 Olivia Hagon-Nicod1
Olivia Hagon-Nicod1 Florence Fellmann2Jan Novy3Sébastien Lebon4Christian Wider3†
Florence Fellmann2Jan Novy3Sébastien Lebon4Christian Wider3† Romain Lazor5Olivier Bonny6,7*
Romain Lazor5Olivier Bonny6,7*- 1Department of Medicine, Service of Internal Medicine, Fribourg State Hospital, Fribourg, Switzerland
- 2The Collaboratory, University of Lausanne, Lausanne, Switzerland
- 3Department of Medicine, Service of Neurology, Lausanne University Hospital, Lausanne, Switzerland
- 4Unit of Pediatric Neurology and Neurorehabilitation, Woman-Mother-Child Department, Lausanne University Hospital, Lausanne, Switzerland
- 5Department of Medicine, Service of Respiratory Medicine, Lausanne University Hospital, Lausanne, Switzerland
- 6Department of Medicine, Service of Nephrology, Fribourg State Hospital, Fribourg, Switzerland
- 7Department of Medicine, Service of Nephrology, Lausanne University Hospital, Lausanne, Switzerland
Aim of the study: Tuberous sclerosis complex (TSC) is a genetic and multisystemic disorder that affects between 1/6’000 and 1/10’000 of newborns. Clinical criteria and/or genetic analysis establish the diagnosis. The mechanistic target of rapamycin (mTOR) inhibitors everolimus or sirolimus reduce the severity of several TSC-related clinical traits. We report here on the epidemiology and management of TSC patients in a large Swiss canton: the canton of Vaud.
Method: We extracted patient files containing the diagnostic code TSC in 2015 at the Lausanne University hospital (tertiary reference center for a population of about 755’000 people) and in specialized neurological institutions of the same region.
Results: We identified 52 patients with a diagnosis of TSC. The majority of the patients with a pathologic result in genetic testing were positive for a pathogenic variant in the TSC2 gene, including five cases of contiguous gene deletion syndrome of TSC2 and PKD1 causing both polycystic kidney disease and TSC. The most frequent clinical manifestations encountered were affecting the skin (87% of patients), the brain (83%), the heart (46%) and the kidneys (46%). Neuropsychiatric disorders were described in 56% of cases. At the time of data collection (2015), there were 2 patients using systemic mTOR inhibitors and 16 patients using topical mTOR inhibitors for dermatological features. Next, we compared this data with those of large published cohorts. While we found fewer cases than expected for the screened population, demographic as well as genetic data were overall similar to the literature. However, we observed that some clinical manifestations (renal, lung and neuropsychiatric disorders) were less frequently described in our cohort.
Conclusion: This work indicates that TSC and some of its clinical manifestations is under-reported. It raises concern that patients with mild manifestations are often not referred to reference centers with dedicated multidisciplinary group. The follow-up by expert board is instrumental in offering systematic screening of all putatively affected organs and to assess the eligibility for targeted treatment.
1 Introduction
Tuberous sclerosis complex (TSC) is a rare genetic disorder that may affect almost any organ. The incidence of the disease is estimated between 1/6’000 and 1/10’000 newborns. The pattern of transmission is autosomal dominant, however, about two thirds of cases are linked to de novo mutations. The disease is caused by pathogenic variants in TSC1 or TSC2 genes that alter the function of proteins, respectively, called hamartin and tuberin. These two proteins form a heterodimer that regulates cell growth and proliferation through the mechanistic target of rapamycin (mTOR) complex. In TSC, the dimer is not functional and the mTOR cascade is constitutively activated, resulting in the formation of multiples benign tumors (also called hamartoma) throughout the body (1–4).
The clinical manifestations of TSC are broad with highly variable degrees of severity but, overall, the morbidity and mortality of this condition are high. The main affected organs are the brain {tubers/migrational defects, epilepsy, subependymal nodules (SENs) or subependymal giant cell astrocytomas (SEGAs)}, the kidneys {angiomyolipomas (AMLs), cysts}, the skin {for example angiofibroma, hypomelanic macules}, the lungs {lymphangioleiomyomatosis (LAM), multifocal micronodular pneumocyte hyperplasia (MMPH)}, the heart {rhabdomyomas} and the eyes {retinal hamartomas}. In addition, the patients often have TSC-associated neuropsychiatric disorders (TAND) such as autism spectrum disorders, intellectual disability, attention deficit/hyperactivity disorders or anxiety disorders (5–9).
For years, only symptomatic treatments were available. However, advances in the understanding of the role of the mTOR pathway in TSC have led to the use of mTOR inhibitors (10, 11). Everolimus is approved for patients with SEGAs when surgery is not possible or when the tumor is invasive, bilateral or multiple (12, 13). Moreover, everolimus is approved for patients with renal AMLs, reducing the tumor size and preventing spontaneous rupture and retroperitoneal bleeding, an event which occurs in large AMLs and may be life-threatening (14–16). In 2018, mTOR inhibitors were approved in Switzerland for patients with intractable epilepsy due to its effect in reducing seizures frequency (17–19). Pulmonary LAM is also stabilized by mTOR inhibitors (20, 21), and an effect of everolimus has also been observed on MMPH (22). For skin lesions, the use of topical mTOR inhibitors is preferred if no systemic indication is warranted (23). However, everolimus is not effective in TAND (24).
The Canton of Vaud in Switzerland has a population of about 755′000 people and its University hospital is a tertiary reference center for the whole state. No data on the number of patients suffering from TSC or on their follow-up and treatment are available in the covered region. We aimed at finding patients followed up in the state and at describing the demography and clinical characteristics. Our second aim was to identify patients that would benefit from mTOR inhibitors treatment.
2 Methods
The Ethics Committee of the State approved the protocol. Given the observational nature of the study, the need for informed consent was waived. The data were anonymized. We asked the archiving center to extract the charts of all patients carrying the diagnosis of TSC found in electronic archives of the Lausanne University hospital in 2015. In parallel, the physicians in charge of TSC patients who are part of the TSC board of the Lausanne University hospital (regrouping specialists of the pediatric and adult ages for each of the organs involved in TSC) contributed to patients identification as well as specialized neurologic institutions of the Canton of Vaud where patients with epilepsy and/or intellectual disability might be living (especially the Institution de Lavigny).
We used the modified criteria of Gomez to ascertain that the patients met the diagnosis of TSC (6). We collected information on gender, age, ethnicity, involved organs, genetic analysis and treatments received.
Descriptive statistics were used as appropriate. For age, we used the median value and standard deviation as the distribution of our population was asymmetrical. The prevalence was calculated according to the population of the state in 2015. When no mention was made in the file of a manifestation or a treatment, we inferred that the patient did not suffer from the said manifestation or received the said treatment.
3 Results
3.1 Demography
We identified 52 patients with a diagnosis of TSC in 2015 in the State of Vaud, Switzerland in which lives a population of about 755′000 persons. Prevalence is thus about 1/15′000. The median age at the time of data collection of the patients was 23 (±16) years (Figure 1) and the majority of patients were females (60%).
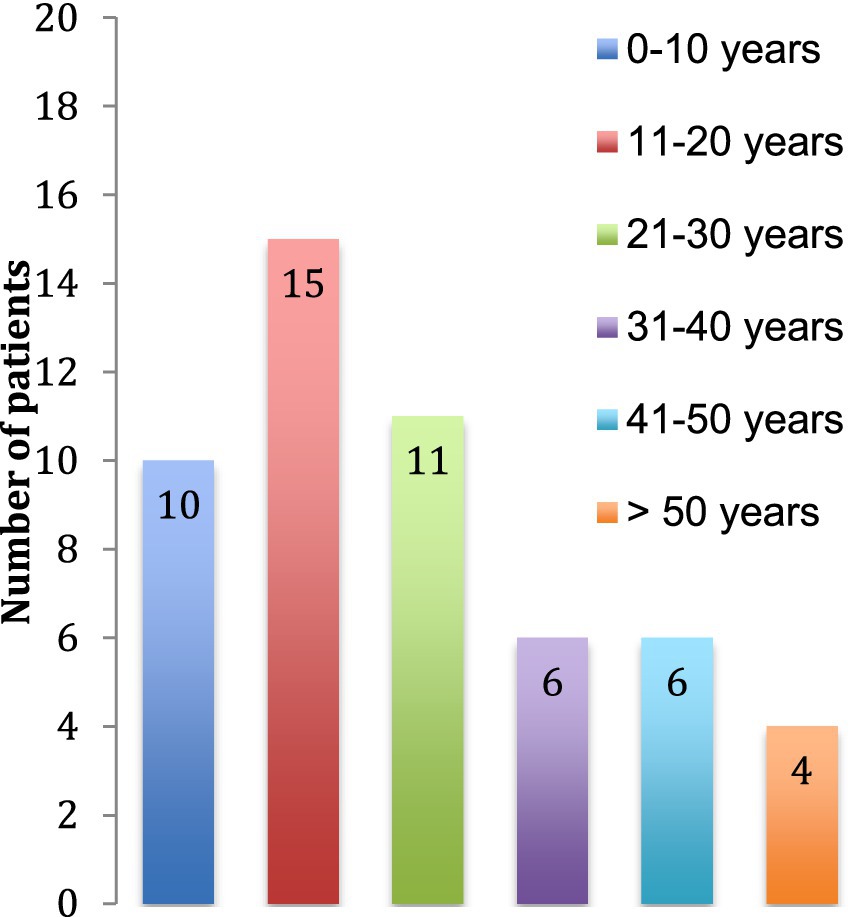
Figure 1. Age of the patients at the time of the study.
3.2 Clinical presentation
The first manifestations of TSC retrieved in the patient charts were epilepsy, cardiac rhabdomyomas and facial angiofibromas (Figure 2). They appeared at a median age of 9 months (± 6 years). The diagnosis of TSC was established at a median age of 5 years (± 9 years) usually when the addition of other features than the initial manifestation raised the suspicion of a systemic and more complex disorder. In 59% of cases, TSC was recognized as the initial diagnosis but in 41% of the cases, patients were first categorized under various forms of epilepsy.
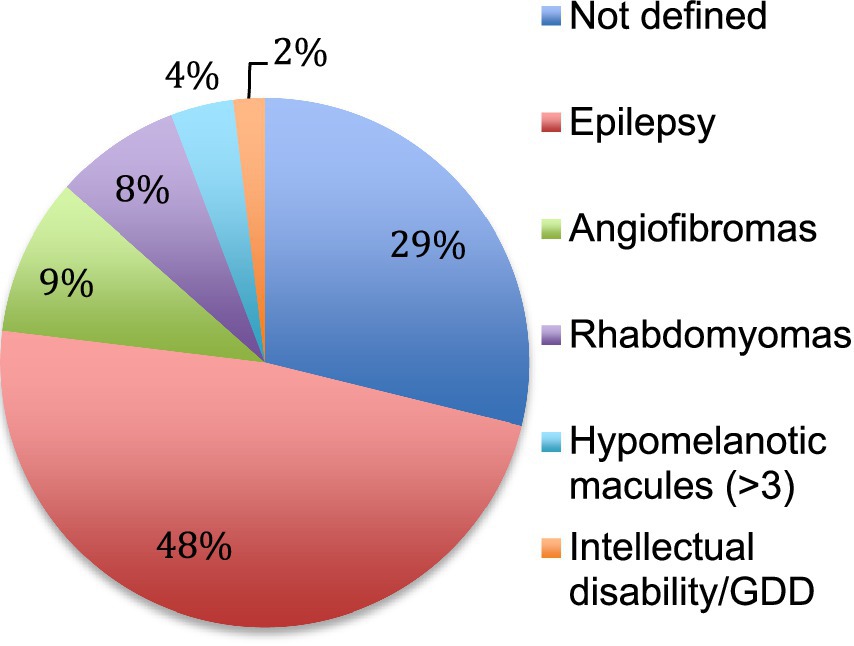
Figure 2. First manifestation of TSC. The first clinical manifestation of TSC is reported in percentage. Not defined means that the initial manifestation was not retrieved in the patient chart. GDD: global developmental delay.
We further analyzed the pattern of organ involvement. The most frequent manifestations were neurologic (47 patients, 90%), including neuropsychiatric disorders, followed by dermatologic (45 patients, 87%), cardiac (24 patients, 46%) and renal (24 patients, 46%). Lung manifestations, found in 13% (7 patients) of the cohort, developed later than the others, at a mean age of 30 years and were reported as LAM, lung cysts, MMPH and, in one case, pneumothorax.
The neurological features consisted in structural brain abnormalities, TAND and epilepsy. On 41 brain MRI available, we found 41 with tubers, 27 with SENs and 10 with SEGAs (Figure 3). The TSC-associated neuropsychiatric disorders we found were intellectual disabilities in 28 patients and autism in 5 patients. Regarding epilepsy, the patients often had focal epilepsy or infantile epileptic spasms syndrome.
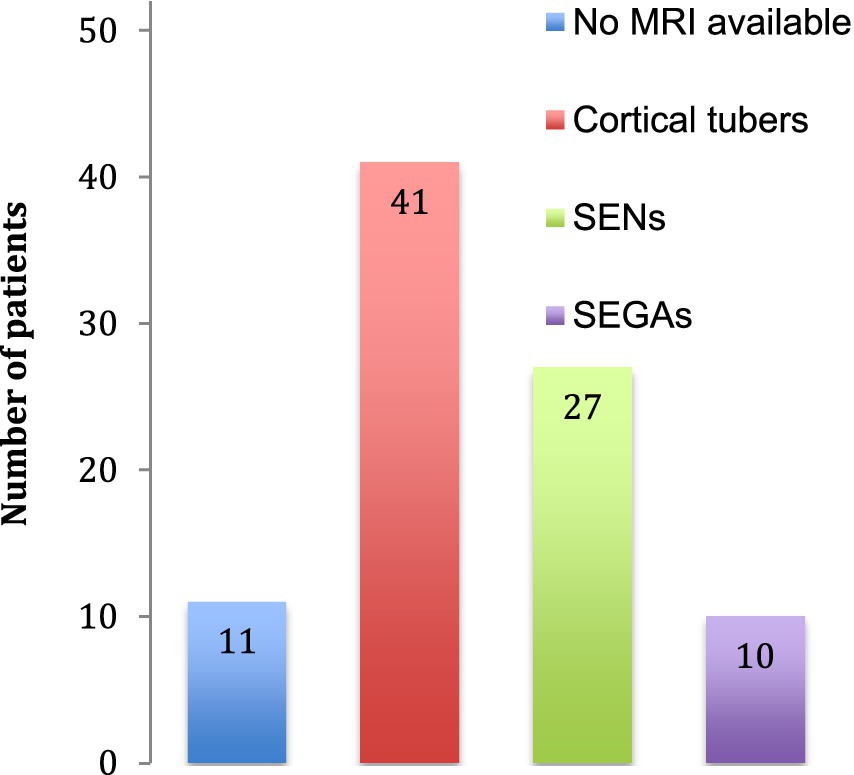
Figure 3. Cerebral abnormalities. The number of patients identified with cerebral abnormalities is indicated. No MRI was available for 11 patients. SENs: subependymal nodules, SEGAs: subependymal giant cell astrocytomas.
Regarding renal phenotypic presentation, high blood pressure was reported in 7 patients of the cohort and chronic renal disease in 6 patients. Ophthalmologic, oral, hepatic and splenic manifestations were also described, but less frequently (Table 1).
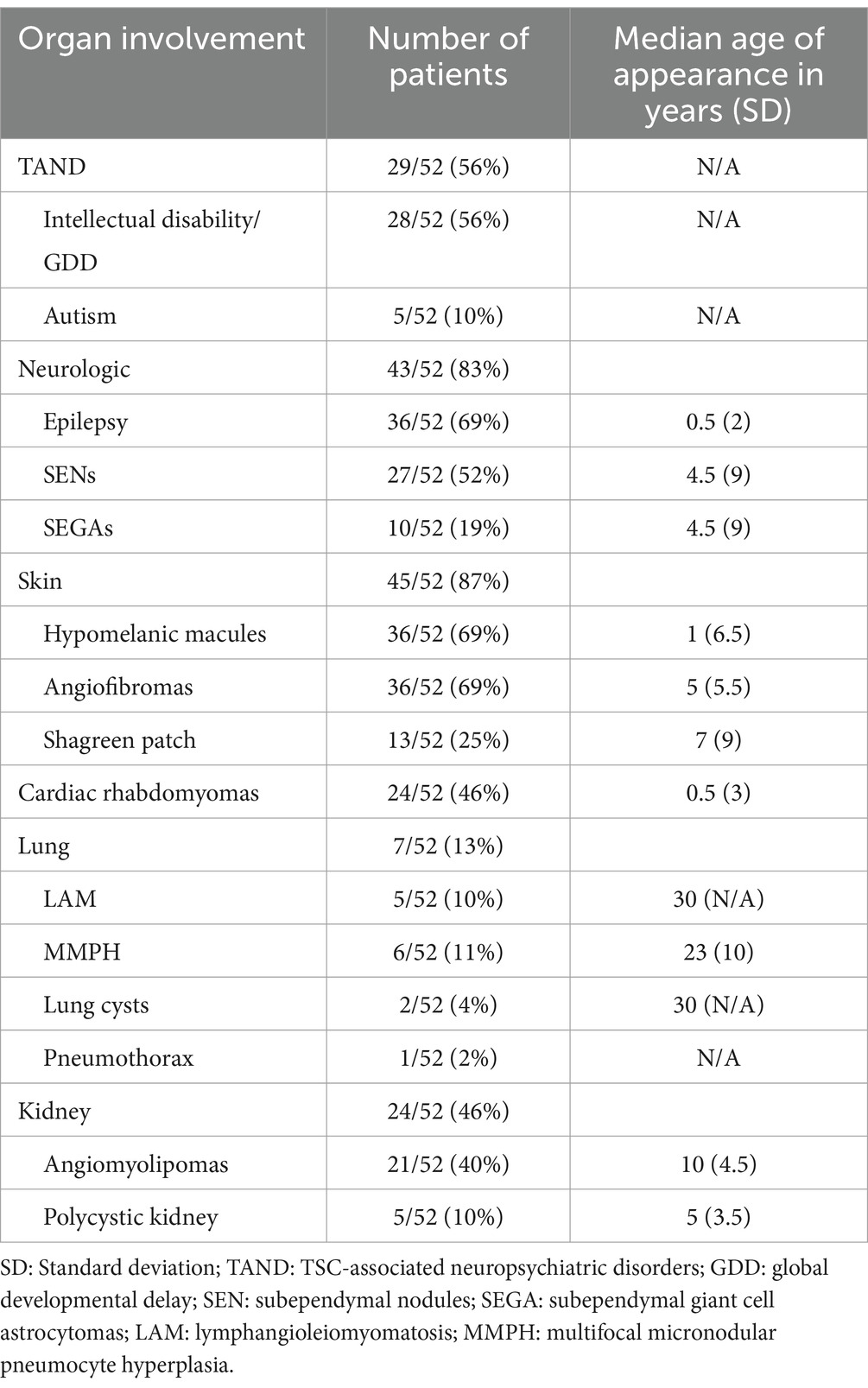
Table 1. Clinical manifestations.
3.3 Treatments
The main treatment category used was antiseizure medication with one to four drug classes combined. At the time of data collection, there were only 2 patients using systemic mTOR inhibitors, but 16 used topical mTOR inhibitors for dermatological purposes. Thirteen patients (25%) had surgery for neurological problems (symptomatic SEGAs or refractory epilepsy) and 6 patients (11%) had selective renal artery embolization or nephrectomy for AML (Figure 4).
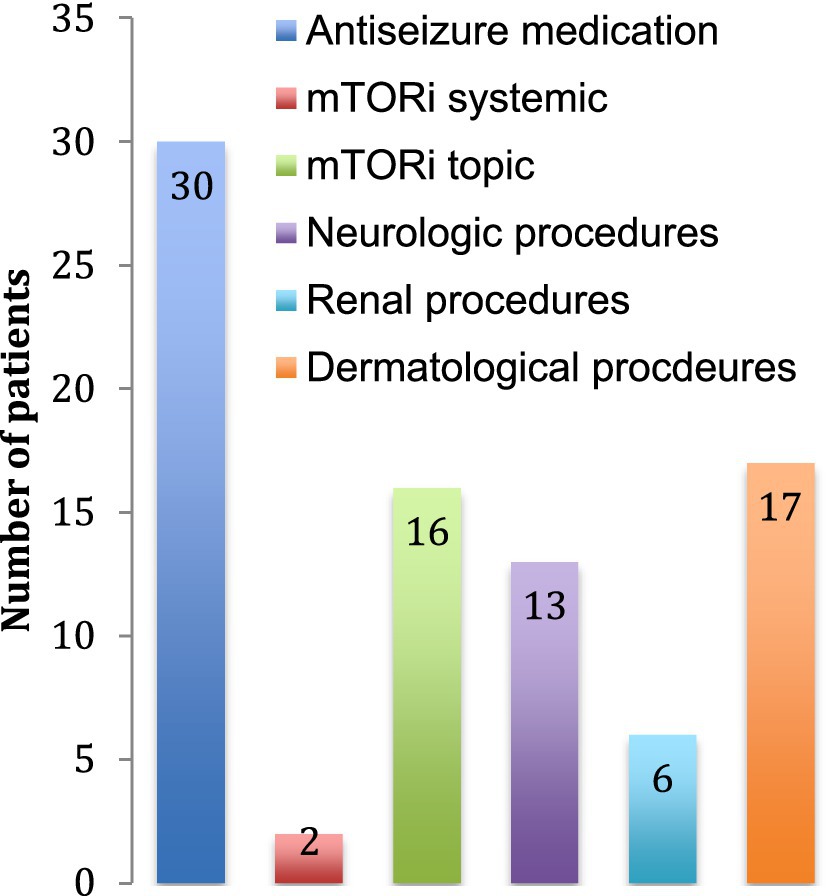
Figure 4. Treatment received. The number of patients receiving each treatment is indicated. Neurological procedures include tumor resections, lobectomies and ventriculo-peritoneal shunts. Renal procedures include arterial embolization, nephrectomy and renal transplantation. Dermatological procedures include laser treatments and excisions of skin tumors.
3.4 Genetics
Genetic analysis was performed in 29 patients (56%). Results were available for 25 patients. In 5 patients, no pathogenic variant was identified. Fifteen pathogenic variants were identified by sequencing analysis, 8 in the TSC2 gene and 7 in the TSC1 gene. In addition, a contiguous gene deletion syndrome (deletion of both TSC2 and PKD1 genes, causing both TSC and polycystic kidney disease) was identified in 5 cases (Figure 5).
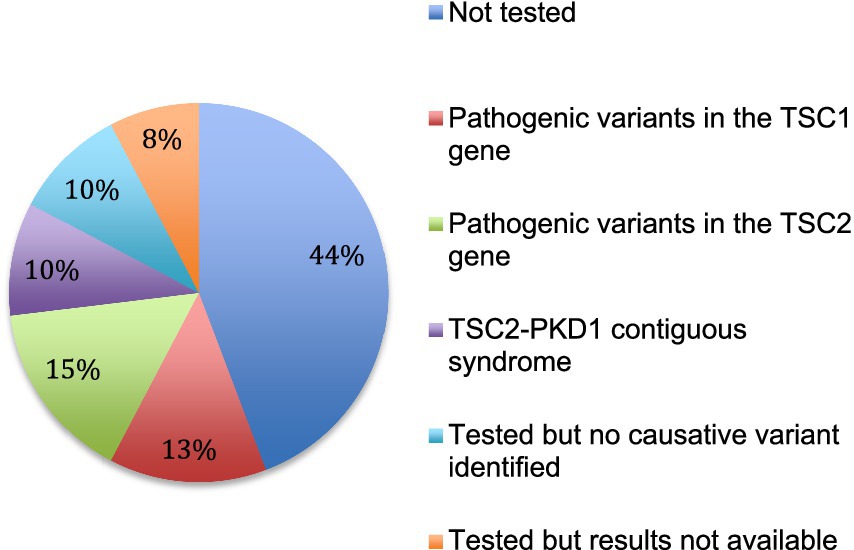
Figure 5. Genetic testing. The results of genetic analysis for the 52 patients are indicated.
Five patients had children at the time of data collection. Of these 5 children, 4 were genetically tested and only one was carrying the parental variant.
4 Discussion
Due to the unfortunate passing of one of the main investigators, publication of our results has been delayed. Nevertheless, we believe that our findings provide an insight of interest into the epidemiology of tuberous sclerosis patients in the canton of Vaud, Switzerland. However, it should be borne in mind that treatment recommendations have changed between 2015 and now, with a broadening of the indications for mTORi that give an explanation to the low number of patients treated by mTORi in this study.
This work is the first one that attempts to identify all patients suffering from TSC in a dedicated state of Switzerland. We searched for TSC patients in a tertiary university hospital and in specialized institutions hosting patients with epilepsy or patients with severe cognitive impairment, thus probably enriching our cohort with the most severe cases.
According to the published estimated prevalence of the disease (1/6′000–1/10′000), we expected to find 75 to 126 patients with TSC in the State of Vaud (according to the total population in 2015), but found only 52. One explanation for this gap might be that some patients might have been missed by our search. We checked for patients in the electronic files of a tertiary university hospital and in one specialized institution. However, some patients might have been classified under other diagnosis. Alternatively, patients with milder forms of TSC might have been followed-up by physicians in private practice (dermatologists, neurologists, pneumologists, and nephrologists) or in other institutions or hospitals. Altogether, it appears that this cohort probably underestimate the real TSC population and might be enriched with the most severe cases. These findings may also be the results of the absence of a centralized management until this assessment, with a proportion of patients being followed by local specialists.
We compared these results obtained in Switzerland with previously published data obtained from the International Tuberous Sclerosis Complex Consensus Conference of 2012, updated in 2021 (6, 7, 25).
Our demographical data matched the previously published data reporting epilepsy as first manifestation, followed by cardiac rhabdomyomas (26–28). A delay between the first suspicion of TSC and the actual TSC diagnosis is reported in the literature (7,14), and was 2 years on average in our cohort. This illustrates the diagnostic wandering that is typical for rare disorders, with an early putative diagnosis of TSC that is confirmed only later with the appearance of additional manifestations.
The number of familial forms found in our cohort is similar to previously published data (3). However, the parent-to-child transmission observed in our population was lower than expected, probably due to the limited size of our cohort. Moreover, some familial cases were probably missed by our survey, as mildly affected parents or cases of mosaicism might have been undiagnosed.
In our cohort, only 29 patients (56%) had genetic analysis. This may be related to the timing of the study, which was when genetic testing was not generalized and was difficult to get reimbursed by health insurances in Switzerland. We infer that if more tests would be done routinely, more diagnosis would be ascertained and more cases with unclear clinical presentation would be amenable to diagnosis.
Genetic analysis confirmed the diagnosis in 80% of cases (20/25), which is similar to the data in the literature (2, 7). However, in our cohort, the proportion of cases linked to a variant of the TSC2 gene was higher than expected. The high proportion of contiguous deletions of the TSC2/PKD1 gene could be linked to the contribution of nephrologists in this work.
As illustrated on Figure 6, the phenotypes identified in our cohort were overall comparable to previously published data on TSC (6, 7). However, we noted a few remarkable exceptions with lung and renal manifestations as well as shagreen patches and autistic disorders that were less often described in our database compared to others. Regarding lung manifestations, the under-reporting may result from the lack of symptoms at early stages of the disease and from the lack of systematic screening at that time (29, 30). The under-reporting of some clinical manifestations reflects the past management of TSC patients who were not screened for all organ manifestations at that time. These data call for improvement in the management of TSC patients and for systematic screening of all organs annually, according to the updated international TSC recommendations (14). After transition from childhood to adulthood, follow-ups seem less frequent and we noticed that later manifestations of TSC were under-represented in our cohort. This might be due to scarce transition at adolescence. Implementation of transition clinics between childhood and adulthood is expected to bring improvements in the follow-up of adult patients.
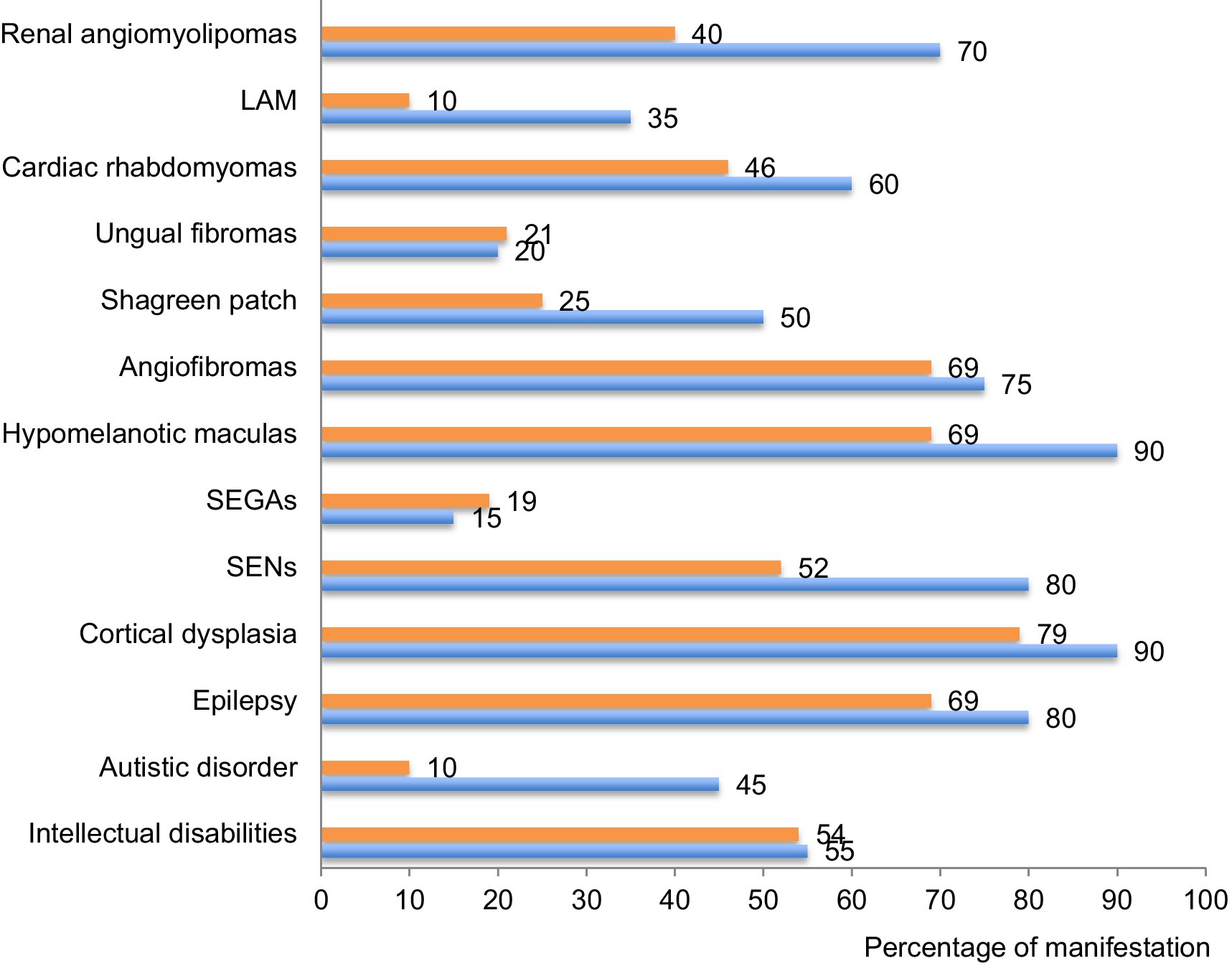
Figure 6. Percentage of each clinical manifestation in our cohort compared to international data. The percentage of patients suffering of each of the described clinical manifestation of the disease is compared between this cohort (in orange) and data provided by the international consortium in 2012 (7) (in blue). TAND: TSC-associated neuropsychiatric disorders; SEN: subependymal nodules; SEGA: subependymal giant cell astrocytomas; LAM: lymphangioleiomyomatosis.
Based on this assessment, the Lausanne University hospital has since then launched a multidisciplinary group of specialists including neurologists, neuropediatricians, nephrologists, nephropediatricians, pneumologists, dermatologists and geneticists. This group holds regular meetings to discuss screening, diagnosis and treatment possibilities for TSC patients. This multidisciplinary and trans-age board should provide a better management of patients and help filling the gap noticed in the systematic screening of all organs putatively affected by the disease.
Two patients were started on mTOR inhibitors for SEGA or AML, but when data were censored, the follow-up was too short to draw conclusions on any positive impacts or adverse events of this treatment. More than 30% of our patients were applying topical rapamycin cream on facial angiofibromas with fair efficiency and tolerance.
No death was reported in the medical charts of the patient identified in our cohort. There are only scarce data about the mortality rate of TSC patients in the literature. A Dutch study performed between 1990 and 2012 reported 29 deaths in a total of 351 TSC patients, i.e., a 8% mortality rate over 15 years of surveillance, i.e., five time higher than a randomly selected group of subjects matched for age and gender (31).
This study has limitations. First, this is a small cohort. This may explain why the numbers presented do not always match those found in larger international cohorts. However, the coverage for most severe cases and the description of their phenotype were probably extensive. Second, this study is only retrospective and cross-sectional. Therefore, longitudinal information about the evolution of the patients is incomplete. Finally, the methodology used to identify patients may have introduced a selection bias. Indeed, patients with a mild spectrum of TSC may not have been referred to our center. Conversely, severely affected patients living in institutions for disabled people may not have been all detected.
5 Conclusion
This study identified TSC patients in one large state of Switzerland and characterized them. It suggested that the diagnosis of milder forms of TSC probably remains a challenge and that all organs potentially involved should be screened for at the time of diagnosis and a regular basis according to recommendations. It highlighted the need for specific multidisciplinary care as it is offered at the Lausanne University Hospital with the dedicated TSC group. In 2024, with the help of a multidisciplinary management, patients are all evaluated for mTORi treatment and followed up over time.
Author’s note
Part of this work was accomplished during a Master Thesis at the University of Lausanne.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Commission cantonale d’éthique de la recherche sur l’être humain. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
OH-N: Writing – original draft, Writing – review & editing. FF: Writing – review & editing. JN: Writing – review & editing. SL: Writing – review & editing. CW: Writing – original draft. RL: Writing – review & editing. OB: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We would like to thank all IT persons and managers from the archives of the Lausanne University Hospital who helped us to create this database. We are thankful to Drs Malin Maeder, Giovanni Foletti, and Andrea Rossetti who helped us identifying patients in several institutions for patients with severe mental and neurologic disabilities. TSC multidisciplinary group of our hospital is acknowledged and can be found at: Orphanet: Tuberous sclerosis multidisciplinary clinic. This study was initiated and led by Dr. Christian Wider, who passed away on July 6, 2016 and is immensely regretted. We would like to dedicate this work to his memory.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Orphanet: Sclérose tubéreuse de Bourneville. [cited 2016 Dec 6]. Available at: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=FR&Expert=805
2. Pfirmann, P, Combe, C, and Rigothier, C. Sclérose tubéreuse de Bourneville: mise au point. Rev Médecine Interne. (2021) 42:714–21. doi: 10.1016/j.revmed.2021.03.003
3. Northrup, H, Koenig, MK, Pearson, DA, and Au, KS. Tuberous sclerosis complex. In: MP Adam, J Feldman, GM Mirzaa, RA Pagon, SE Wallace, and LJ Bean, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2023 Dec 3]. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1220/
4. Zamora, EA, and Aeddula, NR. Tuberous sclerosis. In: StatPearls [internet]. Treasure Island (FL): StatPearls Publishing; (2023) [cited 2023 Dec 2]. Available at: http://www.ncbi.nlm.nih.gov/books/NBK538492/
5. Roach, ES, Gomez, MR, and Northrup, H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. (1998) 13:624–8. doi: 10.1177/088307389801301206
6. Northrup, H, and Krueger, DA. International tuberous sclerosis complex consensus group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational tuberous sclerosis complex consensus conference. Pediatr Neurol. (2013) 49:243–54. doi: 10.1016/j.pediatrneurol.2013.08.001
7. Northrup, H, Aronow, ME, Bebin, EM, Bissler, J, Darling, TN, de Vries, PJ, et al. Updated international tuberous sclerosis complex diagnostic criteria and surveillance and management recommendations. Pediatr Neurol. (2021) 123:50–66. doi: 10.1016/j.pediatrneurol.2021.07.011
8. Kingswood, JC, d’Augères, GB, Belousova, E, Ferreira, JC, Carter, T, Castellana, R, et al. TuberOus SClerosis registry to increase disease awareness (TOSCA) – baseline data on 2093 patients. Orphanet J Rare Dis. (2017) 12:2. doi: 10.1186/s13023-016-0553-5
9. Portocarrero, LKL, Quental, KN, Samorano, LP, De, OZNP, and Dam, R-MMC. Tuberous sclerosis complex: review based on new diagnostic criteria*. An Bras Dermatol. (2018) 93:323–31. doi: 10.1590/abd1806-4841.20186972
10. Curatolo, P, and Moavero, R. mTOR inhibitors in tuberous sclerosis complex. Curr Neuropharmacol. (2012) 10:404–15. doi: 10.2174/157015912804499537
11. Luo, C, Ye, WR, Shi, W, Yin, P, Chen, C, He, YB, et al. Perfect match: mTOR inhibitors and tuberous sclerosis complex. Orphanet J Rare Dis. (2022) 17:106. doi: 10.1186/s13023-022-02266-0
12. Franz, DN, Belousova, E, Sparagana, S, Bebin, EM, Frost, MD, Kuperman, R, et al. Long-term use of Everolimus in patients with tuberous sclerosis complex: final results from the EXIST-1 study. PLoS One. (2016) 11:e0158476. doi: 10.1371/journal.pone.0158476
13. Franz, DN, Leonard, J, Tudor, C, Chuck, G, Care, M, Sethuraman, G, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. (2006) 59:490–8. doi: 10.1002/ana.20784
14. Bissler, JJ, Kingswood, JC, Radzikowska, E, Zonnenberg, BA, Belousova, E, Frost, MD, et al. Everolimus long-term use in patients with tuberous sclerosis complex: four-year update of the EXIST-2 study. PLoS One. (2017) 12:e0180939. doi: 10.1371/journal.pone.0180939
15. Bissler, JJ, Kingswood, JC, Radzikowska, E, Zonnenberg, BA, Frost, M, Belousova, E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. (2013) 381:817–24. doi: 10.1016/S0140-6736(12)61767-X
16. Franz, DN, Budde, K, Kingswood, JC, Belousova, E, Sparagana, S, de Vries, PJ, et al. Effect of everolimus on skin lesions in patients treated for subependymal giant cell astrocytoma and renal angiomyolipoma: final 4-year results from the randomized EXIST-1 and EXIST-2 studies. J Eur Acad Dermatol Venereol JEADV. (2018) 32:1796–803. doi: 10.1111/jdv.14964
17. French, JA, Lawson, JA, Yapici, Z, Ikeda, H, Polster, T, Nabbout, R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet Lond Engl. (2016) 388:2153–63. doi: 10.1016/S0140-6736(16)31419-2
18. Franz, DN, Lawson, JA, Yapici, Z, Ikeda, H, Polster, T, Nabbout, R, et al. Adjunctive everolimus therapy for tuberous sclerosis complex-associated refractory seizures: results from the postextension phase of EXIST-3. Epilepsia. (2021) 62:3029–41. doi: 10.1111/epi.17099
19. Wiegand, G, May, TW, Ostertag, P, Boor, R, Stephani, U, and Franz, DN. Everolimus in tuberous sclerosis patients with intractable epilepsy: a treatment option? Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. (2013) 17:631–8. doi: 10.1016/j.ejpn.2013.06.002
20. McCormack, FX, Inoue, Y, Moss, J, Singer, LG, Strange, C, Nakata, K, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. (2011) 364:1595–606. doi: 10.1056/NEJMoa1100391
21. Goldberg, HJ, Harari, S, Cottin, V, Rosas, IO, Peters, E, Biswal, S, et al. Everolimus for the treatment of lymphangioleiomyomatosis: a phase II study. Eur Respir J. (2015) 46:783–94. doi: 10.1183/09031936.00210714
22. Daccord, C, Nicolas, A, Demicheli, R, Chehade, H, Hottinger, AF, Beigelman, C, et al. Effect of everolimus on multifocal micronodular pneumocyte hyperplasia in tuberous sclerosis complex. Respir Med Case Rep. (2020) 31:101310. doi: 10.1016/j.rmcr.2020.101310
23. Dill, PE, De Bernardis, G, Weber, P, and Lösch, U. Topical everolimus for facial angiofibromas in the tuberous sclerosis complex. A first case report. Pediatr Neurol. (2014) 51:109–13. doi: 10.1016/j.pediatrneurol.2014.02.016
24. Krueger, DA, Sadhwani, A, Byars, AW, de Vries, PJ, Franz, DN, Whittemore, VH, et al. Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann Clin Transl Neurol. (2017) 4:877–87. doi: 10.1002/acn3.494
25. Krueger, DA, and Northrup, H. International tuberous sclerosis complex consensus group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol. (2013) 49:255–65. doi: 10.1016/j.pediatrneurol.2013.08.002
26. Staley, BA, Vail, EA, and Thiele, EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. (2011) 127:e117–25. doi: 10.1542/peds.2010-0192
27. Datta, AN, Hahn, CD, and Sahin, M. Clinical presentation and diagnosis of tuberous sclerosis complex in infancy. J Child Neurol. (2008) 23:268–73. doi: 10.1177/0883073807309250
28. Ebrahimi-Fakhari, D, Mann, LL, Poryo, M, Graf, N, von Kries, R, Heinrich, B, et al. Incidence of tuberous sclerosis and age at first diagnosis: new data and emerging trends from a national, prospective surveillance study. Orphanet J Rare Dis. (2018) 13:117. doi: 10.1186/s13023-018-0870-y
29. Seibert, D, Hong, CH, Takeuchi, F, Olsen, C, Hathaway, O, Moss, J, et al. Recognition of tuberous sclerosis in adult women: delayed presentation with life-threatening consequences. Ann Intern Med. (2011) 154:806–294. doi: 10.7326/0003-4819-154-12-201106210-00008
30. Miyake, M, Tateishi, U, Maeda, T, Kusumoto, M, Satake, M, Arai, Y, et al. Pulmonary lymphangioleiomyomatosis in a male patient with tuberous sclerosis complex. Radiat Med. (2005) 23:525–7.
31. Eijkemans, MJC, van der Wal, W, Reijnders, LJ, Roes, KCB, van Waalwijk van Doorn-Khosrovani, SB, Pelletier, C, et al. Long-term follow-up assessing renal angiomyolipoma treatment patterns, morbidity, and mortality: an observational study in tuberous sclerosis complex patients in the Netherlands. Am J Kidney Dis. (2015) 66:638–45. doi: 10.1053/j.ajkd.2015.05.016
Keywords: tuberous sclerosis complex, mTOR inhibitors, epilepsy, subependymal nodules, subependymal giant cell astrocytomas, angiomyolipomas, lymphangioleiomyomatosis
Citation: Hagon-Nicod O, Fellmann F, Novy J, Lebon S, Wider C, Lazor R and Bonny O (2024) Tuberous sclerosis: a survey in the canton of Vaud, Switzerland. Front. Med. 11:1513619. doi: 10.3389/fmed.2024.1513619
Edited by:
Ovidiu Constantin Baltatu, Anhembi Morumbi University, BrazilReviewed by:
Jianxiang Liao, Shenzhen Children’s Hospital, ChinaClaudio Musetti, Azienda Ospedaliero Universitaria Maggiore della Carità, Italy
Copyright © 2024 Hagon-Nicod, Fellmann, Novy, Lebon, Wider, Lazor and Bonny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olivier Bonny, T2xpdmllci5Cb25ueUBoLWZyLmNo
†Deceased