 Roberto Luisetto1†
Roberto Luisetto1† Anna Scanu2,3*†
Anna Scanu2,3*†- 1Experimental Surgery Research Center, Department of Surgery, Oncology and Gastroenterology-DISCOG, University of Padova, Padova, Italy
- 2Department of Women's and Children's Health-SDB, University of Padova, Padova, Italy
- 3Departement of Neuroscience-DNS, University of Padova, Padova, Italy
The deposition of calcium pyrophosphate (CPP) crystals in joint tissues causes acute and chronic arthritis that commonly affect the adult and elderly population. Experimental calcium pyrophosphate deposition disease (CPPD) models are divided into genetically modified models and crystal-induced inflammation models. The former do not reproduce phenotypes overlapping with the human disease, while in the latter, the direct injection of crystals into the ankles, dorsal air pouch or peritoneum constitutes a useful and reliable methodology that resembles the CPP induced-inflammatory condition in humans. The translational importance of the induced model is also strengthened by the fact that the key molecular and cellular mediators involved in inflammation are shared between humans and laboratory rodents. Although, in vivo models are indispensable tools for studying the pathogenesis of the CPPD and testing new therapies, their development is still at an early stage and major efforts are needed to address this issue. Here, we analyze the strenghts and limitations of each currently available CPPD in vivo model, and critically discuss their translational value.
Introduction
Crystal deposition in the articular and periarticular tissues causes arthropathies that commonly affect the adult and elderly population. Among the most common and well-known of these crystals is calcium pyrophosphate (CPP), which is the causative agent of inflammation and joint damage. The clinical manifestations of calcium pyrophosphate deposition disease (CPPD) can range from asymptomatic tissue calcification to acute or chronic arthritis, and in some cases it may be associated with other conditions, such as osteoarthritis (1).
Although CPPD is a prevalent rheumatic musculoskeletal disease, the precise processes that lead to crystal formation and the molecular mechanisms involved in inflammation remain unclear. The use of experimental murine models has proven to be an indispensable tool to study the pathogenesis of this disease and efficacy of potential treatments; however, their translational value with respect to the corresponding human pathology has to be carefully evaluated considering the strengths, species-specific differences, and possible critical points during the experimental design. Since rodents do not develop crystal deposition diseases spontaneously, experimental modeling is performed either through transgenic strains that reproduce characteristics observed in patients or by exploiting the inflammatory potential of crystals directly injected into joint tissues, peritoneum, artificial dorsal pouches, or bone marrow of wild-type animals (2–5).
Genetically modified models
So far, it has not yet been possible to create laboratory rodents that spontaneously develop CPP crystal-induced arthritis or in which crystal formation is observed.
Since the presence of extracellular inorganic pyrophosphate (ePPi) in cartilage tissues is critical for CPP crystal formation (6, 7), knockout (KO) mice for enzymes or other proteins involved in PPi metabolism and calcification processes were generated. However, despite KO mice for TNAP (Tissue-nonspecific Alkaline Phosphatase, (Akp2−/−)), Phospho1 (phosphoethanolamine/phosphocholine phosphatase 1, (Phospho1−/−)) and osteopontin (OPN) are characterized by high circulating levels of ePPi, they did not show signs of arthritis or a phenotype compatible with CPPD (8–10). The absence of articular involvement was also confirmed in double knock out Akp2−/-OPN−/− and Akp2−/-Phospho1−/− mice, generated to alter the concerted action of ePPi regulators (10, 11). Furthermore, these knock-out models characterized by high levels of ePPi exhibit high embryonic lethality and shortened lifespan, severe disturbances in bone mineralization, and skeletal abnormalities that did not overlap with the human condition (8, 12).
Several hypotheses may be formulated to explain why CPP crystals are not deposited in mice, which should be considered when using genetically conditioned models. For instance, yet unknown molecular mechanisms underlying the regulation of ePPi level, or the thickness of the articular cartilage layers on average 50-fold thinner than in humans, together with different temperatures and tissutal pH could prevent the crystal formation. Furthermore, the relatively short lifespan of the laboratory mouse may not allow enough time for crystal formation to occur.
Unlike the models described above in which mutations are induced artificially, the progressive ankylosis (ank/ank) mouse is a strain with a spontaneous mutation in the Ank gene, that shows progressive impairment of joint mobility and extremely early arthritis with progressive unrelenting ectopic calcification and vertebral fusion conducing to death within 6 months of age (13). The progressive ankylosis gene (ank) encodes a transmembrane protein that regulates the cellular efflux of ATP. Interestingly, several missense mutations have been described in familial CPPD disease and up-regulation of ANK protein expression was found in articular tissues from patients with CPP deposition (14, 15). In particular, this human condition is associated with increases in ePPi and presumed gain-of-function of ANK (14). However, the ank/ank murine model displays important differences compared to human patients with CPPD. Indeed, mice develop a disorder clinically, radiographically, and histologically more similar to human spondyloarthropathies than to CPPD (16), and histopathological evaluation demonstrated significant deposition of hydroxyapatite (HA) crystals but not CPP in the affected joints. Furthermore, this model is characterized by a loss-of-function mutation in ANK, an increase in intracellular PPi (iPPi) that triggers HA crystal deposition, and a decrease in ePPi (2, 17).
Another naturally occurring mutant mouse model linked to altered mineralization is the “tiptoe walking” (ttw/ttw) mouse. This model shows a defective expression of NPP1, the main enzyme that generates PPi, caused by a nonsense mutation in the corresponding gene (18). The ttw/ttw mice exhibit marked postnatal ectopic ossification, including the development of progressive intervertebral ankylosis, arterial calcification, as well as spontaneous increased bone formation process and calcification of articular cartilage in joints (18, 19). However, although calcium crystal deposition was observed in the joint tissues of these mutant mice, the presence of CPP was not evidenced (19, 20). This may be due to reduced levels of ePPi, which have been determined in ttw/ttw and other NPP1-deficient mice (21).
The most widely used genetically modified mouse models to reproduce PPi metabolism and calcification processes are summarized in Table 1.
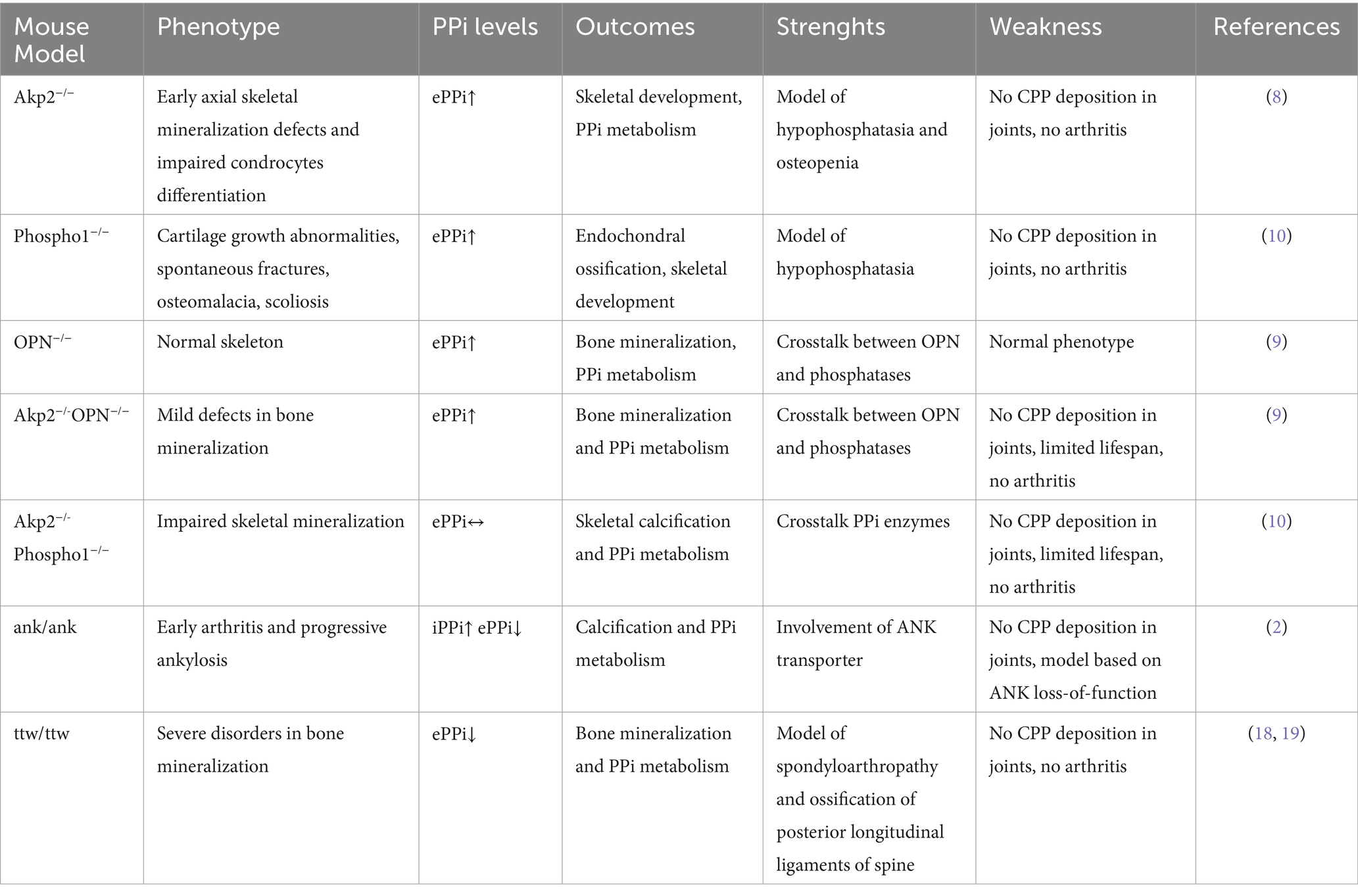
Table 1. Genetically modified mouse models to reproduce PPi metabolism and calcification processes.
Crystal-induced inflammation models
In the absence of spontaneous models, CPP crystal injection can be used to reproduce inflammatory responses similar to those of patients with CPPD. For example, injection of CPP crystals in the ankle causes joint swelling that develops after a few hours, progressively increases, and then declines within a few days (22). Similar conditions are also observed after injection of monosodium urate (MSU) crystals, which are responsible for gout. However, unlike MSU crystal-induced inflammation model in which ankle swelling is maximal at 24 h and completely resolves within 5 days (23), swelling induced by CPP injection peaks after 48 h and persists longer, remaining higher than control even after 6 days (22). Interestingly, this reflects the clinical course in patients. Indeed, untreated gout flares commonly remit spontaneously within a few days, while acute CPP crystal arthritis may persist longer and usually resolve within 1 to 3 weeks (24).
The strength points of this model include: (1) similarity between the human and mouse anatomical joint structure, (2) the acute attack following CPP stimulation induces, in both species, non-specific lesions such as edema, inflammatory infiltrate characterized mainly by neutrophils and macrophages, and (3) in the most severe exacerbations, it is also possible to observe muscle, cartilaginous injury, and synovial hyperplasia (25). Furthermore, swelling of the paw, ankle or knee tissues is extremely rapid and is usually measured using a precision caliber and constitutes a useful parameter for quantifying edema and inflammation. The main limitation of the model is related to the technical difficulty of carrying out the procedure. Crystals are injected into the ankle at the tibio-tarsal joint, but it can be difficult to establish whether it occurred correctly within the joint or in the peri-articular tissues. Of note, despite the importance of the synovial fluid (SF) collection and analysis in clinical diagnosis in human patients (1), these are generally excluded from experimental research designs due to the low amount of SF, which is estimated to be less than 1 μL in mouse knee and ankle joints (26, 27).
The air pouch, crystal-induced peritonitis and pleuritis are experimental models that do not directly involve the joint tissues. In these contexts, the injection of crystals into the cavity triggers a rapid infiltration of neutrophils and macrophages and an increase in the volume of exudate which is also enriched with inflammatory mediators that can be quantified (28–32).
The dorsal air pouch model, described for the first time by Selye, is an easily performed procedure and widely used for the study of several anti-inflammatory and antirheumatic agents because it is able to mimic the synovial environment (30, 33).
However, repeated injection of sterile air into the back of mice may cause microtraumas that trigger an inflammatory response.
CPP crystal-induced inflammation as an experimental model for sterile inflammation
Mice and humans share about 80% of the genetic background and, by comparing genome-wide transcriptional data, Shay et al. demonstrated that both species have essentially the same molecular pathways involved in inflammation and in innate and cellular immune system (34). These findings make experimental models of crystal-induced inflammation a powerful tool for studying sterile inflammation. It is known that crystals, through the activation of inflammasome NLRP3 and TLRs and production of IL-1β, induce massive expression of pro-inflammatory cytokines such as IL-8, CCL2, IL-6, TNF-α as well as other inflammatory mediators (30, 35–39). In this context, several knock-out/knock-in and transgenic mice have been produced for several factors involved in the NRLP3 inflammatory pathway. Generally, knock-out animals are resistant to inflammation induced by crystal injection, making them a useful system for pathophysiological studies rather than for testing new therapies. For example, mice deficient in various key proteins in the inflammasome complex NRLP3 or the IL-1 receptor (IL-1R) are fertile and viable, but did not show inflammatory changes when isolated macrophages were stimulated, or animals were intraperitoneally injected with CPP crystals (38). Interestingly, opposite conditions were observed after injection of BCP crystals, which are frequently found in OA joints. Indeed, i.a. administration of BCP crystals in the knee of NLRP3 and ASC, IL-1β or IL-1α deficient mice showed similar inflammation compared to WT mice (40), thus suggesting that different crystals can trigger distinct inflammatory pathways and careful consideration should be taken when choosing the most appropriate animal model.
TLR-4 is another key factor shared between the two species. Indeed, this is mainly expressed in myeloid cells in both humans and mice and has high sequence homology in the promoter sequence and receptor transcript (41). However, unlike humans, murine TLR-4 is also significantly expressed in skeletal muscles, where CPP can accumulate during experimental procedures; this may explain the reason of persistent edema observed after injection of crystals into the ankle of mice (22, 42).
The use of murine models of crystal-induced inflammation is further corroborated by the fact that mice and humans do not differ in total leukocyte count in peripheral blood. Furthermore, although in healthy mice lymphocytes are the predominant leukocyte making up 70 to 80%, while neutrophils generally comprise 20 to 30% of the white blood cell differential count, in acute inflammatory conditions the lymphocyte/neutrophil ratio is rapidly reversed, thus reproducing the typical pattern of acute inflammation observed in humans (43–45).
These events occur despite the fact that the homolog of human IL-8, the main chemokine regulating neutrophil recruitment, has not been detected in rodents (46). The absence of IL-8 in mice seems compensated for by the ligands CXCL1/KC, which bind the murine CXCR2 receptor and attract immune cells to inflamed tissues (30, 47).
Conclusion
In conclusion, animal models are needed to understand the pathophysiology of CPPD, but their development is still at an early stage. In this context, some general critical issues have to be considered in the creation of animal models that may interfere with their translational value. For example, in vivo experiments to study CPPD involve the use of healthy and pathogen-free animals of inbred strains with many homozygous mutations, whereas the human population is highly heterogeneous in terms of genetic background and individual clinical history. Furthermore, there are often significant age- and sex-related biases in exclusively using young and male mice that scarcely overlap with the epidemiology of CPPD. It usually affects elderly subjects and is slightly more common in women than in men.
Currently available genetically conditioned models contribute to understanding of some aspects of disease but are far to mimic the human CPPD pathology.
CPP crystal-induced inflammation models reproduce some important aspects of the disorder and provide key information on the molecular pathophysiology of human disease (Figure 1). They are relatively simple, fast to realize, and useful for testing new therapeutical approaches. However, they only reproduce what the crystals cause but not their formation, thus not providing useful information on the mechanisms of the early stage of the disease. Furthermore, no experimental studies have been conducted to characterize the specific differences in the various clinical manifestations associated with CPP deposition. Finally, none of the models used to date consider the frequent presence of osteoarthritis or associated conditions such as hyperparathyroidism, hemochromatosis, or hypomagnesaemia observed in CPPD patients. Therefore, one of future efforts should be to develop animal models that reproduce the impact of these concomitant diseases to investigate their role in CPPD.
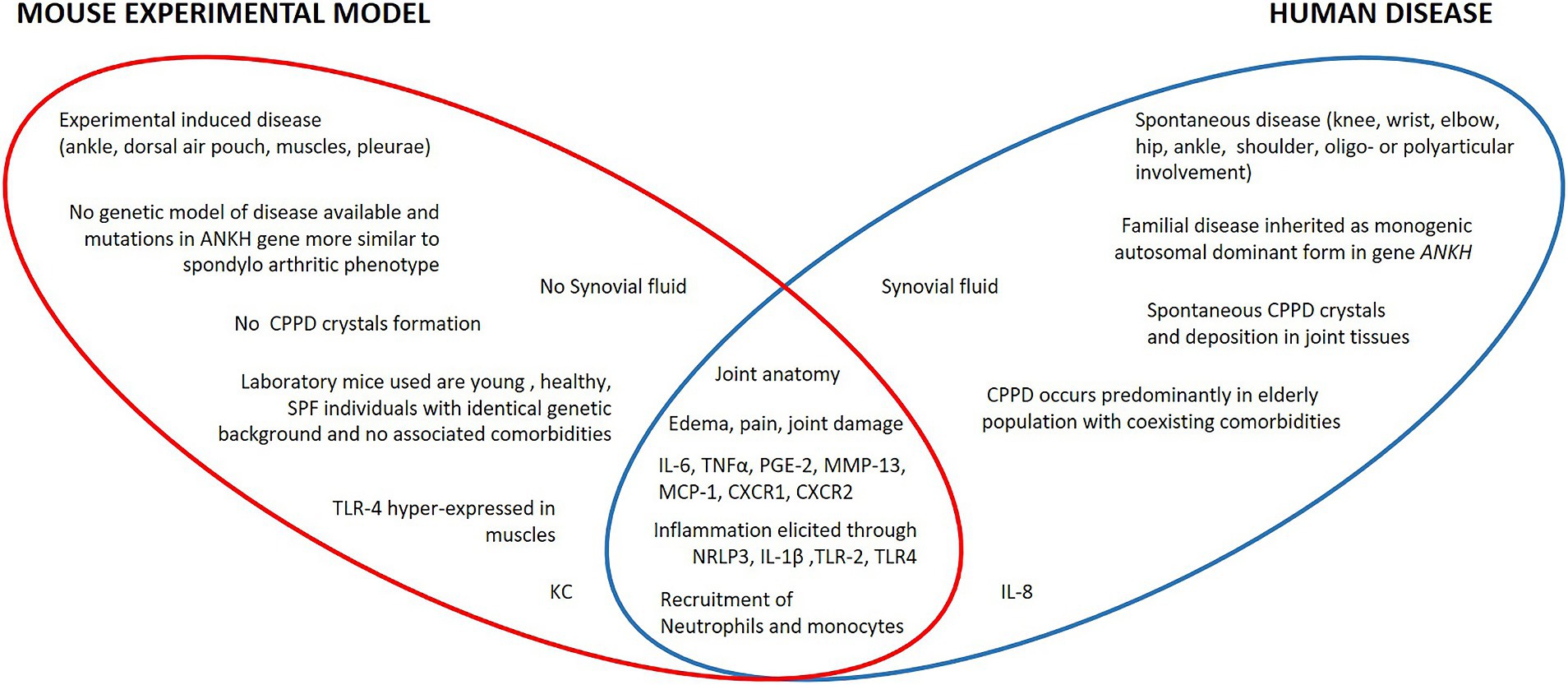
Figure 1. Specific and shared characteristics of human CPPD and corresponding mouse models. In mice, no spontaneous CPPD and CPP crystals formation has ever been observed. In humans, inflammation occurs at the joint level; in mice, induced inflammation is carried out in the ankle or in extra-articular anatomical districts such as the dorsal air pouch or in the intraperitoneal visceral compartment that has no homologies with the human pathology. Furthermore, laboratory rodent strains differ significantly for clinical conditions from the human population affected by CPPD. Apart from some species-specific molecular mediators such as IL-8 or KC and differences in the basal composition of leukocytes, humans and mice share the same molecular and histopathological mechanisms of CPP-induced inflammation.
Author contributions
RL: Conceptualization, Writing – original draft. AS: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Open Access funding provided by Università degli Studi di Padova (University of Padua), Open Science Committee.
Acknowledgments
The authors thank Giovanna Luisetto for the linguistic revision of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zhang, W, Doherty, M, Bardin, T, Barskova, V, Guerne, PA, Jansen, TL, et al. European league against rheumatism recommendations for calcium pyrophosphate deposition. Part I: terminology and diagnosis. Ann Rheum Dis. (2011) 70:563–70. doi: 10.1136/ard.2010.139105
2. Ho, AM, Johnson, MD, and Kingsley, DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. (2000) 289:265–70. doi: 10.1126/science.289.5477.265
3. Onai, N, and Ogasawara, C. Calcium pyrophosphate dihydrate crystals increase the granulocyte/monocyte progenitor (GMP) and enhance granulocyte and monocyte differentiation in vivo. Int J Mol Sci. (2020) 22:262. doi: 10.3390/ijms22010262
4. Reginato, AM, and Olsen, BR. Genetics and experimental models of crystal-induced arthritis. Lessons learned from mice and men: is it crystal clear? Curr Opin Rheumatol. (2007) 19:134–45. doi: 10.1097/BOR.0b013e328040c00b
5. Zaka, R, and Williams, CJ. Genetics of chondrocalcinosis. Osteoarthr Cartil. (2005) 13:745–50. doi: 10.1016/j.joca.2005.04.006
6. Abhishek, A, and Doherty, M. Pathophysiology of articular chondrocalcinosis—role of ANKH. Nat Rev Rheumatol. (2011) 7:96–104. doi: 10.1038/nrrheum.2010.182
7. Williams, CJ, and Rosenthal, AK. Pathogenesis of calcium pyrophosphate deposition disease. Best Pract Res Clin Rheumatol. (2021) 35:101718. doi: 10.1016/j.berh.2021.101718
8. Fedde, KN, Blair, L, Silverstein, J, Coburn, SP, Ryan, LM, Weinstein, RS, et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. (1999) 14:2015–26. doi: 10.1359/jbmr.1999.14.12.2015
9. Harmey, D, Johnson, KA, Zelken, J, Camacho, NP, Hoylaerts, MF, Noda, M, et al. Elevated skeletal osteopontin levels contribute to the hypophosphatasia phenotype in Akp2(−/−) mice. J Bone Miner Res. (2006) 21:1377–86. doi: 10.1359/jbmr.060619
10. Yadav, MC, Simão, AM, Narisawa, S, Huesa, C, McKee, MD, Farquharson, C, et al. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification. J Bone Miner Res. (2011) 26:286–97. doi: 10.1002/jbmr.195
11. Addison, WN, Azari, F, Sørensen, ES, Kaartinen, MT, and McKee, MD. Pyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, up-regulating osteopontin, and inhibiting alkaline phosphatase activity. J Biol Chem. (2007) 282:15872–83. doi: 10.1074/jbc.M701116200
12. Zhou, X, Cui, Y, Zhou, X, and Han, J. Phosphate/pyrophosphate and MV-related proteins in mineralisation: discoveries from mouse models. Int J Biol Sci. (2012) 8:778–90. doi: 10.7150/ijbs.4538
13. Sweet, HO, and Green, MC. Progressive ankylosis, a new skeletal mutation in the mouse. J Hered. (1981) 72:87–93. doi: 10.1093/oxfordjournals.jhered.a109459
14. Mitton-Fitzgerald, E, Gohr, CM, Bettendorf, B, and Rosenthal, AK. The role of ANK in calcium pyrophosphate deposition disease. Curr Rheumatol Rep. (2016) 18:25. doi: 10.1007/s11926-016-0574-z
15. Uzuki, M, Sawai, T, Ryan, LM, Rosenthal, AK, and Masuda, I. Upregulation of ANK protein expression in joint tissue in calcium pyrophosphate dihydrate crystal deposition disease. J Rheumatol. (2014) 41:65–74. doi: 10.3899/jrheum.111476
16. Krug, HE, Mahowald, ML, and Clark, C. Progressive ankylosis (ank/ank) in mice: an animal model of spondyloarthropathy. III. Proliferative spleen cell response to T cell mitogens. Clin Exp Immunol. (1989) 78:97–101.
17. Masuda, I, and Hirose, J. Animal models of pathologic calcification. Curr Opin Rheumatol. (2002) 14:287–91. doi: 10.1097/00002281-200205000-00016
18. Okawa, A, Nakamura, I, Goto, S, Moriya, H, Nakamura, Y, and Ikegawa, S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet. (1998) 19:271–3. doi: 10.1038/956
19. Bertrand, J, Nitschke, Y, Fuerst, M, Hermann, S, Schäfers, M, Sherwood, J, et al. Decreased levels of nucleotide pyrophosphatase phosphodiesterase 1 are associated with cartilage calcification in osteoarthritis and trigger osteoarthritic changes in mice. Ann Rheum Dis. (2012) 71:1249–53. doi: 10.1136/annrheumdis-2011-200892
20. Sakamoto, M, Hosoda, Y, Kojimahara, K, Yamazaki, T, and Yoshimura, Y. Arthritis and ankylosis in twy mice with hereditary multiple osteochondral lesions: with special reference to calcium deposition. Pathol Int. (1994) 44:420–7. doi: 10.1111/j.1440-1827.1994.tb01705.x
21. Nitschke, Y, Yan, Y, Buers, I, Kintziger, K, Askew, K, and Rutsch, F. ENPP1-fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp Mol Med. (2018) 50:1–12. doi: 10.1038/s12276-018-0163-5
22. Oliviero, F, Galozzi, P, Scanu, A, Galuppini, F, Lazzarin, V, Brocco, S, et al. Polydatin prevents calcium pyrophosphate crystal-induced arthritis in mice. Nutrients. (2021) 13:929. doi: 10.3390/nu13030929
23. Reber, LL, Marichal, T, Sokolove, J, Starkl, P, Gaudenzio, N, Iwakura, Y, et al. Contribution of mast cell-derived interleukin-1β to uric acid crystal-induced acute arthritis in mice. Arthritis Rheumatol. (2014) 66:2881–91. doi: 10.1002/art.38747
24. Abhishek, A, and Doherty, M. Update on calcium pyrophosphate deposition. Clin Exp Rheumatol. (2016) 34:32–8.
25. Rosenthal, AK, and Ryan, LM. Calcium pyrophosphate deposition disease. N Engl J Med. (2016) 374:2575–84. doi: 10.1056/NEJMra1511117
26. Long, PH, and Leininger, JR. Bones, joints, and synovia In: R Maronpot, G Boorman, and BW Gaul, editors. Pathology of the mouse. St Louis: Cache River Press (1999)
27. Seifer, DR, Furman, BD, Guilak, F, Olson, SA, Brooks, SC 3rd, and Kraus, VB. Novel synovial fluid recovery method allows for quantification of a marker of arthritis in mice. Osteoarthr Cartil. (2008) 16:1532–8. doi: 10.1016/j.joca.2008.04.013
28. Campillo-Gimenez, L, Renaudin, F, Jalabert, M, Gras, P, Gosset, M, Rey, C, et al. Inflammatory potential of four different phases of calcium pyrophosphate relies on NF-κB activation and MAPK pathways. Front Immunol. (2018) 9:2248. doi: 10.3389/fimmu.2018.02248
29. Moore, AR. Pleural models of inflammation: immune and nonimmune. Methods Mol Biol. (2003) 225:123–8. doi: 10.1385/1-59259-374-7:123
30. Scanu, A, Luisetto, R, Oliviero, F, Gruaz, L, Sfriso, P, Burger, D, et al. High-density lipoproteins inhibit urate crystal-induced inflammation in mice. Ann Rheum Dis. (2015) 74:587–94. doi: 10.1136/annrheumdis-2013-203803
31. Scanu, A, Luisetto, R, Oliviero, F, Galuppini, F, Lazzarin, V, Pennelli, G, et al. Bactericidal/permeability-increasing protein downregulates the inflammatory response in in vivo models of arthritis. Int J Mol Sci. (2022) 23:13066. doi: 10.3390/ijms232113066
32. Willoughby, DA, Dunn, CJ, Yamamoto, S, Capasso, F, Deporter, DA, and Giroud, JP. Calcium pyrophosphate-induced pleurisy in rats: a new model of acute inflammation. Agents Actions. (1975) 5:35–8. doi: 10.1007/BF02027156
33. Selye, H. On the mechanism through which hydrocortisone affects the resistance of tissues to injury; an experimental study with the granuloma pouch technique. JAMA J Am Med Assoc. (1953) 152:1207–13. doi: 10.1001/jama.1953.63690130001006
34. Shay, T, Jojic, V, Zuk, O, Rothamel, K, Puyraimond-Zemmour, D, Feng, T, et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc Natl Acad Sci USA. (2013) 110:2946–51. doi: 10.1073/pnas.1222738110
35. Joosten, LA, Netea, MG, Mylona, E, Koenders, MI, Malireddi, RK, Oosting, M, et al. Engagement of fatty acids with toll-like receptor 2 drives interleukin-1β production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum. (2010) 62:3237–48. doi: 10.1002/art.27667
36. Liu-Bryan, R, Scott, P, Sydlaske, A, Rose, DM, and Terkeltaub, R. Innate immunity conferred by toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. (2005) 52:2936–46. doi: 10.1002/art.21238
37. Martin, WJ, Walton, M, and Harper, J. Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum. (2009) 60:281–9. doi: 10.1002/art.24185
38. Martinon, F, Pétrilli, V, Mayor, A, Tardivel, A, and Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. (2006) 440:237–41. doi: 10.1038/nature04516
39. Punzi, L, Scanu, A, Spinella, P, Galozzi, P, and Oliviero, F. Gout: one year in review 2018. Clin Exp Rheumatol. (2019) 37:1–11. doi: 10.55563/clinexprheumatol/uhyzcr
40. Ea, HK, Chobaz, V, Nguyen, C, Nasi, S, van Lent, P, Daudon, M, et al. Pathogenic role of basic calcium phosphate crystals in destructive arthropathies. PLoS One. (2013) 8:e57352. doi: 10.1371/journal.pone.0057352
41. Rehli, M. Of mice and men: species variations of toll-like receptor expression. Trends Immunol. (2002) 23:375–8. doi: 10.1016/s1471-4906(02)02259-7
42. Qureshi, ST, Larivière, L, Leveque, G, Clermont, S, Moore, KJ, Gros, P, et al. Endotoxin-tolerant mice have mutations in toll-like receptor 4 (Tlr4). J Exp Med. (1999) 189:615–25. doi: 10.1084/jem.189.4.615
43. Hackert, NS, Radtke, FA, Exner, T, Lorenz, HM, Müller-Tidow, C, Nigrovic, PA, et al. Human and mouse neutrophils share core transcriptional programs in both homeostatic and inflamed contexts. Nat Commun. (2023) 14:8133. doi: 10.1038/s41467-023-43573-9
44. McGarry, MP, Protheroe, CA, and Lee, JJ. Mouse hematology: a laboratory manual. New York: Cold Spring Harbor Laboratory Press (2010).
45. O'Connell, KE, Mikkola, AM, Stepanek, AM, Vernet, A, Hall, CD, Sun, CC, et al. Practical murine hematopathology: a comparative review and implications for research. Comp Med. (2015) 65:96–113.
46. Modi, WS, and Yoshimura, T. Isolation of novel GRO genes and a phylogenetic analysis of the CXC chemokine subfamily in mammals. Mol Biol Evol. (1999) 16:180–93. doi: 10.1093/oxfordjournals.molbev.a026101
47. Rovai, LE, Herschman, HR, and Smith, JB. The murine neutrophil-chemoattractant chemokines LIX, KC, and MIP-2 have distinct induction kinetics, tissue distributions, and tissue-specific sensitivities to glucocorticoid regulation in endotoxemia. J Leukoc Biol. (1998) 64:494–502. doi: 10.1002/jlb.64.4.494
Keywords: calcium pyrophosphate, pyrophosphate, arthritis, mouse models, translational models, inflammation, CPPD
Citation: Luisetto R and Scanu A (2024) The translational value of calcium pyrophosphate deposition disease experimental mouse models. Front. Med. 11:1417318. doi: 10.3389/fmed.2024.1417318
Edited by:
Georgios Filippou, IRCCS Istituto Ortopedico Galeazzi, ItalyReviewed by:
Sonia Nasi, Centre Hospitalier Universitaire Vaudois (CHUV), SwitzerlandAnn Rosenthal, Medical College of Wisconsin, United States
Copyright © 2024 Luisetto and Scanu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Scanu, YW5uYS5zY2FudUB1bmlwZC5pdA==
†These authors have contributed equally to this work and share first authorship