 Ankita Mitra
Ankita Mitra Dan Yi
Dan Yi Zhiyu Dai
Zhiyu Dai Vinicio de Jesus Perez
Vinicio de Jesus Perez- 1Division of Pulmonary and Critical Care, Stanford University, Palo Alto, CA, United States
- 2Department of Internal Medicine, University of Arizona College of Medicine Phoenix, Phoenix, AZ, United States
- 3Department of Medicine, Washington University School of Medicine in St. Louis (WashU), St. Louis, MO, United States
Pulmonary arterial hypertension (PAH) is characterized by pulmonary vascular remodeling with high pulmonary pressure, which ultimately leads to right heart failure and premature death. Emerging evidence suggests that both hypoxia and epigenetics play a pivotal role in the pathogenesis of PAH development. In this review article, we summarize the current developments in regulation of hypoxia inducible factor (HIF) isoforms in PAH vascular remodeling and the development of suitable animal models for discovery and testing of HIF pathway-targeting PAH therapeutics. In addition, we also discuss the epigenetic regulation of HIF-dependent isoforms in PAH and its therapeutic potential from a new perspective which highlights the importance of HIF isoform-specific targeting as a novel salutary strategy for PAH treatment.
1 Introduction
Pulmonary arterial hypertension (PAH) is a rare, life-threatening disorder associated with progressive elevation in pulmonary arterial pressure due to increased pulmonary vasculature resistance that leads to right heart failure and death (1–3). Pulmonary vascular remodeling is the key hallmark of PAH, which is driven by a combination of vasoconstriction, proliferation, inflammation, vascular stiffness, and thrombosis resulting in pulmonary vascular resistance (3). Despite extensive research in this area, the underlying mechanisms of PAH progression are incompletely understood. Current therapies are limited to the targeted pathways that only control vasocontraction, i.e., nitric oxide, prostacyclin and endothelin signaling (4). However, even a combination of therapies only improves symptoms and hemodynamics but fails to alleviate eventual right heart failure. Further understanding of vascular remodeling pathways can facilitate the development of appropriate vascular animal models for drug testing and accelerate the search for novel therapeutics against PAH.
PAH development can be linked to a multitude of pulmonary vascular insults such as hypoxia, genetic factors, environmental factors, drugs and toxins, etc. (4, 5). In addition, several molecular pathways have been reported to be associated with PAH development (5). It is, therefore, essential to identify specific ‘master’ pathways that are key triggers for PAH. Among all, Hypoxia inducible factor (HIF) signaling has been shown to be a crucial pathway in PAH pathogenesis (6, 7). Since the HIF pathway contains an abundance of key signaling molecules, a detailed and systematic analysis of the molecules and regulatory mechanisms involved is vital to facilitate identification of specific therapeutic targets (8). In addition, apart from critical molecular targets it is also necessary to identify cellular phenotypes that contribute to PAH pathogenesis and understand their association with the HIF pathway.
In this review, we will further our understanding of the HIF pathway and its epigenetic regulation while discussing the development of suitable animal models for discovery and testing of HIF pathway-targeting PAH therapeutics.
2 HIF in PAH
2.1 Biology of HIF signaling; role of hypoxia and inflammation in triggering HIF signaling
HIF is one of the major transcription factors and a master regulator for detecting and adapting to cellular oxygen levels, thereby transcriptionally activating the genes that modulate oxygen homeostasis and metabolic activation. It acts as a heterodimeric complex, composed of the oxygen-sensitive HIF-α subunit which includes HIF-1α, HIF-2α (EPAS1) and HIF-3α, and oxygen-insensitive HIF-β subunits including HIF-1β [aryl hydrocarbon receptor nuclear translocator (ARNT1, ARNT2, and ARNT3)] (9). Under normoxia, E3 ligase VHL protein binds to the HIF-α subunits upon hydroxylation by Prolyl Hydroxylase Domain proteins (PHDs) and Factor Inhibiting HIF (FIH), thus activating the ubiquitin ligase system, and leading to proteasomal degradation of HIF-α. However, under hypoxia, the PHDs are inactivated, leading to the attenuation of HIF-a stabilization and dimerization of HIF-1b, which forms an active HIF complex. Upon complex formation, HIF translocate to the nucleus and binds to the E-box-like hypoxia response elements (HREs) to induce gene expression which are involved in various cellular processes such as angiogenesis, erythropoiesis, regulation of vascular tone, cellular metabolism, proliferation, cell survival (9–11) (Figure 1). In pulmonary vasculature, angiogenesis is a repair program where endothelial cells (ECs) form new vessels by angiogenic sprouting which usually occurs after primary vascular plexus formation (9). VEGF is known as a master regulator of angiogenesis and under hypoxia, both VEGF and HIF-1α/HIF-2α are transcriptionally upregulated, promoting angiogenesis (12–14).
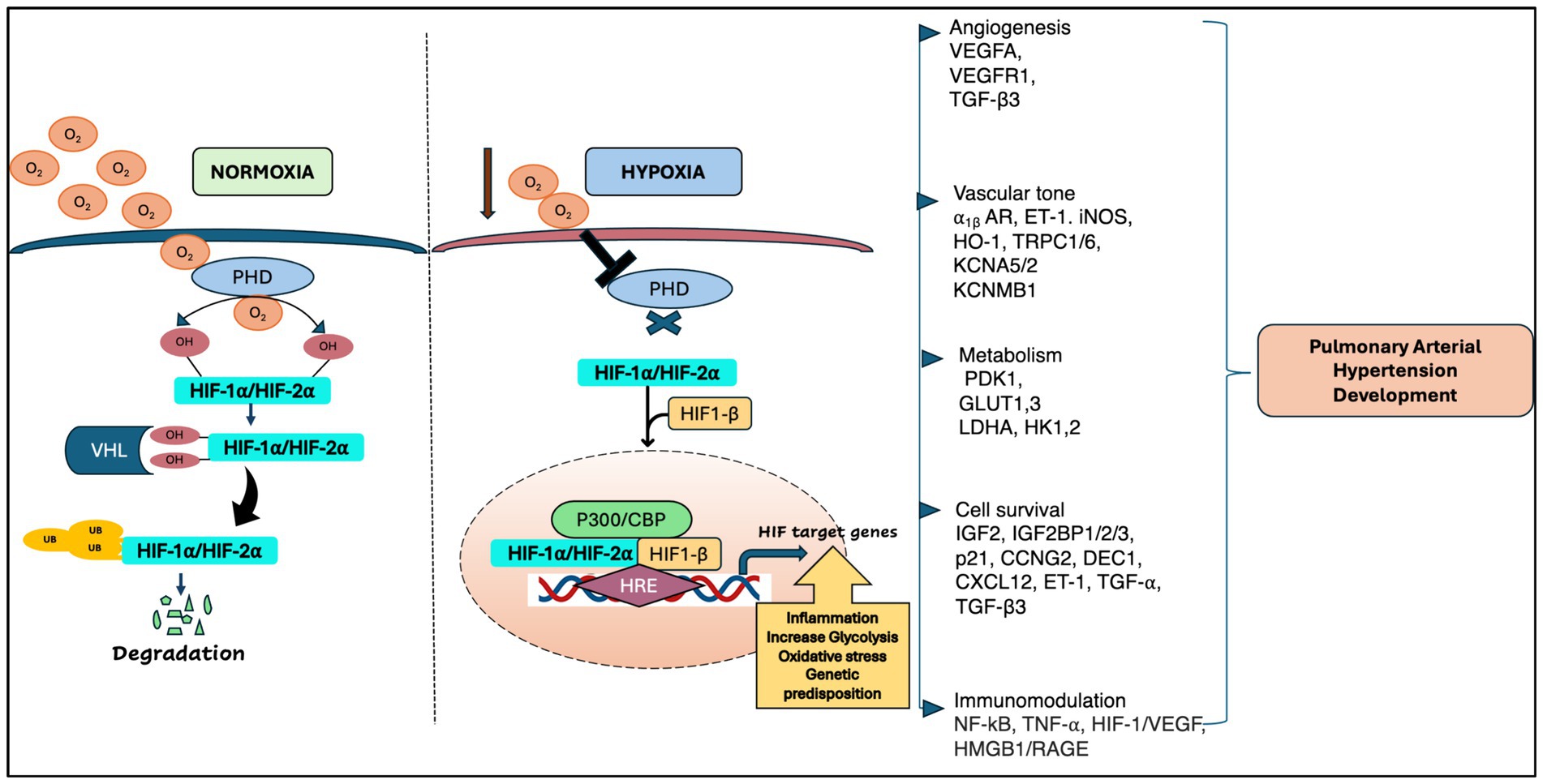
Figure 1. A schematic representation of HIF regulation in pulmonary arterial hypertension [adapted from Wan et al. (133) and Pullamsetti et al. (7)].
Besides oxygen-dependent HIF activity, HIF is also activated by inflammatory cytokines, bacterial products, and growth factors under normoxia conditions (15). To date, among all HIF-α, HIF-1α is predominantly expressed in both innate and adaptive immune populations including neutrophils, macrophages, dendritic cells, and lymphocytes (15, 16). In addition, HIF-2α expression is limited to certain range of endothelial cells and tumor associated macrophages including CD8+ T cells under hypoxia conditions (17). The orchestrated processes of HIF regulation modulate inflammation and govern the plethora of signaling pathways and gene expression in numerous physiological responses to hypoxia and the pathogenesis of various lung vascular diseases, including lung cancer, chronic obstructive pulmonary disease (COPD), pulmonary fibrosis, and pulmonary arterial hypertension (PAH).
2.2 HIF contribution to PAH: clinical evidence, role in metabolic reprogramming, inflammation, vascular cell senescence, vascular biology, remodeling
Chronic hypoxia is one of the major contributing factors to the development of pathological condition in PAH. It encompasses inflammation, metabolic reprogramming and vascular cell senescence which are governed by gene expression and has an adverse impact on pulmonary vascular remodeling. Several studies support the crucial role of HIFs in chronic hypoxia-induced PAH (7, 18). In PAH, HIFs are involved in the regulation of cell proliferation, migration, and pulmonary vascular remodeling. Activation of HIFs is evident in different categories of PH, including group I PH and PH-associated chronic lung diseases (such as COPD, pulmonary fibrosis, and chronic high-altitude exposure) (19–24). Studies reported that compared to HIF-1α, HIF-2α significantly impacts pulmonary arterial remodeling and the development of PAH (8, 25, 26). Preclinical studies on HIF-2α; inhibitors PT7567 and C76 showed that they reduced the severity of PH in Sugen/hypoxia rats via reducing hemodynamic parameters (8, 27, 28). Recently, overexpression of HIF-2α was reported in lung pericytes of PAH patients (29), highlighting the importance of HIF-2α in pulmonary vasculature remodeling.
Studies show that in PAH, HIF orchestrates the immune/inflammatory dysregulation in response to hypoxia condition which profoundly changes pulmonary arterial endothelial cells (PAECs) and pulmonary arterial smooth muscle cells (PASMCs) phenotype in vascular remodeling (25, 30). The release of inflammatory mediators and inflammatory cells are accelerated by HIF (17). For example, in hypoxia-induced PAH, upon invasion of macrophages and neutrophils, HIF-1α is known to promote macrophage proliferation, chemotaxis and infiltration and induces the release of cytokines (17). In addition, studies reported that HIF and nuclear factor-κB (NF-κB) are the key transcriptional regulator which are jointly involved in the initiation of inflammation of PAH vasculopathy under hypoxia (30, 31). HIF-1α expression is upregulated by the p65 and p50 subunits of NF-κB which bind to the HIF-1α promoter (26). In hypoxia-induced PAH, studies show that inhibition of the TNF-α/NF-κB/HIF signaling pathway inhibits angiogenesis via decreasing the HIF-dependent activation (32). Studies also show that increased CD146 expression through NF-κB/HIF mediated cascades promotes synthetic changes in PASMCs which are associated with PAH (27). Taken together, HIF signaling plays crucial roles in regulation of inflammation, metabolic reprogramming, and vascular remodeling in PAH.
Metabolic aberrations have also been identified as a crucial component involved in the disease progression of PAH (28, 29, 33). HIF is the master regulator that controls the metabolic reprogramming in response to hypoxia (33). Studies show that in PAH PAECs, both HIF-1α and HIF-2α contribute to modify the metabolic phenotypes by regulating the expression of mitochondrial enzymes such as pyruvate dehydrogenase kinase 1 (PDK1), lactate dehydrogenase A (LDHA), Hexokinase 1,2 (HK1,2) and Glucose transporter 1,3 (GLUT1,3) to regulate Warburg effect (aerobic glycolysis) and anerobic glycolysis (33, 34). A study reported that increased expression HIF-1α modulates the metabolic shift in the endothelial cells (ECs) of IPAH patients due to decreased nitric oxide (NO) levels with reduced superoxide dismutase activity (SOD) (35). In addition, various studies identified that in PAH, HIF-1α has a close association with mitochondria. It is reported that activation of HIF-1α by cobalt chloride or deferoxamine can lead to mitochondrial fission and subsequent modulation of mitochondrial plasticity in PAH SMCs (36).
In addition, iron deficiency is also one of the metabolic factors reported in PAH population (37). A transcriptional target of HIF, microRNA-210 (miR-210) was found to cause iron deficiency in PAH PAEC via hypoxic repression of iron–sulfur (Fe-S) cluster assembly protein 1 and 2 (ISCU) (33, 38). In contrast, upregulation of miR-210 by HIF-1α increases survival of PASMCs via targeting of the E2F3 transcription factor (39). With regards to HIF-2α, it was observed that patients with Chuvash polycythemia showed HIF-2α gain-of-function mutations associated with PAH development and symptoms such as elevated heart rate and pulmonary ventilation related to metabolic aberrations (40–42). In addition, studies show that octamer-binding transcription factor (OCT4) expression is driven by HIF-2α through miR-130/131-mediated downregulation of peroxisome proliferator-activated receptor-γ (PPARγ), resulting in increased proliferation of PAECs and PASMCs in PAH (43).
Another factor which also involved significantly in vascular remodeling in PAH is cellular senescence. Cellular senescence is having a crucial contribution in several vascular diseases, such as coronary artery disease, stroke, myocardial infraction and PAH (44, 45). Mostly the senescent vascular endothelial cells are not identified in the normal lesions; they are predominantly present in the plaque of human atherosclerosis which also leads to the endothelial dysfunction and resulting PAH (46). It has been established that vascular cell senescence plays a major role in the contribution of vascular remodeling and PAH development (47–49). So far, the primarily senescence marker in PAH mainly focuses on p21, p16, p53 and BCl2 and their expression is well studied in animal models as well as in PAH patients (45). However, only few studies reported that HIF signaling also contributes to the vascular senescence in PAH (50). Among HIFs, the crucial role of HIF-1α is primarily observed in endothelial cell (EC) senescence phenotype development and the progression of atherosclerosis (51–54). Emerging evidence reported that HIF-1α and mir-126 plays critical role in EC senescence and it has been proposed that they are new markers of EC senescence progression (55). In addition, other studies revealed that differential expression of HIF-1α/HIF-2α and P53 was identified in PAEC and PASMC in hypoxia-induced PAH animal models, which demonstrated the HIF pathway and P53 crosstalk in the vascular cell senescence and PAH development (50). Hence, based on the above evidence we can demonstrate how HIF pathway is crucial for vascular remodeling and can be used for developing a new drug for the treatment of PAH by targeting HIF pathway and specific HIF animal and cell specific animal models.
2.3 HIF-1α vs. HIF-2α: common vs. antagonistic roles in PAH, evidence from animal and cell-specific models
Considering the multitude of cellular and mechanistic roles of HIF contribution in PAH, it is evident that the use of HIF knockout mouse models is a practical approach to gain valuable insights on the HIF pathway involved in hypoxic adaptation of the pulmonary vasculature and PAH development (Table 1). Previously, studies reported that the HIF pathway genes are playing a critical role in embryonic development; however, biallelic deletion in these genes is embryonically lethal (14, 56, 57). For example, double deletion of Egln1−/−, Hif2a−/−, and Vhl−/− mice are all embryonically lethal. In contrast, adult mice with heterogenous deletion of Hif1α and Hif2α are protected from hypoxia-induced PH development (58, 59). One study from Hu et al. reported that the mice with global deletion of Hif1α and Hif2α failed to survive under hypoxia-induced PH (60).
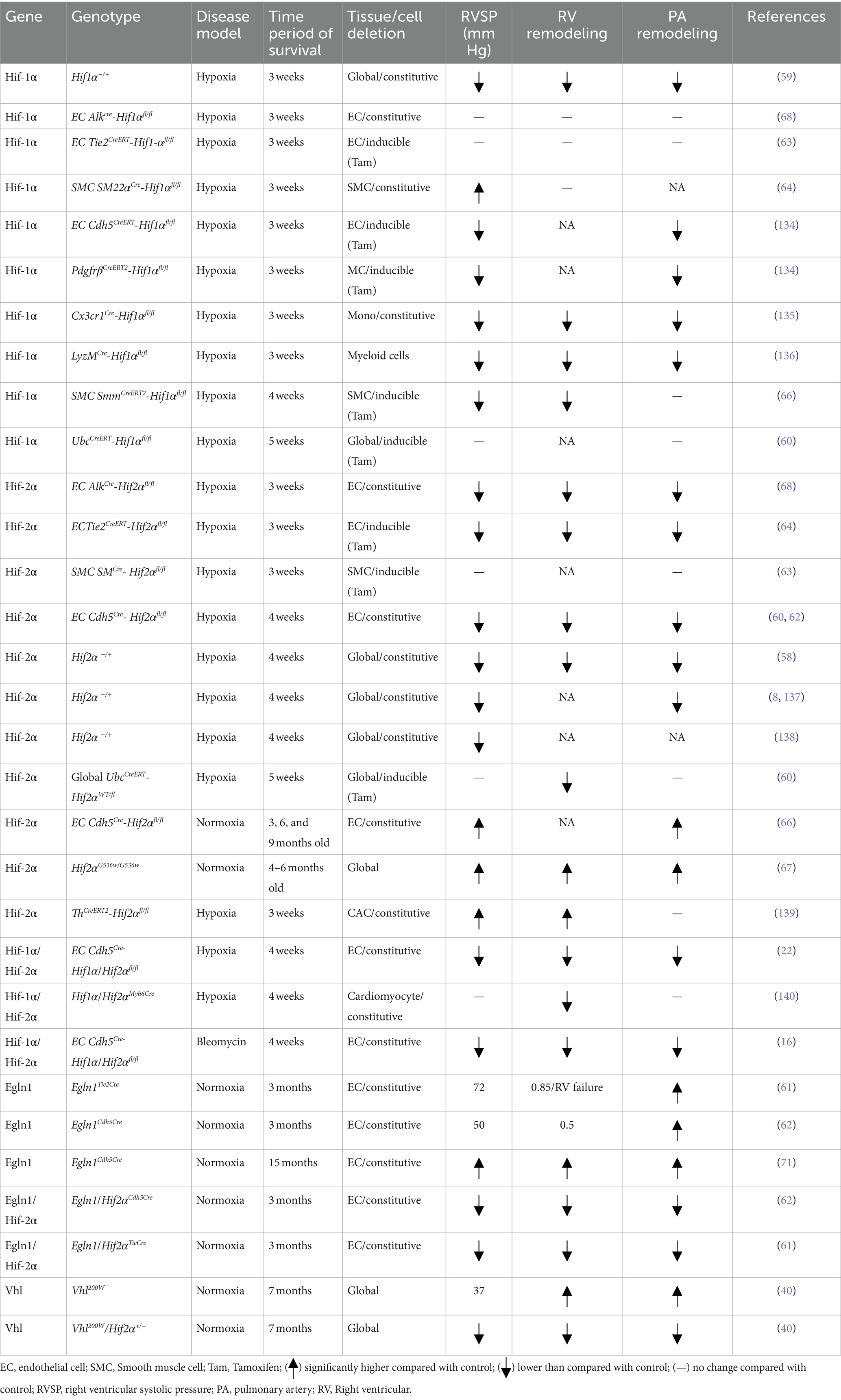
Table 1. A summary table for HIF signaling related animal model in PH.
To further dissect the cell-specific role of HIF pathway components in PAH pathogenesis, numerous studies have used cell-specific knockout mouse models to study vascular remodeling. For instance, the PHD2, encoded by EGLN1, is a crucial isoenzyme in normoxia conditions and is a participant in hypoxia related processes such as angiogenesis and cardiac function. Dai et al. and others reported that Egln1 endothelial conditional knockout mice (Egln1EC−/−) can spontaneously develop PAH with severe pulmonary vascular remodeling and occlusive pulmonary vascular lesions even in normoxia conditions (61–63), whereas heterogenous Egln1EC+/− mice shows mild PAH symptoms (63). With regards to Hif-1α, a study from Kim et al. shows that mice with constitutive smooth muscle cell (SMC) Hif-1α deletion aggravated hypoxia-induced PH (64). Interestingly, another study reported that in mice with SMC-specific Hif-1α (inducible) deletion attenuated PH but did not show RV hypertrophy (65).
There is significant progress in the understanding of the role of HIF-2α in the pathogenesis of PAH vascular remodeling (8, 61, 62, 66). Studies report that mice with HIF2α G536W (gain-of-functions mutations) develop spontaneous PAH with right ventricle systolic pressure of 66 mm Hg (67). In addition, recent studies reported that endothelial Hif2α plays a more significant role in regulating vascular remodeling and PH caused by Vhl or Egln1 deficiency compared to Hif-1α (68, 69). The impact of endothelial Hif-2α deletion has also been observed on mice with chronic hypoxia-induced PH or Vhl or Egln1 deficiency-induced PH (61, 68, 69). Mice models with VhlR200W loss of function mutations showed increased susceptibility to PH (40). Similarly, Egln1Tie2Cre mice progressively develop severe PH at the age of 3.5 months and show 80% mortality by the age of 6 months with increased RV hypertrophy, RV fibrosis, and RV failure (69, 70). In comparison, other studies show that Egln1Cdh5Cre mice display weaker PH pathogenesis (62, 71).
Interestingly, it is observed that the heterozygous deletion of Hif-2α, but not Hif-1α, in mice with VhlR200W (loss of function) mutation rescued them from PH development (40). However, other authors show that endothelial deletion of only Hif-2α and not Hif1α can alleviate PH development in Egln1Tie2Cre or Egln1Cdh5Cre mice (61, 62). Although these studies revealed the association of HIF-2α with PH development, the mechanism by which HIF-2α exerts its effect is unclear. Moreover, it is also reported that partial deletion of both HIF-1α and HIF-2α show similar protection from PH development (59). In contrast, Skuti et al. showed that EC Hif2a−/− using Cdh5Cre in mice at 6–9 months ago developed PH (66). One possible reason for this contradictory result May be the upregulation of HIF-1α activating factors in smooth muscle by endothelial HIF-2α (59). Another contributing factor could be the differences in the roles of HIF-1α and HIF-2α over a period, evidenced by the reduction of PH rescue in partial HIF-1α deletion mice from 3 months to 6 months (61, 62). However, the lack of supporting mechanistic evidence for these observations indicates that there May be additional co-modifiers involved in regulating the activities of HIF-1α and HIF-2α independently, such as epigenetic factors. However, development of mouse models which incorporate other factors for generating long-lasting and/or irreversible PAH pathology in mice (72). Improved animal models, which well recapitulate irreversible clinical pathology in patients and facilitate development of better therapeutic development and approaches.
3 Overview of epigenetic mechanisms and their relevance to PAH
Epigenetic mechanisms play a critical role in PAH, influencing gene expression and phenotype without altering the DNA sequence. Various epigenetic alterations, including DNA methylation, histone modifications, and non-coding RNA dysregulation, contribute to the onset and progression of PAH (73, 74).
One key epigenetic modification observed in PAH is the dysregulation of DNA methylation, which is a process where a methyl group is added to the cytosine residues of DNA, often resulting in gene silencing. In PAH, dysregulation of DNA methylation can lead to the aberrant expression of genes involved in crucial cellular processes. One significant example is the decreased expression of superoxide dismutase 2 (SOD2) due to CpG island methylation. This epigenetic change disrupts redox signaling, leading to the activation of HIF-1α even under normoxic conditions. This inappropriate activation of HIF-1α promotes excessive cell proliferation, which is a hallmark of PAH (75). The chronic proliferation of PASMCs and PAECs contributes to vascular remodeling and the narrowing of the pulmonary arteries, increasing vascular resistance and pressure.
Histone modifications, such as acetylation and methylation, play a significant role in the regulation of chromatin structure and PAH pathogenesis. Histone acetylation and methylation alter the accessibility of the DNA to transcriptional machinery. Changes in histone levels and the expression of histone-modifying enzymes, such as histone deacetylases (HDACs) and bromodomain-containing protein 4 (BRD4), contribute to the abnormal proliferation and resistance to apoptosis in vascular cells. HDACs, for instance, remove acetyl groups from histones, leading to a more condensed chromatin structure and reduced gene expression. In PAH, increased HDAC activity has been linked to the repression of genes that inhibit cell proliferation and promote apoptosis. Abnormal histone modifications can lead to the persistent activation of proliferative pathways and the suppression of apoptotic signals (76). This imbalance contributes to the pathologic remodeling of pulmonary arteries, which is characteristic of PAH progression. Histone modifications can also affect the expression of genes involved in inflammation, fibrosis, and vascular tone, further exacerbating the disease.
Non-coding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), are crucial regulators of gene expression in PAH. miRNAs are small RNA molecules that can inhibit the translation of target mRNAs or lead to their degradation. Dysregulated miRNAs in PAH can affect pathways related to inflammation, fibrosis, and vascular remodeling. For example, certain miRNAs are known to target and regulate genes involved in SMC proliferation and migration, EC function, and extracellular matrix production. lncRNAs can modulate gene expression at various levels, including chromatin remodeling, transcription, and post-transcriptional processing. In PAH, lncRNAs influence the abnormal proliferation, migration, and survival of pulmonary vascular cells. Some lncRNAs have been found to interact with chromatin-modifying complexes, affecting the expression of genes involved in cell cycle regulation, apoptosis, and inflammation (77). Dysregulation of these lncRNAs contributes to abnormal proliferation, migration, and survival of pulmonary vascular cells. So, targeting epigenetic modifiers holds promise as a therapeutic strategy for PAH.
Understanding the complex interplay of epigenetic mechanisms in PAH provides insights into disease pathogenesis and offers potential avenues for developing novel diagnostic and therapeutic approaches. By elucidating the epigenetic landscape of PAH, it is possible to uncover new treatment strategies that could improve outcomes for patients with this life-threatening condition.
3.1 Epigenetic regulation of HIF-dependent genes in PAH
HIF serves as a central regulator in the metabolic reprogramming and mitochondrial dynamics observed in PAH. It exerts control over metabolic enzymes such as pyruvate kinase (PK), pyruvate dehydrogenase kinase (PDK), and pyruvate dehydrogenase (PDH), orchestrating shifts in cellular energy metabolism (78). This metabolic rewiring plays a pivotal role in PAH pathogenesis, influencing cell proliferation and survival. Notably, studies indicate a bidirectional relationship between metabolism and epigenetics in PAH. Epigenetic modifications, including alterations in DNA methylation and histone modifications, can precede changes in metabolism by modulating gene expression, including genes involved in the HIF pathway (79). The stability and transactivity of HIF-α are further modulated by its acetylation and methylation. Under hypoxic conditions, histone-modifying enzymes dynamically change the chromatin structure. Some Histone Methyltransferases (HMT) and histone deacetylases (HDACs) induce repressive histone marks. Histone demethylases (HDT), and Histone acetyltransferase (HATs) induce activating marks in chromatin leading to the activation of hypoxia-related genes, including those associated with glycolysis, angiogenesis (80). In the context of PAH, there have been a few studies identified miRNA regulate HIF-1α and HIF-1β, PHD2 leads to dysregulation of HIF signaling (81–83). IRP1, an iron response protein, specifically inhibits HIF-2α via repressing HIF-2α mRNA translation (84). Glycolysis related metabolite and gene, Acetyl-CoA, and PKM2 promote p300, meditate histone acetylation, and promote HIF signaling (85).
In PAH, DNA methylation, particularly hypermethylation, plays a crucial role in vascular pathology. Studies have linked DNA hypermethylation to abnormal cell proliferation and resistance to cell death in the small pulmonary arteries. For instance, Hypermethylation in specific regions of the SOD2 gene was identified, which encodes an enzyme critical for neutralizing harmful superoxide radicals in cells (86). Decreased SOD2 levels are observed in PH, leading to increased oxidative stress and activation of HIF-1α signaling, which promotes abnormal vascular responses such as angiogenesis and inflammation (87). Recent studies have extended this understanding to include epigenetic metabolic changes in right ventricular fibroblasts in PAH. These studies reveal that epigenetic alterations impair mitochondrial function, leading to a pseudo-hypoxic state and activation of HIF-1α, even in normoxic conditions. This dysregulation is associated with abnormal levels of pyruvate dehydrogenase kinase (PDK) isoforms, which promote a metabolic shift known as the Warburg effect. Additionally, upregulation of DNMT1 suppresses SOD2 expression, exacerbating mitochondrial dysfunction and HIF-1α activation. Ultimately, this cascade contributes to increased inflammation and fibrosis in the right ventricle (74).
Hypermethylation of promoters or enhancers of genes encoding key regulators of HIF activity, such as prolyl hydroxylase domain enzymes (PHDs) or von Hippel–Lindau (VHL) tumor suppressor, can impair their expression, leading to sustained HIF activation even under normoxic conditions. Conversely, hypomethylation of HIF-dependent genes May enhance their expression, contributing to pathological processes in PAH. In PAH, pyruvate dehydrogenase kinase 1 (PDK1) inhibits the activity of pyruvate dehydrogenase (PDH), driving a metabolic shift known as the Warburg effect (88). This shift involves a preference for glycolysis over oxidative phosphorylation, leading to increased lactate production even in the presence of oxygen. Additionally, HIF-1α promotes this Warburg shift, further enhancing glycolysis. As a result, there is an increase in the production of inflammatory cytokines like connective tissue growth factor (CTGF) (89) and transforming growth factor beta (TGF-β1) (71, 90) contributing to PAH pathology.
3.2 Role of HIF in triggering miRNA and lncRNA
HIF plays a pivotal role in orchestrating the expression of various non-coding RNAs (ncRNAs), including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), in the context of PAH (91) and other hypoxia-related conditions (92). These ncRNAs, in turn, regulate critical pathways involved in cellular responses to hypoxia, contributing to the progression of PAH and other diseases. One such example is the lncRNA STEAP3-AS1, whose transcription is induced by HIF-1α under hypoxic conditions. STEAP3-AS1, acting as a positive regulator, enhances the expression of STEAP3, thereby promoting colorectal cancer progression (93, 94). This regulatory axis highlights the role of HIF-mediated lncRNA regulation in cancer pathogenesis (93). Furthermore, HIF-1α has been associated with the modulation of exosomal cargo, including miRNAs, which can serve as biomarkers for endothelial senescence progression (95). Dysregulated miRNAs like miR-125a-5p and miR-139-5p have been implicated in EC senescence under hypoxia, suggesting their involvement in PAH pathogenesis (96, 97). Additionally, HIF-1α serves as a target for certain miRNAs, such as miR-155, which regulates fibroblast behavior, impacting apoptosis, migration, and proliferation (98). Moreover, intricate networks involving miRNAs, lncRNAs, and core genes like PDGFRB and HIF-1α have been identified, shedding light on their roles in the development of CTEPH (24).
In PAH, dysregulated ncRNAs, including lncRNAs like LINC00963 (99) and 5031425E22Rik/KMT2E-AS1 (100), contribute to disease progression by affecting cellular processes such as proliferation, migration, and metabolic reprogramming. These ncRNAs interact with HIF-mediated pathways, further emphasizing the importance of HIF in regulating ncRNA expression and function in PAH. Overall, the intricate interplay between HIF and ncRNAs underscores their significance in PAH pathogenesis and highlights their potential as therapeutic targets for this debilitating condition.
The dysregulation of HIF signaling is implicated in various pathways relevant to PAH pathology, including vasoconstriction, vascular cell proliferation, metabolic alterations, and inflammation. Among the HIF isoforms, HIF-1α emerges as a key mediator of these effects. It modulates the expression of numerous microRNAs (miRNAs) and, in turn, is reciprocally regulated by miRNAs. For instance, miR-138, whose upregulation is HIF-1α-dependent, is implicated in hypoxia-induced EC dysfunction by regulating nitric oxide (NO) expression via S100A1 (101). Additionally, miR-138 inhibits the hypoxia-induced proliferation of endothelial progenitor cells. These findings underscore the intricate interplay between HIF-1α signaling and miRNA regulation in the context of PAH pathogenesis (78). Moreover, HIF-2α facilitates the expression of nearby genes, specifically the long noncoding RNA (lncRNA) histone lysine N-methyltransferase 2E-antisense 1 (KMT2E-AS1) and histone lysine N-methyltransferase 2E (KMT2E). KMT2E-AS1 helps stabilize the KMT2E protein, leading to an increase in the epigenetic mark histone 3 lysine 4 trimethylation (H3K4me3), which drives HIF-2α-dependent metabolic and pathological endothelial functions. Additionally, this lncRNA pathway boosts HIF-2α expression through epigenetic, transcriptional, and posttranscriptional mechanisms, establishing a positive feedback loop that further enhances HIF-2α activity (102).
3.3 Role of HIF in regulating histone modifications and DNA methylation
HIF plays a crucial role in regulating histone modifications and DNA methylation, thereby influencing gene expression under hypoxic conditions. Specifically, HIFs target the expression of proteins associated with modifications of histones, such as histone acetylation, and regulate the balance between DNA methylation and acetylation (103).
In the nucleus, HIFs regulate the expression of numerous genes involved in cellular adaptation to hypoxia, including those encoding proteins associated with histone modifications and DNA methylation (80). HIFs directly target the expression of enzymes involved in histone modifications, such as histone methyltransferases and histone demethylases, as well as enzymes involved in DNA methylation, such as DNA methyltransferases (DNMTs). HIFs influence the balance between histone methylation and acetylation by regulating the expression of histone acetyltransferases (HATs) and histone deacetylases (HDACs). This dynamic regulation of histone modifications by HIFs plays a crucial role in modulating gene expression patterns in response to hypoxia (104, 105).
In the context of ECs, epigenetic modifications, including histone acetylation, contribute to the regulation of placental growth factor (Plgf) expression under hypoxic conditions (106). HIF-1α binds to HREs located in the second intron of Plgf. This binding facilitates the spatial association between the transcriptional start site and the regulatory site within the Plgf gene, potentially enabling efficient gene expression (106). Recent research suggests that transcription factors like HIF-1α can facilitate the formation of chromatin loops, allowing distant regions of DNA to interact effectively (103). This process May occur within specialized nuclear structures known as transcription factories, where genes are organized and looped out from chromosome territories. However, further investigations are needed to fully elucidate the mechanisms by which HIF-1α regulates the three-dimensional chromatin structure of Plgf regulatory regions and how this impacts gene expression in response to hypoxia (7).
Understanding the role of HIF in regulating histone modifications and DNA methylation provides insights into the epigenetic mechanisms underlying cellular responses to hypoxia. By targeting these processes, researchers can develop novel therapeutic strategies for diseases characterized by aberrant hypoxic signaling, such as PAH. Overall, HIFs play a central role in coordinating the cellular response to hypoxia by directly and indirectly regulating histone modifications and DNA methylation. This regulatory network contributes to the adaptation of cells to hypoxic conditions and has implications for various pathological processes, including cancer and PAH (Table 2).
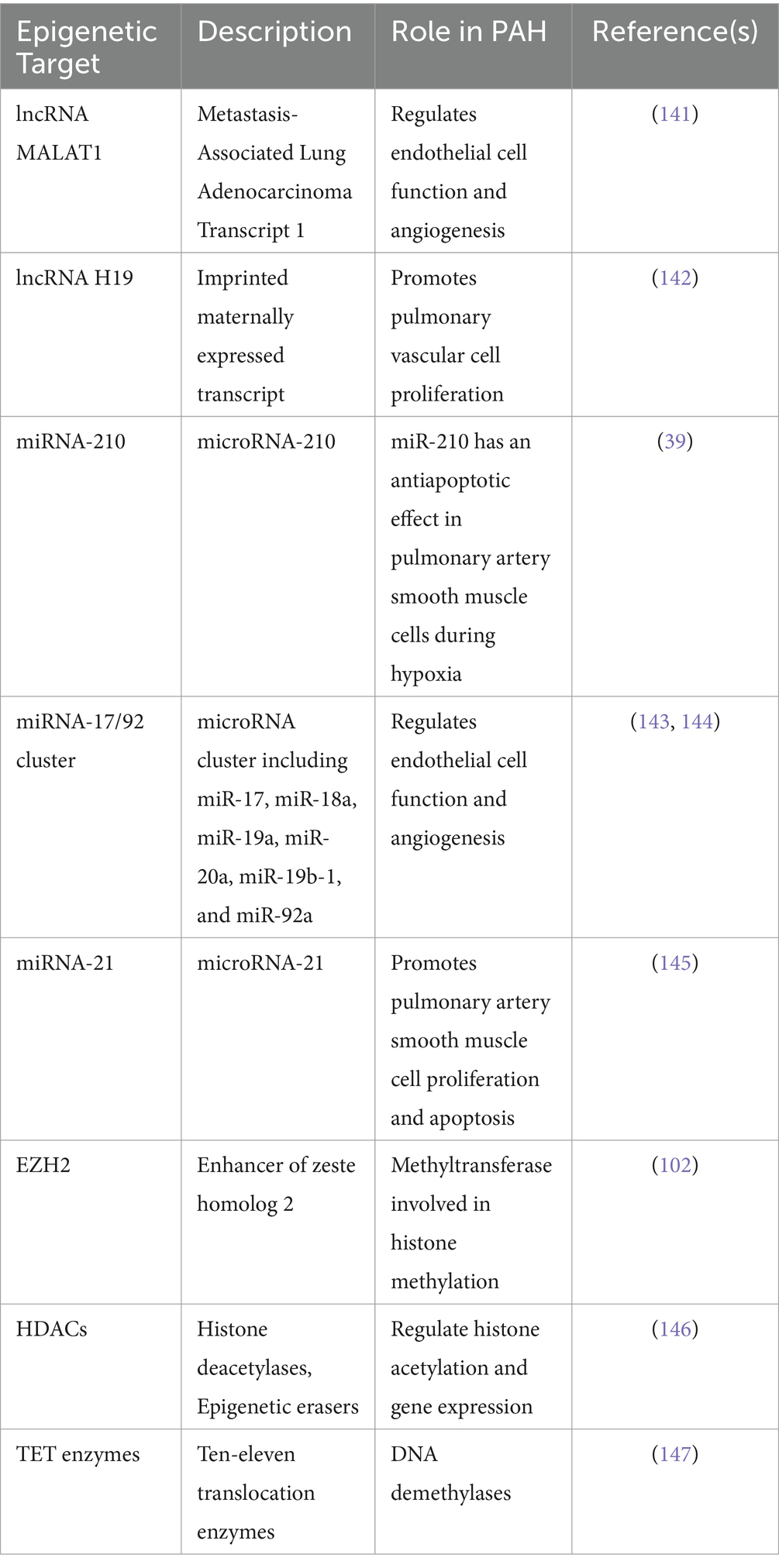
Table 2. Known epigenetic downstream targets of HIF-1α, HIF-2α: focus on lncRNA, miRNAs and histone/DNA modifier enzymes discussed in the section.
These epigenetic targets have been implicated in PAH pathogenesis and are regulated by HIF-1α and/or HIF-2α signaling. They play crucial roles in modulating gene expression, chromatin structure, and cellular processes relevant to PAH, such as proliferation, apoptosis, and angiogenesis.
4 Potential therapeutics targeting HIF signaling
4.1 Targeting HIF as a therapeutic target in PAH
Targeting HIF signaling holds considerable promise as a therapeutic strategy for PAH and other hypoxia-related conditions. Several potential therapeutics have been proposed, leveraging insights from cancer research (107) and focusing on modulating epigenetic processes (76), metabolomics (108), and antioxidant systems (109) (Table 3). However, controversies surrounding the long-term safety, potential adverse effects, and off-target actions of these drugs highlight the need for further research to elucidate their therapeutic potential and optimize their use in PAH treatment.
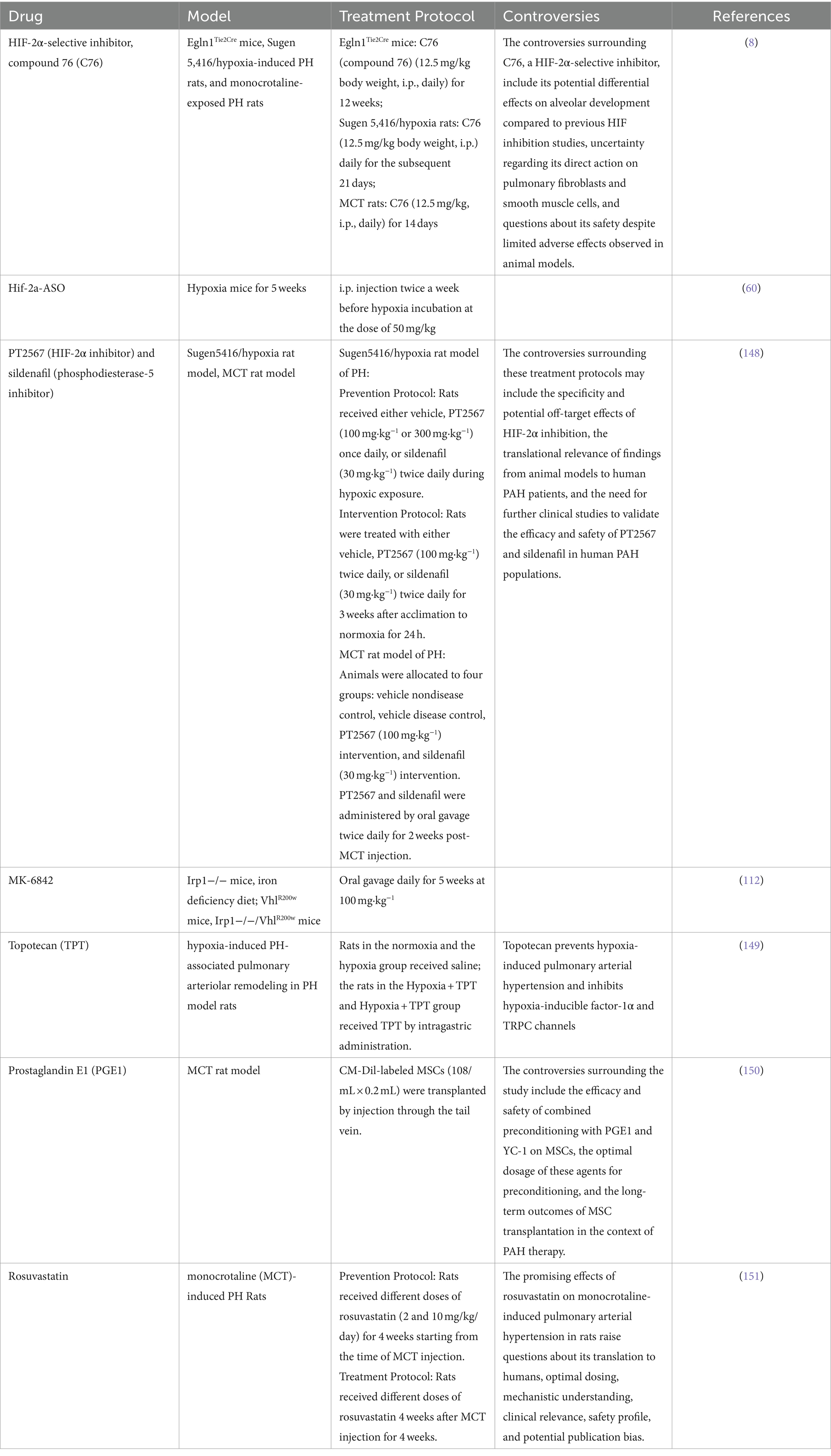
Table 3. Summary the detail about the HIF targeted drugs, model and treatment protocol.
Multiple preclinical trials have been conducted using PH mouse and rat models (see Table 3). Dai et al. were the first to demonstrate that pharmacological inhibition of HIF-2α, using the translational inhibitor C76, effectively prevents obliterative pulmonary vascular remodeling and right heart failure in three distinct rodent models of severe PH: Egln1Tie2Cre mice, monocrotaline (MCT)-rats, and sugen5416 plus hypoxia (SuHx)-rats. They also observed clear survival benefits in two of these models through HIF-2α inhibition for the first time (110). Hu et al. showed that antisense oligonucleotides targeting Hif2a reduced pulmonary vascular muscularization and right ventricular hypertrophy in hypoxia-exposed mice. The same group also demonstrated that a small molecule HIF2 inhibitor, PT2567, significantly reduced monocyte recruitment, vascular cell proliferation, vessel muscularization, and PH development in hypoxia-exposed rats (60). Macias et al. further supported these findings, showing that global inhibition of HIF-2α reduced pulmonary vascular hemodynamics and remodeling in both Su5416/hypoxia prevention and intervention models, as well as in MCT-exposed rodents (111). Additionally, Ghosh et al. demonstrated that inactivating HIF-2α with the second-generation allosteric inhibitor MK-6482, a FDA approved drug for renal carcinoma, attenuated polycythemia and PH in VhlR200W mice, Irp1-KO mice, and double mutant VhlR200W;Irp1-KO mice with PH. Overall, these studies suggest that inhibiting HIF-2 signaling is a promising therapeutic approach for PAH (112).
4.1.1 Epigenetic modifiers
Similar to anticancer therapies, modifying epigenetic processes in pulmonary vascular cells shows promise for treating PAH (113). This approach involves targeting the epigenetic regulation of genes involved in HIF signaling and other pathways implicated in PAH pathogenesis (74, 114). However, challenges related to targeting specificity and managing off-target effects need to be addressed to maximize the effectiveness of epigenetic modifiers in PAH treatment.
4.1.2 Metabolomics-based interventions
Metabolomics approaches offer opportunities for early detection, personalized dietary interventions, and advanced drug therapies in PAH (115). By targeting metabolic pathways regulated by HIF and other factors under hypoxic conditions, metabolomics-based interventions aim to restore metabolic homeostasis and mitigate disease progression in PAH patients (116).
4.1.3 Antioxidant systems modulation
Antioxidant systems play a crucial role in regulating HIF signaling by modulating levels of reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) (117, 118). Strategies aimed at modulating antioxidant systems, such as superoxide dismutase 2 (SOD2), can influence HIF-1α activity and downstream signaling pathways. For instance, siSOD2 has been shown to activate HIF-1α, highlighting the potential of targeting antioxidant systems to modulate HIF signaling in PAH (86).
Despite the promising therapeutic prospects, several challenges need to be addressed to translate these approaches into effective treatments for PAH. These include ensuring target specificity, minimizing off-target effects, understanding cell-specific differences in metabolic activity, and optimizing the magnitude of therapeutic interventions to achieve desired outcomes. Collaborative efforts integrating insights from basic research, clinical studies, and computational modeling will be essential for overcoming these challenges and developing effective therapeutics targeting HIF signaling in PAH (119).
4.2 Future perspectives on therapeutic targeting of HIF isoforms in PAH
In PAH, dysregulation of HIF signaling is implicated in vascular remodeling and pulmonary vascular dysfunction. Targeting specific HIF isoforms to promote a favorable epigenetic landscape in PAH represents a promising avenue for research and potential treatment development (7). Selective targeting of HIF isoforms could potentially modulate the epigenetic landscape in PAH, leading to beneficial effects such as improved vascular function and inhibition of pathological remodeling. HIF isoforms, particularly HIF-1α and HIF-2α, play critical roles in cellular responses to hypoxia by regulating genes involved in angiogenesis, metabolism, and cell proliferation (7).
Researchers have explored targeting HIF isoforms in cancer therapy, aiming to disrupt tumor growth and metastasis (120, 121). Strategies include small molecule inhibitors, gene silencing techniques, and immunotherapy approaches (122–124). There are several examples from other diseases where HIF inhibitors have shown promise, suggesting potential applicability in treating PAH (125). In certain cancers, aberrant HIF-1α or HIF-2α signaling contributes to tumor progression by promoting angiogenesis and metabolic reprogramming. For example, HIF-1 inhibitors are extensively studied in cancer treatment because of their role in reducing tumor growth, metastasis, and hypoxia-induced drug resistance. For example, PX-478 was tested in a Phase I dose-escalation study involving cancer patients (NCT00522652), demonstrating effective inhibition of HIF-1α and a reasonable safety profile (126). These inhibitors could be useful in PAH by targeting similar pathways of hypoxia and cellular proliferation in pulmonary arteries. In diseases such as idiopathic pulmonary fibrosis (IPF) (110), liver fibrosis (111), and renal fibrosis (127), HIF-1 inhibitors have demonstrated potential in reducing fibrotic tissue formation. Given that PAH involves vascular remodeling and fibrosis, HIF-1 inhibitors could help mitigate these processes. Additionally, HIF-1 is implicated in chronic kidney disease (CKD) progression by promoting inflammation and fibrosis (128, 129). HIF-1 inhibitors have been explored to reduce these effects, suggesting that similar mechanisms might help in PAH, which also involves inflammatory and fibrotic processes. In conditions like myocardial infarction, stroke (130), and ischemic diseases (131, 132), HIF-1 inhibitors have been investigated for their ability to modulate hypoxia responses, potentially reducing tissue damage and improving outcomes. Belzutifan (PT2977), a HIF-2α inhibitor, has been approved for advanced renal cell carcinoma and showed promising efficacy in preclinical PH models. Taken together, HIF inhibitors might be beneficial for patients with PAH.
Given these examples, HIF inhibitors might offer new therapeutic avenues for PAH by targeting hypoxia, inflammation, and fibrosis—key aspects of PAH pathology. However, rigorous preclinical and clinical studies are necessary to confirm their efficacy and safety in PAH. Translating these strategies to PAH would involve understanding the specific roles of HIF isoforms in pulmonary vascular cells and identifying interventions that can selectively target these isoforms without causing adverse effects. Additionally, considering the complex interplay between HIF signaling and other pathways implicated in PAH pathogenesis could provide further insights into potential therapeutic targets and combination therapies. Overall, while there May not yet be direct examples of targeting specific HIF isoforms in PAH, insights from other diseases and ongoing research into HIF signaling hold promise for developing novel therapeutic approaches. These approaches could modulate the epigenetic landscape and improve outcomes in PAH.
Author contributions
AM: Writing – original draft, Writing – review & editing. DY: Writing – original draft, Writing – review & editing. ZD: Writing – original draft. VJ: Writing – original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by an NIH R01 HL134776, R01 HL134776-02, R01 HL159886-01 to V. de Jesus Perez.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lai, YC, Potoka, KC, Champion, HC, Mora, AL, and Gladwin, MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res. (2014) 115:115–30. doi: 10.1161/CIRCRESAHA.115.301146
2. Condon, DF, Nickel, NP, Anderson, R, Mirza, S, and de Jesus Perez, VA. The 6th world symposium on pulmonary hypertension: what's old is new. F1000Res. (2019) 8:8. doi: 10.12688/f1000research.18811.1
3. Galie, N, Palazzini, M, and Manes, A. Pulmonary arterial hypertension: from the kingdom of the near-dead to multiple clinical trial meta-analyses. Eur Heart J. (2010) 31:2080–6. doi: 10.1093/eurheartj/ehq152
4. Humbert, M, Lau, EM, Montani, D, Jais, X, Sitbon, O, and Simonneau, G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. (2014) 130:2189–208. doi: 10.1161/CIRCULATIONAHA.114.006974
5. de Jesus Perez, VA . Molecular pathogenesis and current pathology of pulmonary hypertension. Heart Fail Rev. (2016) 21:239–57. doi: 10.1007/s10741-015-9519-2
6. Zhu, J, Zhao, L, Hu, Y, Cui, G, Luo, A, Bao, C, et al. Hypoxia-inducible factor 2-alpha mediated gene sets differentiate pulmonary arterial hypertension. Front Cell Dev Biol. (2021) 9:701247. doi: 10.3389/fcell.2021.701247
7. Pullamsetti, SS, Mamazhakypov, A, Weissmann, N, Seeger, W, and Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J Clin Invest. (2020) 130:5638–51. doi: 10.1172/JCI137558
8. Dai, Z, Zhu, MM, Peng, Y, Machireddy, N, Evans, CE, Machado, R, et al. Therapeutic targeting of vascular remodeling and right heart failure in pulmonary arterial hypertension with a HIF-2alpha inhibitor. Am J Respir Crit Care Med. (2018) 198:1423–34. doi: 10.1164/rccm.201710-2079OC
9. Krock, BL, Skuli, N, and Simon, MC. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. (2011) 2:1117–33. doi: 10.1177/1947601911423654
10. Majmundar, AJ, Wong, WJ, and Simon, MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. (2010) 40:294–309. doi: 10.1016/j.molcel.2010.09.022
11. Semenza, GL . Hypoxia-inducible factors in physiology and medicine. Cell. (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
12. Liu, Y, Cox, SR, Morita, T, and Kourembanas, S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5′ enhancer. Circ Res. (1995) 77:638–43. doi: 10.1161/01.RES.77.3.638
13. Forsythe, JA, Jiang, BH, Iyer, NV, Agani, F, Leung, SW, Koos, RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. (1996) 16:4604–13. doi: 10.1128/MCB.16.9.4604
14. Compernolle, V, Brusselmans, K, Acker, T, Hoet, P, Tjwa, M, Beck, H, et al. Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med. (2002) 8:702–10. doi: 10.1038/nm721
15. McGettrick, AF, and O'Neill, LAJ. The role of HIF in immunity and inflammation. Cell Metab. (2020) 32:524–36. doi: 10.1016/j.cmet.2020.08.002
16. Bhandari, T, and Nizet, V. Hypoxia-inducible factor (HIF) as a pharmacological target for prevention and treatment of infectious diseases. Infect Dis Ther. (2014) 3:159–74. doi: 10.1007/s40121-014-0030-1
17. Palazon, A, Goldrath, AW, Nizet, V, and Johnson, RS. HIF transcription factors, inflammation, and immunity. Immunity. (2014) 41:518–28. doi: 10.1016/j.immuni.2014.09.008
18. Waypa, GB, and Schumacker, PT. Roles of HIF1 and HIF2 in pulmonary hypertension: it all depends on the context. Eur Respir J. (2019) 54:1901929. doi: 10.1183/13993003.01929-2019
19. Wilkins, MR, Ghofrani, HA, Weissmann, N, Aldashev, A, and Zhao, L. Pathophysiology and treatment of high-altitude pulmonary vascular disease. Circulation. (2015) 131:582–90. doi: 10.1161/CIRCULATIONAHA.114.006977
20. Fu, X, and Zhang, F. Role of the HIF-1 signaling pathway in chronic obstructive pulmonary disease. Exp Ther Med. (2018) 16:4553–61. doi: 10.3892/etm.2018.6785
21. Aquino-Galvez, A, Gonzalez-Avila, G, Jimenez-Sanchez, LL, Maldonado-Martinez, HA, Cisneros, J, Toscano-Marquez, F, et al. Dysregulated expression of hypoxia-inducible factors augments myofibroblasts differentiation in idiopathic pulmonary fibrosis. Respir Res. (2019) 20:130. doi: 10.1186/s12931-019-1100-4
22. Bryant, AJ, Carrick, RP, McConaha, ME, Jones, BR, Shay, SD, Moore, CS, et al. Endothelial HIF signaling regulates pulmonary fibrosis-associated pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2016) 310:L249–62. doi: 10.1152/ajplung.00258.2015
23. Garcia-Morales, LJ, Chen, NY, Weng, T, Luo, F, Davies, J, Philip, K, et al. Altered hypoxic-adenosine axis and metabolism in group III pulmonary hypertension. Am J Respir Cell Mol Biol. (2016) 54:574–83. doi: 10.1165/rcmb.2015-0145OC
24. Wang, M, Gu, S, Liu, Y, Yang, Y, Yan, J, Zhang, X, et al. miRNA-PDGFRB/HIF1A-lncRNA CTEPHA1 network plays important roles in the mechanism of chronic thromboembolic pulmonary hypertension. Int Heart J. (2019) 60:924–37. doi: 10.1536/ihj.18-479
25. Nicolls, MR, Mizuno, S, Taraseviciene-Stewart, L, Farkas, L, Drake, JI, Husseini, AA, et al. New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis. Pulm Circ. (2012) 2:434–42. doi: 10.4103/2045-8932.105031
26. Jiang, Y, Zhu, Y, Wang, X, Gong, J, Hu, C, Guo, B, et al. Temporal regulation of HIF-1 and NF-κB in hypoxic hepatocarcinoma cells. Oncotarget. (2015) 6:9409–19. doi: 10.18632/oncotarget.3352
27. Luo, Y, Teng, X, Zhang, L, Chen, J, Liu, Z, Chen, X, et al. CD146-HIF-1α hypoxic reprogramming drives vascular remodeling and pulmonary arterial hypertension. Nat Commun. (2019) 10:3551. doi: 10.1038/s41467-019-11500-6
28. Saygin, D, Highland, KB, Farha, S, Park, M, Sharp, J, Roach, EC, et al. Metabolic and functional evaluation of the heart and lungs in pulmonary hypertension by gated 2-[18F]-Fluoro-2-deoxy-D-glucose positron emission tomography. Pulm Circ. (2017) 7:428–38. doi: 10.1177/2045893217701917
29. Xu, W, Koeck, T, Lara, AR, Neumann, D, DiFilippo, FP, Koo, M, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA. (2007) 104:1342–7. doi: 10.1073/pnas.0605080104
30. Ye, Y, Xu, Q, and Wuren, T. Inflammation and immunity in the pathogenesis of hypoxic pulmonary hypertension. Front Immunol. (2023) 14:1162556. doi: 10.3389/fimmu.2023.1162556
31. D'Ignazio, L, Batie, M, and Rocha, S. Hypoxia and inflammation in cancer, focus on HIF and NF-κB. Biomedicines. (2017) 5:21. doi: 10.3390/biomedicines5020021
32. Jin, F, Zheng, X, Yang, Y, Yao, G, Ye, L, Doeppner, TR, et al. Impairment of hypoxia-induced angiogenesis by LDL involves a HIF-centered signaling network linking inflammatory TNFα and angiogenic VEGF. Aging (Albany NY). (2019) 11:328–49. doi: 10.18632/aging.101726
33. Chan, SY, and Rubin, LJ. Metabolic dysfunction in pulmonary hypertension: from basic science to clinical practice. Eur Respir Rev. (2017) 26:170094. doi: 10.1183/16000617.0094-2017
34. Marin-Hernandez, A, Gallardo-Perez, JC, Ralph, SJ, Rodriguez-Enriquez, S, and Moreno-Sanchez, R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. (2009) 9:1084–101. doi: 10.2174/138955709788922610
35. Fijalkowska, I, Xu, W, Comhair, SA, Janocha, AJ, Mavrakis, LA, Krishnamachary, B, et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol. (2010) 176:1130–8. doi: 10.2353/ajpath.2010.090832
36. Marsboom, G, Toth, PT, Ryan, JJ, Hong, Z, Wu, X, Fang, YH, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. (2012) 110:1484–97. doi: 10.1161/CIRCRESAHA.111.263848
37. Rhodes, CJ, Howard, LS, Busbridge, M, Ashby, D, Kondili, E, Gibbs, JSR, et al. Iron deficiency and raised hepcidin in idiopathic pulmonary arterial hypertension: clinical prevalence, outcomes, and mechanistic insights. J Am Coll Cardiol. (2011) 58:300–9. doi: 10.1016/j.jacc.2011.02.057
38. Chan, SY, Zhang, YY, Hemann, C, Mahoney, CE, Zweier, JL, and Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. (2009) 10:273–84. doi: 10.1016/j.cmet.2009.08.015
39. Gou, D, Ramchandran, R, Peng, X, Yao, L, Kang, K, Sarkar, J, et al. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am J Physiol Lung Cell Mol Physiol. (2012) 303:L682–91. doi: 10.1152/ajplung.00344.2011
40. Hickey, MM, Richardson, T, Wang, T, Mosqueira, M, Arguiri, E, Yu, H, et al. The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J Clin Invest. (2010) 120:827–39. doi: 10.1172/JCI36362
41. Culley, MK, and Chan, SY. Mitochondrial metabolism in pulmonary hypertension: beyond mountains there are mountains. J Clin Invest. (2018) 128:3704–15. doi: 10.1172/JCI120847
42. Formenti, F, Beer, PA, Croft, QP, Dorrington, KL, Gale, DP, Lappin, TRJ, et al. Cardiopulmonary function in two human disorders of the hypoxia-inducible factor (HIF) pathway: von Hippel-Lindau disease and HIF-2alpha gain-of-function mutation. FASEB J. (2011) 25:2001–11. doi: 10.1096/fj.10-177378
43. Bertero, T, Lu, Y, Annis, S, Hale, A, Bhat, B, Saggar, R, et al. Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J Clin Invest. (2014) 124:3514–28. doi: 10.1172/JCI74773
44. Childs, BG, Baker, DJ, Wijshake, T, Conover, CA, Campisi, J, and van Deursen, JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. (2016) 354:472–7. doi: 10.1126/science.aaf6659
45. Roger, I, Milara, J, Belhadj, N, and Cortijo, J. Senescence alterations in pulmonary hypertension. Cells. (2021) 10:12. doi: 10.3390/cells10123456
46. Minamino, T, Yoshida, T, Tateno, K, Miyauchi, H, Zou, Y, Toko, H, et al. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation. (2003) 108:2264–9. doi: 10.1161/01.CIR.0000093274.82929.22
47. Tian, XL, and Li, Y. Endothelial cell senescence and age-related vascular diseases. J Genet Genomics. (2014) 41:485–95. doi: 10.1016/j.jgg.2014.08.001
48. Ramadhiani, R, Ikeda, K, Miyagawa, K, Ryanto, GRT, Tamada, N, Suzuki, Y, et al. Endothelial cell senescence exacerbates pulmonary hypertension by inducing juxtacrine notch signaling in smooth muscle cells. iScience. (2023) 26:106662. doi: 10.1016/j.isci.2023.106662
49. Born, E, Lipskaia, L, Breau, M, Houssaini, A, Beaulieu, D, Marcos, E, et al. Eliminating senescent cells can promote pulmonary hypertension development and progression. Circulation. (2023) 147:650–66. doi: 10.1161/CIRCULATIONAHA.122.058794
50. Wang, Z, Yang, K, Zheng, Q, Zhang, C, Tang, H, Babicheva, A, et al. Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L216–l228. doi: 10.1152/ajplung.00538.2017
51. Alique, M, Luna, C, Carracedo, J, and Ramírez, R. LDL biochemical modifications: a link between atherosclerosis and aging. Food Nutr Res. (2015) 59:29240. doi: 10.3402/fnr.v59.29240
52. Alique, M, Ramírez-Carracedo, R, Bodega, G, Carracedo, J, and Ramírez, R. Senescent microvesicles: a novel advance in molecular mechanisms of atherosclerotic calcification. Int J Mol Sci. (2018) 19:2003. doi: 10.3390/ijms19072003
53. Katsuumi, G, Shimizu, I, Yoshida, Y, and Minamino, T. Vascular senescence in cardiovascular and metabolic diseases. Front Cardiovasc Med. (2018) 5:18. doi: 10.3389/fcvm.2018.00018
54. Wang, JC, and Bennett, M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. (2012) 111:245–59. doi: 10.1161/CIRCRESAHA.111.261388
55. Alique, M, Bodega, G, Giannarelli, C, Carracedo, J, and Ramírez, R. MicroRNA-126 regulates hypoxia-inducible factor-1α which inhibited migration, proliferation, and angiogenesis in replicative endothelial senescence. Sci Rep. (2019) 9:7381. doi: 10.1038/s41598-019-43689-3
56. Gnarra, JR, Ward, JM, Porter, FD, Wagner, JR, Devor, DE, Grinberg, A, et al. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci USA. (1997) 94:9102–7. doi: 10.1073/pnas.94.17.9102
57. Takeda, K, Ho, VC, Takeda, H, Duan, LJ, Nagy, A, and Fong, GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. (2006) 26:8336–46. doi: 10.1128/MCB.00425-06
58. Brusselmans, K, Compernolle, V, Tjwa, M, Wiesener, MS, Maxwell, PH, Collen, D, et al. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest. (2003) 111:1519–27. doi: 10.1172/JCI15496
59. Yu, AY, Shimoda, LA, Iyer, NV, Huso, DL, Sun, X, McWilliams, R, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. (1999) 103:691–6. doi: 10.1172/JCI5912
60. Hu, CJ, Poth, JM, Zhang, H, Flockton, A, Laux, A, Kumar, S, et al. Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur Respir J. (2019) 54:1900378. doi: 10.1183/13993003.00378-2019
61. Dai, Z, Li, M, Wharton, J, Zhu, MM, and Zhao, YY. Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces Obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2alpha. Circulation. (2016) 133:2447–58. doi: 10.1161/CIRCULATIONAHA.116.021494
62. Kapitsinou, PP, Rajendran, G, Astleford, L, Michael, M, Schonfeld, MP, Fields, T, et al. The endothelial prolyl-4-hydroxylase domain 2/hypoxia-inducible factor 2 axis regulates pulmonary artery pressure in mice. Mol Cell Biol. (2016) 36:1584–94. doi: 10.1128/MCB.01055-15
63. Tang, H, Babicheva, A, McDermott, KM, Gu, Y, Ayon, RJ, Song, S, et al. Endothelial HIF-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L256–75. doi: 10.1152/ajplung.00096.2017
64. Kim, YM, Barnes, EA, Alvira, CM, Ying, L, Reddy, S, and Cornfield, DN. Hypoxia-inducible factor-1alpha in pulmonary artery smooth muscle cells lowers vascular tone by decreasing myosin light chain phosphorylation. Circ Res. (2013) 112:1230–3. doi: 10.1161/CIRCRESAHA.112.300646
65. Ball, MK, Waypa, GB, Mungai, PT, Nielsen, JM, Czech, L, Dudley, VJ, et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1alpha. Am J Respir Crit Care Med. (2014) 189:314–24. doi: 10.1164/rccm.201302-0302OC
66. Skuli, N, Liu, L, Runge, A, Wang, T, Yuan, L, Patel, S, et al. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood. (2009) 114:469–77. doi: 10.1182/blood-2008-12-193581
67. Tan, Q, Kerestes, H, Percy, MJ, Pietrofesa, R, Chen, L, Khurana, TS, et al. Erythrocytosis and pulmonary hypertension in a mouse model of human HIF2A gain of function mutation. J Biol Chem. (2013) 288:17134–44. doi: 10.1074/jbc.M112.444059
68. Cowburn, AS, Crosby, A, Macias, D, Branco, C, Colaço, RDDR, Southwood, M, et al. HIF2alpha-arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci USA. (2016) 113:8801–6. doi: 10.1073/pnas.1602978113
69. Dai, Z . Translational potential of hypoxia-inducible factor-2α signaling in pulmonary hypertension In: Y. Zhao editor. Lung biology and pathophysiology. Boca Raton, FL USA: CRC Press (2024). 87–100.
70. Dai, Z, and Zhao, YY. Discovery of a murine model of clinical PAH: mission impossible? Trends Cardiovasc Med. (2017) 27:229–36. doi: 10.1016/j.tcm.2016.12.003
71. Wang, S, Zeng, H, Xie, XJ, Tao, YK, He, X, Roman, RJ, et al. Loss of prolyl hydroxylase domain protein 2 in vascular endothelium increases pericyte coverage and promotes pulmonary arterial remodeling. Oncotarget. (2016) 7:58848–61. doi: 10.18632/oncotarget.11585
72. Park, CS, Kim, SH, Yang, HY, Kim, JH, Schermuly, RT, Cho, YS, et al. <i>Sox17</i> deficiency promotes pulmonary arterial hypertension via HGF/c-met signaling. Circ Res. (2022) 131:792–806. doi: 10.1161/CIRCRESAHA.122.320845
73. Ho, L, Hossen, N, Nguyen, T, Vo, A, and Ahsan, F. Epigenetic mechanisms as emerging therapeutic targets and microfluidic chips application in pulmonary arterial hypertension. Biomedicines. (2022) 10:170. doi: 10.3390/biomedicines10010170
74. Dave, J, Jagana, V, Janostiak, R, and Bisserier, M. Unraveling the epigenetic landscape of pulmonary arterial hypertension: implications for personalized medicine development. J Transl Med. (2023) 21:477. doi: 10.1186/s12967-023-04339-5
75. Kim, GH, Ryan, JJ, Marsboom, G, and Archer, SL. Epigenetic mechanisms of pulmonary hypertension. Pulm Circ. (2011) 1:347–56. doi: 10.4103/2045-8932.87300
76. Chelladurai, P, Seeger, W, and Pullamsetti, SS. Epigenetic mechanisms in pulmonary arterial hypertension: the need for global perspectives. Eur Respir Rev. (2016) 25:135–40. doi: 10.1183/16000617.0036-2016
77. Bisserier, M, Janostiak, R, Lezoualc'h, F, and Hadri, L. Targeting epigenetic mechanisms as an emerging therapeutic strategy in pulmonary hypertension disease. Vasc Biol. (2020) 2:R17–r34. doi: 10.1530/VB-19-0030
78. Wu, D, Dasgupta, A, Read, AD, Bentley, RET, Motamed, M, Chen, KH, et al. Oxygen sensing, mitochondrial biology and experimental therapeutics for pulmonary hypertension and cancer. Free Radic Biol Med. (2021) 170:150–78. doi: 10.1016/j.freeradbiomed.2020.12.452
79. Ranasinghe, A, and Schwarz, MA. Integrating epigenetics and metabolomics to advance treatments for pulmonary arterial hypertension. Biochem Pharmacol. (2022) 204:115245. doi: 10.1016/j.bcp.2022.115245
80. Kim, J, Lee, H, Yi, SJ, and Kim, K. Gene regulation by histone-modifying enzymes under hypoxic conditions: a focus on histone methylation and acetylation. Exp Mol Med. (2022) 54:878–89. doi: 10.1038/s12276-022-00812-1
81. Chen, T, Zhou, Q, Tang, H, Bozkanat, M, Yuan, JXJ, Raj, JU, et al. miR-17/20 controls prolyl hydroxylase 2 (PHD2)/hypoxia-inducible factor 1 (HIF1) to regulate pulmonary artery smooth muscle cell proliferation. J Am Heart Assoc. (2016) 5:e004510. doi: 10.1161/JAHA.116.004510
82. Deng, B, Du, J, Hu, R, et al. MicroRNA-103/107 is involved in hypoxia-induced proliferation of pulmonary arterial smooth muscle cells by targeting HIF-1β. Life Sci. (2016) 147:117–24. doi: 10.1016/j.lfs.2016.01.043
83. Yue, J, Guan, J, Wang, X, Zhang, L, Yang, Z, Ao, Q, et al. MicroRNA-206 is involved in hypoxia-induced pulmonary hypertension through targeting of the HIF-1α/Fhl-1 pathway. Lab Investig. (2013) 93:748–59. doi: 10.1038/labinvest.2013.63
84. Anderson, SA, Nizzi, CP, Chang, YI, Deck, KM, Schmidt, PJ, Galy, B, et al. The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. (2013) 17:282–90. doi: 10.1016/j.cmet.2013.01.007
85. Luo, W, Hu, H, Chang, R, Zhong, J, Knabel, M, O'Meally, R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. (2011) 145:732–44. doi: 10.1016/j.cell.2011.03.054
86. Archer, SL, Marsboom, G, Kim, GH, Zhang, HJ, Toth, PT, Svensson, EC, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation. (2010) 121:2661–71. doi: 10.1161/CIRCULATIONAHA.109.916098
87. Tian, L, Wu, D, Dasgupta, A, Chen, KH, Mewburn, J, Potus, F, et al. Epigenetic metabolic reprogramming of right ventricular fibroblasts in pulmonary arterial hypertension: a pyruvate dehydrogenase kinase-dependent shift in mitochondrial metabolism promotes right ventricular fibrosis. Circ Res. (2020) 126:1723–45. doi: 10.1161/CIRCRESAHA.120.316443
88. Di, R, Yang, Z, Xu, P, and Xu, Y. Silencing PDK1 limits hypoxia-induced pulmonary arterial hypertension in mice via the Akt/p70S6K signaling pathway. Exp Ther Med. (2019) 18:699–704. doi: 10.3892/etm.2019.7627
89. Higgins, DF, Biju, MP, Akai, Y, Wutz, A, Johnson, RS, and Haase, VH. Hypoxic induction of Ctgf is directly mediated by Hif-1. Am J Physiol Renal Physiol. (2004) 287:F1223–32. doi: 10.1152/ajprenal.00245.2004
90. Xia, XD, Peng, YP, Lei, D, and Chen, WQ. Hypercapnia downregulates hypoxia-induced lysyl oxidase expression in pulmonary artery smooth muscle cells via inhibiting transforming growth factor β(1) signalling. Cell Biochem Funct. (2019) 37:193–202. doi: 10.1002/cbf.3390
91. Ma, C, Xu, Q, Huang, S, Song, J, Sun, M, Zhang, J, et al. The HIF-1α/miR-26a-5p/PFKFB3/ULK1/2 axis regulates vascular remodeling in hypoxia-induced pulmonary hypertension by modulation of autophagy. FASEB J. (2023) 37:e22906. doi: 10.1096/fj.202200699RR
92. Zheng, F, Chen, J, Zhang, X, Wang, Z, Chen, J, Lin, X, et al. The HIF-1α antisense long non-coding RNA drives a positive feedback loop of HIF-1α mediated transactivation and glycolysis. Nat Commun. (2021) 12:1341. doi: 10.1038/s41467-021-21535-3
93. Zhou, L, Jiang, J, Huang, Z, Jin, P, Peng, L, Luo, M, et al. Hypoxia-induced lncRNA STEAP3-AS1 activates Wnt/β-catenin signaling to promote colorectal cancer progression by preventing m(6)A-mediated degradation of STEAP3 mRNA. Mol Cancer. (2022) 21:168. doi: 10.1186/s12943-022-01638-1
94. Na, H, Li, X, Zhang, X, Xu, Y, Sun, Y, Cui, J, et al. lncRNA STEAP3-AS1 modulates cell cycle progression via affecting CDKN1C expression through STEAP3 in colon cancer. Mol Ther Nucleic Acids. (2020) 21:480–91. doi: 10.1016/j.omtn.2020.06.011
95. Shan, Y, Hou, B, Wang, J, Chen, A, and Liu, S. Exploring the role of exosomal MicroRNAs as potential biomarkers in preeclampsia. Front Immunol. (2024) 15:1385950. doi: 10.3389/fimmu.2024.1385950
96. Zhao, H, Duan, R, Wang, Q, Hu, X, Zhao, Q, Wu, W, et al. MiR-122-5p as a potential regulator of pulmonary vascular wall cell in idiopathic pulmonary arterial hypertension. Heliyon. (2023) 9:e22922. doi: 10.1016/j.heliyon.2023.e22922
97. Wade, SM, Ohnesorge, N, McLoughlin, H, Biniecka, M, Carter, SP, Trenkman, M, et al. Dysregulated miR-125a promotes angiogenesis through enhanced glycolysis. EBioMedicine. (2019) 47:402–13. doi: 10.1016/j.ebiom.2019.08.043
98. Bruning, U, Cerone, L, Neufeld, Z, Fitzpatrick, SF, Cheong, A, Scholz, CC, et al. MicroRNA-155 promotes resolution of hypoxia-inducible factor 1alpha activity during prolonged hypoxia. Mol Cell Biol. (2011) 31:4087–96. doi: 10.1128/MCB.01276-10
99. Yang, C, Rong, R, Li, Y, Cheng, M, and Luo, Y. Decrease in LINC00963 attenuates the progression of pulmonary arterial hypertension via microRNA-328-3p/profilin 1 axis. J Clin Lab Anal. (2022) 36:e24383. doi: 10.1002/jcla.24383
100. Tai, YY, Yu, Q, Tang, Y, Sun, W, Kelly, NJ, Okawa, S, et al. Allele-specific control of rodent and human lncRNA KMT2E-AS1 promotes hypoxic endothelial pathology in pulmonary hypertension. Sci Transl Med. (2024) 16:eadd2029. doi: 10.1126/scitranslmed.add2029
101. Pleger, ST, Harris, DM, Shan, C, Vinge, LE, Chuprun, JK, Berzins, B, et al. Endothelial S100A1 modulates vascular function via nitric oxide. Circ Res. (2008) 102:786–94. doi: 10.1161/CIRCRESAHA.108.172031
102. Habbout, K, Omura, J, Awada, C, Bourgeois, A, Grobs, Y, Krishna, V, et al. Implication of EZH2 in the pro-proliferative and apoptosis-resistant phenotype of pulmonary artery smooth muscle cells in PAH: a transcriptomic and proteomic approach. Int J Mol Sci. (2021) 22:2957. doi: 10.3390/ijms22062957
103. Yfantis, A, Mylonis, I, Chachami, G, Nikolaidis, M, Amoutzias, GD, Paraskeva, E, et al. Transcriptional response to hypoxia: the role of HIF-1-associated co-regulators. Cells. (2023) 12:798. doi: 10.3390/cells12050798
104. Li, G, Tian, Y, and Zhu, WG. The roles of histone deacetylases and their inhibitors in cancer therapy. Front Cell Dev Biol. (2020) 8:576946. doi: 10.3389/fcell.2020.576946
105. Xu, Y, and Zhu, Q. Histone modifications represent a key epigenetic feature of epithelial-to-mesenchyme transition in pancreatic cancer. Int J Mol Sci. (2023) 24:4820. doi: 10.3390/ijms24054820
106. Tudisco, L, Della Ragione, F, Tarallo, V, Apicella, I, D'Esposito, M, Matarazzo, MR, et al. Epigenetic control of hypoxia inducible factor-1α-dependent expression of placental growth factor in hypoxic conditions. Epigenetics. (2014) 9:600–10. doi: 10.4161/epi.27835
107. Wigerup, C, Påhlman, S, and Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. (2016) 164:152–69. doi: 10.1016/j.pharmthera.2016.04.009
108. Chumak, V, Rajacharya, GH, and Singh, PK. Metabolomic investigations into hypoxia-mediated metabolic reprogramming of pancreatic cancer cells. Methods Mol Biol. (2024) 2755:191–200. doi: 10.1007/978-1-0716-3633-6_14
109. Iacobini, C, Vitale, M, Haxhi, J, Pesce, C, Pugliese, G, and Menini, S. Mutual regulation between redox and hypoxia-inducible factors in cardiovascular and renal complications of diabetes. Antioxidants (Basel). (2022) 11:2183. doi: 10.3390/antiox11112183
110. Brereton, CJ, Yao, L, Davies, ER, Zhou, Y, Vukmirovic, M, Bell, JA, et al. Pseudohypoxic HIF pathway activation dysregulates collagen structure-function in human lung fibrosis. eLife. (2022) 11:11. doi: 10.7554/eLife.69348
111. Cai, J, Hu, M, Chen, Z, and Ling, Z. The roles and mechanisms of hypoxia in liver fibrosis. J Transl Med. (2021) 19:186. doi: 10.1186/s12967-021-02854-x
112. Ghosh, MC, Zhang, DL, Ollivierre, WH, Noguchi, A, Springer, DA, Linehan, WM, et al. Therapeutic inhibition of HIF-2α reverses polycythemia and pulmonary hypertension in murine models of human diseases. Blood. (2021) 137:2509–19. doi: 10.1182/blood.2020009138
113. Boucherat, O, Vitry, G, Trinh, I, Paulin, R, Provencher, S, and Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm Circ. (2017) 7:285–99. doi: 10.1177/2045893217701438
114. Chen, D, Gao, W, Wang, S, Ni, B, and Gao, Y. Critical effects of epigenetic regulation in pulmonary arterial hypertension. Cell Mol Life Sci. (2017) 74:3789–808. doi: 10.1007/s00018-017-2551-8
115. Bassareo, PP, and D'Alto, M. Metabolomics in pulmonary hypertension-a useful tool to provide insights into the dark side of a tricky pathology. Int J Mol Sci. (2023) 24:13227. doi: 10.3390/ijms241713227
116. Qiu, S, Cai, Y, Yao, H, Lin, C, Xie, Y, Tang, S, et al. Small molecule metabolites: discovery of biomarkers and therapeutic targets. Signal Transduct Target Ther. (2023) 8:132. doi: 10.1038/s41392-023-01399-3
117. Espinosa-Diez, C, Miguel, V, Mennerich, D, Kietzmann, T, Sánchez-Pérez, P, Cadenas, S, et al. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. (2015) 6:183–97. doi: 10.1016/j.redox.2015.07.008
118. Yan, Q, Liu, S, Sun, Y, Chen, C, Yang, S, Lin, M, et al. Targeting oxidative stress as a preventive and therapeutic approach for cardiovascular disease. J Transl Med. (2023) 21:519. doi: 10.1186/s12967-023-04361-7
119. Rai, N, Shihan, M, Seeger, W, Schermuly, RT, and Novoyatleva, T. Genetic delivery and gene therapy in pulmonary hypertension. Int J Mol Sci. (2021) 22:1179. doi: 10.3390/ijms22031179
120. Fallah, J, and Rini, BI. HIF inhibitors: status of current clinical development. Curr Oncol Rep. (2019) 21:6. doi: 10.1007/s11912-019-0752-z
121. Qiu, GZ, Jin, MZ, Dai, JX, Sun, W, Feng, JH, and Jin, WL. Reprogramming of the tumor in the hypoxic niche: the emerging concept and associated therapeutic strategies. Trends Pharmacol Sci. (2017) 38:669–86. doi: 10.1016/j.tips.2017.05.002
122. Rani, S, Roy, S, Singh, M, and Kaithwas, G. Regulation of transactivation at C-TAD domain of HIF-1α by factor-inhibiting HIF-1α (FIH-1): a potential target for therapeutic intervention in cancer. Oxidative Med Cell Longev. (2022) 2022:1–21. doi: 10.1155/2022/2407223
123. Lafleur, VN, Halim, S, Choudhry, H, Ratcliffe, PJ, and Mole, DR. Multi-level interaction between HIF and AHR transcriptional pathways in kidney carcinoma. Life Sci Alliance. (2023) 6:e202201756. doi: 10.26508/lsa.202201756
124. Infantino, V, Santarsiero, A, Convertini, P, Todisco, S, and Iacobazzi, V. Cancer cell metabolism in hypoxia: role of HIF-1 as key regulator and therapeutic target. Int J Mol Sci. (2021) 22:5703. doi: 10.3390/ijms22115703
125. Camuzi, D, de Amorim, ÍSS, Ribeiro Pinto, LF, Oliveira Trivilin, L, Mencalha, AL, and Soares Lima, SC. Regulation is in the air: the relationship between hypoxia and epigenetics in cancer. Cells. (2019) 8:300. doi: 10.3390/cells8040300
126. Tibes, R, Falchook, GS, von Hoff, DD, Weiss, GJ, Iyengar, T, Kurzrock, R, et al. Results from a phase I, dose-escalation study of PX-478, an orally available inhibitor of HIF-1α. J Clin Oncol. (2010) 28:3076. doi: 10.1200/jco.2010.28.15_suppl.3076
127. Wei, X, Hou, Y, Long, M, Jiang, L, and Du, Y. Molecular mechanisms underlying the role of hypoxia-inducible factor-1 α in metabolic reprogramming in renal fibrosis. Front Endocrinol (Lausanne). (2022) 13:927329. doi: 10.3389/fendo.2022.927329
128. Locatelli, F, Minutolo, R, De Nicola, L, and Del Vecchio, L. Evolving strategies in the treatment of anaemia in chronic kidney disease: the HIF-prolyl hydroxylase inhibitors. Drugs. (2022) 82:1565–89. doi: 10.1007/s40265-022-01783-3
129. Cochrane Kidney and Transplant GroupNatale, P, Palmer, SC, Jaure, A, Hodson, EM, Ruospo, M, et al. Hypoxia-inducible factor stabilisers for the anaemia of chronic kidney disease. Cochrane Database Syst Rev. (2022) 2022:CD013751. doi: 10.1002/14651858.CD013751.pub2
130. He, Q, Ma, Y, Liu, J, Zhang, D, Ren, J, Zhao, R, et al. Biological functions and regulatory mechanisms of hypoxia-inducible factor-1α in ischemic stroke. Front Immunol. (2021) 12:801985. doi: 10.3389/fimmu.2021.801985
131. Dong, P, Li, Q, and Han, H. HIF-1α in cerebral ischemia (review). Mol Med Rep. (2022) 25:41. doi: 10.3892/mmr.2021.12557
132. Zhang, J, Qin, Y, Martinez, M, Flores-Bellver, M, Rodrigues, M, Dinabandhu, A, et al. HIF-1α and HIF-2α redundantly promote retinal neovascularization in patients with ischemic retinal disease. J Clin Invest. (2021) 131:e139202. doi: 10.1172/JCI139202
133. Wan, JJ, Yi, J, Wang, FY, Zhang, C, and Dai, AG. Expression and regulation of HIF-1a in hypoxic pulmonary hypertension: focus on pathological mechanism and pharmacological treatment. Int J Med Sci. (2024) 21:45–60. doi: 10.7150/ijms.88216
134. Sheikh, AQ, Saddouk, FZ, Ntokou, A, Mazurek, R, and Greif, DM. Cell autonomous and non-cell autonomous regulation of SMC progenitors in pulmonary hypertension. Cell Rep. (2018) 23:1152–65. doi: 10.1016/j.celrep.2018.03.043
135. Yu, YA, Malakhau, Y, Yu, CA, et al. Nonclassical monocytes sense hypoxia, regulate pulmonary vascular remodeling, and promote pulmonary hypertension. J Immunol. (2020) 204:1474–85. doi: 10.4049/jimmunol.1900239
136. Kojima, H, Tokunou, T, Takahara, Y, Sunagawa, K, Hirooka, Y, Ichiki, T, et al. Hypoxia-inducible factor-1 alpha deletion in myeloid lineage attenuates hypoxia-induced pulmonary hypertension. Physiol Rep. (2019) 7:e14025. doi: 10.14814/phy2.14025
137. Peng, Y, Cui, C, He, Y, Ouzhuluobu,, Zhang, H, Yang, D, et al. Down-regulation of EPAS1 transcription and genetic adaptation of Tibetans to high-altitude hypoxia. Mol Biol Evol. (2017) 34:818–30. doi: 10.1093/molbev/msw280
138. Song, D, Navalsky, BE, Guan, W, Ingersoll, C, Wang, T, Loro, E, et al. Tibetan PHD2, an allele with loss-of-function properties. Proc Natl Acad Sci USA. (2020) 117:12230–8. doi: 10.1073/pnas.1920546117
139. Macias, D, Cowburn, AS, Torres-Torrelo, H, Ortega-Saenz, P, Lopez-Barneo, J, and Johnson, RS. HIF-2alpha is essential for carotid body development and function. eLife. (2018) 7:7. doi: 10.7554/eLife.38781
140. Smith, KA, Waypa, GB, Dudley, VJ, Budinger, GRS, Abdala-Valencia, H, Bartom, E, et al. Role of hypoxia-inducible factors in regulating right ventricular function and remodeling during chronic hypoxia-induced pulmonary hypertension. Am J Respir Cell Mol Biol. (2020) 63:652–64. doi: 10.1165/rcmb.2020-0023OC
141. He, M, Shen, J, Zhang, C, Chen, Y, Wang, W, and Tao, K. Long-chain non-coding RNA metastasis-related lung adenocarcinoma transcript 1 (MALAT1) promotes the proliferation and migration of human pulmonary artery smooth muscle cells (hPASMCs) by regulating the MicroRNA-503 (miR-503)/toll-like receptor 4 (TLR4) signal axis. Med Sci Monit. (2020) 26:e923123. doi: 10.12659/MSM.923123
142. Su, H, Xu, X, Yan, C, Shi, Y, Hu, Y, Dong, L, et al. LncRNA H19 promotes the proliferation of pulmonary artery smooth muscle cells through AT(1)R via sponging let-7b in monocrotaline-induced pulmonary arterial hypertension. Respir Res. (2018) 19:254. doi: 10.1186/s12931-018-0956-z
143. Xu, J, Linneman, J, Zhong, Y, Yin, H, Xia, Q, Kang, K, et al. MicroRNAs in pulmonary hypertension, from pathogenesis to diagnosis and treatment. Biomol Ther. (2022) 12:496. doi: 10.3390/biom12040496
144. Caruso, P, MacLean, MR, Khanin, R, McClure, J, Soon, E, Southgate, M, et al. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol. (2010) 30:716–23. doi: 10.1161/ATVBAHA.109.202028
145. Sarkar, J, Gou, D, Turaka, P, Viktorova, E, Ramchandran, R, and Raj, JU. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am J Physiol Lung Cell Mol Physiol. (2010) 299:L861–71. doi: 10.1152/ajplung.00201.2010
146. António, T, Soares-da-Silva, P, Pires, NM, and Gomes, P. Salt-inducible kinases: new players in pulmonary arterial hypertension? Trends Pharmacol Sci. (2022) 43:806–19. doi: 10.1016/j.tips.2022.06.008
147. D'Addario, CA, Lanier, GM, Jacob, C, Bauer, N, Hewes, JL, Bhadra, A, et al. Differences in the expression of DNA methyltransferases and demethylases in leukocytes and the severity of pulmonary arterial hypertension between ethnic groups. Physiol Rep. (2022) 10:e15282. doi: 10.14814/phy2.15282
148. Macias, D, Moore, S, Crosby, A, Southwood, M, du, X, Tan, H, et al. Targeting HIF2α-ARNT hetero-dimerisation as a novel therapeutic strategy for pulmonary arterial hypertension. Eur Respir J. (2021) 57:1902061. doi: 10.1183/13993003.02061-2019
149. Jiang, Y, Zhou, Y, Peng, G, Liu, N, Tian, H, Pan, D, et al. Topotecan prevents hypoxia-induced pulmonary arterial hypertension and inhibits hypoxia-inducible factor-1α and TRPC channels. Int J Biochem Cell Biol. (2018) 104:161–70. doi: 10.1016/j.biocel.2018.09.010
150. Jiang, DT, Tuo, L, Bai, X, Bing, WD, Qu, QX, Zhao, X, et al. Prostaglandin E1 reduces apoptosis and improves the homing of mesenchymal stem cells in pulmonary arterial hypertension by regulating hypoxia-inducible factor 1 alpha. Stem Cell Res Ther. (2022) 13:316. doi: 10.1186/s13287-022-03011-x
Keywords: hypoxia inducible factor, epigenetics, pulmonary arterial hypertension, animal model, therapeutics
Citation: Mitra A, Yi D, Dai Z and de Jesus Perez V (2024) Unraveling the role of HIF and epigenetic regulation in pulmonary arterial hypertension: implications for clinical research and its therapeutic approach. Front. Med. 11:1460376. doi: 10.3389/fmed.2024.1460376
Edited by:
Francois Potus, Laval University, CanadaReviewed by:
Stephen Y. Chan, University of Pittsburgh Medical Center, United StatesInjune Kim, Korea Advanced Institute of Science and Technology (KAIST), Republic of Korea
Copyright © 2024 Mitra, Yi, Dai and de Jesus Perez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinicio de Jesus Perez, dmRlamVzdXNAc3RhbmZvcmQuZWR1; Zhiyu Dai, ei5kYWlAd3VzdGwuZWR1
†These authors have contributed equally to this work