M, Werner U, Haack T, Brun P, et al. Oral delivery of diabetes peptides—comparing standard formulations incorporating functional excipients and nanotechnologies in the translational context. Adv Drug Deliv Rev. (2016) 106:196–222. doi: 10.1016/j.addr.2016.02.011
99. Jun CS, Xu S, Ming WH, Ling Y, Dong J, Dong XR, et al. Nanoparticles: oral delivery for protein and peptide drugs. AAPS Pharm Sci Tech. (2019) 20:1–11. doi: 10.1208/s12249-019-1325-z
Keywords: oral route, antimicrobial peptides (AMPs), infectiology, pharmaceutical forms, drug delivery systems (DDS)
Citation: Deshayes C, Arafath MN, Apaire-Marchais V and Roger E (2022) Drug Delivery Systems for the Oral Administration of Antimicrobial Peptides: Promising Tools to Treat Infectious Diseases. Front. Med. Technol. 3:778645. doi: 10.3389/fmedt.2021.778645
Received: 17 September 2021; Accepted: 06 December 2021;
Published: 25 January 2022.
Edited by:
Ana Beloqui, Catholic University of Louvain, BelgiumReviewed by:
Inmaculada Conejos-Sanchez, Principe Felipe Research Center (CIPF), SpainCopyright © 2022 Deshayes, Arafath, Apaire-Marchais and Roger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emilie Roger, ZW1pbGllLnJvZ2VyQHVuaXYtYW5nZXJzLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.