 Wei Huang
Wei Huang Juan Zheng1†
Juan Zheng1† Ling-Yao Du
Ling-Yao Du Lang Bai
Lang Bai Hong Tang
Hong Tang- 1Center of Infectious Diseases, West China Hospital of Sichuan University, Chengdu, China
- 2Laboratory of Infectious and Liver Diseases, Institute of Infectious Diseases, West China Hospital of Sichuan University, Chengdu, China
Organ fibrosis is a pathological process characterized by the inability of normal tissue cells to regenerate sufficiently to meet the dynamic repair demands of chronic injury, resulting in excessive extracellular matrix deposition and ultimately leading to organ dysfunction. Despite the increasing depth of research in the field of organ fibrosis and a more comprehensive understanding of its pathogenesis, effective treatments for fibrosis-related diseases are still lacking. Melatonin, a neuroendocrine hormone synthesized by the pineal gland, plays a crucial role in regulating biological rhythms, sleep, and antioxidant defenses. Recent studies have shown that melatonin may have potential in inhibiting organ fibrosis, possibly due to its functions in anti-oxidative stress, anti-inflammation, remodeling the extracellular matrix (ECM), inhibiting epithelial-mesenchymal transition (EMT), and regulating apoptosis, thereby alleviating fibrosis. This review aims to explore the therapeutic potential of melatonin in fibrosis-related human diseases using findings from various in vivo and in vitro studies. These discoveries should provide important insights for the further development of new drugs to treat fibrosis.
1 Introduction
Organ fibrosis is the ultimate outcome of various chronic diseases, characterized by the deposition of extracellular matrix (ECM) leading to scar tissue formation as the primary pathological change (1). Continuous progression can result in structural damage and loss of function in organs, ultimately leading to death, as seen in end-stage liver, kidney, lung, and heart diseases (2). Organ fibrosis significantly impacts global morbidity and mortality rates. In developed countries, nearly 45% of human deaths are associated with fibrosis-related diseases, and this proportion may be even higher in developing countries (3). Despite ongoing research into the occurrence and development of fibrosis, current methods for anti-fibrotic treatment are limited and often ineffective, making it crucial to search for effective therapeutic approaches.
Melatonin (N-Acetyl-5-methoxytryptamine) is a methoxylated indole produced in peripheral tissues such as the retina, gastrointestinal tract and bone marrow and primarily synthesized and secreted by the pineal gland under normal light–dark conditions during the night in vertebrates (4). Melatonin was first isolated from the bovine pineal gland by Lerner et al. (5). Melatonin synthesis begins with the hydroxylation of tryptophan to form 5-hydroxytryptophan, followed by decarboxylation to produce 5-hydroxytryptamine (5-HT). This is then acetylated to form N-acetyl-5-hydroxytryptamine, which is finally methylated to produce N-acetyl-5-methoxytryptamine (6). The production of melatonin is typically influenced by various factors, and it is currently understood that the secretion rhythm of melatonin relies on an intrinsic circadian structure triggered by light signals received by the retina. This rhythm originates from the suprachiasmatic nucleus (SCN), where light exposure transmits neural impulses to the SCN via the retinohypothalamic tract, synchronizing the activity of the SCN and the nocturnal secretion rhythm of melatonin with the 24-h light/dark cycle. Norepinephrine (NE) serves as a mediator in the effect of light on melatonin synthesis and secretion. Light can inhibit the release of NE through specific pathways, whereas in darkness, sympathetic neuronal activity within the pineal gland significantly increases, resulting in higher NE release. NE binds to receptors on the pineal gland cell membrane, promoting the entry of tryptophan into the cells. Once NE binds to its receptors, cyclic adenosine monophosphate (cAMP) facilitates the synthesis of serotonin and melatonin (7). Additionally, light is an important external factor influencing melatonin secretion. Various non-environmental factors, such as sleep quality and duration, hunger, and physical activity, may also affect melatonin secretion (8–10).
After melatonin is synthesized, it is immediately released into the cerebrospinal fluid and blood, distributing throughout the body via systemic circulation. Its plasma concentration exhibits a distinct circadian rhythm, being higher at night than during the day (11). Based on the affinity for binding sites, melatonin receptors can be classified into three subtypes: melatonin receptor 1A (MT1) (12), melatonin receptor 1B (MT2) (13), and melatonin receptor 1C (MT3) (14). As members of the G protein-coupled receptor superfamily, MT1 and MT2 are both primarily located in the brain and other extra-pineal tissues, including the liver, skeletal muscle, and retina. MT3 is found in the liver, kidneys, heart, adipose tissue, and brain (15). By binding to different melatonin receptors, melatonin rapidly activates various signal transduction cascades, thereby exerting its biological effects in the body. Melatonin is widely recognized as a potent antioxidant and is also involved in mediating various biological effects such as anti-inflammatory responses, circadian rhythms, cellular metabolism, and immune regulation (16). Recent research suggests that exogenous melatonin supplementation can alleviate organ fibrosis, including liver fibrosis, lung fibrosis, heart fibrosis, and kidney fibrosis, indicating that melatonin may be a potential anti-fibrotic agent.
This review begins by outlining the process of liver fibrosis development. It then summarizes the possible mechanisms by which melatonin inhibits fibrosis development. Finally, it discusses the roles melatonin may play in different organ fibrosis scenarios. It is hoped that this article will provide a new perspective for subsequent experimental research and offer more theoretical support for the clinical recommendation and widespread application of melatonin.
2 Pathogenesis of organ fibrosis
Typically, when tissue is injured, wound healing undergoes three main phases: the inflammatory phase, the proliferative phase, and the remodeling/maturation phase (2, 17). These phases serve different functions and overlap in time (18) (Figure 1).
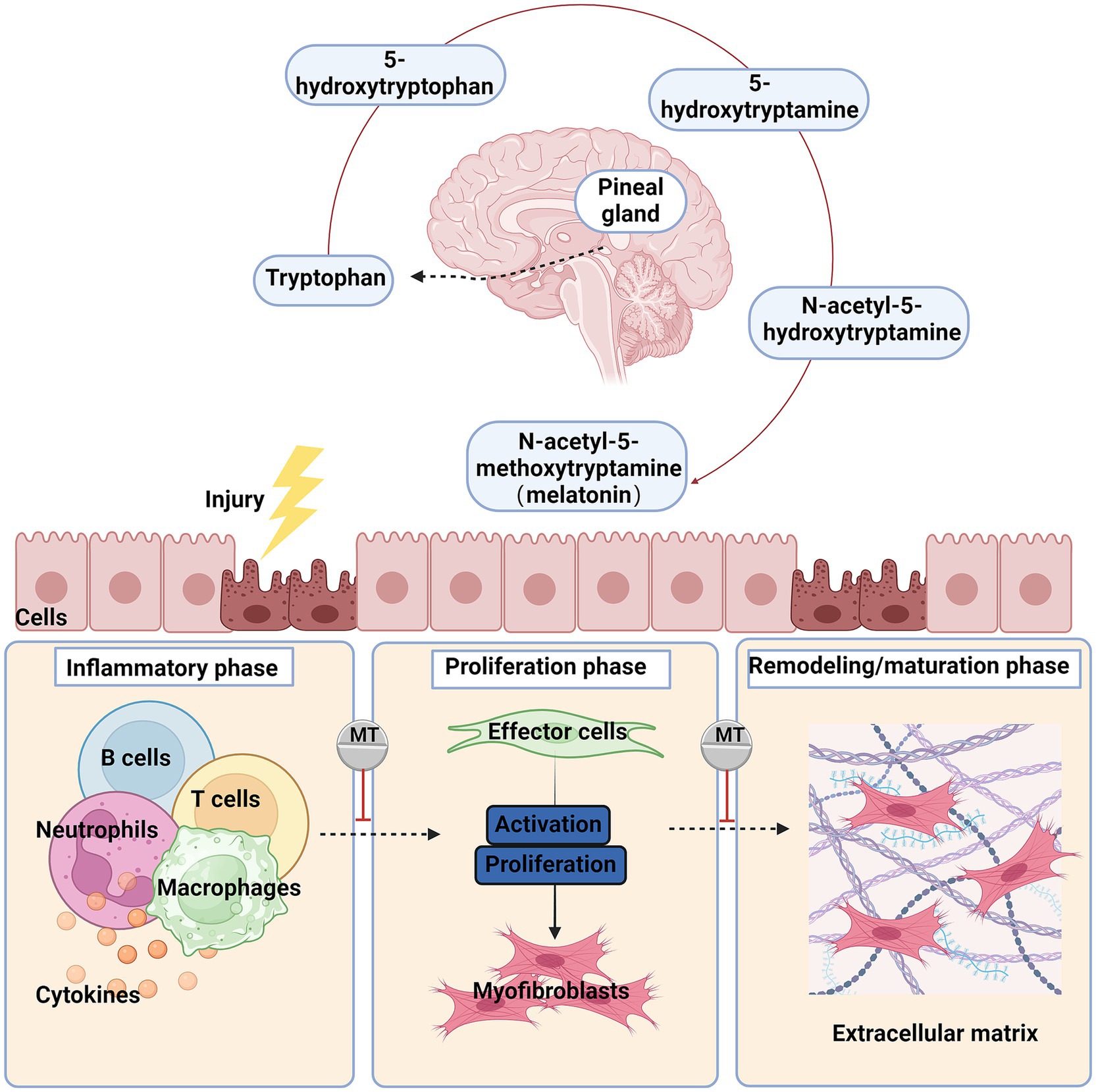
Figure 1. The primary synthesis process and anti-fibrotic effects of melatonin after organ injury, the damaged cells secrete chemokines and cytokines, recruiting and activating inflammatory cells such as macrophages, monocytes, and lymphocytes. The signals released by these cells activate fibroblasts, which then transform into myofibroblasts and synthesize extracellular matrix (ECM). Chronic injury and inflammation lead to persistent myofibroblast activation, excessive ECM deposition, and ultimately fibrosis. Red blunted line: an inhibitory effect of melatonin.
Under normal circumstances, after an injury, the formation of a platelet plug and provisional ECM stops the bleeding, accompanied by an increased threshold of the inflammatory response and recruitment of immune cells. This process initiates the first phase of healing, the inflammatory phase. During this phase, a large number of neutrophils and macrophages infiltrate the tissue to combat potential infection and clear debris (19). In the inflammatory phase, damaged endothelial/epithelial cells and myofibroblasts produce aberrant matrix metalloproteinases (MMPs), disrupting the basement membrane of local tissues and releasing various cytokines and chemokines, thereby recruiting and activating more immune cells, including neutrophils, macrophages, T lymphocytes, and B lymphocytes (3, 20).
Activated leukocytes release mediators such as pro-inflammatory, vasoactive, and pro-fibrotic effectors, inducing precursor cells to differentiate into myofibroblasts. Myofibroblasts rapidly produce large amounts of ECM to maintain the integrity of damaged tissue during the repair process and promote cell proliferation to form granulation tissue. Myofibroblasts are the primary effector cells in fibrosis (21).These cells can secrete large amounts of ECM proteins (mainly collagen I, collagen III, and fibronectin), increase the production of tissue inhibitors of metalloproteinases, and express alpha smooth muscle actin (α-SMA), which confers contractility (22, 23). The sources of myofibroblasts may vary across different tissues. Currently, it is believed that the activation of resident fibroblasts in the tissue is the main source of myofibroblasts (24). Under certain conditions, epithelial and endothelial cells also can differentiate into myofibroblasts through epithelial/endothelial-mesenchymal transition (EMT/EndoMT) (25–27), and mesothelial cells can differentiate into myofibroblasts through mesothelial-mesenchymal transition (28, 29).
In the final tissue remodeling/maturation phase, activated myofibroblasts cause wound contraction; dynamic ECM degradation and remodeling restore the parenchymal tissue structure. However, persistent inflammation, necrotic cells, ongoing fibroblast activation, and excessive ECM deposition lead to abnormal tissue reconstruction, resulting in organ fibrosis.
To date, various mediators have been identified that can activate fibroblasts and promote the occurrence and progression of fibrosis, including transforming growth factor-β (TGF-β) (30), interleukins (IL-1β, IL-6, IL-13, IL-33, IL-11, IL-17, etc.), tumor necrosis factor-α (TNF-α), platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF) (31), angiotensin-II (Ang-II) (32), heat shock protein (33), endothelin-1 (34), integrins (35), reactive oxygen species (ROS), and hypoxia. TGF-β is a cytokine with diverse physiological functions, playing a key role in regulating cell proliferation, differentiation, apoptosis, and immune responses (36). Currently, three isoforms of TGF-β are known: TGF-β1, TGF-β2, and TGF-β3. Among these, TGF-β1 is the most abundant isoform in humans, widely expressed in most cell types, with platelets serving as a significant source (37). Although these three isoforms share similar bioactive regions and can bind to the same type I and type II TGF-β receptor complexes, TGF-β is widely recognized as the most potent pro-fibrotic factor, primarily acting through the Smad (Smad-2, −3, −4, etc.) signaling pathway to promote fibrosis development (38, 39). In addition, TGF-β can also facilitate fibrosis through multiple non-Smad-dependent pathways, such as those involving c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (p38 MAPK), extracellular signal-regulated kinase (ERK1/2), and phosphatidylinositol 3-kinase/Akt (PI3K/Akt) or Rho-like GTPases (30, 40). During TGF-β signaling, activated Smad complexes can induce the expression of various transcription factors in the nucleus, thereby promoting the activation of fibroblasts. Research indicates that the Smad pathway can induce the expression of the JunD transcription factor, and knocking out this gene results in a reduction in the number of myofibroblasts and decreased collagen release (41). Additionally, the Smad pathway interacts with the Snail family of transcription factors (42) and zinc finger E-box-binding homeobox (ZEB) transcription factor family to enhance myofibroblast differentiation through EMT (43). IL-1β-treated fibroblasts differentiate into myofibroblasts, leading to increased ECM deposition. IL-13 promotes liver fibrosis formation by directly inducing the expression of liver fibrosis-related genes such as collagen and CTGF. TNF-α is a critical inflammatory signal molecule in fibrosis, mainly secreted by macrophages. By binding to specific receptors, TNF-α initiates signal cascades, activating nuclear factor-kappa B (NF-κB) to regulate inflammatory responses, and triggers cell activation, differentiation, cytokine production, and apoptosis, thus contributing to fibrosis progression. NF-κB is an inducible transcription factor family responsible for regulating the initiation and progression of inflammatory responses. The activation of NF-κB occurs primarily through two pathways: the canonical and the non-canonical pathways. The canonical pathway is activated when signals such as pro-inflammatory cytokines or pathogen-associated molecular patterns activate cell surface receptors, including pattern recognition receptors, Toll-like receptors (TLR), and T cell receptors. In contrast, the non-classical signaling pathway is primarily activated by TNF and its corresponding TNF receptors (44). Studies have shown that during the NF-κB signaling process, its inhibitors can suppress fibroblast activity in a concentration-dependent manner (45).
3 The mechanisms of melatonin alleviating fibrosis
Melatonin was initially discovered and named for its ability to lighten the skin of amphibians (5, 46). As research progressed, melatonin was identified as a lipid-soluble hormone that can act on almost every cell in the organism, crossing all biological barriers. Its primary function is to regulate and reset circadian rhythms (47), but it also has anti-cancer (48), immune-modulating (49) and many other functions. Melatonin exerts its anti-fibrotic effects, and the mechanisms may involve reducing oxidative stress, inhibiting inflammatory responses, ECM remodeling, suppressing EMT, and preventing apoptosis (Figure 2).
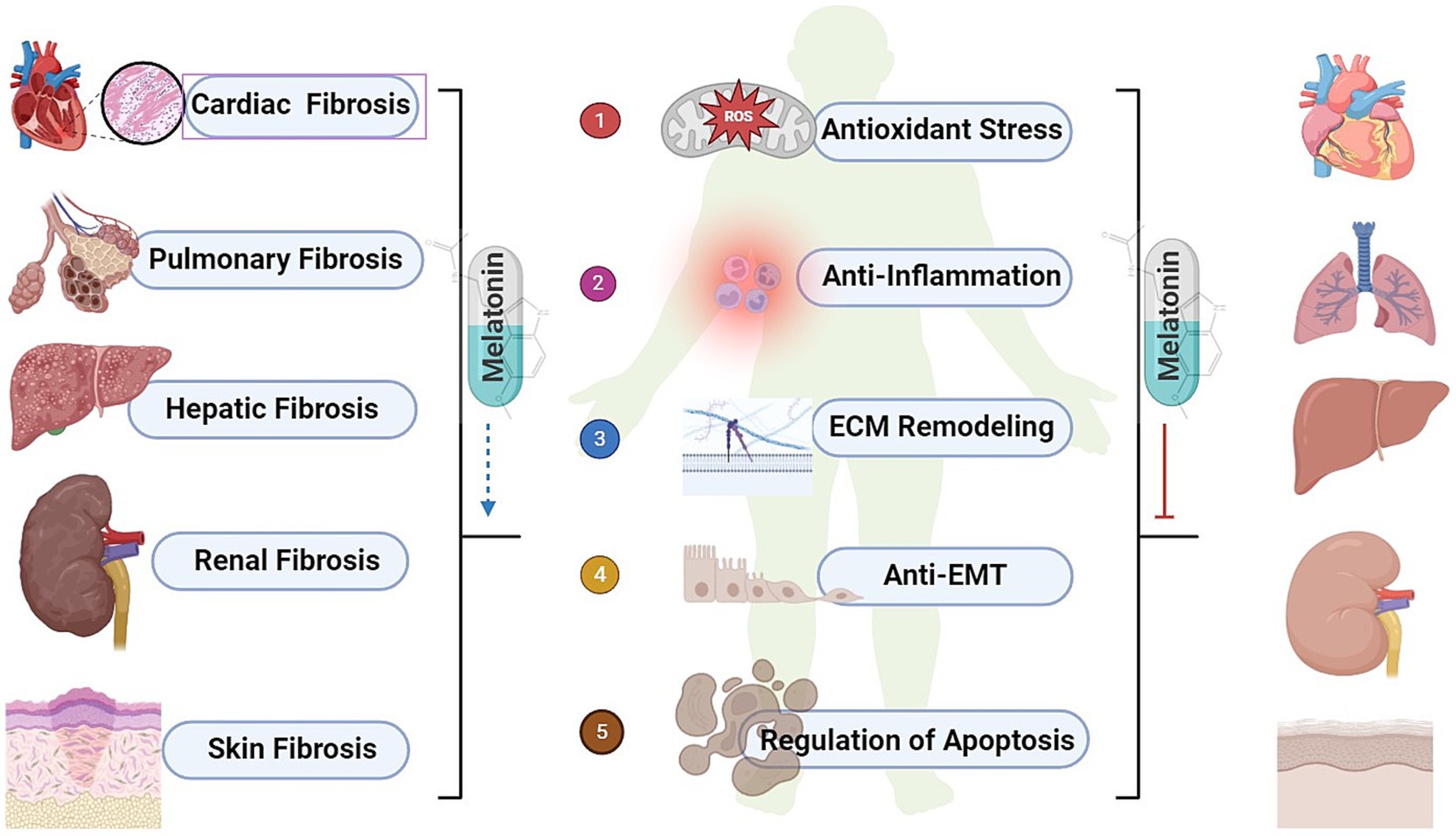
Figure 2. The biological effects of melatonin and common organ fibrosis. ROS, reactive oxygen species; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition.
3.1 Antioxidant stress
Oxidative stress arises from cells’ inefficient utilization of molecular oxygen (50). ROS include superoxide anion radicals, hydroxyl radicals, hydrogen peroxide, and singlet oxygen, which are byproducts of cellular respiration and other metabolic processes (51). Additionally, there are highly destructive nitrogen-based substances such as nitric oxide, especially peroxynitrite anion (52). Oxidative stress is considered a major cause of fibrosis as it damages the structures of cellular macromolecules like DNA, proteins, and lipids, ultimately leading to cell damage and promoting fibrosis (53).Studies have shown that melatonin can enhance mitochondrial adenosine triphosphate synthesis and reduce the production of reactive oxygen species (54). Furthermore, melatonin not only directly interacts with various ROS, reactive nitrogen species, and organic radicals to exert a direct scavenging effect, but also upregulates the activities of antioxidant enzymes such as superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione, (GSH), glutathione reductase (GRd), and glucose-6-phosphate dehydrogenase (52, 55). Additionally, melatonin downregulates the activities of pro-oxidant enzymes like nitric oxide synthase (NOS) and lipoxygenase, thereby exerting an indirect antioxidant effect (52). Furthermore, melatonin, similar to metallothioneins, can bind to heavy metals such as aluminum, cadmium, copper, iron, lead, and zinc, thus mitigating oxidative stress (56). As a lipid-soluble free radical scavenger, melatonin can easily cross the blood–brain barrier, replacing or supplementing metallothioneins in the brain as the primary binding agents for transition metals (57).
3.2 Anti-inflammation
Recruited inflammatory cells and damaged epithelial cells eventually transform into fibrogenic effector cells or induce the activation of precursor cells, which is a crucial component of the fibrosis process. Recent studies have found that melatonin can inhibit the infiltration of inflammatory cells by reducing myeloperoxidase activity (58, 59). Melatonin effectively lowers the levels of pro-inflammatory factors and fibrosis markers, demonstrating anti-inflammatory activity in various diseases. In human blood cells, melatonin can reduce lipopolysaccharide (LPS)-induced TNF-α levels (60, 61). In patients with osteoarthritis, researchers observed that melatonin effectively inhibits the production of pro-inflammatory cytokines during inflammation by suppressing the Erk and PI3K/Akt signaling pathways, significantly reducing the expression of TNF-α, IL-8, and vascular endothelial growth factor in synovial fibroblasts (62). The NF-κB transcription factor family is considered a central mediator in the inflammatory process (63), and studies have shown that melatonin can inhibit NF-κB signaling and activate the antioxidant regulators nuclear erythroid 2-related factor 2 (Nrf2) (63, 64). Furthermore, melatonin exerts its anti-inflammatory effects by inhibiting the expression of the inflammasome NLR family pyrin domain containing 3 (NLRP3) and the activation of NF-κB while upregulating the expression of the transcription factor Nrf2 (65). Melatonin also significantly reduces the expression levels of nitric oxide and malondialdehyde (MDA), which are closely associated with inflammation (59, 66).
3.3 ECM remodeling
Under physiological conditions, the ECM is a highly dynamic structure present in all tissues, constantly undergoing balanced remodeling. This remodeling process is primarily mediated by specific enzymes, particularly MMPs (67). When tissue is damaged, the synthesis and degradation of ECM become progressively imbalanced, eventually leading to ECM deposition and, consequently, tissue fibrosis (68). During ECM deposition, a large amount of MMPs is activated, breaking down the cells and their surrounding basement membrane matrix, allowing leukocytes to migrate into the tissue (69). Additionally, MMPs influence the secretion of various chemokines and cytokines, creating a strong chemokine gradient that leads to the recruitment of inflammatory cells to the site of injury (70, 71). Melatonin has a regulatory effect on MMP gene expression and catalytic activity (72). MMP-9 is a zinc metalloproteinase that, as a matrix protein, is released from intracellular stores and becomes active extracellularly (73, 74). Studies have found that in mice with liver fibrosis treated with melatonin, MMP-9 activity is reduced and Nrf2 expression is increased (75). In gastric adenocarcinoma cell lines, melatonin inhibits both the induction and catalytic activity of MMP-9 (73). Collagen and glycosaminoglycan (GAG) are major components of the ECM, and research has shown that melatonin can effectively reduce the amounts of collagen (76–78) and GAG (79), thereby alleviating tissue fibrosis to some extent.
3.4 Anti-EMT
EMT is an important process in normal embryonic development and tissue repair, during which differentiated epithelial cells undergo phenotypic transformation to acquire a mesenchymal phenotype (80). This is a reversible biological process. The EMT process involves the induction and regulation of multiple signal transduction pathways, primarily including TGF-β, NF-κB, Wnt, Notch and et. pathways (67). These pathways converge on the EMT-related transcription factor families, triggering extensive cellular transcriptional reprogramming. The main EMT-related transcription factors include members of the SNAIL, ZEB, TWIST, and PRRX families, as well as various EMT effectors (81). In the context of persistent chronic injury, the role of EMT shifts from promoting tissue repair to contributing to tissue degeneration, leading to irreversible dedifferentiation and cell cycle arrest (82). Moreover, EMT can enhance the secretion of TGF-β and inflammatory cytokines by damaged epithelial cells, which, respectively, promote the differentiation of fibroblasts into myofibroblasts and trigger immune inflammation, ultimately resulting in repair failure and tissue fibrosis (83, 84). Research indicates that melatonin may inhibit the activity of the Notch signaling pathway, thereby blocking the migration, invasion, and EMT of normal and endometriosis epithelial cells induced by 17β-estradiol (85). Research indicates that melatonin can inhibit the secretion of IL-1β, IL-6, and TGF-β induced by LPS both in vivo and in vitro (86). Additionally, melatonin can reverse LPS-induced EMT in peritoneal mesothelial cells by inhibiting the TLR4/ JNK and TLR4/NF-κB-Snail signaling pathways (87).
3.5 Regulation of apoptosis
Apoptosis is an orderly process of programmed cell death controlled by genes, involving the activation, expression, and regulation of genes (88). It is a proactive response of cells to adapt to their living environment. The regulation of apoptosis mainly occurs through the intrinsic mitochondrial pathway and the extrinsic death receptor pathway (89). The intrinsic pathway is initiated by the insertion of Bax/Bak into the mitochondrial membrane, followed by the release of cytochrome c (Cyt C) (90). Cyt C then combines with Apaf-1 and procaspase-9 to form the apoptosome, which subsequently activates caspase-3, initiating the apoptotic cascade (91). The extrinsic pathway is triggered by external stimuli or ligand molecules, primarily involving death receptors (92). Research indicates that melatonin can reduce cisplatin-induced apoptosis in renal tubular cells by inhibiting the activation of caspase-3 (93). In thymocytes induced by glucocorticoids, melatonin exerts an anti-apoptotic effect by regulating the levels of Bax protein (94). During fibrosis, the abnormal proliferation of fibrotic effector cells and their resistance to apoptosis are key factors. Interestingly, the regulation of apoptosis by melatonin depends on the cell type. In human hypertrophic scar tissue, melatonin inhibits fibroblast proliferation and induces apoptosis by regulating the gene expression of cyclin E, p53, and Fas (95). The alleviation of fibrosis by melatonin may be related to the reduction of fibroblast migration through decreased chloride channel activity mediated by protein kinase C (96).
4 Protective effects of melatonin in fibrotic diseases
Numerous studies have shown that melatonin provides comprehensive protection for various organs and tissues, including the heart, lungs, liver, and kidneys, by preventing fibrosis and aiding in the repair of damage to these organs and tissues (Table 1).

Table 1. Summary of anti-fibrotic effects and underlying mechanisms of melatonin.
4.1 Melatonin and cardiac fibrosis
Cardiac fibrosis is the ultimate pathological outcome of various cardiovascular diseases, commonly seen in myocardial infarction (MI), hypertension, myocarditis, cardiomyopathy, arrhythmias, diabetes and radiation exposure (97). Its main pathological features include excessive proliferation of cardiac fibroblasts and ECM protein deposition, leading to progressive diastolic and systolic dysfunction, which eventually results in chronic heart failure, and sudden cardiac arrest (98, 99). Numerous studies have shown that melatonin affects the cardiovascular system. In cardiovascular diseases, levels of melatonin are found to be reduced (100–102). Melatonin therapy can effectively alleviate fibrosis following these diseases. Melatonin can mitigate cardiac dysfunction and fibrosis induced by fine particulate matter (PM2.5) in mice by inhibiting the phenotypic transformation of cardiac fibroblasts into myofibroblasts. This mechanism may involve the suppression of mitochondrial oxidative damage and regulation of sirtuins 3 (SIRT3)-mediated SOD2 deacetylation (103). MiR-141 is an upstream factor in cardiac fibrosis, involved in regulating NLRP3 and TGF-β1. Melatonin can improve cardiac function and reduce collagen production in diabetic mice by inhibiting the lncR-MALAT1/miR-141 mediated activation of the NLRP3 inflammasome and TGF-β1/Smads signaling pathway (104).Histone deacetylases (HDAC) are involved in several processes related to cardiovascular diseases, including cardiac hypertrophy, remodeling, and fibrosis (105). Melatonin treatment can exert anti-fibrotic effects by inhibiting HDAC expression (106). Additionally, melatonin treatment can alleviate cardiac injury and fibrosis in diabetic mice following MI by inhibiting the JNK/p53-mediated apoptotic pathway. This inhibition reduces the levels of p53, caspase-3, and Bax, while increasing the level of Bcl-2 (107). It is well known that Notch1 plays a significant role in cardiac injury repair (108), and melatonin enhances Notch1/Hes1/Akt signaling in a receptor-dependent manner, rescuing the intracellular thioredoxin (Trx) system, reducing infarct size, cardiomyocyte apoptosis, and oxidative stress, thereby improving myocardial ischemia–reperfusion injury (109).
4.2 Melatonin and pulmonary fibrosis
Pulmonary fibrosis is an end-stage lung condition caused by various factors, characterized by fibroblast proliferation and extensive ECM deposition, leading to the destruction of lung tissue structure (110). Pulmonary fibrosis is a major clinical outcome of most chronic respiratory diseases, such as chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), acute respiratory distress syndrome (ARDS), radiation and those caused by environmental exposures [e.g., inorganic mineral dust, carbon tetrachloride (CCl4), and bleomycin]. Numerous studies have shown that melatonin can alleviate pulmonary fibrosis caused by various factors. In bleomycin-induced pulmonary fibrosis, melatonin reduces the infiltration and accumulation of inflammatory cells in the alveolar walls (111), as well as the expression of inflammatory mediators such as cyclooxygenase 2 (COX-2) (78). Additionally, melatonin inhibits the increase in hydroxyproline content and the decrease in CAT activity in lung tissue, preventing fibrosis progression by suppressing protein and lipid peroxidation reactions (112). As a key protective regulator in lung diseases, research has found that melatonin also increases the expression of Apelin 13 and inhibits the production of ROS, thereby restoring mitochondrial function and reducing cell apoptosis and senescence during lung injury (113). Melatonin can also effectively inhibit LPS-induced EMT in human type II alveolar epithelial cells (AECs II) by activating the PI3K/Akt signaling pathway, leading to glycogen synthase kinase 3β (GSK-3β) phosphorylation and Nrf2 protein upregulation (114).Furthermore, the Wnt/β-catenin pathway may be involved in various pathological processes of IPF, such as inducing EMT in damaged AECs II and fibrotic cell migration (115, 116). Melatonin inhibits TGF-β1-induced EMT in alveolar epithelial cells by downregulating the Wnt/β-catenin and Smad signaling pathways (117).
4.3 Melatonin and hepatic fibrosis
Hepatic fibrosis is a pathological process resulting from liver damage and inflammatory responses caused by various factors such as chronic viral infection, excessive alcohol consumption, exposure to toxic substances, biliary diseases, autoimmune hepatitis (AIH), and non-alcoholic steatohepatitis (NASH). This leads to the activation of hepatic stellate cells (HSCs) and excessive ECM deposition (118). Continued progression of liver fibrosis results in severe complications such as cirrhosis and liver cancer and commonly needs liver transplantation (119).
Numerous in vivo and in vitro experiments have demonstrated that melatonin treatment has a significant effect on alleviating liver fibrosis (120).As the key effector cells in the process of liver fibrosis, HSCs can be activated under conditions of inflammation and oxidative stress, leading to excessive proliferation and transdifferentiation into myofibroblasts. These myofibroblasts express large amounts of α-SMA and ECM proteins, promoting liver fibrosis (121). Melatonin not only reversed the PM2.5-induced increase in MDA and ROS levels and the decrease in GSH levels by activating Nrf2 but also reduced the expression levels of inflammatory factors such as IL-1β, TNF-α, NF-κB, and NLRP3, thereby improving liver function and inhibiting HSCs activation to exert anti-fibrotic effects (122). Melatonin can increase GPx activity and reduce serum markers of liver fibrosis, including hyaluronic acid (HA), hydroxyproline (HYP), and laminin (LN), thereby alleviating CCl4-induced liver dysfunction and fibrosis in rats (123).Additionally, melatonin reduces the expression of collagen I and III, TGF-β, PDGF, CTGF, doublecortin-like kinase, and phosphorylated Smad3, as well as MMP-9 activity, while increasing Nrf2 expression. By limiting multiple pro-fibrotic gene pathways, melatonin alleviates CCl4-induced liver fibrosis in mice (75). In bile duct-ligated rats, melatonin exerts antioxidant effects by increasing SOD and GSH activities, while also reducing hepatocyte apoptosis, thereby alleviating cholestatic liver injury, bile duct proliferation, and fibrosis (124).
4.4 Melatonin and renal fibrosis
Renal fibrosis is caused by damage to intrinsic cells (e.g., glomerular podocytes, endothelial cells, mesangial cells, and proximal tubule epithelial cells) due to various pathogenic factors such as ischemia, infection, obstruction, autoimmune diseases, drug injury, hypertension, and diabetes. This damage progresses to extensive collagen deposition and accumulation, leading to the gradual hardening of the renal parenchyma, scar formation, and eventually the complete loss of kidney function (125). It is estimated that chronic kidney disease (CKD) affects 13% of the global population, imposing a significant economic burden on society (126). Renal fibrosis is a common progressive and irreversible pathological feature of CKD, characterized by glomerulosclerosis, tubular atrophy, chronic interstitial inflammation and fibrosis, and vascular rarefaction (127). Currently, there are no effective treatments for renal fibrosis. Research indicates that melatonin can exert antioxidant effects by increasing the activities of SOD, GSH, and CAT and reducing MDA levels in damaged renal tissue. It also exerts anti-inflammatory effects by decreasing the expression of TNF-α and inducible NOS (iNOS), and anti-apoptotic effects by downregulating the expression of caspase-3 and Bax and upregulating the expression of Bcl-2. Consequently, melatonin inhibits the expression of fibrosis-related genes and improves renal damage caused by cadmium exposure in mice (128). Melatonin significantly reduced hydroxyproline levels and the ratio and total amount of collagen I and III in the glomeruli and tubular interstitium in rat with lactoferrin-induced renal fibrosis (129). Additionally, melatonin can inhibit ROS in a receptor-independent manner by suppressing both Smad and non-Smad signaling cascades. This prevents the TGF-β1-induced transdifferentiation of renal interstitial fibroblasts into myofibroblasts, thereby alleviating renal fibrosis (130).
4.5 Melatonin and skin fibrosis
Skin fibrosis is characterized by the excessive proliferation of fibroblasts and the deposition of excessive ECM in the dermis, with abnormal cross-linking types, leading to skin hardening and damage (131). Progressive fibrosis mediated by myofibroblasts can invade the skin, causing skin fibrotic diseases, including hypertrophic scar (HS), keloids, localized scleroderma (LS), and systemic sclerosis (SSc), chronic graft-versus-host disease (GVHD) (132). The global impact of skin fibrosis is significant, affecting over 100 million people annually in developed countries (133). Research has found that melatonin enhances autophagy by inhibiting PI3K/Akt/mTOR signaling through the MT2 receptor, thereby inhibiting the migration and contractility of HS fibroblasts (HSFs), as well as the production of collagen and α-SMA, thus playing a role in the prevention and treatment of HS (134).Furthermore, melatonin promotes the apoptosis of keloid fibroblasts (KFs) through the Erk and Smad signaling pathways, inhibiting their proliferation, migration, invasion, contractility, and collagen production, thereby altering the cellular functions of KFs (135).
5 Clinical applications of melatonin
Dysfunction of melatonin can lead to a range of health issues, including sleep disorders, depression, and decreased immune function (136–138). This, in turn, may increase the risk of developing various conditions such as obesity, diabetes, cardiovascular diseases, and even cancer (139–141). Oral administration of melatonin follows first-order kinetics with a relatively low bioavailability of approximately 15%, which exhibits significant inter-individual variability (142, 143). Melatonin is primarily metabolized in the liver, where it is first hydroxylated to 6-hydroxymelatonin by cytochrome P450 enzymes CYP1A1 and 1A2 enzymes. The majority of this metabolite is then conjugated with sulfate, while a smaller portion is conjugated with glucuronic acid, ultimately being excreted in the urine (144). The absorption half-life is about 6 min, with serum peak concentrations reached on average 40.8 min post-administration, where the maximum concentration can reach 3550.5 pg./mL, thousands of times higher than physiological nighttime levels (125). The average elimination half-life is approximately 53.7 min (125). The low bioavailability and short half-life of melatonin pose significant challenges for its clinical application. Currently, there are melatonin-related compounds, including remelteon, being developed that can last up to 6 h (145).
The incidence of adverse reactions to melatonin is low and its effects are generally mild. Studies have shown positive effects at doses ranging from 2 to 500 mg/day, without any toxic effects (146); even doses of 100 mg/day sustained over a month have shown no substantial negative impact (147). Additionally, administration via various routes has proven to be safe (147).
While there are currently no approved melatonin or derived products for the treatment of fibrotic diseases, numerous cell and animal studies, as mentioned earlier, suggest that melatonin may have potential benefits in managing such conditions. Based on this, a few relevant clinical trials have also been initiated to clarify melatonin’s potential positive influences on fibrotic diseases. For instance, a daily supplementation of 3 mg of melatonin effectively enhanced pulmonary rehabilitation in patients with COPD (148). Similarly, in hypertensive patients, continuous use of a 1 mg/day melatonin supplement for one year effectively improved arterial stiffness (149). However, there are also reports from clinical trials that yielded contrary results. For example, administering melatonin via coronary and intravenous routes did not improve myocardial salvage index in patients with ST-segment elevation myocardial infarction (150). The timing of melatonin usage, the route of administration, and the treatment duration seem to contribute to these conflicting results. Further clinical research is needed to establish the dose–response relationship of melatonin across different diseases and to elucidate the primary mechanisms by which melatonin exerts its effects in these conditions.
6 Conclusion
Melatonin, as a natural neuroendocrine hormone, has good biocompatibility and safety. This review summarizes the latest advances in melatonin research and treatment for fibrosis. Increasing experimental data indicate that melatonin exerts potential antifibrotic effects in multiple tissues and organs, including the heart, lungs, liver, kidneys, and skin. Its mechanisms of action may involve reducing oxidative stress, inhibiting inflammatory responses, ECM remodeling, suppressing EMT, and regulation of apoptosis. We have identified several common molecular signaling pathways involving melatonin in fibrosis across different organs. The TGF-β pathway is crucial in nearly all types of fibrosis, as it not only has complex regulatory effects on fibrotic effector cells but also interacts with other cellular signaling pathways (97). Additionally, the NF-κB transcription factor family, as a key player in the inflammatory process, plays an important role in fibrosis across different organs (151, 152). Furthermore, the activation of the PI3K/Akt signaling pathway also contributes to the activation of various cytokines involved in fibrosis in different organs (153). Melatonin may inhibit the fibrotic process by regulating these pathways. However, the antifibrotic effects of melatonin are mostly derived from animal studies and fibrotic cell models, lacking clinical trial evidence. Despite some current research limitations, melatonin remains a promising therapeutic target with broad application prospects. Future studies should further elucidate its mechanisms of action in different organ fibrosis and develop corresponding therapeutic strategies, providing new insights and methods for treating fibrotic diseases.
Author contributions
WH: Investigation, Writing – original draft, Writing – review & editing. JZ: Investigation, Writing – original draft. MW: Supervision, Writing – review & editing. L-YD: Visualization, Writing – review & editing. LB: Conceptualization, Funding acquisition, Writing – review & editing. HT: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key Research and Development Program of China (No. 2022YFC2304800) and 1.3.5 project for disciplines of excellence-Clinical Research Incubation Project, West China Hospital, Sichuan University (ZYJC21014).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ECM, Extracellular matrix; 5-HT, 5-hydroxytryptamine; SCN, Suprachiasmatic nucleus; NE, Norepinephrine; cAMP, Cyclic adenosine monophosphate; MT1, Melatonin receptor 1A; MT2, Melatonin receptor 1B; MT3, Melatonin receptor 1C; MMPs, Matrix metalloproteinases; α-SMA, Alpha smooth muscle actin; EMT, Epithelial mesenchymal transition; EndoMT, Endothelial mesenchymal transition; TGF-β, Transforming growth factor; IL, Interleukin; TNF-α, Tumor necrosis factor-α; PDGF, Platelet-derived growth factor; CTGF, Connective tissue growth factor; Ang-II, Angiotensin-II; ROS, Reactive oxygen species; JNK, c-Jun N-terminal kinase; ERK, Extracellular signal-regulated kinase; PI3K/Akt, Phosphatidylinositol 3-kinase/Akt; ZEB, Zinc finger E-box-binding homeobox; NF-κB, Nuclear factor-kappa B; SOD, Superoxide dismutase; CAT, Catalase; GPx, Glutathione peroxidase; GSH, Glutathione; GRd, Glutathione reductase; NOS, Nitric oxide synthase; LPS, Lipopolysaccharide; Nrf2, Nuclear erythroid 2-related factor 2; NLRP3, NLR family pyrin domain containing 3; MDA, Malondialdehyde; GAG, Glycosaminoglycan; TLR, Toll-like receptor; Cyt C, Cytochrome c; MI, Myocardial infarction; PM2.5, Particulate matter; SIRT3, Sirtuins 3; HDAC, Histone deacetylases; Trx, Thioredoxin; COPD, Chronic obstructive pulmonary disease; IPF, Idiopathic pulmonary fibrosis; ARDS, Acute respiratory distress syndrome; CCl4, Carbon tetrachloride; COX2, Cyclooxygenase 2; AECs, Alveolar epithelial cells; GSK-3β, Glycogen synthase kinase 3β; AIH, Autoimmune hepatitis; NASH, Non-alcoholic steatohepatitis; HSCs, Hepatic stellate cells; HA, Hyaluronic acid; HYP, Hydroxyproline; LN, Laminin; CKD, Chronic kidney disease; iNOS, Inducible NOS; HS, Hypertrophic scar; LS, Localized scleroderma; SSc, Systemic sclerosis; GVHD, Chronic graft-versus-host disease; HSFs, HS fibroblasts; KFs, Keloid fibroblasts.
References
1. Rosenbloom, J, Castro, SV, and Jimenez, SA. Narrative review: fibrotic diseases: cellular and molecular mechanisms and novel therapies. Ann Intern Med. (2010) 152:159–66. doi: 10.7326/0003-4819-152-3-201002020-00007
2. Rockey, DC, Bell, PD, and Hill, JA. Fibrosis--A common pathway to organ injury and failure. N Engl J Med. (2015) 373:96. doi: 10.1056/NEJMc1504848
3. Wynn, T. Cellular and molecular mechanisms of fibrosis. J Pathol. (2008) 214:199–210. doi: 10.1002/path.2277
4. Esposito, E, and Cuzzocrea, S. Antiinflammatory activity of melatonin in central nervous system. Curr Neuropharmacol. (2010) 8:228–42. doi: 10.2174/157015910792246155
5. Lerner, AB, Case, JD, Takahashi, Y, Lee, TH, and Mori, W. Isolation of melatonin, the pineal gland factor that lightens MELANOCYTES1. J Am Chem Soc. (1958) 80:2587–7. doi: 10.1021/ja01543a060
6. Amaral, FGD, and Cipolla-Neto, J. A brief review about melatonin, a pineal hormone. Arch Endocrinol Metab. (2018) 62:472–9. doi: 10.20945/2359-3997000000066
7. Gorman, MR. Temporal organization of pineal melatonin signaling in mammals. Mol Cell Endocrinol. (2020) 503:110687. doi: 10.1016/j.mce.2019.110687
8. Hajak, G, Rodenbeck, A, Staedt, J, Huether, G, Rüther, E, and Rüther, E. Nocturnal plasma melatonin levels in patients suffering from chronic primary insomnia. J Pineal Res. (1995) 19:116–22. doi: 10.1111/j.1600-079X.1995.tb00179.x
9. Fernández-Durán, B, Ruibal, C, Polakof, S, Ceinos, RM, Soengas, JL, and Míguez, JM. Evidence for arylalkylamine N-acetyltransferase (AANAT2) expression in rainbow trout peripheral tissues with emphasis in the gastrointestinal tract. Gen Comp Endocrinol. (2007) 152:289–94. doi: 10.1016/j.ygcen.2006.12.008
10. Buxton, OM, L'Hermite-Balériaux, M, Hirschfeld, U, and van Cauter, E. Acute and delayed effects of exercise on human melatonin secretion. J Biol Rhythm. (1997) 12:568–74. doi: 10.1177/074873049701200611
11. Cassone, VM. Effects of melatonin on vertebrate circadian systems. Trends Neurosci. (1990) 13:457–64. doi: 10.1016/0166-2236(90)90099-V
12. Reppert, SM, Weaver, DR, and Ebisawa, T. Cloning and characterization of a mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron. (1994) 13:1177–85. doi: 10.1016/0896-6273(94)90055-8
13. Reppert, SM, Godson, C, Mahle, CD, Weaver, DR, Slaugenhaupt, SA, and Gusella, JF. Molecular characterization of a second melatonin receptor expressed in human retina and brain: the Mel1b melatonin receptor. Proc Natl Acad Sci. (1995) 92:8734–8. doi: 10.1073/pnas.92.19.8734
14. Nosjean, O, Ferro, M, Cogé, F, Beauverger, P, Henlin, JM, Lefoulon, F, et al. Identification of the melatonin-binding SiteMT 3 as the Quinone reductase 2*. J Biol Chem. (2000) 275:31311–7. doi: 10.1074/jbc.M005141200
15. Luo, C, Yang, Q, Liu, Y, Zhou, S, Jiang, J, Reiter, RJ, et al. The multiple protective roles and molecular mechanisms of melatonin and its precursor N-acetylserotonin in targeting brain injury and liver damage and in maintaining bone health. Free Radic Biol Med. (2019) 130:215–33. doi: 10.1016/j.freeradbiomed.2018.10.402
16. Ashrafizadeh, M, Najafi, M, Kavyiani, N, Mohammadinejad, R, Farkhondeh, T, and Samarghandian, S. Anti-inflammatory activity of melatonin: a focus on the role of NLRP3 Inflammasome. Inflammation. (2021) 44:1207–22. doi: 10.1007/s10753-021-01428-9
17. Gurtner, GC, Werner, S, Barrandon, Y, and Longaker, MT. Wound repair and regeneration. Nature. (2008) 453:314–21. doi: 10.1038/nature07039
18. Li, X, Mo, N, and Li, Z. Ginsenosides: potential therapeutic source for fibrosis-associated human diseases. J Ginseng Res. (2020) 44:386–98. doi: 10.1016/j.jgr.2019.12.003
19. Wynn, TA, and Vannella, KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. (2016) 44:450–62. doi: 10.1016/j.immuni.2016.02.015
20. Stramer, BM, Mori, R, and Martin, P. The inflammation–fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol. (2007) 127:1009–17. doi: 10.1038/sj.jid.5700811
21. Hinz, B, Phan, SH, Thannickal, VJ, Galli, A, Bochaton-Piallat, ML, and Gabbiani, G. The Myofibroblast: one function, multiple origins. Am J Pathol. (2007) 170:1807–16. doi: 10.2353/ajpath.2007.070112
22. Kirk, TZ, Mark, ME, Chua, CC, Chua, BH, and Mayes, MD. Myofibroblasts from scleroderma skin synthesize elevated levels of collagen and tissue inhibitor of metalloproteinase (TIMP-1) with two forms of TIMP-1. J Biol Chem. (1995) 270:3423–8. doi: 10.1074/jbc.270.7.3423
23. Hinz, B, Phan, SH, Thannickal, VJ, Prunotto, M, Desmoulière, A, Varga, J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. (2012) 180:1340–55. doi: 10.1016/j.ajpath.2012.02.004
24. Mcanulty, RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. (2007) 39:666–71. doi: 10.1016/j.biocel.2006.11.005
25. Karasek, MA. Does transformation of microvascular endothelial cells into myofibroblasts play a key role in the etiology and pathology of fibrotic disease? Med Hypotheses. (2007) 68:650–5. doi: 10.1016/j.mehy.2006.07.053
26. Piera-Velazquez, S, Mendoza, FA, and Jimenez, SA. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of human fibrotic diseases. J Clin Med. (2016) 5:45. doi: 10.3390/jcm5040045
27. Lamouille, S, Xu, J, and Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. (2014) 15:178–96. doi: 10.1038/nrm3758
28. du, Y, Ma, J, Fan, Y, Wang, X, Zheng, S, Feng, J, et al. Naringenin: A promising therapeutic agent against organ fibrosis. Oxidative Med Cell Longev. (2021) 2021:1210675. doi: 10.1155/2021/1210675
29. Lua, I, and Asahina, K. The role of mesothelial cells in liver development, injury, and regeneration. Gut Liver. (2016) 10:166–76. doi: 10.5009/gnl15226
30. Kim, KK, Sheppard, D, and Chapman, HA. TGF-β1 signaling and tissue fibrosis. Cold Spring Harb Perspect Biol. (2018) 10:a022293. doi: 10.1101/cshperspect.a022293
31. Isshiki, T, Naiel, S, Vierhout, M, Otsubo, K, Ali, P, Tsubouchi, K, et al. Therapeutic strategies to target connective tissue growth factor in fibrotic lung diseases. Pharmacol Ther. (2024) 253:108578. doi: 10.1016/j.pharmthera.2023.108578
32. Kaschina, E, Lauer, D, Lange, C, and Unger, T. Angiotensin AT(2) receptors reduce inflammation and fibrosis in cardiovascular remodeling. Biochem Pharmacol. (2024) 222:116062. doi: 10.1016/j.bcp.2024.116062
33. Zhang, X, Zhang, X, Huang, W, and Ge, X. The role of heat shock proteins in the regulation of fibrotic diseases. Biomed Pharmacother. (2021) 135:111067. doi: 10.1016/j.biopha.2020.111067
34. Swigris, JJ, and Brown, KK. The role of endothelin-1 in the pathogenesis of idiopathic pulmonary fibrosis. BioDrugs. (2010) 24:49–54. doi: 10.2165/11319550-000000000-00000
35. Li, R, and Frangogiannis, NG. Integrins in cardiac fibrosis. J Mol Cell Cardiol. (2022) 172:1–13. doi: 10.1016/j.yjmcc.2022.07.006
36. Morikawa, M, Derynck, R, and Miyazono, KJCSHPIB. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Perspect Biol. (2016) 8:a021873. doi: 10.1101/cshperspect.a021873
37. Ong, CH, Tham, CL, Harith, HH, Firdaus, N, and Israf, DA. TGF-β-induced fibrosis: A review on the underlying mechanism and potential therapeutic strategies. Eur J Pharmacol. (2021) 911:174510. doi: 10.1016/j.ejphar.2021.174510
38. Kamato, D, Burch, ML, Piva, TJ, Rezaei, HB, Rostam, MA, Xu, S, et al. Transforming growth factor-β signalling: role and consequences of Smad linker region phosphorylation. Cell Signal. (2013) 25:2017–24. doi: 10.1016/j.cellsig.2013.06.001
39. Hu, HH, Chen, DQ, Wang, YN, Feng, YL, Cao, G, Vaziri, ND, et al. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem Biol Interact. (2018) 292:76–83. doi: 10.1016/j.cbi.2018.07.008
40. Heldin, CH, and Moustakas, A. Signaling receptors for TGF-β family members. Cold Spring Harb Perspect Biol. (2016) 8:a022053. doi: 10.1101/cshperspect.a022053
41. Palumbo, K, Zerr, P, Tomcik, M, Dees, C, Akhmetshina, A, Avouac, J, et al. The transcription factor JunD mediates transforming growth factor -induced fibroblast activation and fibrosis in systemic sclerosis. Ann Rheum Dis. (2011) 70:1320–6. doi: 10.1136/ard.2010.148296
42. Wang, Y-L, Zhao, X-M, Shuai, Z-F, Li, CY, Bai, QY, and Yu, XW. Snail promotes epithelial-mesenchymal transition and invasiveness in human ovarian cancer cells. Int J Clin Exp Med. (2015) 8:7388.
43. Chang, YC, Tsai, CH, Lai, YL, Yu, CC, Chi, WY, Li, JJ, et al. Arecoline‐induced myofibroblast transdifferentiation from human buccal mucosal fibroblasts is mediated byZEB1. J Med Biochem. (2014) 18:698–708. doi: 10.1111/jcmm.12219
44. White, S, Lin, L, and Hu, K. NF-κB and tPA signaling in kidney and other diseases. Cells. (2020) 9:1348. doi: 10.3390/cells9061348
45. Jaffar, J, Glaspole, I, Symons, K, and Westall, G. Inhibition of NF-κB by ACT001 reduces fibroblast activity in idiopathic pulmonary fibrosis. Biomed Pharmacother. (2021) 138:111471. doi: 10.1016/j.biopha.2021.111471
46. Brzezinski, A. Melatonin in humans. N Engl J Med. (1997) 336:186–95. doi: 10.1056/NEJM199701163360306
47. Armstrong, SM. Melatonin and circadian control in mammals. Experientia. (1989) 45:932–8. doi: 10.1007/BF01953050
48. Jung, B, and Ahmad, N. Melatonin in cancer management: progress and promise. Cancer Res. (2006) 66:9789–93. doi: 10.1158/0008-5472.CAN-06-1776
49. Szczepanik, M. Melatonin and its influence on immune system. J Physiol Pharmacol. (2007) 58:115–24. doi: 10.2170/physiolsci.SC008107
50. Reiter, RJ, Tan, DX, Gitto, E, Sainz, RM, Mayo, JC, Leon, J, et al. Pharmacological utility of melatonin in reducing oxidative cellular and molecular damage. Pol J Pharmacol. (2004) 56:159–70.
51. Kozina, LS, Arutjunyan, AV, and Khavinson, VK. Antioxidant properties of geroprotective peptides of the pineal gland. Arch Gerontol Geriatr. (2007) 44:213–6. doi: 10.1016/j.archger.2007.01.029
52. Tengattini, S, Reiter, RJ, Tan, D-X, Terron, MP, Rodella, LF, and Rezzani, R. Cardiovascular diseases: protective effects of melatonin. J Pineal Res. (2008) 44:16–25. doi: 10.1111/j.1600-079X.2007.00518.x
53. Urtasun, R, Conde De La Rosa, L, and Nieto, N. Oxidative and nitrosative stress and fibrogenic response. Clin Liver Dis. (2008) 12:769–790, viii. doi: 10.1016/j.cld.2008.07.005
54. Lei, X, Xu, Z, Huang, L, Huang, Y, Tu, S, Xu, L, et al. The potential influence of melatonin on mitochondrial quality control: a review. Front Pharmacol. (2023) 14:1332567. doi: 10.3389/fphar.2023.1332567
55. Li, D, Pan, JH, Huang, XF, Liao, YQ, Ling, YJ, and Luo, JY. Effect of melatonin on oxidative stress indicators in animal models of fibrosis: A systematic review and meta-analysis. Free Radic Biol Med. (2023) 195:158–77. doi: 10.1016/j.freeradbiomed.2022.12.094
56. Limson, J, Nyokong, T, and Daya, S. The interaction of melatonin and its precursors with aluminium, cadmium, copper, iron, lead, and zinc: an adsorptive voltammetric study. J Pineal Res. (1998) 24:15–21. doi: 10.1111/j.1600-079X.1998.tb00361.x
57. Reiter, RJ, Mayo, JC, Tan, D-X, Sainz, RM, Alatorre-Jimenez, M, and Qin, L. Melatonin as an antioxidant: under promises but over delivers. J Pineal Res. (2016) 61:253–78. doi: 10.1111/jpi.12360
58. Paskaloğlu, K, Şener, G, Kapucu, C, and Ayanoğlu-Dülger, G. Melatonin treatment protects against sepsis-induced functional and biochemical changes in rat ileum and urinary bladder. Life Sci. (2004) 74:1093–104. doi: 10.1016/j.lfs.2003.07.038
59. Sener, G, Toklu, H, Kapucu, C, Ercan, F, Erkanli, G, Kaçmaz, A, et al. Melatonin protects against oxidative organ injury in a rat model of sepsis. Surg Today. (2005) 35:52–9. doi: 10.1007/s00595-004-2879-1
60. Fjaerli, O, Lund, T, and Osterud, B. The effect of melatonin on cellular activation processes in human blood. J Pineal Res. (1999) 26:50–5. doi: 10.1111/j.1600-079X.1999.tb00566.x
61. Silva, SO, Rodrigues, MR, Ximenes, VF, Bueno-da-Silva, AEB, Amarante-Mendes, GP, and Campa, A. Neutrophils as a specific target for melatonin and kynuramines: effects on cytokine release. J Neuroimmunol. (2004) 156:146–52. doi: 10.1016/j.jneuroim.2004.07.015
62. Liu, SC, Tsai, CH, Wang, YH, Su, CM, Wu, HC, Fong, YC, et al. Melatonin abolished proinflammatory factor expression and antagonized osteoarthritis progression in vivo. Cell Death Dis. (2022) 13:215. doi: 10.1038/s41419-022-04656-5
63. Negi, G, Kumar, A, and Sharma, SS. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: effects on NF-κB and Nrf2 cascades. J Pineal Res. (2011) 50:124–31. doi: 10.1111/j.1600-079X.2010.00821.x
64. Ismail, IA, El-Bakry, HA, and Soliman, SS. Melatonin and tumeric ameliorate aging-induced changes: implication of immunoglobulins, cytokines, DJ-1/NRF2 and apoptosis regulation. Int J Physiol Pathophysiol Pharmacol. (2018) 10:70–82.
65. Hardeland, R. Melatonin and inflammation—story of a double-edged blade. J Pineal Res. (2018) 65:e12525. doi: 10.1111/jpi.12525
66. Bilici, D, Akpinar, E, and Kiziltunç, A. Protective effect of melatonin in carrageenan-induced acute local inflammation. Pharmacol Res. (2002) 46:133–9. doi: 10.1016/S1043-6618(02)00089-0
67. Su, S-C, Hsieh, M-J, Yang, W-E, Chung, WH, Reiter, RJ, and Yang, SF. Cancer metastasis: mechanisms of inhibition by melatonin. J Pineal Res. (2017) 62:e12370. doi: 10.1111/jpi.12370
68. Jun, J-I, and Lau, LFJTJOCI. Resolution of organ fibrosis. Clin Res. (2018) 128:97–107. doi: 10.1172/JCI93563
69. Gill, SE, and Parks, WC. Metalloproteinases and their inhibitors: regulators of wound healing. Int J Biochem Cell Biol. (2008) 40:1334–47. doi: 10.1016/j.biocel.2007.10.024
70. Parks, WC, Wilson, CL, and López-Boado, YSJNRI. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immun. (2004) 4:617–29. doi: 10.1038/nri1418
71. Butler, GS, and Overall, CMJP. Matrix metalloproteinase processing of signaling molecules to regulate inflammation. Periodontol. (2013) 63:123–48. doi: 10.1111/prd.12035
72. Swarnakar, S, Paul, S, Singh, LP, and Reiter, RJ. Matrix metalloproteinases in health and disease: regulation by melatonin. J Pineal Res. (2011) 50:8–20. doi: 10.1111/j.1600-079X.2010.00812.x
73. Rudra, DS, Pal, U, Maiti, NC, Reiter, RJ, and Swarnakar, S. Melatonin inhibits matrix metalloproteinase-9 activity by binding to its active site. J Pineal Res. (2013) 54:398–405. doi: 10.1111/jpi.12034
74. Kapoor, C, Vaidya, S, Wadhwan, V, Hitesh,, Kaur, G, and Pathak, A. Seesaw of matrix metalloproteinases (MMPs). J Cancer Res Ther. (2016) 12:28–35. doi: 10.4103/0973-1482.157337
75. Crespo, I, San-Miguel, B, Fernández, A, Ortiz de Urbina, J, González-Gallego, J, and Tuñón, MJ. Melatonin limits the expression of profibrogenic genes and ameliorates the progression of hepatic fibrosis in mice. Transl Res. (2015) 165:346–57. doi: 10.1016/j.trsl.2014.10.003
76. Simko, F, Pechanova, O, Pelouch, V, Krajcirovicova, K, Mullerova, M, Bednarova, K, et al. Effect of melatonin, captopril, spironolactone and simvastatin on blood pressure and left ventricular remodelling in spontaneously hypertensive rats. J Hypertens Suppl. (2009) 27:S5–S10. doi: 10.1097/01.hjh.0000358830.95439.e8
77. Marquez, E, Sánchez-Fidalgo, S, Calvo, JR, Lastra, CA, and Motilva, V. Acutely administered melatonin is beneficial while chronic melatonin treatment aggravates the evolution of TNBS-induced colitis. J Pineal Res. (2006) 40:48–55. doi: 10.1111/j.1600-079X.2005.00275.x
78. Karimfar, MH, Rostami, S, Haghani, K, Bakhtiyari, S, and Noori-Zadeh, A. Melatonin alleviates bleomycin-induced pulmonary fibrosis in mice. J Biol Regul Homeost Agents. (2015) 29:327–34.
79. Drobnik, J, Slotwinska, D, Olczak, S, Tosik, D, Pieniazek, A, Matczak, K, et al. Pharmacological doses of melatonin reduce the glycosaminoglycan level within the infarcted heart scar. J Physiol Pharmacol. (2011) 62:29–35.
80. Shook, D, and Keller, R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev. (2003) 120:1351–83. doi: 10.1016/j.mod.2003.06.005
81. Heerboth, S, Housman, G, Leary, M, Longacre, M, Byler, S, Lapinska, K, et al. EMT and tumor metastasis. Clin Transl Med. (2015) 4:6. doi: 10.1186/s40169-015-0048-3
82. Lovisa, S, LeBleu, VS, Tampe, B, Sugimoto, H, Vadnagara, K, Carstens, JL, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. (2015) 21:998–1009. doi: 10.1038/nm.3902
83. Youssef, KK, and Nieto, MA. Epithelial-mesenchymal transition in tissue repair and degeneration. Nat Rev Mol Cell Biol. (2024) 25:720–39. doi: 10.1038/s41580-024-00733-z
84. López-Novoa, JM, and Nieto, MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. (2009) 1:303–14. doi: 10.1002/emmm.200900043
85. Qi, S, Yan, L, Liu, Z, Mu, YL, Li, M, Zhao, X, et al. Melatonin inhibits 17β-estradiol-induced migration, invasion and epithelial-mesenchymal transition in normal and endometriotic endometrial epithelial cells. Reprod Biol Endocrinol. (2018) 16:62. doi: 10.1186/s12958-018-0375-5
86. Qin, T, Feng, D, Zhou, B, Bai, L, and Yin, Y. Melatonin suppresses LPS-induced oxidative stress in dendritic cells for inflammatory regulation via the Nrf2/HO-1 Axis. Antioxidants (Basel). (2022) 11:2012. doi: 10.3390/antiox11102012
87. Shi, S, Zhang, Y, Wen, W, Zhao, Y, and Sun, L. Molecular mechanisms of melatonin in the reversal of LPS-induced EMT in peritoneal mesothelial cells. Mol Med Rep. (2016) 14:4342–8. doi: 10.3892/mmr.2016.5744
88. Hetts, SW. To die or not to die: an overview of apoptosis and its role in disease. JAMA. (1998) 279:300–7. doi: 10.1001/jama.279.4.300
89. Jan, R, and Chaudhry, GE. Understanding apoptosis and apoptotic pathways targeted Cancer therapeutics. Adv Pharm Bull. (2019) 9:205–18. doi: 10.15171/apb.2019.024
90. Kim, R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer. (2005) 103:1551–60. doi: 10.1002/cncr.20947
91. Jin, Z, and El-Deiry, WS. Overview of cell death signaling pathways. Cancer Biol Ther. (2005) 4:139–63. doi: 10.4161/cbt.4.2.1508
92. Kumar, R, Herbert, PE, and Warrens, AN. An introduction to death receptors in apoptosis. Int J Surg. (2005) 3:268–77. doi: 10.1016/j.ijsu.2005.05.002
93. Kim, JW, Jo, J, Kim, JY, Choe, M, Leem, J, and Park, JH. Melatonin attenuates cisplatin-induced acute kidney injury through dual suppression of apoptosis and necroptosis. Biology (Basel). (2019) 8:64. doi: 10.3390/biology8030064
94. Hoijman, E, Rocha Viegas, L, Keller Sarmiento, MI, Rosenstein, RE, and Pecci, A. Involvement of Bax protein in the prevention of glucocorticoid-induced thymocytes apoptosis by melatonin. Endocrinology. (2004) 145:418–25. doi: 10.1210/en.2003-0764
95. Xie, YF, Zhang, JC, Liu, SJ, Dai, LB, and du, GW. Effect of melatonin on proliferation and apoptosis of fibroblasts in human hypertrophic scar. Zhonghua Shao Shang Za Zhi. (2011) 27:422–6. doi: 10.3760/cma.j.issn.1009-2587.2011.06.005
96. Ben Soussia, I, Mies, F, Naeije, R, and Shlyonsky, V. Melatonin down-regulates volume-sensitive chloride channels in fibroblasts. Pflugers Arch. (2012) 464:273–85. doi: 10.1007/s00424-012-1139-2
97. Rockey, DC, Bell, PD, and Hill, JA. Fibrosis — A common pathway to organ injury and failure. N Engl J Med. (2015) 372:1138–49. doi: 10.1056/NEJMra1300575
98. Berk, BC, Fujiwara, K, and Lehoux, S. ECM remodeling in hypertensive heart disease. J Clin Invest. (2007) 117:568–75. doi: 10.1172/JCI31044
99. Nguyen, TP, Qu, Z, and Weiss, JN. Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol. (2014) 70:83–91. doi: 10.1016/j.yjmcc.2013.10.018
100. Misaka, T, Yoshihisa, A, Yokokawa, T, Sato, T, Oikawa, M, Kobayashi, A, et al. Plasma levels of melatonin in dilated cardiomyopathy. J Pineal Res. (2019) 66:e12564. doi: 10.1111/jpi.12564
101. Lusardi, P, Piazza, E, and Fogari, R. Cardiovascular effects of melatonin in hypertensive patients well controlled by nifedipine: a 24-hour study. Br J Clin Pharmacol. (2000) 49:423–7. doi: 10.1046/j.1365-2125.2000.00195.x
102. Dominguez-Rodriguez, A, Abreu-Gonzalez, P, Arroyo-Ucar, E, and Reiter, RJ. Decreased level of melatonin in serum predicts left ventricular remodelling after acute myocardial infarction. J Pineal Res. (2012) 53:319–23. doi: 10.1111/j.1600-079X.2012.01001.x
103. Jiang, J, Liang, S, Zhang, J, du, Z, Xu, Q, Duan, J, et al. Melatonin ameliorates PM(2.5) -induced cardiac perivascular fibrosis through regulating mitochondrial redox homeostasis. J Pineal Res. (2021) 70:e12686. doi: 10.1111/jpi.12686
104. Che, H, Wang, Y, Li, H, Li, Y, Sahil, A, Lv, J, et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-β1/Smads signaling in diabetic cardiomyopathy. FASEB J. (2020) 34:5282–98. doi: 10.1096/fj.201902692R
105. Bagchi, RA, and Weeks, KL. Histone deacetylases in cardiovascular and metabolic diseases. J Mol Cell Cardiol. (2019) 130:151–9. doi: 10.1016/j.yjmcc.2019.04.003
106. Wu, Y, Si, F, Luo, L, Jing, F, Jiang, K, Zhou, J, et al. The effect of melatonin on cardio fibrosis in juvenile rats with pressure overload and deregulation of HDACs. Korean J Physiol Pharmacol. (2018) 22:607–16. doi: 10.4196/kjpp.2018.22.6.607
107. Lu, L, Ma, J, Sun, M, Wang, X, Gao, E, Lu, L, et al. Melatonin ameliorates MI-induced cardiac remodeling and apoptosis through a JNK/p53-dependent mechanism in diabetes mellitus. Oxidative Med Cell Longev. (2020) 2020:1–14. doi: 10.1155/2020/1535201
108. Li, Y, Hiroi, Y, and Liao, JK. Notch signaling as an important mediator of cardiac repair and regeneration after myocardial infarction. Trends Cardiovasc Med. (2010) 20:228–31. doi: 10.1016/j.tcm.2011.11.006
109. Yu, L, Fan, C, Li, Z, Zhang, J, Xue, X, Xu, Y, et al. Melatonin rescues cardiac thioredoxin system during ischemia-reperfusion injury in acute hyperglycemic state by restoring Notch1/Hes1/Akt signaling in a membrane receptor-dependent manner. J Pineal Res. (2017) 62:1–17. doi: 10.1111/jpi.12375
110. Wuyts, WA, Agostini, C, Antoniou, KM, Bouros, D, Chambers, RC, Cottin, V, et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. (2013) 41:1207–18. doi: 10.1183/09031936.00073012
111. Genovese, T, Paola, RD, Mazzon, E, Muià, C, Caputi, AP, and Cuzzocrea, S. Melatonin limits lung injury in bleomycin treated mice. J Pineal Res. (2005) 39:105–12. doi: 10.1111/j.1600-079X.2005.00229.x
112. Yildirim, Z, Kotuk, M, Erdogan, H, Iraz, M, Yagmurca, M, Kuku, I, et al. Preventive effect of melatonin on bleomycin-induced lung fibrosis in rats. J Pineal Res. (2006) 40:27–33. doi: 10.1111/j.1600-079X.2005.00272.x
113. Zhang, L, Li, F, Su, X, Li, Y, Wang, Y, Fang, R, et al. Melatonin prevents lung injury by regulating apelin 13 to improve mitochondrial dysfunction. Exp Mol Med. (2019) 51:1–12. doi: 10.1038/s12276-019-0273-8
114. Ding, Z, Wu, X, Wang, Y, Ji, S, Zhang, W, Kang, J, et al. Melatonin prevents LPS-induced epithelial-mesenchymal transition in human alveolar epithelial cells via the GSK-3β/Nrf2 pathway. Biomed Pharmacother. (2020) 132:110827. doi: 10.1016/j.biopha.2020.110827
115. Henderson, WR, Chi, EY, Ye, X, Nguyen, C, Tien, YT, Zhou, B, et al. Inhibition of Wnt/β-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. PNAS Nexus. (2010) 107:14309–14. doi: 10.1073/pnas.1001520107
116. Lam, AP, and Gottardi, CJ. β-Catenin signaling: a novel mediator of fibrosis and potential therapeutic target. Curr Opin Rheumatol. (2011) 23:562–7. doi: 10.1097/BOR.0b013e32834b3309
117. Yu, N, Sun, YT, Su, XM, He, M, Dai, B, and Kang, J. Melatonin attenuates TGFβ1-induced epithelial-mesenchymal transition in lung alveolar epithelial cells. Mol Med Rep. (2016) 14:5567–72. doi: 10.3892/mmr.2016.5950
118. Ginès, P, Cárdenas, A, Arroyo, V, and Rodés, J. Management of cirrhosis and ascites. N Engl J Med. (2004) 350:1646–54. doi: 10.1056/NEJMra035021
119. Bataller, R, and Brenner, DA. Liver fibrosis. J Clin Invest. (2005) 115:209–18. doi: 10.1172/JCI24282
120. Hosseinzadeh, A, Pourhanifeh, MH, Amiri, S, Sheibani, M, Irilouzadian, R, Reiter, RJ, et al. Therapeutic potential of melatonin in targeting molecular pathways of organ fibrosis. Pharmacol Rep. (2024) 76:25–50. doi: 10.1007/s43440-023-00554-5
121. Elpek, G. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: an update. World J Gastroenterol. (2014) 20:7260–76. doi: 10.3748/wjg.v20.i23.7260
122. Zhu, L, Zhang, Q, Hua, C, and Ci, X. Melatonin alleviates particulate matter-induced liver fibrosis by inhibiting ROS-mediated mitophagy and inflammation via Nrf2 activation. Ecotoxicol Environ Saf. (2023) 268:115717. doi: 10.1016/j.ecoenv.2023.115717
123. Hong, RT, Xu, JM, and Mei, Q. Melatonin ameliorates experimental hepatic fibrosis induced by carbon tetrachloride in rats. World J Gastroenterol. (2009) 15:1452–8. doi: 10.3748/wjg.15.1452
124. Aktas, C, Kanter, M, Erboga, M, Mete, R, and Oran, M. Melatonin attenuates oxidative stress, liver damage and hepatocyte apoptosis after bile-duct ligation in rats. Toxicol Ind Health. (2014) 30:835–44. doi: 10.1177/0748233712464811
125. Tampe, D, and Zeisberg, M. Potential approaches to reverse or repair renal fibrosis. Nat Rev Nephrol. (2014) 10:226–37. doi: 10.1038/nrneph.2014.14
126. Ammirati, AL. Chronic kidney disease. Rev Assoc Med Bras. (2020) 66:s03–9. doi: 10.1590/1806-9282.66.s1.3
127. Djudjaj, S, and Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol Asp Med. (2019) 65:16–36. doi: 10.1016/j.mam.2018.06.002
128. Yang, Z, He, Y, Ma, Q, Wang, H, and Zhang, Q. Alleviative effect of melatonin against the nephrotoxicity induced by cadmium exposure through regulating renal oxidative stress, inflammatory reaction, and fibrosis in a mouse model. Ecotoxicol Environ Saf. (2023) 265:115536. doi: 10.1016/j.ecoenv.2023.115536
129. Repova, K, Stanko, P, Baka, T, Krajcirovicova, K, Aziriova, S, Hrenak, J, et al. Lactacystin-induced kidney fibrosis: protection by melatonin and captopril. Front Pharmacol. (2022) 13:978337. doi: 10.3389/fphar.2022.978337
130. Kim, JY, Park, JH, Jeon, EJ, Leem, J, and Park, KK. Melatonin prevents transforming growth factor-β1-stimulated Transdifferentiation of renal interstitial fibroblasts to Myofibroblasts by suppressing reactive oxygen species-dependent mechanisms. Antioxidants (Basel). (2020) 9:39. doi: 10.3390/antiox9010039
131. Nguyen, JK, Austin, E, Huang, A, Mamalis, A, and Jagdeo, J. The IL-4/IL-13 axis in skin fibrosis and scarring: mechanistic concepts and therapeutic targets. Arch Dermatol Res. (2020) 312:81–92. doi: 10.1007/s00403-019-01972-3
132. Griffin, MF, Desjardins-Park, HE, Mascharak, S, Borrelli, MR, and Longaker, MT. Understanding the impact of fibroblast heterogeneity on skin fibrosis. Dis Model Mech. (2020) 13:dmm044164. doi: 10.1242/dmm.044164
133. Bayat, A, Mcgrouther, DA, and Ferguson, MW. Skin scarring. BMJ. (2003) 326:88–92. doi: 10.1136/bmj.326.7380.88
134. Dong, Y, Cao, X, Huang, J, Hu, Z, Chen, C, Chen, M, et al. Melatonin inhibits fibroblast cell functions and hypertrophic scar formation by enhancing autophagy through the MT2 receptor-inhibited PI3K/Akt /mTOR signaling. Biochim Biophys Acta Mol basis Dis. (2024) 1870:166887. doi: 10.1016/j.bbadis.2023.166887
135. Huang, S, Deng, W, Dong, Y, Hu, Z, Zhang, Y, Wang, P, et al. Melatonin influences the biological characteristics of keloid fibroblasts through the Erk and Smad signalling pathways. Burns. Trauma. (2023) 11:tkad005. doi: 10.1093/burnst/tkad005
136. Comai, S, and Gobbi, G. Melatonin, melatonin receptors and sleep: moving beyond traditional views. J Pineal Res. (2024) 76:e13011. doi: 10.1111/jpi.13011
137. Yousefi, T, Yousef Memar, M, Ahmadi Jazi, A, Zand, S, Reiter, RJ, Amirkhanlou, S, et al. Molecular pathways and biological roles of melatonin and vitamin D; effects on immune system and oxidative stress. Int Immunopharmacol. (2024) 143:113548. doi: 10.1016/j.intimp.2024.113548
138. He, S, Yu, Y, Chen, PQ, Sun, HM, Gao, XR, Sun, HZ, et al. Insufficient plasma melatonin and its association with neuropsychiatric impairments in patients with T2DM. J Diabetes Res. (2024) 2024:5661751. doi: 10.1155/2024/5661751
139. Zambrano, C, Garitaonaindia, MT, Salmerón, D, Pérez-Sanz, F, Tchio, C, Picinato, MC, et al. Melatonin decreases human adipose tissue insulin sensitivity. J Pineal Res. (2024) 76:e12965. doi: 10.1111/jpi.12965
140. Song, Y-J, Zhong, C-B, and Wu, W. Cardioprotective effects of melatonin: focusing on its roles against diabetic cardiomyopathy. Biomed Pharmacother. (2020) 128:110260. doi: 10.1016/j.biopha.2020.110260
141. Blask, DE, Brainard, GC, Dauchy, RT, Hanifin, JP, Davidson, LK, Krause, JA, et al. Melatonin-depleted blood from premenopausal women exposed to light at night stimulates growth of human breast cancer xenografts in nude rats. Cancer Res. (2005) 65:11174–84. doi: 10.1158/0008-5472.CAN-05-1945
142. Andersen, LP, Werner, MU, Rosenkilde, MM, Harpsøe, NG, Fuglsang, H, Rosenberg, J, et al. Pharmacokinetics of oral and intravenous melatonin in healthy volunteers. BMC Pharmacol Toxicol. (2016) 17:8. doi: 10.1186/s40360-016-0052-2
143. Harpsøe, NG, Andersen, LP, Gögenur, I, and Rosenberg, J. Clinical pharmacokinetics of melatonin: a systematic review. Eur J Clin Pharmacol. (2015) 71:901–9. doi: 10.1007/s00228-015-1873-4
144. Lu, KH, Lin, RC, Yang, JS, Yang, WE, Reiter, RJ, and Yang, SF. Molecular and cellular mechanisms of melatonin in osteosarcoma. Cells. (2019) 8:1618. doi: 10.3390/cells8121618
145. Williams, WP, McLin, DE III, Dressman, MA, and Neubauer, DN. Comparative Review of approved melatonin agonists for the treatment of circadian rhythm sleep-wake disorders. Pharmacotherapy. (2016) 36:1028–41. doi: 10.1002/phar.1822
146. Reiter, RJ, Sharma, R, Simko, F, Dominguez-Rodriguez, A, Tesarik, J, Neel, RL, et al. Melatonin: highlighting its use as a potential treatment for SARS-CoV-2 infection. Cell Mol Life Sci. (2022) 79:143. doi: 10.1007/s00018-021-04102-3
147. Nordlund, JJ, and Lerner, AB. The effects of oral melatonin on skin color and on the release of pituitary hormones. J Clin Endocrinol Metab. (1977) 45:768–74. doi: 10.1210/jcem-45-4-768
148. Viana, S, de Bruin, VMS, Vasconcelos, RS, Nogueira, ANC, Mesquita, R, and de Bruin, PFC. Melatonin supplementation enhances pulmonary rehabilitation outcomes in COPD: a randomized, double-blind, placebo-controlled study. Respir Med. (2023) 220:107441. doi: 10.1016/j.rmed.2023.107441
149. Franco, C, Sciatti, E, Favero, G, Bonomini, F, Vizzardi, E, and Rezzani, R. Essential hypertension and oxidative stress: novel future perspectives. Int J Mol Sci. (2022) 23:14489. doi: 10.3390/ijms232214489
150. Ekeloef, S, Halladin, N, Fonnes, S, Jensen, SE, Zaremba, T, Rosenberg, J, et al. Effect of intracoronary and intravenous melatonin on myocardial salvage index in patients with ST-elevation myocardial infarction: a randomized placebo controlled trial. J Cardiovasc Transl Res. (2017) 10:470–9. doi: 10.1007/s12265-017-9768-7
151. Yamamoto, Y, and Gaynor, RB. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J Clin Invest. (2001) 107:135–42. doi: 10.1172/JCI11914
152. Franceschetti, L, Bonomini, F, Rodella, LF, and Rezzani, R. Critical role of NFκB in the pathogenesis of non-alcoholic fatty liver disease: A widespread key regulator. Curr Mol Med. (2021) 21:495–505. doi: 10.2174/1566524020666201026162343
Keywords: melatonin, fibrosis, therapeutics, protective effects, mechanisms
Citation: Huang W, Zheng J, Wang M, Du L-Y, Bai L and Tang H (2024) The potential therapeutic role of melatonin in organ fibrosis: a comprehensive review. Front. Med. 11:1502368. doi: 10.3389/fmed.2024.1502368
Edited by:
Yan-Ling Wu, Yanbian University, ChinaReviewed by:
Ratnakar Tripathi, University of Missouri, United StatesLorenzo Franceschetti, University of Milan, Italy
Copyright © 2024 Huang, Zheng, Wang, Du, Bai and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lang Bai, cGFuZ2JhaWxhbmdAMTYzLmNvbQ==; Hong Tang, dGFuZ2hvbmc2MTk4QHdjaHNjdS5jbg==
†These authors have contributed equally to this work