 Natsuki Higa1
Natsuki Higa1 Takaaki Hayashi1,2*†
Takaaki Hayashi1,2*† Kei Mizobuchi2†Maki Iwasa3Shingo Kubota4
Kei Mizobuchi2†Maki Iwasa3Shingo Kubota4 Kazuki Kuniyoshi5Shuhei Kameya6
Kazuki Kuniyoshi5Shuhei Kameya6 Hiroyuki Kondo7Mineo Kondo8Tadashi Nakano2
Hiroyuki Kondo7Mineo Kondo8Tadashi Nakano2- 1Department of Ophthalmology, Katsushika Medical Center, The Jikei University School of Medicine, Tokyo, Japan
- 2Department of Ophthalmology, The Jikei University School of Medicine, Tokyo, Japan
- 3Department of Ophthalmology, Shiga University of Medical Science, Otsu, Japan
- 4Kubota Eye Clinic, Kusatsu, Shiga, Japan
- 5Department of Ophthalmology, Kindai University Faculty of Medicine, Osaka-sayama, Japan
- 6Kameya Eye Clinic, Chiba, Japan
- 7Department of Ophthalmology, University of Occupational and Environmental Health, Fukuoka, Japan
- 8Department of Ophthalmology, Mie University Graduate School of Medicine, Tsu City, Japan
Introduction: In Japan, inherited retinal dystrophy caused by biallelic variants of the RPE65 gene is exceedingly rare. The purpose of this study was to describe a Japanese male patient with a novel variant in RPE65 associated with Leber congenital amaurosis (LCA).
Case report: The patient, diagnosed with LCA, exhibited infantile nystagmus and reported experiencing night blindness since early childhood. At 27 years of age, the patient underwent an ophthalmologically evaluation. Corrected visual acuity was Snellen equivalent 20/133 in the right eye and Snellen equivalent 20/100 in the left eye. Fundus examination revealed alterations in the retinal pigment epithelium characterized by hypopigmentation and narrowing of retinal vessels. Fundus autofluorescence imaging demonstrated a generally diminished autofluorescent signal. Full-field electroretinography identified a generalized dysfunction of both rod and cone systems in each eye. Whole exome sequencing identified a novel missense variant in RPE65 (NM_000329.3): c.1172C > A p.(Ala391Asp), which was classified as pathogenic, as well as a recurrent variant p.(Arg515Trp).
Conclusion: This study provides valuable insights into the genotype–phenotype correlation of RPE65-associated LCA in Japanese patients, with critical implications for enhanced diagnostic accuracy and informed therapeutic decisions.
1 Introduction
Inherited retinal dystrophies (IRDs) constitute a diverse group of genetically and clinically heterogeneous inherited retinal disorders characterized by progressive retinal degeneration that culminates in legal blindness. A particularly severe form of IRDs, Leber congenital amaurosis (LCA), manifests with profound congenital or early-onset vision loss and nystagmus (1). Previous reports have indicated that pathogenic variants in genes such as CEP290, GUCY2D, CRB1, RDH12, and RPE65 are predominantly associated with LCA and early-onset severe retinal dystrophy (EOSRD) (1, 2).
The RPE65 gene (NM_000329.3) is located on the short arm of chromosome 1 (1p31.3) and spans 14 exons over 21.1 kilobase pairs. The encoded RPE65 protein, composed of 533 amino acids (NP_000320.1), functions within the retinal pigment epithelium, catalyzing the conversion of all-trans-retinyl fatty acid esters into 11-cis-retinol, a crucial step in visual chromophore regeneration (1). Biallelic pathogenic variants in RPE65 were first identified in LCA/EOSRD patients in 1997 (3, 4). Such variants in RPE65 account for approximately 11.4% of cases in early-onset retinal dystrophies in Western populations (5). These variants are also implicated in autosomal recessive retinitis pigmentosa (RP) (6). However, RPE65-associated retinal dystrophies are reported to be relatively rare in Japan (7, 8), with few documented cases detailing their clinical progression (9–11).
In 2018, the FDA approved Voretigene neparvovec (Luxturna®) as the inaugural gene therapy for LCA/EOSRD with biallelic RPE65 variants, following positive results from phase 3 clinical trial (12). Subsequently, Voretigene neparvovec was approved in Japan in 2023, post phase 3 clinical trials (NCT04516369). Despite the therapeutic potential of gene therapy, its implementation in Japan faces challenges. First, the progression of RPE65-LCA/EOSRD in Japanese patients is poorly documented. Additionally, the incidence of such dystrophies in Japan is considerably lower than reported in other populations. The second challenge pertains to the exceptionally low incidence rates among previously published reports on IRDs (7, 8). This report introduces a case of LCA in a young Japanese patient with biallelic RPE65 variants and reviews the literature on Japanese patients with biallelic variants in RPE65.
2 Case report
A male patient, identified as JU2139, exhibited infantile nystagmus noticed by his parents before he was 1 year old. He has reported experiencing night blindness since early childhood, although no systemic diseases have been diagnosed. There was no history of consanguinity among his unaffected parents. The patient recalled having relatively good visual acuity upon entering elementary school. His first ophthalmological consultation occurred at 13 years of age at a local clinic, where he was observed for 3 years. Initial fundus photography indicated mild coarsening of the retinal pigment epithelium (RPE) bilaterally (OU). The patient was eventually diagnosed with LCA. By the age of 24, he achieved a decimal best-corrected visual acuity (BCVA) of 0.5 (Snellen equivalent 20/40) in the right eye (OD) and 0.4 (Snellen equivalent 20/50) in the left eye (OS).
At 27 years of age, the patient was referred to the Department of Ophthalmology at The Jikei University Hospital for evaluation of a suspected inherited retinal dystrophy. The examination revealed a decimal BCVA of 0.1 (Snellen equivalent 20/200) OD and 0.2 (Snellen equivalent 20/100) OS, with respective refractive corrections of +2.50 diopters sphere (DS) with −2.00 diopters cylinder (DC) at 180 degrees OD and +1.75 DS with −3.00 DC at 175 degrees OS. Intraocular pressure was measured at 17 mmHg OD and 14 mmHg OS. Slit-lamp examination of the anterior segment showed no remarkable abnormalities. Examination of the posterior segment through fundus photography disclosed alterations in the RPE characterized by hypopigmentation, as well as narrowing of retinal vessels OU (Figure 1A). Both conventional and ultra-widefield fundus autofluorescence (FAF) imaging revealed areas of hyper-autofluorescence corresponding to the lesions and a generally diminished FAF signal within and beyond the vascular arcades (Figure 1B). Optical coherence tomography (OCT) performed using a Cirrus 5000 system (Carl Zeiss Meditec AG, Dublin, CA, United States), revealed disruption of ellipsoid zone (EZ), interdigitation zone and external limiting membrane OD. In contrast, the left eye exhibited a faintly preserved EZ in the foveal region (Figure 1C). Full-field electroretinograms (ERGs) were obtained under both dark-adapted (DA) and light-adapted (LA) conditions with dilated pupils, induced by a combination of tropicamide and phenylephrine hydrochloride eye drops, in accordance with the International Society for Clinical Electrophysiology of Vision protocols (13). In the ERGs, the recorded amplitudes of the rod (DA 0.01 cd s m−2) b-waves, standard/bright-flash (DA 3.0/10.0 cd s m−2) a- and b-waves, cone (LA 3.0 cd s m−2) a- and b-waves and LA 30-Hz flicker responses were undetectable (Figure 2), signifying a generalized dysfunction of both rod and cone systems.
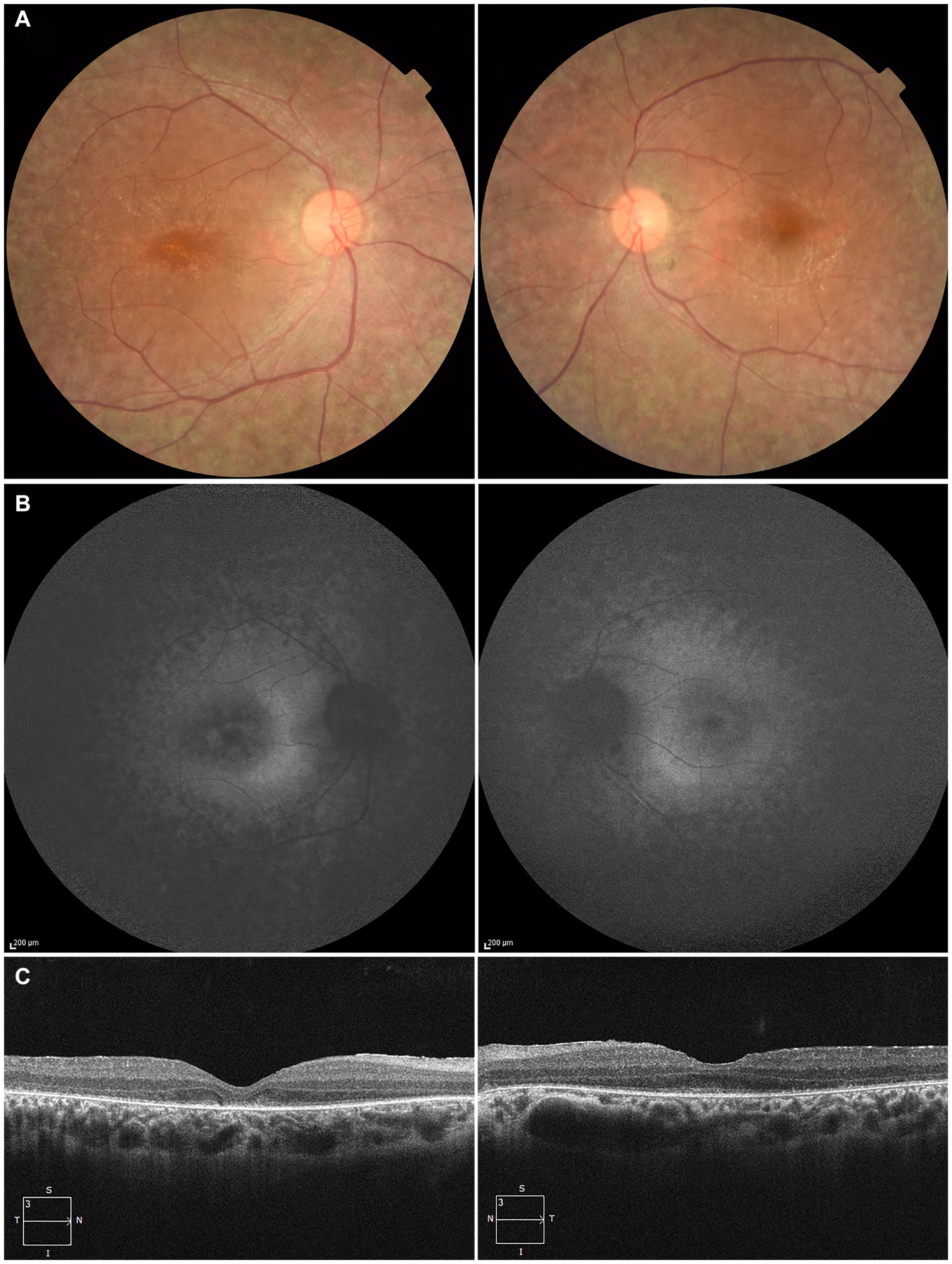
Figure 1. Multimodal imaging of the right eye (left column) and the left eye (right column) of the patient at the age of 27. (A) Fundus photography shows alterations in the retinal pigment epithelium characterized by hypopigmentation and narrowing of retinal vessels in both eyes. (B) Fundus autofluorescence imaging reveals areas of hyper-autofluorescence corresponding to the lesions and a generally diminished autofluorescent signal within and beyond the vascular arcades. (C) Optical coherence tomography reveals disruption of ellipsoid zone (EZ), interdigitation zone and external limiting membrane in the right eye. In contrast, the left eye exhibited a faintly preserved EZ in the foveal region.
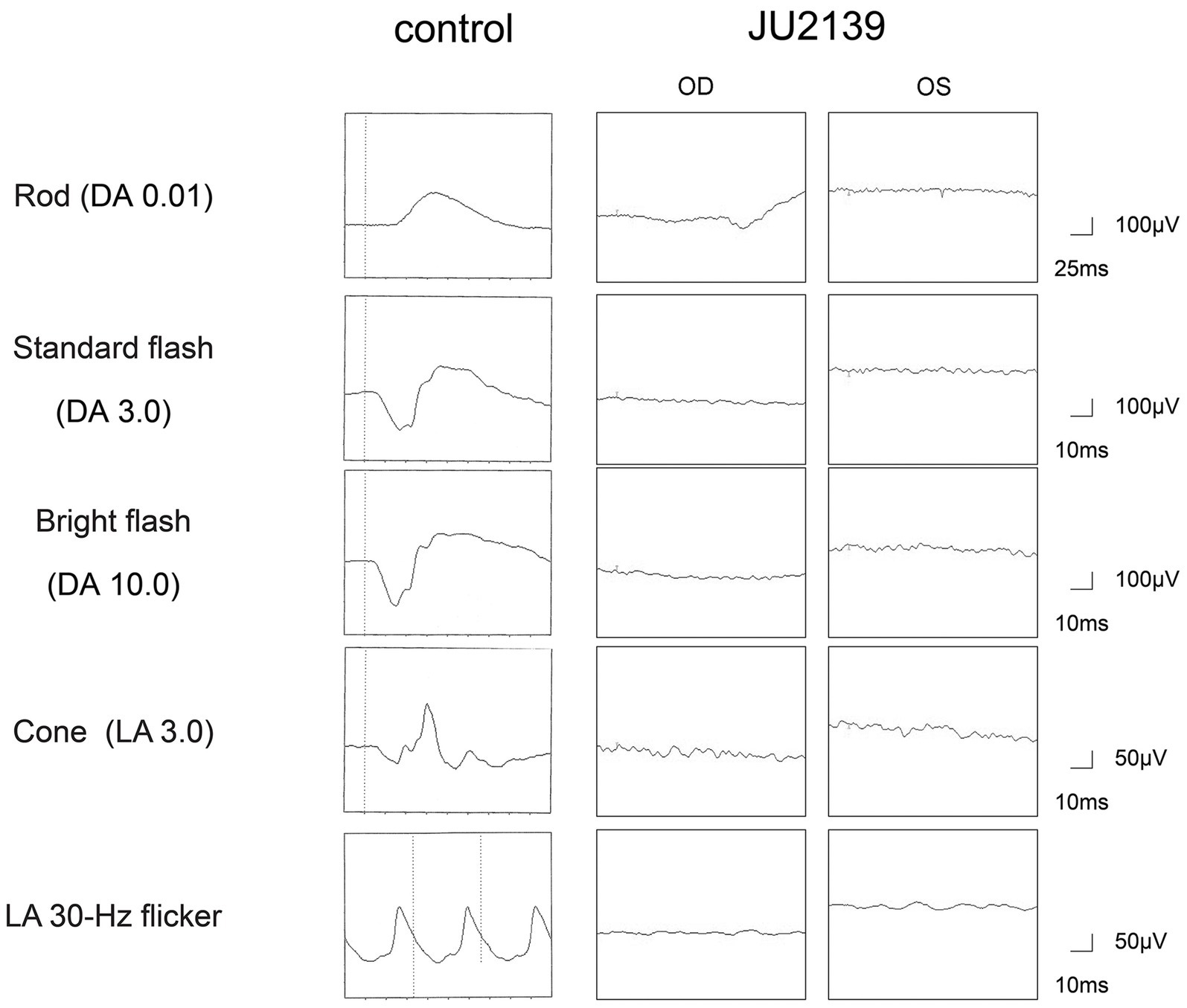
Figure 2. Full-field electroretinograms in an age-matched control and the patient (JU2139). The electroretinograms are recorded under both dark-adapted (DA) and light-adapted (LA) conditions in the right eye (OD) and in the left eye (OS). The recorded amplitudes of the rod (DA 0.01 cd s m−2) b-waves, standard/bright-flash (DA 3.0/10.0 cd s m−2) a- and b-waves, cone (LA 3.0 cd s m−2) a- and b-waves and LA 30-Hz flicker responses are undetectable in the patient, signifying a generalized dysfunction of both rod and cone systems.
For the purpose of genetic testing, the protocol of our study received approval from the Institutional Review Board of The Jikei University School of Medicine (approval numbers: 24-2316997). Informed consent was procured from both the patient and his parents. Genomic DNA was initially extracted from peripheral blood samples. Subsequent whole-exome sequencing (WES) and bioinformatics analyses were conducted to identify disease causing variants in the patient’s genome. The methodologies for WES have been elaborated upon in our previous publications (8, 14, 15). The Genome Reference Consortium Human Genome Build 38 (GRCh38) served as the reference sequence. Through a series of filtering steps, two missense variants in RPE65 (NM_000329.3) were detected: c.1172C > A (chr1:68431542G > T, GRCh38.p14, rs753540419); p.(Ala391Asp) in exon 11 and c.1543C > T (chr1:68429835G > A, GRCh38.p14, rs121917745);p.(Arg515Trp) in exon 14. These variants were visualized using the Integrative Genomic Viewer (IGV version 2.16.2, Broad Institute, MA, United States) software, as depicted in Figure 3A.
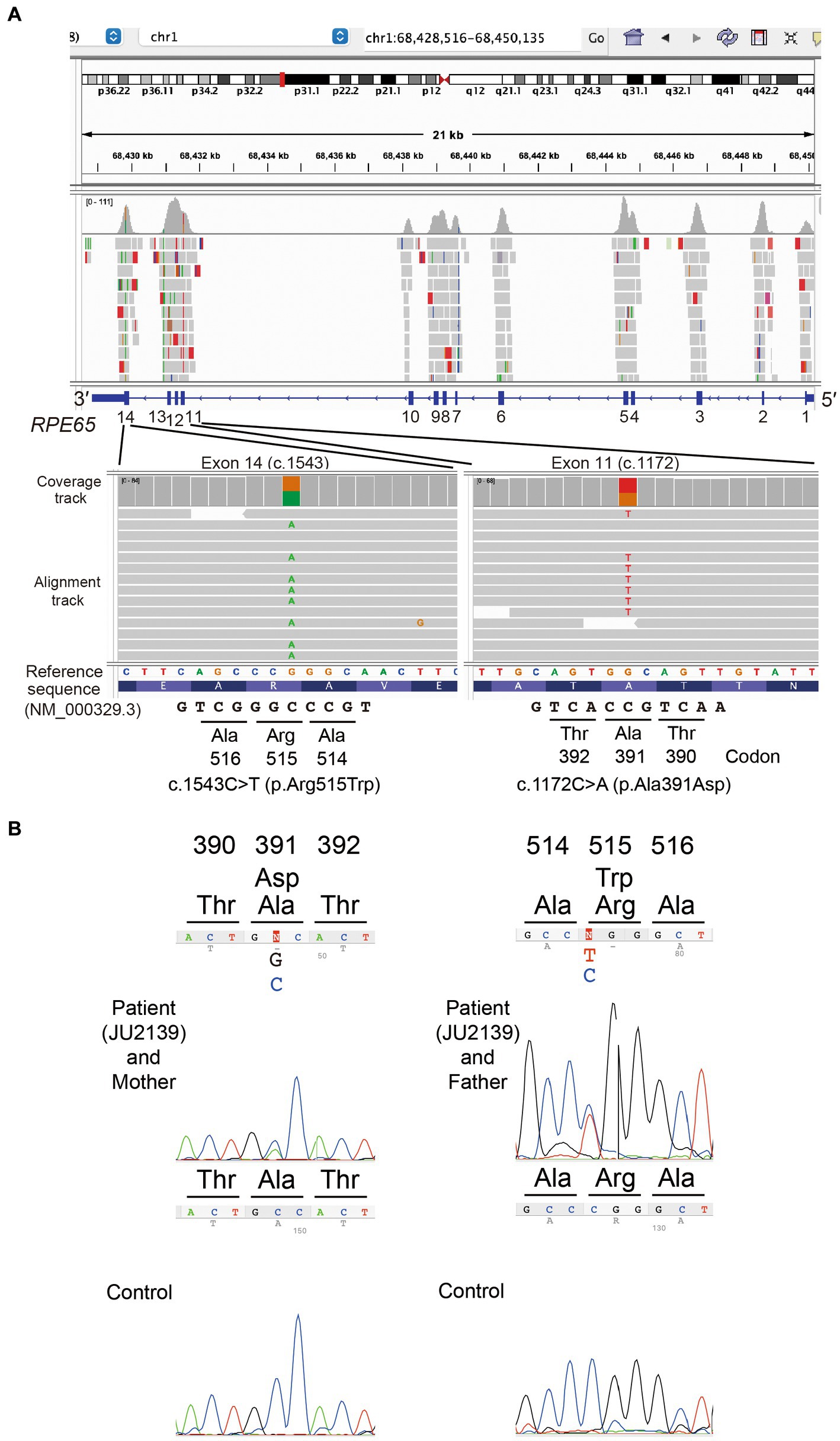
Figure 3. Molecular genetic analysis data. (A) In the patient, the Integrative Genomics Viewer screenshots of whole exome sequencing validation data show a heterozygous missense variant c.1172C > A; p.(Ala391Asp) in exon 11 and a heterozygous missense variant c.1543C > T; p.(Arg515Trp) in exon 14 of the RPE65 gene (NM_000329.3). (B) Sanger sequencing chromatograms of RPE65. Partial nucleotide sequences demonstrate that the patient (JU2139) carries the compound heterozygous variants (c.1172C > A; p.(Ala391Asp) and c.1543C > T; p.(Arg515Trp)), whereas his mother and father carry the heterozygous variants p.(Ala391Asp) and p.(Arg515Trp), respectively.
Sanger sequencing was performed for validation of the variants and for cosegregation analysis. The following primer sets were used for RPE65: exon 11: forward primer (RPE65-11bF) 5′-GAGCCAAGACTTAAGAACTC-3′ and reverse primer (RPE65-11bR) 5′-GACTAGCATATACTCAAAGCAC-3′, and exon 14 forward primer (RPE65-14F) 5′-TTAGAGCTTACAGTGTAGGTAG-3′ and reverse primer (RPE65-14R) 5′-AAGCCATTTAGTAAGTCCAC-3′ were used. His mother carried the p.(Ala391Asp) variant heterozygously, while his father carried the p.(Arg515Trp) variant heterozygously (Figure 3B).
The p.(Ala391Asp) variant has never been previously reported, as confirmed by searches in the Human Gene Mutation Database Professional (HGMD)1 as of February 2023, Leiden Open Variation Database 3.0 (LOVD)2 and ClinVar,3 thus designating it as a novel missense variant. The alanine residue at position 391, within the dimer-mediating region (16, 17) composed of amino-acid residues 371-404, is highly conserved across vertebrate species. The allele frequency of the p.(Ala391Asp) variant in the Genome Aggregation Database (gnomAD)4 is exceedingly rare at 0.000004 (1 in 250,644 alleles). This variant is absent in the Tohoku Medical Megabank Organization’s 54 K Japanese Genome Variation Database [ToMMo 54KJPN; (18)]5, which includes data from 54,267 Japanese individuals. According to the American College of Medical Genetics standards, the p.(Ala391Asp) was determined to be “pathogenic” based on the following criteria: PM1 (within the dimer-mediating region), PM2 (not found in controls), PM3 (detected in trans), PP3 (computational evidence for pathogenicity), and PP4 (highly specific for the LCA phenotype). The other p.(Arg515Trp) variant, classified as pathogenic in ClinVar, has been documented in Japanese patients diagnosed with LCA (Table 1). Within the ToMMo 54KJPN, this variant’s allele frequency stands at 0.0008 (86 in 108,534 alleles), which is notably 80-fold higher than the frequency observed in gnomAD, where it is 0.00001 (4 in 280,264 alleles).
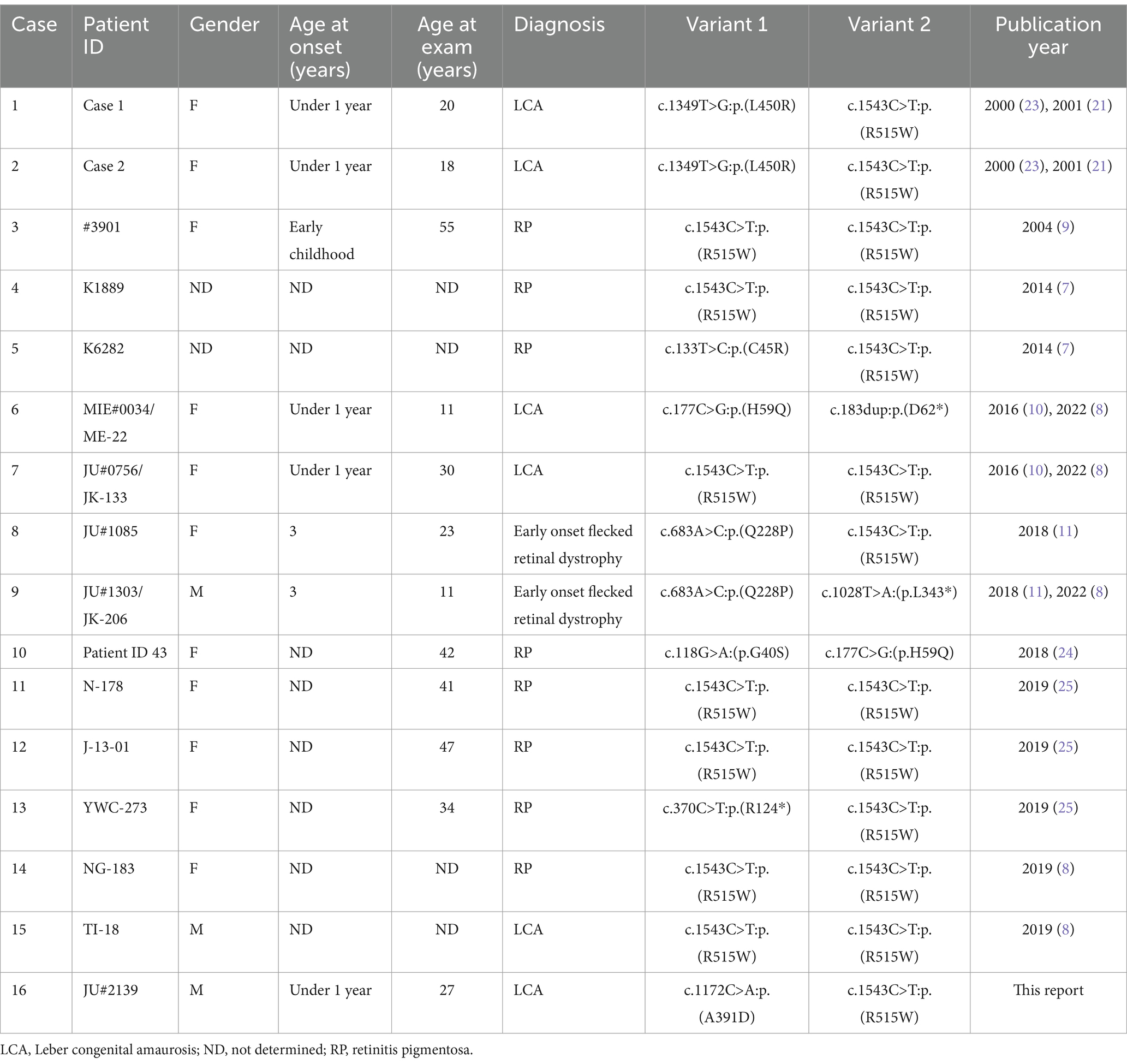
Table 1. Summary of Japanese patients with inherited retinal dystrophy, who carry biallelic variants in the RPE65 gene (NM_000329.3).
3 Discussion
This study documents the clinical and genetic characteristics of LCA associated with biallelic RPE65 variants in a Japanese patient, emphasizing the implications for genetic diagnosis and potential gene therapy in Japan.
Our patient exhibited early symptoms such as nystagmus and night blindness, which are typical of LCA. The progression of his visual acuity loss and the characteristic ophthalmological findings, including FAF and OCT (Figures 1B,C), are consistent with those described in RPE65-associated LCA/EOSRD. The absence of responses in the ERGs (Figure 2) confirms the generalized dysfunction of both rod and cone systems, indicative of LCA.
Through whole exome sequencing, we identified two missense variants in RPE65: the novel p.(Ala391Asp) variant and the p.(Arg515Trp) variant, which has been previously reported in Japanese patients with RPE65-associated LCA/EOSRD (Figures 3A,B). The novel p.(Ala391Asp) variant’s rarity is highlighted by its extremely low or non-existent allele frequency in large databases such as gnomAD and ToMMo 54KJPN (18). In contrast, the allele frequency of the recurrent p.(Arg515Trp) variant is significantly higher in the Japanese population, underscoring the importance of localized genetic databases for accurate variant interpretation and the necessity for population-specific approaches in genetic research and diagnostics.
Our report provides an opportunity to discuss the genotype–phenotype correlation, particularly in the context of the Japanese population. Although genotype–phenotype correlations in patients with RPE65 variants are not well-defined, truncating variants appear to result in more severe clinical phenotypes (19, 20). We have compiled previous reports of RPE65-associated RP/EOSRD/LCA from Japan (8–11, 21–25). Including our case, only 16 cases of patients with biallelic RPE65 variants have been documented (Table 1). Clinical diagnoses ranged from RP, EOSRD to LCA. Most patients had biallelic missense variants. Notably, the p.(Arg515Trp) variant was detected at a high frequency, accounting for 20 out of 32 alleles (62.5%) (Table 1). This aligns with allele frequency data from the Japanese database, ToMMo 54KJPN (18). When considering RPE65-associated LCA/EOSRD in Japanese patients, there is a high probability of encountering this frequent p.(Arg515Trp) variant. Our case also harbored the p.(Arg515Trp) variant heterozygously. Seven cases have been reported with the p.(Arg515Trp) variant in the homozygous state, with five diagnosed with RP (Case # 3, 4, 11, 12, and 14) and two with LCA (Case # 7 and 15) (Table 1). Phenotypic variability may be present even in cases with the p.(Arg515Trp) variants homozygously. On the other hand, the novel p.(Ala391Asp) variant is located within the dimer-mediating region (16, 17) of the RPE65 protein, which forms a dimer of two symmetrical enzymatically independent subunits. Therefore, the alanine at position 391 is likely to be a functionally important amino acid residue.
In 2023, Japan’s Ministry of Health, Labour and Welfare approved Voretigene neparvovec as the first gene therapy for LCA/EOSRD with biallelic RPE65 variants in Japan. However, the lower incidence of RPE65-associated LCA/EOSRD in the Japanese population may influence the feasibility and prioritization of such therapies. A thorough understanding of the disease’s natural history in this demographic is crucial for initiating treatment appropriately.
This report underlines the significance of comprehensive genetic testing and detailed clinical characterization in this rare disease. It highlights potential avenues for future research into the pathophysiology of RPE65-associated LCA/EOSRD and emphasizes the necessity for extensive genetic studies within the Japanese population to elucidate the spectrum of RPE65 variants and their clinical presentations.
4 Conclusion
This study provides valuable insights into the phenotype and genetic basis of RPE65-associated LCA/EOSRD in Japanese patients, establishing a foundation for improved diagnostic accuracy, informed therapeutic decisions, and future research in IRDs.
Data availability statement
The datasets for this article are not publicly available in order to protect patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Institutional Review Board of The Jikei University School of Medicine (approval numbers: 24-231 6997). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
NH: Writing – original draft, Writing – review & editing. TH: Conceptualization, Data curation, Investigation, Writing – original draft, Writing – review & editing. KM: Writing – original draft, Writing – review & editing. MI: Writing – original draft, Writing – review & editing. SKu: Writing – original draft, Writing – review & editing. KK: Writing – original draft, Writing – review & editing. SKa: Writing – original draft, Writing – review & editing. HK: Supervision, Writing – original draft, Writing – review & editing. MK: Supervision, Writing – original draft, Writing – review & editing. TN: Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by Grants-in-Aid for Scientific Research (KAKENHI; grant numbers 22K09825 and 24K12771), the Health and Labor Sciences Research Grants (23FC1043) and research funds from Alcon (Tokyo, Japan), Johnson and Johnson Vision, AMO (Tokyo, Japan), Chugai (Tokyo, Japan), Mitsubishi Tanabe Pharma (Osaka, Japan), Senju (Osaka, Japan), Bayer (Osaka, Japan), Santen (Osaka, Japan). The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Acknowledgments
We acknowledge the assistance of the patient and his parents, and express our gratitude to Ritsuko Nakayama for her help with the genetic analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
2. ^https://www.lovd.nl/3.0/home
3. ^https://www.ncbi.nlm.nih.gov/clinvar/
References
1. den, A, Roepman, R, Koenekoop, RK, and Cremers, FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. (2008) 27:391–419. doi: 10.1016/j.preteyeres.2008.05.003
2. Kumaran, N, Moore, AT, Weleber, RG, and Michaelides, M. Leber congenital Amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. (2017) 101:1147–54. doi: 10.1136/bjophthalmol-2016-309975
3. Gu, SM, Thompson, DA, Srikumari, CR, Lorenz, B, Finckh, U, Nicoletti, A, et al. Mutations in Rpe65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet. (1997) 17:194–7. doi: 10.1038/ng1097-194
4. Marlhens, F, Bareil, C, Griffoin, JM, Zrenner, E, Amalric, P, Eliaou, C, et al. Mutations in RPE65 cause Leber’s congenital Amaurosis. Nat Genet. (1997) 17:139–41. doi: 10.1038/ng1097-139
5. Thompson, DA, Gyurus, P, Fleischer, LL, Bingham, EL, McHenry, CL, Apfelstedt-Sylla, E, et al. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest Ophthalmol Vis Sci. (2000) 41:4293–9.
6. Morimura, H, Fishman, GA, Grover, SA, Fulton, AB, Berson, EL, and Dryja, TP. Mutations in the RPE65 gene in patients with autosomal recessive retinitis Pigmentosa or Leber congenital Amaurosis. Proc Natl Acad Sci USA. (1998) 95:3088–93. doi: 10.1073/pnas.95.6.3088
7. Oishi, M, Oishi, A, Gotoh, N, Ogino, K, Higasa, K, Iida, K, et al. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis Pigmentosa and usher syndrome patients by next-generation sequencing. Invest Ophthalmol Vis Sci. (2014) 55:7369–75. doi: 10.1167/iovs.14-15458
8. Suga, A, Yoshitake, K, Minematsu, N, Tsunoda, K, Fujinami, K, Miyake, Y, et al. Genetic characterization of 1210 Japanese pedigrees with inherited retinal diseases by whole-exome sequencing. Hum Mutat. (2022) 43:2251–64. doi: 10.1002/humu.24492
9. Kondo, H, Qin, M, Mizota, A, Kondo, M, Hayashi, H, Hayashi, K, et al. A homozygosity-based search for mutations in patients with autosomal recessive retinitis Pigmentosa, using microsatellite markers. Invest Ophthalmol Vis Sci. (2004) 45:4433–9. doi: 10.1167/iovs.04-0544
10. Katagiri, S, Hayashi, T, Kondo, M, Tsukitome, H, Yoshitake, K, Akahori, M, et al. RPE65 mutations in two Japanese families with Leber congenital Amaurosis. Ophthalmic Genet. (2016) 37:161–9. doi: 10.3109/13816810.2014.991931
11. Katagiri, S, Hosono, K, Hayashi, T, Kurata, K, Mizobuchi, K, Matsuura, T, et al. Early onset flecked retinal dystrophy associated with new compound heterozygous RPE65 variants. Mol Vis. (2018) 24:286–96.
12. Russell, S, Bennett, J, Wellman, JA, Chung, DC, Yu, ZF, Tillman, A, et al. Efficacy and safety of Voretigene Neparvovec (Aav2-Hrpe65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. (2017) 390:849–60. doi: 10.1016/S0140-6736(17)31868-8
13. Robson, AG, Frishman, LJ, Grigg, J, Hamilton, R, Jeffrey, BG, Kondo, M, et al. Iscev standard for full-field clinical electroretinography (2022 update). Doc Ophthalmol. (2022) 144:165–77. doi: 10.1007/s10633-022-09872-0
14. Hayashi, T, Mizobuchi, K, Kameya, S, Ueno, S, Matsuura, T, and Nakano, T. A mild form of POC1B-associated retinal dystrophy with relatively preserved cone system function. Doc Ophthalmol. (2023) 147:59–70. doi: 10.1007/s10633-023-09936-9
15. Morohashi, T, Hayashi, T, Mizobuchi, K, Nakano, T, and Morioka, I. Bardet-Biedl syndrome associated with novel compound heterozygous variants in BBS12 gene. Doc Ophthalmol. (2023) 146:165–71. doi: 10.1007/s10633-022-09915-6
16. Kiser, PD, Farquhar, ER, Shi, W, Sui, X, Chance, MR, and Palczewski, K. Structure of RPE65 isomerase in a Lipidic matrix reveals roles for phospholipids and Iron in catalysis. Proc Natl Acad Sci USA. (2012) 109:E2747–56. doi: 10.1073/pnas.1212025109
17. Poli, G, Barravecchia, I, Demontis, GC, Sodi, A, Saba, A, Rizzo, S, et al. Predicting potentially pathogenic effects of hRPE65 missense mutations: a computational strategy based on molecular dynamics simulations. J Enzyme Inhib Med Chem. (2022) 37:1765–72. doi: 10.1080/14756366.2022.2090547
18. Tadaka, S, Kawashima, J, Hishinuma, E, Saito, S, Okamura, Y, Otsuki, A, et al. Jmorp: Japanese multi-omics reference panel update report 2023. Nucleic Acids Res. (2024) 52:D622–32. doi: 10.1093/nar/gkad978
19. Gao, FJ, Wang, DD, Li, JK, Hu, FY, Xu, P, Chen, F, et al. Frequency and phenotypic characteristics of RPE65 mutations in the Chinese population. Orphanet J Rare Dis. (2021) 16:174. doi: 10.1186/s13023-021-01807-3
20. Lopez-Rodriguez, R, Lantero, E, Blanco-Kelly, F, Avila-Fernandez, A, Martin Merida, I, Del Pozo-Valero, M, et al. RPE65-related retinal dystrophy: mutational and phenotypic Spectrum in 45 affected patients. Exp Eye Res. (2021) 212:108761. doi: 10.1016/j.exer.2021.108761
21. Wada, Y, and Tamai, M. RPE65 gene mutations and retinal degeneration: Leber congenital Amaurosis [in Japanese]. Rinsho Ganka (Jpn J Clin Ophthalmol). (2001) 55:401–3. doi: 10.11477/mf.1410907225
22. Wada, Y, Itabashi, T, Sukegawa, M, Tada, A, Sato, H, Imai, E, et al. Prevalence of mutations in seven candidate genes in Japanese patients with Leber’s congenital Amaurosis (Arvo annual meeting abstract). Invest Ophthalmol Vis Sci. (2006) 47:1691.
23. Wada, Y, Nakazawa, T, Abe, T, Fuse, N, and Tamai, M. Clinical Varibility of patients associated with gene mutations of visual cycle protein; Arrestin, RPE65 and RDH5 genes. (Arvo annual meeting abstract). Invest Ophthalmol Vis Sci. (2000) 41:S617.
24. Maeda, A, Yoshida, A, Kawai, K, Arai, Y, Akiba, R, Inaba, A, et al. Development of a molecular diagnostic test for retinitis Pigmentosa in the Japanese population. Jpn J Ophthalmol. (2018) 62:451–7. doi: 10.1007/s10384-018-0601-x
Keywords: inherited retinal dystrophy, Leber congenital amaurosis, electroretinography, RPE65 gene mutation, whole exome sequencing
Citation: Higa N, Hayashi T, Mizobuchi K, Iwasa M, Kubota S, Kuniyoshi K, Kameya S, Kondo H, Kondo M and Nakano T (2024) A novel RPE65 variant p.(Ala391Asp) in Leber congenital amaurosis: a case report and literature review in Japan. Front. Med. 11:1442107. doi: 10.3389/fmed.2024.1442107
Edited by:
Ryo Mukai, Fukushima Medical University, JapanReviewed by:
Atsushi Mizota, Teikyo University, JapanDong Ho Park, Kyungpook National University Hospital, Republic of Korea
Manuel Soliño, University of Buenos Aires, Argentina
Copyright © 2024 Higa, Hayashi, Mizobuchi, Iwasa, Kubota, Kuniyoshi, Kameya, Kondo, Kondo and Nakano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takaaki Hayashi, dGFrYUBqaWtlaS5hYy5qcA==
†ORCID: Takaaki Hayashi, http://orcid.org/0000-0002-1535-0279
Kei Mizobuchi, http://orcid.org/0000-0001-5389-6507