 Yang Ma1†
Yang Ma1† Xiaoming Fan
Xiaoming Fan- 1Department of Human Anatomy, School of Basic Medicine, Guilin Medical University, Guilin, China
- 2School of Public Health, Guilin Medical University, Guilin, China
The incidence of metabolic dysfunction-associated steatotic liver disease (MASLD) is continuously rising, evolving into a global health challenge. Concurrently, cases of hepatocellular carcinoma (HCC) associated with MASLD are also on the increase. Although traditional risk factors such as age, gender, and metabolic factors play significant roles in the development of HCC, it cannot be overlooked that MASLD, triggered by changes in modern lifestyle and dietary habits, may also exacerbate the risk of HCC, and this phenomenon is common even among non-obese individuals. Regrettably, MASLD often fails to receive timely diagnosis, resulting in a limited number of patients receiving HCC surveillance. Moreover, there is currently a lack of clear definition for the target population for surveillance beyond patients with cirrhosis. Consequently, MASLD-related HCC is often detected at a late stage, precluding the optimal timing for curative treatment. However, our understanding of the pathogenesis and progression of HCC remains limited. Therefore, this paper reviews relevant literature from recent years, delving into multiple dimensions such as pathogenesis, surveillance and diagnosis, prevention, and treatment, aiming to provide new ideas and directions for the prevention and treatment of MASLD-related HCC.
1 Introduction
Primary hepatic carcinoma ranks as the sixth most common cancer and the third leading cause of cancer deaths globally. Hepatocellular carcinoma (HCC) accounts for 75%−85% of these cases. In 2020, ~900,000 new cases of primary liver cancer were reported worldwide, leading to 830,000 deaths, indicating a close proximity between the incidence and mortality rates (1). Multiple risk factors contribute to HCC, including viral hepatitis, aflatoxin exposure, exposure to certain chemicals, and metabolic dysfunction-associated steatotic liver disease (MASLD) (2). In the past, viral hepatitis was the primary risk factor for HCC. However, with improvements in lifestyle and living conditions, the prevalence of MASLD has been increasing, posing a significant global public health challenge (3). Concurrently, the incidence of MASLD-related HCC is also on the rise. Although the incidence of MASLD-related HCC is lower compared to other causes such as hepatitis B-related HCC, the prevalence of MASLD surpasses that of other liver diseases. Furthermore, MASLD-related HCC is often diagnosed at an advanced stage, making curative treatment unfeasible (4). Therefore, there is an urgent need to raise global awareness and conduct thorough research on the underlying mechanisms to mitigate the impending burden of MASLD-related HCC.
2 Epidemiological investigation and association analysis of MASLD and HCC
The worldwide occurrence and frequency of MASLD are rapidly on the rise, with a global estimation of ~25% for its prevalence (5). Notably, the majority of metabolic dysfunction-associated steatohepatitis (MASH) cases are expected to increase by 56% between 2016 and 2030 in China, France, Germany, Italy, Japan, Spain, the United Kingdom, and the U.S. (6). MASLD is characterized as a clinicopathological syndrome marked by hepatic steatosis, associated with factors such as insulin resistance (IR) (7), autophagy (8), and gut microbiota (9), and obesity (10). These factors collectively contribute to chronic inflammation, altered lipid metabolism, and ultimately, the promotion of HCC development. Given that primary liver cancer ranks as the sixth most common cancer and the third leading cause of cancer death worldwide (11), these findings highlight the substantial risk of liver cancer. Studies suggest that the incidence of MASLD-related HCC may increase alongside modern living standards and lifestyle changes. For instance, 35% of the U.S. population was obese in 2012, with projections estimating that 48.9% will be obese by 2030 (12). This obesity epidemic is likely to parallel an increase in the incidence of MASLD-related HCC. A similar crisis is unfolding in China, where the prevalence of MASLD is expected to rise in tandem with the obesity and diabetes epidemics. Although most patients with MASLD present with simple steatosis without advanced liver disease, a subset of these patients may progress to MASH, leading to cirrhosis and HCC. From 2016 to 2030, an 82% increase in HCC incidence is projected for China (6). Additionally, a significant proportion of MASLD patients without cirrhosis may develop HCC (13), underscoring the importance of further examining the link between cirrhotic MASLD, non-cirrhotic MASLD, and HCC in our discussion.
2.1 The association between MASLD and HCC in cirrhosis
It is now widely recognized that cirrhosis serves as a critical risk factor for the emergence of HCC in MASLD patients, with an annual incidence rate of 10.6 per 1,000 person-years among those with cirrhosis caused by MASH (14). The primary cause of MASLD is overnutrition, resulting in the enlargement of fat deposits and ectopic fat accumulation (15). In this scenario, the infiltration of macrophages into visceral adipose tissue compartments gives rise to a proinflammatory state, promotes insulin resistance, and imbalances in lipid metabolism, leading to the formation of lipotoxic lipids. These lipids contribute to cellular stress, including oxidative stress and endoplasmic reticulum stress, and trigger inflammasome activation, apoptotic cell death, inflammatory stimulation, tissue regeneration, and fibrosis, ultimately leading to cirrhosis (16). Notably, the large vesicular steatosis observed in hepatocytes disappears in advanced cirrhosis, and the underlying mechanism for this fat disappearance remains under investigation (17). A prevailing hypothesis suggests that the physiological characteristics of cirrhosis may either directly or indirectly affect the ability of hepatocytes to uptake triglycerides, resulting in abnormalities in the flow of fat-forming molecules to the subendothelial hepatocytes, thus impacting hepatocyte permeability (18). Nevertheless, the precise mechanisms linking MASLD and HCC in the context of cirrhosis remain elusive and require further intensive investigation.
2.2 The association between non-cirrhotic MASLD and HCC
In contrast to hepatitis virus-driven HCC, a significant proportion of patients with MASH develop HCC in the absence of cirrhosis (19, 20). The exact mechanisms underlying this phenomenon remain partly unclear but are thought to be related to pathophysiological changes associated with lipotoxicity, bile acid (B.A.) signaling, and inflammation (21). It has been shown that serum B.A. concentrations are elevated in patients with advanced MASH, and B.A. accumulation can induce parenchymal injury (22). This, in turn, promotes the progression of MASLD to HCC. Among patients without cirrhosis who progress to HCC, the highest risk is observed in males over 65 years old with a history of smoking, type 2 diabetes, and elevated levels of alanine aminotransferase (ALT) (23). Furthermore, heightened levels of ALT and indicators of liver inflammation have been independently correlated with an increased risk of HCC [hazard ratio (HR) 6.80, 95% CI: 3.00–15.42; p < 0.001] among individuals with non-cirrhotic MASLD. This increased risk is attributed to the influence of a proliferative environment and inflammation on tumor development (24). This highlights the critical role of metabolic and inflammatory pathways in the pathogenesis of HCC in patients without cirrhosis. However, the exploration of other potential mechanisms remains an essential area for further research.
3 The mechanisms underlying the advancement of MASLD to HCC
The progression of MASLD to HCC is facilitated by a range of mechanisms, as illustrated in Figure 1.
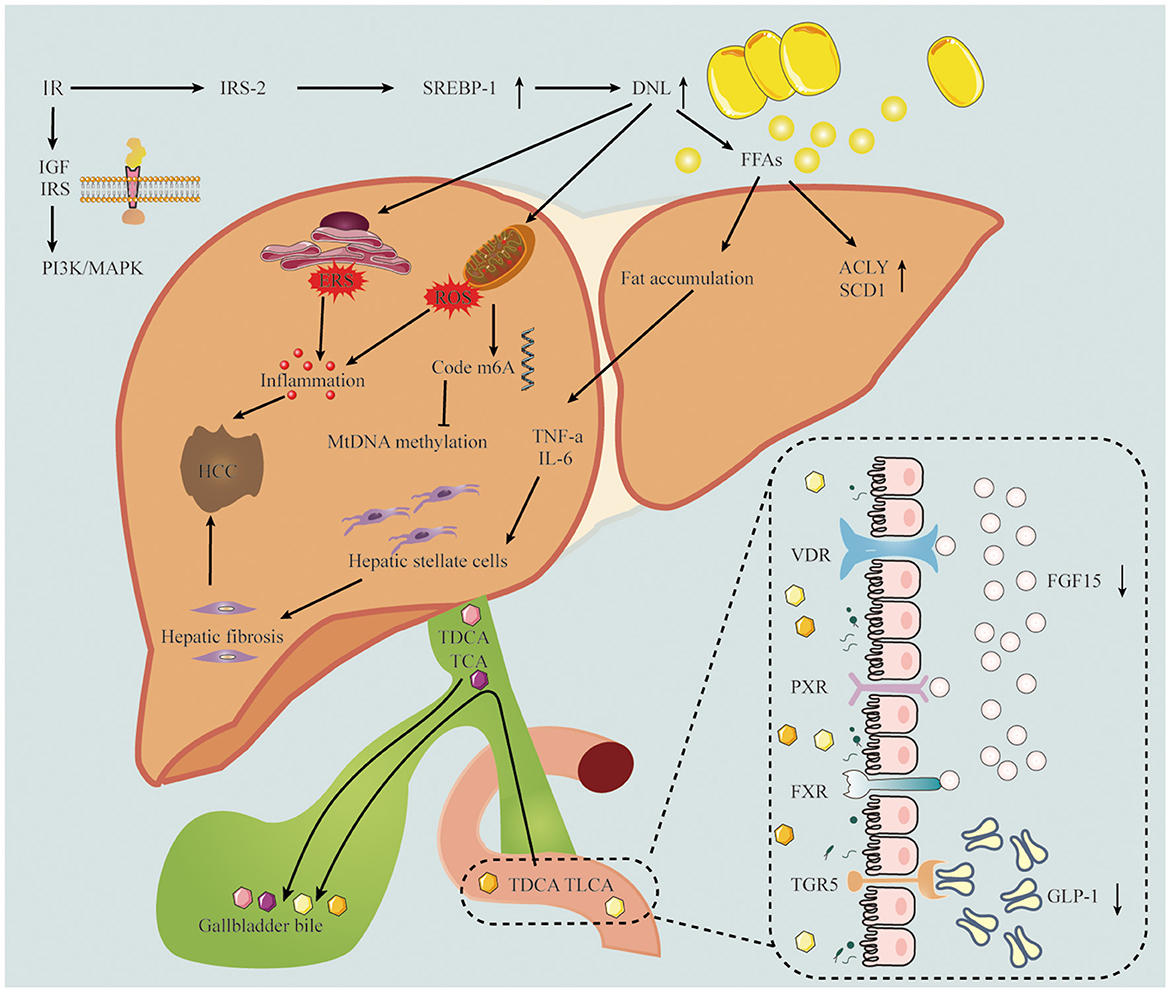
Figure 1. The pathogenesis of MASLD-related HCC includes insulin resistance, inflammation, oxidative stress and gut microbiota. The down regulation of IRS-2 leads to the over expression of SREBP-1 and the up-regulation of DNL, which leads to inflammatory response and oxidative stress. TDCA and TLCA in intestinal epithelial cells interact with bile acid receptors, resulting in FGF-15 and GLP-1 decline. IRS-2, insulin receptor substrate 2; SREBR-1, sterol regulatory element-binding protein 1; DNL, de novo lipogenesis; FFAs, free fatty acids; IGF, insulin-like growth factor; ACLY, ATP citrate lyase; SCD1, stearoyl coenzyme A desaturase-1; TNF-a, tumor necrosis factor alpha; IL-6, Interleukin 6; TDCA, taurodeoxycholic acid; TLCA, taurolithocholic acid; TCA, taurocholic acid; VDR, vitamin D receptor; PXR, pregnane X receptor; FXR, Farnesoid X nuclear receptor; TGR5, transmembrane G protein-coupled receptor 5; FGF15, fibroblast growth factor 15; GLP-1, glucagon-like peptide-1.
3.1 Insulin resistance
The initiation of MASLD/MASH is marked by the accumulation of fat in the liver, with IR being the pivotal pathogenic event leading to hepatic steatosis. Insulin typically promotes anabolic processes in visceral adipose tissue; however, adipocyte de novo lipogenesis is diminished during insulin resistance (25). Furthermore, the expression of carbohydrate response element binding protein beta (ChREBP beta), which positively regulates de novo lipogenesis, is notably low in the visceral adipose tissue of individuals with obesity or those with both obesity and type 2 diabetes. Moreover, the level of ChREBP beta in visceral adipose tissue is inversely associated with the severity of hepatic steatosis in these individuals (26). In the context of IR, the downregulation of insulin receptor substrate 2 (IRS-2) contributes to the overexpression of sterol regulatory element-binding protein 1 (SREBP-1) (27). This condition leads to an upregulation of de novo lipogenesis (DNL), crucial for promoting lipid storage in the liver. Moreover, β-oxidation of FFAs is inhibited in states of IR, further exacerbating hepatic lipid accumulation (28). Insulin and insulin-like growth factor-1 (IGF-1) are growth factors known to inhibit cell apoptosis. The synthesis and bioactivity of IGF-1 increase in the presence of IR, and elevated levels of IGF-1 stimulate cell proliferation while inhibiting apoptosis. This effect significantly elevates the risk of hepatocellular carcinogenesis (29). Insulin receptor substrate (IRS) molecules, upon binding to insulin or IGF, activate the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) pathway and the mitogen-activated protein kinase (MAPK) pathway, promoting hepatocarcinogenesis (30). This intricate interplay of metabolic pathways underscores the complex relationship between insulin resistance and the progression of MASLD/MASH to HCC.
3.2 Inflammatory response
The inflammatory response is a critical hallmark of MASLD, with inflammation being driven by multiple intra- and extrahepatic factors (31, 32). A high-calorie diet, obesity, certain lifestyles, and genetic predispositions all contribute to the risk of developing MASLD. Hepatocyte overload and an increase in de novo lipogenesis lead to excessive lipid accumulation within hepatocytes. This lipid overload induces lipotoxicity, resulting in endoplasmic reticulum (E.R.) stress, oxidative stress, reactive oxygen species (ROS) production, and mitochondrial damage. In response to stress, hepatocytes release pro-inflammatory mediators and damage-associated molecular patterns (DAMPs), leading to the activation and infiltration of immune cells, which further exacerbates hepatocyte damage. Various forms of cell death, along with hepatocyte senescence, trigger a more pronounced immune response (33). Bile duct cells may also contribute to the inflammatory milieu by releasing inflammatory mediators. Moreover, hepatic inflammation extends to several extrahepatic systems, including adipose tissue, the intestine, skeletal muscle, and bone marrow (34). Necroinflammation, hepatocyte death, and oxidative stress significantly increase the risk of hepatocarcinogenesis. For instance, oxidative stress in hepatocytes activates inflammatory pathways, such as those mediated by STAT1 and STAT3, which are known pro-inflammatory transcription factors. Elevated levels of STAT1 are implicated in inducing MASH-related inflammation, while high levels of STAT3 independently drive the development of HCC in mice. This illustrates the intricate connection between inflammation and carcinogenesis in the progression from MASH to HCC (35). Such findings highlight the complex interplay between metabolic dysregulation and immune responses in the pathogenesis of MASLD and its progression to HCC.
3.3 Oxidative stress
Oxidative stress (O.S.) is defined as a condition where the production of reactive oxygen species (ROS) by the body surpasses the detoxification ability of antioxidants. In simpler terms, it represents a pathological state characterized by an imbalance in the body's antioxidant system, leading to increased oxidation (36). Oxidative stress stands as a pivotal hallmark of MASLD. The free radicals responsible for inducing O.S. play a crucial role in the advancement of MASLD, primarily through disrupting the process of lipid peroxidation via fatty acid mitochondrial oxidation, alongside instigating the release of cytokines, which subsequently ignite inflammation (37). Consequently, O.S. inflicts damage upon hepatocytes. The lipid peroxidation process initiates when free radical ROS abstract hydrogen atoms from unsaturated fatty acids, sparking a deleterious chain reaction. This chain reaction not only leads to the rupture of cell membranes but also results in the generation of reactive metabolites capable of causing cellular dysfunction. Furthermore, lipid peroxidation and its ensuing products stimulate hepatic stellate cells and promote the upregulation of pro-inflammatory cytokines (38). Hepatic iron overload is commonly observed in patients with MASLD. Due to the Fenton reaction catalyzed by iron, highly reactive hydroxyl radicals are generated from hydrogen peroxide (39, 40). Excessive hepatic iron can exacerbate oxidative stress and inflammation, and elevated iron levels can lead to increased ROS production. The occurrence of ferroptosis largely depends on the interconnected signals among subcellular organelles such as mitochondria, lysosomes, and peroxisomes. These organelles coordinate the regulation of ROS production and lipid oxidation during ferroptosis (41). Additionally, these processes have the potential to trigger cellular pathways leading to necrosis and apoptosis (42), which could culminate in liver fibrosis and, ultimately, HCC. This intricate cascade underscores the significant role oxidative stress plays in the progression from MASLD to more severe hepatic conditions.
3.4 Gut microbiota
The transport of blood from the gut to the liver, despite an efficient multi-layered intestinal barrier, is accompanied by metabolites and products of the intestinal microbiota, a process often referred to as microbiota-associated molecular patterns (43). In the liver, Kupffer cells serve as an effective “bacterial firewall,” averting bacterial infection while maintaining low levels of inflammation under physiological conditions (44). Additionally, the expression of specific bile acid synthases is influenced by the gut microbiota. Among MASLD's crucial bile acid receptors are the Farnesoid X nuclear receptor (FXR) and the transmembrane G protein-coupled receptor 5 (TGR5) (45). FXR has the capability to bind to ligands and form dimers with the retinoid X receptor, thereby regulating the transcription of target genes (46). These target genes encompass a variety of functions, including bile acid metabolism/transport, lipid metabolism, and glucose metabolism (47). Emerging studies have shown that gut flora may precipitate MASLD by modulating bile acid metabolism, thereby playing a role in the progression from MASLD to HCC (48–50). Increased intestinal permeability is a characteristic of cirrhosis, which subjects the liver to a substantial load of bacteria and bacterial components. Furthermore, within the gut microbiota-bile acid axis, secondary bile acids can enhance Toll-like receptor 2 (TLR2) expression on hepatic stellate cells and elevate levels of the TLR2 agonist lysophosphatidic acid. This, in turn, promotes the secretion of senescence-associated secretory phenotype (SASP) factors, thereby accelerating tumor progression (51). Additionally, the metabolism of bile acids by gut microbiota can influence liver tumor growth by modulating the hepatic expression of CXCL16, which is instrumental in recruiting natural killer T (NKT) cells (52). This intricate interplay between gut microbiota and liver pathology underscores the complex mechanisms by which gut-liver axis dysregulation contributes to the pathogenesis and progression of liver diseases, including MASLD and HCC.
3.5 Genetics and epigenetics
Polygenic risk scores based on genetic background are becoming fundamental for monitoring MASLD and assessing clinical risk, particularly for HCC. The PNPLA3, TM6SF2, and MBOAT7 genes play crucial roles in the development and progression of MASLD. These genes affect lipid droplet accumulation, mitochondrial functionality, and metabolic reprogramming, leading to HCC (53). Epigenetics involves alterations in gene expression levels due to changes that do not affect the DNA sequence itself, primarily through the regulation of gene transcription or translation processes, thereby influencing their function and properties. The metabolic ecology of tumors is intricate, involving reprogramming of metabolism to re-establish the tumor microenvironment conducive for survival and proliferation. This reprogramming encompasses DNA methylation, histone acetylation, and N6-methyladenosine (m6A) modifications. Among these, epigenetic alterations, such as mitochondrial DNA (mtDNA) methylation, may occur during the development of MASLD. Mitochondria serve as both a major source and target of reactive oxygen species (ROS), with the mitochondrially encoded NADH dehydrogenase six genes being a site of mitochondrial methylation in MASLD (54). m6A is recognized as the most prevalent internal RNA modification in eukaryotes, playing a pivotal role in lipid metabolism, hepatocyte inflammation, and the progression of MASLD to liver tumorigenesis and metastasis. Excessive m6A modification can lead to increased expression of citrate lyase (ACLY) and stearoyl-CoA desaturase (SCD1). Furthermore, in vitro targeting of METTL3/14 has been shown to elevate the protein levels of ACLY and SCD1, resulting in increased production of triglycerides and cholesterol and accumulation of lipid droplets (55). A comparative study identified that 44 miRNAs were differentially expressed in the liver of MASLD patients vs. a healthy liver. Notably, research in MASH consistently observed a downregulation of liver-specific miR-122 and an upregulation of miR-34a (56). These changes in miRNA-targeted genes play crucial roles in hepatic energy metabolism, inflammation, cell regeneration, and fibrotic signaling, driving the progression from MASLD to hepatic fibrosis and subsequently to HCC (57). This highlights the significant impact of epigenetic mechanisms in the pathogenesis and progression of liver diseases, underlining the importance of understanding these processes for developing targeted therapies. The pathogenesis of MASLD-related HCC were showed in Table 1.
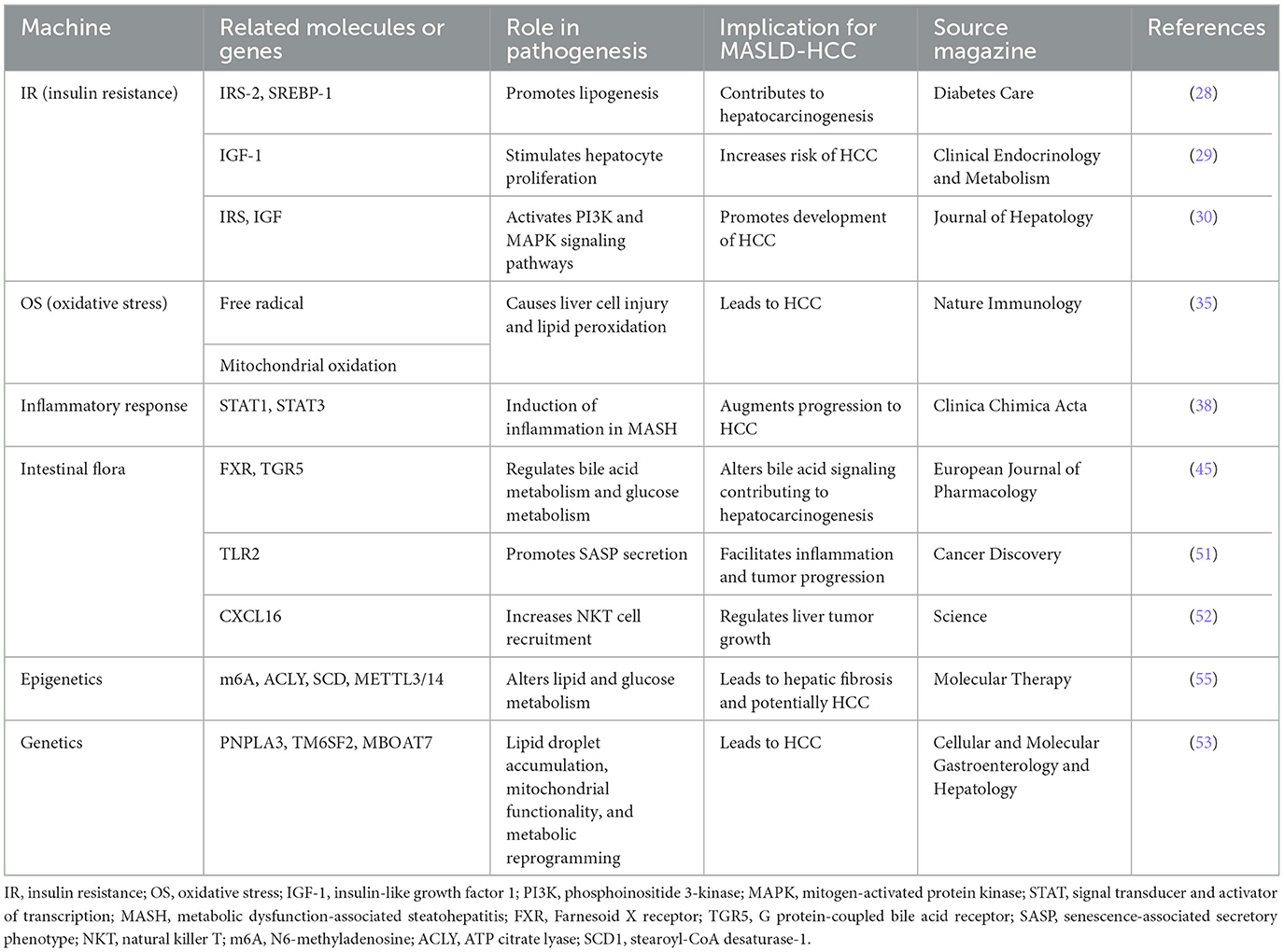
Table 1. The pathogenesis of MASLD-related HCC.
4 Monitoring the evolution and diagnosing MASLD-HCC
MASLD represents a potentially severe liver disease, leading to significant healthcare costs, economic losses, and diminished health-related quality of life at the societal level (58). MASH, a severe form of MASLD, is a leading cause of progression to cirrhosis and HCC, becoming an increasingly common indication for liver transplantation (59). Liver cancer ranks as the second leading cause of life expectancy loss among all cancers worldwide (60), underscoring the critical need for effective progression monitoring of MASLD to HCC. However, the surveillance of MASLD-HCC progression presents challenges. Firstly, the incidence of MASLD-HCC is lower than that of viral HCC, which impacts the cost-effectiveness of screening programs (14). Secondly, MASLD is typically asymptomatic in its early stages, with clinical symptoms becoming apparent only as the condition progresses to more severe stages, such as cirrhosis or HCC. Thirdly, the presence of subcutaneous fat and hepatic steatosis in obese patients can impede the effectiveness of ultrasound imaging (61). Loomba et al. (62), after reviewing the available literature, recommended monitoring patients with advanced liver fibrosis and cirrhosis associated with MASLD but advised against monitoring MASLD patients without signs of advanced liver fibrosis. In scenarios where ultrasound conditions are challenging, CT or MRI scans are recommended as alternative imaging modalities. Specifically, non-enhanced MRI is favored over ultrasound for its brief time requirement, absence of contrast agent need, and superior sensitivity and specificity (63). The current Japanese guidelines for MASLD/MASH suggest a two-step risk stratification process, which includes screening for fibrosis through serum markers or platelet count, the Fibrosis-4 index, or the MASLD fibrosis score (64). Depending on the risk of fibrosis, subsequent liver elastography or biopsy is suggested. The 2024 Chinese Standard for the Diagnosis and Treatment of Primary Liver Cancer recommends the combination of ultrasound and serum AFP levels to screen high-risk groups early. This approach is especially significant in diagnosing patients with metabolic dysfunction-associated cirrhosis, facilitating the early detection of high-risk HCC cases (65). Despite these recommendations, appropriate monitoring strategies are still lacking, leading to MASLD-HCC often being diagnosed at a late stage. This situation highlights the urgent need for improved diagnostic and monitoring approaches to address this growing public health concern effectively. The references for diagnosing and monitoring fibrosis in MASLD were presented in Table 2. The references for diagnosis and monitoring of HCC at any stage (Table 3).
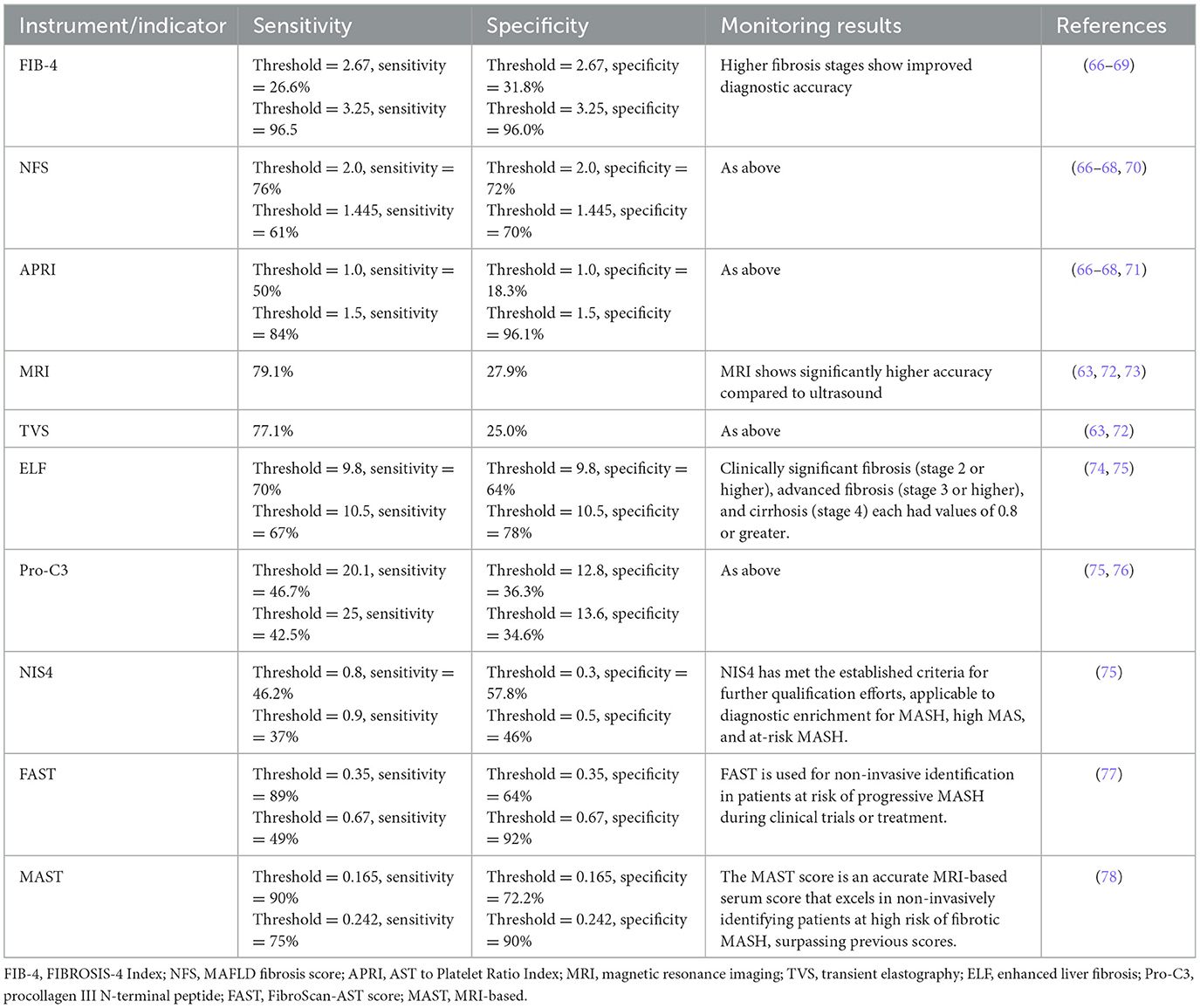
Table 2. Diagnosis and monitoring of fibrosis in MASLD.
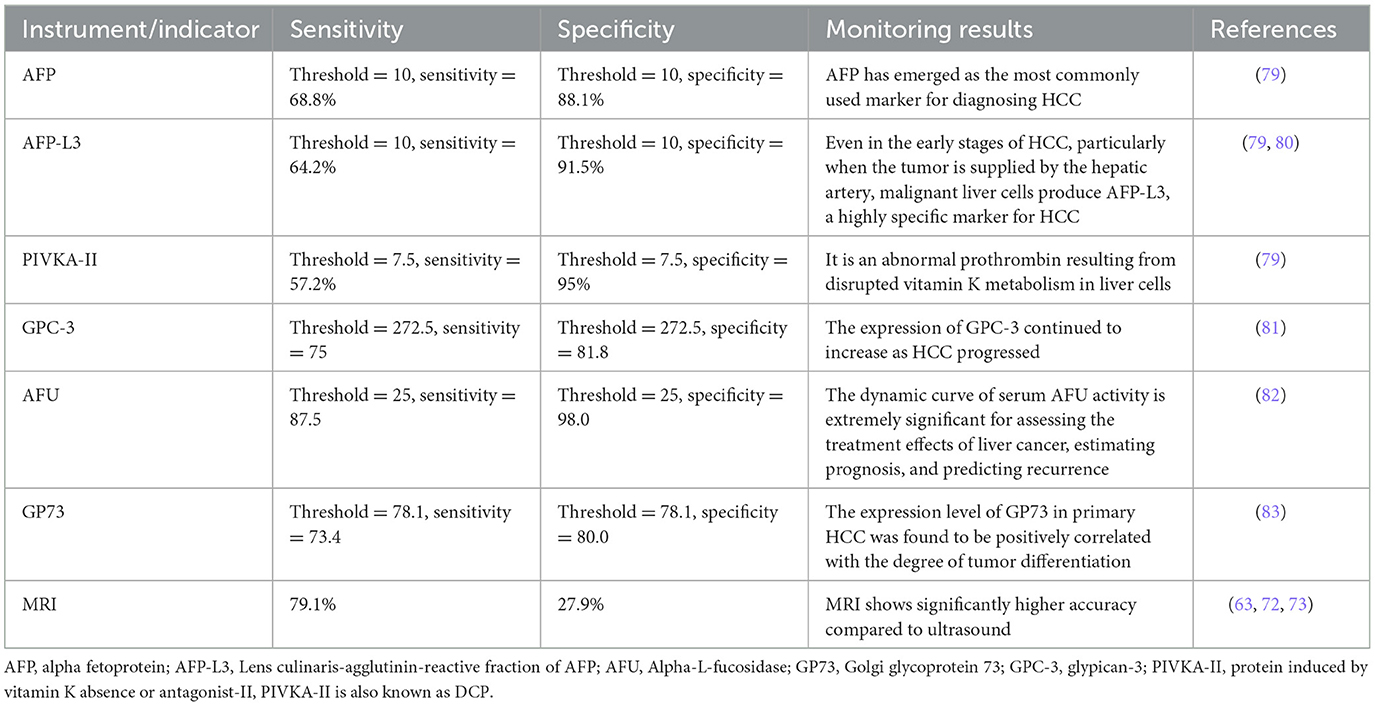
Table 3. Diagnosis and monitoring of HCC at any stage.
5 Treatment of MASLD-HCC
The treatment options for HCC can be broadly categorized into surgical resection and non-surgical treatments. Liver resection is the standard treatment modality for HCC patients without cirrhosis. For cirrhotic patients with HCC who meet the Milan criteria and are not candidates for primary surgical resection, liver transplantation (LT) is recommended. However, the availability of LT is constrained by the scarcity of donor organs. Current international guidelines advocate for a Barcelona Clinic Liver Cancer (BCLC)-based treatment algorithm for managing HCC (84). Despite these established guidelines, the specific impact of MASLD as an etiology on the outcomes of HCC patients remains relatively underexplored. However, it is essential to consider the unique pathophysiological mechanisms underlying MASLD-related HCC when determining treatment strategies. This approach allows for tailoring interventions according to the mechanism of disease occurrence, potentially improving patient outcomes by addressing the distinct characteristics of HCC that arises in the context of MASLD. Incorporating the understanding of MASLD's role in HCC development into treatment planning is crucial for optimizing therapeutic efficacy and patient care (Figure 2).
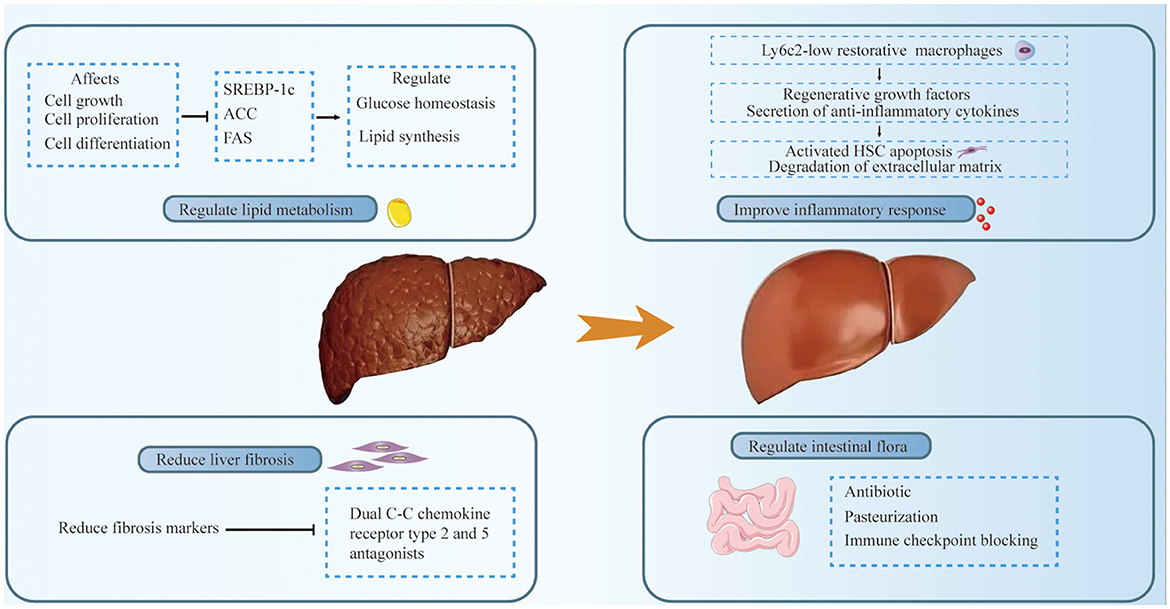
Figure 2. Treatment of MASLD-related HCC includes the following aspects: regulation of lipid metabolism, improvement of inflammatory response, mitigation of liver fibrosis, and intestinal microbia-related therapy. Glucose homeostasis and lipid synthesis were regulated by down-regulating SREBP-1C and ACC. Ly6c2 low-expressing restorative macrophages express regenerative growth factors and secrete anti-inflammatory cytokines. These cytokines may promote the apoptosis of activated hepatic stellate cells (HSCs) and the degradation of the extracellular matrix, thereby facilitating the resolution of inflammation. A dual C-C chemokine receptor type 2 and 5 antagonists significantly improved liver fibrosis, and antibiotics and pasteurization are used to regulate gut microbiota.
5.1 Regulation of lipid metabolism
Prevention and control of liver diseases, including HCC, can be effectively managed by regulating lipid metabolism. Changes in lipid metabolism play a significant role in the progression of many tumor cells by influencing critical cellular processes such as growth, proliferation, differentiation, and migration. Agonists that target the thyroid hormone receptor (THR)-β, predominantly located in the liver, have been shown to enhance lipophagy, mitochondrial biogenesis, and mitophagy. This stimulation leads to increased β-oxidation of fatty acids in the liver, which reduces the accumulation of lipotoxic lipids (85). Additionally, these agonists facilitate the uptake of low-density lipoprotein (LDL) and positively influence lipid profiles. Several natural compounds have been identified as potent regulators of lipid metabolism, particularly through their impact on the expression of genes related to adipogenesis (86). For instance, compounds such as lignocaine (87), oleanolic acid (88), and oxymatrine (89) have been shown to regulate glucose homeostasis and lipid synthesis by decreasing the expression of SREBP-1c, ACC, and FAS. Furthermore, betaine has demonstrated significant improvements in hepatic steatosis in C57BL/6J mice by activating AMPK and downregulating SREBP-1c. Moreover, betaine reduces hepatic triglyceride accumulation by lowering methylation of the PPARα promoter and enhancing PPARα expression, offering a promising approach to mitigating liver disease progression through the modulation of lipid metabolism (90). In the context of MASLD, research indicates that metformin modulates the ATP/AMP ratio to activate AMPK, which in turn regulates lipid metabolism (91). In HepG2 cells exposed to palmitate, metformin orchestrates the regulation of caspase-3, eukaryotic initiation factor-2a (eIF2a), and insulin receptor substrate-1 (IRS-1), thereby mitigating endoplasmic reticulum (ER) stress (92). These findings highlight the therapeutic potential of targeting lipid metabolism pathways in the prevention and management of liver diseases, including HCC, through natural compounds.
5.2 Modulating the inflammatory response
Inflammation plays a critical role in MASH and is considered a key factor in the progression from MASH to HCC. Consequently, anti-inflammatory strategies may serve as vital components in the treatment of both MASH and MASH-related HCC. Recent data suggest that targeting anti-inflammatory and regressive pathways shows promise for treating MASH. Evidence from patients with chronic liver diseases and murine model studies have conclusively demonstrated that fibrosis, even at advanced stages, can regress following the elimination of the underlying cause (93, 94). The activation of Mucosal-Associated Invariant T (MAIT) cells and the subsequent phenotypic shift of liver macrophages are marked pathological features of liver fibrosis, which can be targeted through anti-fibrogenic therapy (95). Specifically, the role of Kupffer cells (K.C.s) and infiltrating macrophages in mediating local liver inflammation repair is noteworthy (96). Within the liver microenvironment, K.C.s can be categorized into two primary phenotypes: pro-inflammatory M1 macrophages and anti-inflammatory M2 macrophages. Upon cessation of liver injury, hepatic macrophages transition into Ly6c2-low restorative macrophages, characterized by the expression of regenerative growth factors and the secretion of anti-inflammatory cytokines. These cytokines may promote the apoptosis and extracellular matrix degradation of activated hepatic stellate cells (HSCs), thus facilitating the resolution of inflammation (97). FGF21 inhibits hepatic lipid influx and accumulation through combined endocrine and autocrine signaling pathways, thereby preventing Kupffer cell activation and reducing the presence of lipid-associated and scar-associated macrophages to suppress fibrogenesis (98). The timely and appropriate modulation of macrophage phenotype is essential for successfully resolving inflammation and fostering liver tissue repair. Moreover, there is a notable link between anti-inflammatory approaches and antioxidants, suggesting that antioxidant treatment can also be beneficial. For example, rhodopsin has been shown to inhibit LPS-induced expression of pro-inflammatory cytokines in macrophages, reducing inflammatory cell infiltration and ameliorating liver function. Additionally, flavonoids have been found to significantly mitigate inflammation and thereby prevent hyperlipidemia and liver injury. This is achieved by enhancing antioxidant enzyme activity and inhibiting the secretion of inflammatory cytokines (TNF-α, IL-1, IL-6) in high fructose-induced obesity mice (99). These findings underscore the potential of integrating anti-inflammatory and antioxidant therapies in managing MASH and preventing its progression to HCC.
5.3 Reducing the progression of liver fibrosis
Fibrosis is a critical and often inevitable process in the progression of MASLD to HCC, with longitudinal studies highlighting an elevated risk of HCC and mortality in MASLD/MASH patients with severe liver fibrosis compared to those with milder forms of liver fibrosis (100). Although the progression from MASH to HCC does not invariably require liver fibrosis, the onset of excessive fibrosis almost certainly leads to irreversible liver damage. Thus, preventing and controlling the development of liver fibrosis is of paramount importance. Cysteine aspartate protease has been identified as a protease linked to hepatic apoptosis and inflammation. However, treatments with pan-cysteine protease inhibitors have not shown efficacy in improving fibrosis or in the regression of MASH (101). In the 2-year phase IIb CENTAUR trial, a dual C-C chemokine receptor type 2 and 5 antagonists demonstrated a fibrosis improvement of ≥1 stage without worsening. Interestingly, a significant proportion (60%) of patients who showed a fibrosis response after 1 year of treatment compared with placebo maintained this fibrosis reduction into the second year. Despite these promising results, the long-term effects of this treatment on fibrosis remain to be fully elucidated (102). Treatment may exert a protective effect against fibrosis by reducing the expression of fibrosis markers in mice with steatosis induced by a combination of a high-fat diet (HFD) and carbon tetrachloride (CCl4). This emerging evidence underscores the potential of novel therapeutic agents in mitigating fibrosis and highlights the necessity for continued research into effective treatments for liver fibrosis in the context of MASLD and MASH.
5.4 Gut microbiota interventions
The critical role of gut microbiota in the pathogenesis of MASH and its progression to hepatocellular carcinoma (MASH-HCC) has been increasingly recognized, prompting investigations into various therapeutic approaches. These include the use of antibiotics such as rivastigmine, probiotics, farnesoid X receptor (FXR) agonists, Toll-like receptor (TLR) antagonists, and inhibitors of bacterial metabolites (103). Furthermore, recent studies have explored the gut microbiome's potential role in modulating responses to immune checkpoint inhibitor therapy, highlighting a novel area of therapeutic intervention. An intriguing outcome emerged from a clinical trial involving overweight or obese insulin-resistant participants. The daily administration of pasteurized bacteria over 3 months was found to be safe, well-tolerated, and resulted in improved insulin sensitivity and lipid profiles among the treated patients. While this study did not directly demonstrate a clinical benefit in reducing HCC risk, the approach suggests a possible efficacy in lowering HCC risk among patients with MASLD (104). This finding introduces a promising avenue for leveraging gut microbiota modulation to enhance metabolic health and potentially reduce cancer risk. Additionally, a pioneering clinical trial that combines vancomycin therapy with immune checkpoint blockade has been initiated at the National Cancer Institute (NCT03785210). This trial aims to investigate whether the integration of checkpoint inhibition with selective manipulation of the microbiota yields benefits for patients with HCC (105). Immune checkpoint inhibitors, which target immunomodulatory molecules (or their ligands) on T cell surfaces to amplify antitumor immune responses, have gained approval for treating HCC (106). This innovative approach underscores the intersection of microbiome science and immunotherapy in crafting novel treatment strategies for HCC, marking a significant step forward in our understanding and management of this complex disease.
5.5 Metabolic pathway interventions
There is an acknowledged link between metabolic disorders and HCC. This recognition has spurred interest in therapeutic approaches that target metabolic pathways as potential strategies for HCC treatment. It has been proposed that type 2 diabetes and hypertension, hyperlipidemia, obesity, indicated by body mass index (BMI), are key contributors to severe liver disease (107). Consequently, approaches focusing on insulin resistance and diabetes may be effective in mitigating metabolic imbalances in HCC. Contemporary antidiabetic medications that also promote weight loss could help alleviate HCC symptoms. Notable among these medications are the agonists of glucagon-like peptide-1 (GLP-1), which enhance insulin secretion in response to glucose levels (108). Metformin, an oral antidiabetic drug, is the preferred treatment for type 2 diabetes due to its high efficacy and low cost (109). It reduces the risk of HCC in patients with diabetes-related chronic liver disease by inhibiting the progression of HCC. Dipeptidyl peptidase-4 inhibitors (DPP-4 inhibitors), another class of oral antidiabetic drugs based on incretin hormones, have been shown to decrease the risk of HCC in patients with type 2 diabetes and chronic HCV infection (110). Other therapeutic targets of interest are FGF19 and FGF21, which have garnered attention for their hepatoprotective properties and beneficial effects on inflammation and fibrosis, making them promising candidates for drug development. The FGF21 analog, pegbelfermin, has demonstrated favorable metabolic effects and reduced liver fat content in MASH patients after 16 weeks of treatment (111). Similarly, the FGF19 analog, NGM282, appears to be highly effective in reducing hepatic steatosis (112). In HCC, mutations within the C-terminal domains of specific receptor tyrosine kinases lead to their persistent activation. Inhibiting this phosphorylation has dual benefits: it suppresses tumor growth and inhibits angiogenesis in HCC. Tyrosine kinase inhibitors (TKIs), which serve as multi-kinase inhibitors, are primarily used in treatment to target key receptors such as VEGF, PDGF, RAF, FGF, KIT, and RET (113). A vital pathophysiological approach includes using TKIs to curb both angiogenesis in the tumor's microenvironment and the proliferation of cells (113, 114). The main receptors affected in angiogenesis signaling by these inhibitors include VEGF, PDGF, and FGF receptors (115). Peroxisome proliferator-activated receptors (PPARs) are crucial regulators of metabolism and inflammation. In various independent experimental mouse models, the pan-PPAR agonist lanifibranor has shown improvements across multiple facets of MASLD (116). In a phase 2b trial involving MASH patients, a 1,200 mg dose of lanifibranor significantly outperformed a placebo, as evidenced by at least a two-point reduction in the SAF-A score and a notably higher percentage of patients who did not experience worsening of fibrosis (117). Exploring metabolic pathways in the management of HCC has highlighted the substantial potential for therapeutic interventions. Focusing on antidiabetic drugs and agents targeting specific metabolic receptors presents a promising approach to reducing both the incidence and severity of HCC (Table 4). Further clinical trials and research are required to fully understand the effectiveness and safety of these treatment methods across various patient populations.
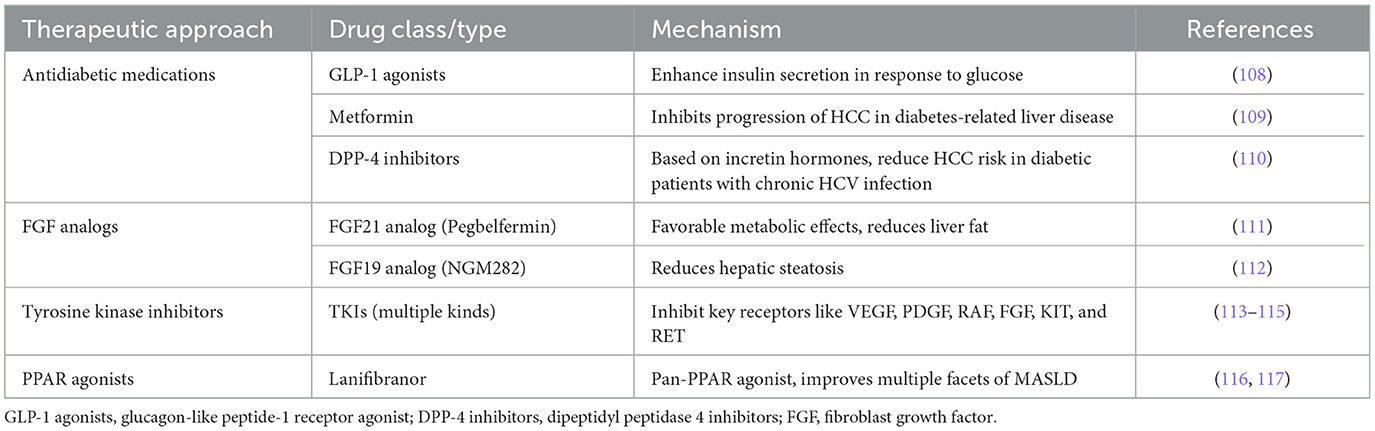
Table 4. Potential of metabolic pathway interventions in HCC related drug research.
6 Preventing the occurrence of MASLD-HCC
Implementing healthy lifestyle changes, encompassing both diet and physical activity, is recognized as the most effective and cost-effective strategy for managing MASLD. This approach is pivotal not only in correcting MASLD but also in delaying its progression to HCC. Moreover, these lifestyle modifications play a crucial role in addressing other complications associated with metabolic syndrome, such as hypertension, insulin resistance, and cardiovascular risk. Additionally, adopting a healthier lifestyle can mitigate the risk of obesity-related cancers. This holistic approach underscores the significance of lifestyle interventions in the comprehensive management of MASLD and its associated comorbidities, highlighting the interconnectedness of liver health with overall metabolic and cardiovascular wellbeing. Through the promotion of dietary improvements and increased physical activity, individuals can significantly enhance their health outcomes, thereby reducing the burden of MASLD and preventing its progression to more severe liver diseases and other metabolic conditions (Table 5; Figure 3).
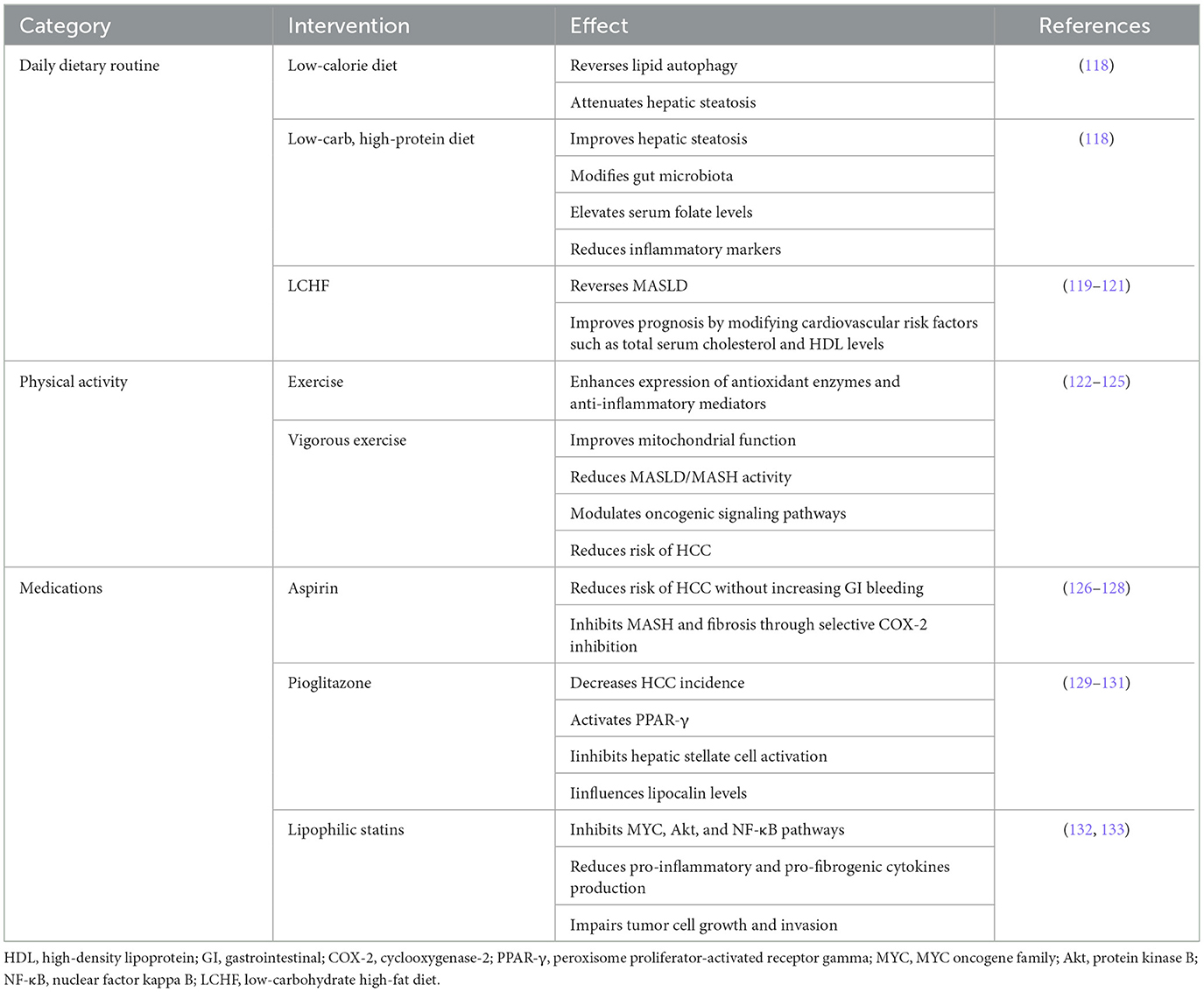
Table 5. Prevention of the progression of MASLD-HCC.
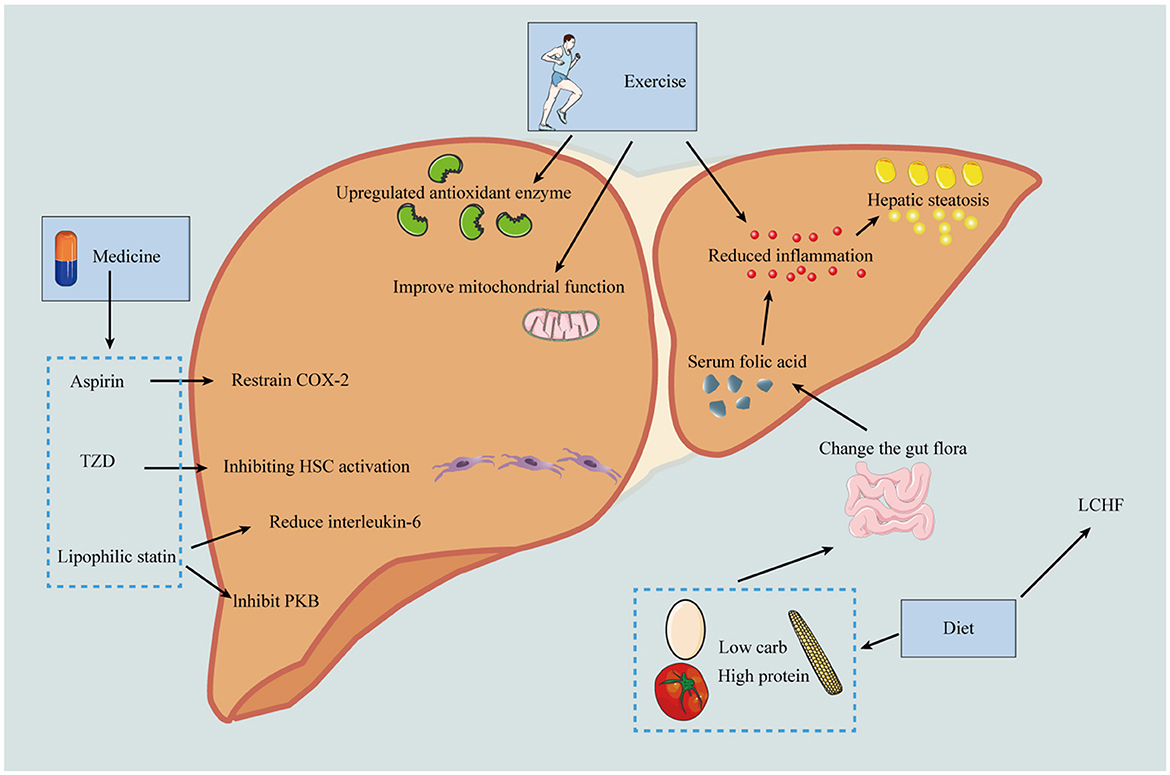
Figure 3. Prevention of the progression of MASLD-HCC should be carried out from the following three aspects: including diet, exercise and drugs. In terms of diet, low calorie diet can reduce the production of fat; Low carbohydrate and high protein diets reduce inflammation by improving microbiota and thereby serum folate levels, and the LCHF diet can reverse MASLD. Exercise can up-regulate antioxidant enzymes and anti-inflammatory mediators, improve mitochondrial protection, and thus reduce the risk of HCC. In terms of drugs, aspirin can selectively inhibit COX-2 and thus reduce liver fibrosis; pioglitazone can inhibit HSC activation and thus reduce HCC incidence; lipophilic statins can inhibit PKB pathway and reduce IL-6 production. HDL, high-density lipoprotein; GI, gastrointestinal; COX-2, cyclooxygenase-2; PPAR-γ, peroxisome proliferator-activated receptor gamma; MYC, MYC oncogene family; Akt, protein kinase B; NF-κB, nuclear factor kappa B; LCHF, low-carbohydrate high-fat diet.
6.1 Daily dietary routine
The cornerstone of dietary intervention for managing MASLD focuses on controlling dietary caloric intake and modifying the dietary structure. Consumption of a high-fat diet has been linked to significant activation of Akt and increased mTOR phosphorylation, leading to a marked reduction in lipid autophagy. This adverse effect, however, can be mitigated through the adoption of a low-calorie diet (118). Implementing a low-carbohydrate, high-protein diet may further enhance hepatic steatosis management by modifying the gut microbiota, elevating serum folate levels, and reducing inflammatory markers. Such dietary adjustments underscore the importance of a holistic approach to MASLD management, targeting both hepatic and metabolic pathways. Moreover, cardiovascular events represent the primary cause of mortality among individuals with MASLD. A low-carbohydrate, high-fat (LCHF) diet may not only facilitate the reversal of MASLD (121) but also improve the prognosis of MASLD patients by modifying risk factors associated with cardiovascular events. These modifications include improvements in total serum cholesterol and high-density lipoprotein levels (119, 120). This dual benefit highlights the critical role of dietary management in addressing the multifaceted challenges posed by MASLD, emphasizing the potential for dietary interventions to concurrently tackle liver health and cardiovascular risk. Through strategic dietary changes, individuals can significantly impact their health outcomes, underlining the importance of diet in the comprehensive management of MASLD and its associated comorbidities.
6.2 Physical activity
Oxidative stress, triggered by reactive oxygen species, alongside inflammation, plays a crucial role in hepatocellular damage among patients with MASLD. Exercise emerges as a potent intervention for MASLD by enhancing the expression of antioxidant enzymes and anti-inflammatory mediators. Notably, exercise has been shown to exert a beneficial effect on MASLD (125), underlining the intrinsic value of physical activity in liver health management. Furthermore, exercise contributes to reducing the risk of HCC. This protective effect is thought to be mediated through several mechanisms, including the improvement of mitochondrial function (exemplified by enhanced mitochondrial biosynthesis and autophagy) reduction of MASLD/MASH activity, and modulation of oncogenic signaling pathways (122). The link between regular physical activity and a decreased risk of HCC was substantiated by findings from the European Prospective Investigation into Cancer and Nutrition (EPIC) study, which revealed that individuals engaging in at least 2 h of vigorous exercise weekly had a reduced HCC risk (123). This association was further supported by a meta-analysis of prospective studies, demonstrating a significantly lower risk of HCC in individuals with high levels of physical activity compared to those with low activity levels (124). Given these insights, it is advised for patients who experience improvements in fatty liver conditions due to exercise to commit to at least 12 weeks of moderate-intensity physical activity. Following this period, it becomes particularly insightful to assess the extent of hepatic steatosis, highlighting the importance of sustained exercise regimens in the effective management of MASLD and the prevention of its progression to more severe liver conditions like HCC. This recommendation underscores the critical role of exercise not only as a therapeutic strategy for MASLD but also as a preventive measure against its progression to HCC.
6.3 Medications
Pharmacological prevention emerges as a viable option when lifestyle interventions fail to yield desired outcomes, with several drugs demonstrating the ability to modulate risk factors and carcinogenic pathways associated with MASLD/MASH-induced HCC. A notable national registry study in Sweden provided evidence that regular intake of aspirin at doses below 160 mg/day for a duration of at least 5 years significantly reduced the risk of HCC without escalating the risk of gastrointestinal bleeding (126). Further, in a cohort of 361 patients with biopsy-proven MASLD, daily aspirin administration was significantly associated with a reduced odds ratio (OR) for MASH and fibrosis. This protective effect of aspirin against MASH and fibrosis is thought to be mediated through selective inhibition of cyclooxygenase-2 (127). Based on a systematic review and meta-analysis by Abdelmalak et al., the use of aspirin in patients without cirrhosis is associated with an ~30% reduction in the risk of HCC. However, in patients with cirrhosis, this protective effect did not reach statistical significance (adjusted HR 0.96, 95% CI 0.84–1.09). Furthermore, the study also noted an increased risk of bleeding associated with aspirin use (adjusted HR 1.11, 95% CI 1.02–1.22) (128). Given the inherently higher risk of bleeding in cirrhotic patients, this risk may be more pronounced. Therefore, the pharmacological prevention of HCC should be carefully case-selected, particularly considering the stage of liver disease and the individual bleeding risk, to ensure an optimal risk-benefit ratio. Pioglitazone, an activator of the peroxisome proliferator-activated receptor-gamma (PPAR-γ), renowned for its insulin-sensitizing properties, has been observed to decrease the incidence of HCC in both a hospital-based case-control study and a population-based cohort study (129). The anticancer effects of pioglitazone are postulated to arise from the inhibition of hepatic stellate cell activation, as suggested by in vitro studies (130). Moreover, pioglitazone has shown to positively influence lipocalin levels, which is linked to the prevention of carcinogenesis (131). Lipophilic statins, known for their higher lipid solubility and membrane permeability, enable them to exert cholesterol-dependent effects on HCC development (132). These effects potentially include the inhibition of key oncogenic pathways such as MYC, protein kinase B (Akt), and NF-κB, along with a reduction in the production of pro-inflammatory and pro-fibrogenic cytokines such as interleukin-6, tumor necrosis factor-alpha, and transforming growth factor-beta1. Furthermore, simvastatin has been documented to impair tumor cell growth and disrupt the adhesion of tumor cells to endothelial cell monolayers, consequently hindering tumor cell invasion (133). Especially for patients with decompensated cirrhosis and liver failure, considering the increased risk of myopathy side effect, current evidence suggests that statins should be used cautiously and only at low doses (max. 20 mg) to balance therapeutic benefits and potential adverse effects (134). This array of pharmacological interventions highlights the nuanced approach required in the management of MASLD/MASH and the prevention of its progression to HCC, where medication can play a critical role alongside lifestyle modifications.
7 Progress and prospects
The clinical management of HCC still faces major challenges, and many unanswered questions remain to be addressed by the scientific community. In particular, there are challenges and problems in the area of MASLD-HCC, where some of the pathogenesis is understood, but much remains to be learned. Although current treatments can target known mechanisms, completely halting the pathogenesis through pharmacological or surgical interventions remains elusive. Consequently, the emphasis on prevention (particularly through diet and exercise) is paramount. This review delves into the pathogenesis, diagnosis, and monitoring of MASLD-HCC, alongside treatment options and preventative measures. Presently, the underlying mechanisms of MASLD-HCC are still under investigation, with a notable deficiency in effective screening tools, preventive measures, and curative medications. Hence, it is imperative for both patients and physicians to prioritize the management and control of relevant risk factors in clinical practice. Such diligence is essential to offer precise medication guidance and support patients in achieving a more favorable prognosis. Emphasizing preventative strategies, including lifestyle modifications, can play a crucial role in mitigating the risk of MASLD progressing to HCC. This approach underscores the need for continued research and development in the field, aiming to enhance our understanding, refine diagnostic tools, and discover more effective treatments for this complex disease.
Author contributions
YM: Conceptualization, Writing—original draft, Writing—review & editing, Visualization. JW: Writing—original draft, Data curation, Methodology, Formal analysis, Investigation, Visualization, Writing—review & editing. WX: Data curation, Writing—original draft, Formal analysis, Methodology, Visualization. XF: Conceptualization, Funding acquisition, Supervision, Validation, Writing—review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Guangxi Province Natural Science Foundation Surface Project [2023JJA141287]. 2022 Guilin Self-funded Science and Technology Project (No. 20220129z).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Shah PA, Patil R, Harrison SA. NAFLD-related hepatocellular carcinoma: the growing challenge. Hepatology. (2023) 77:323–38. doi: 10.1002/hep.32542
3. Younossi ZM, Golabi P, Paik JM, Henry A, Van Dongen C, Henry L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology. (2023) 77:1335–47. doi: 10.1097/HEP.0000000000000004
4. Seif El Dahan K, Daher D, Singal AG. Hepatocellular carcinoma surveillance in patients with non-alcoholic fatty liver disease. Clin Mol Hepatol. (2023) 29(Suppl):S207–19. doi: 10.3350/cmh.2022.0247
5. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. (2016) 64:73–84. doi: 10.1002/hep.28431
6. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in china, France, Germany, Italy, Japan, Spain, united kingdom, and united states for the period 2016–2030. J Hepatol. (2018) 69:896–904. doi: 10.1016/j.jhep.2018.05.036
7. Lonardo A, Adinolfi LE, Loria P, Carulli N, Ruggiero G, Day CP. Steatosis and hepatitis C virus: mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology. (2004) 126:586–97. doi: 10.1053/j.gastro.2003.11.020
8. Ren Q, Sun Q, Fu J. Dysfunction of autophagy in high-fat diet-induced non-alcoholic fatty liver disease. Autophagy. (2024) 20:221–41. doi: 10.1080/15548627.2023.2254191
9. Fang J, Yu CH, Li XJ, Yao JM, Fang ZY, Yoon SH, et al. Gut dysbiosis in nonalcoholic fatty liver disease: pathogenesis, diagnosis, and therapeutic implications. Front Cell Infect Microbiol. (2022) 12:997018. doi: 10.3389/fcimb.2022.997018
10. Marchesini G, Marchignoli F, Petta S. Evidence-based medicine and the problem of healthy volunteers. Ann Hepatol. (2017) 16:832–4. doi: 10.5604/01.3001.0010.5272
11. Donne R, Lujambio A. The liver cancer immune microenvironment: therapeutic implications for hepatocellular carcinoma. Hepatology. (2023) 77:1773–96. doi: 10.1002/hep.32740
12. Ward ZJ, Bleich SN, Cradock AL, Barrett JL, Giles CM, Flax C, et al. Projected US state-level prevalence of adult obesity and severe obesity. N Engl J Med. (2019) 381:2440–50. doi: 10.1056/NEJMsa1909301
13. Tateishi R, Uchino K, Fujiwara N, Takehara T, Okanoue T, Seike M, et al. A nationwide survey on non-B, non-C hepatocellular carcinoma in japan: 2011–2015 update. J Gastroenterol. (2019) 54:367–76. doi: 10.1007/s00535-018-1532-5
14. Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, Richardson PA, et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology. (2018) 155:1828–37. doi: 10.1053/j.gastro.2018.08.024
15. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
16. Sanyal AJ. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. (2019) 16:377–86. doi: 10.1038/s41575-019-0144-8
17. van der Poorten D, Samer CF, Ramezani-Moghadam M, Coulter S, Kacevska M, Schrijnders D, et al. Hepatic fat loss in advanced nonalcoholic steatohepatitis: are alterations in serum adiponectin the cause? Hepatology. (2013) 57:2180–8. doi: 10.1002/hep.26072
18. Vucenik I, Stains JP. Obesity and cancer risk: evidence, mechanisms, and recommendations. Ann N Y Acad Sci. (2012) 1271:37–43. doi: 10.1111/j.1749-6632.2012.06750.x
19. Mittal S, El-Serag HB, Sada YH, Kanwal F, Duan Z, Temple S, et al. Hepatocellular carcinoma in the absence of cirrhosis in united states veterans is associated with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. (2016) 14:124–31. doi: 10.1016/j.cgh.2015.07.019
20. Dongiovanni P, Meroni M, Longo M, Fargion S, Fracanzani AL. Genetics, immunity and nutrition boost the switching from NASH to HCC. Biomedicines. (2021) 9:1524. doi: 10.3390/biomedicines9111524
21. Farrell GC, Haczeyni F, Chitturi S. Pathogenesis of NASH: how metabolic complications of overnutrition favour lipotoxicity and pro-inflammatory fatty liver disease. Adv Exp Med Biol. (2018) 1061:19–44. doi: 10.1007/978-981-10-8684-7_3
22. Mendez-Sanchez N, Cruz-Ramon V, Ramirez-Perez O, Hwang JP, Barranco-Fragoso B, Cordova-Gallardo J. New aspects of lipotoxicity in nonalcoholic steatohepatitis. Int J Mol Sci. (2018) 19:2034. doi: 10.3390/ijms19072034
23. Pinyopornpanish K, Khoudari G, Saleh MA, Angkurawaranon C, Pinyopornpanish K, Mansoor E, et al. Hepatocellular carcinoma in nonalcoholic fatty liver disease with or without cirrhosis: a population-based study. BMC Gastroenterol. (2021) 21:394. doi: 10.1186/s12876-021-01978-0
24. Lee T-Y, Wu J-C, Yu S-H, Lin J-T, Wu M-S, Wu C-Y. The occurrence of hepatocellular carcinoma in different risk stratifications of clinically noncirrhotic nonalcoholic fatty liver disease. Int J Cancer. (2017) 141:1307–14. doi: 10.1002/ijc.30784
25. Santoro A, McGraw TE, Kahn BB. Insulin action in adipocytes, adipose remodeling, and systemic effects. Cell Metab. (2021) 33:748–57. doi: 10.1016/j.cmet.2021.03.019
26. Eissing L, Scherer T, Tödter K, Knippschild U, Greve JW, Buurman WA, et al. De novo lipogenesis in human fat and liver is linked to ChREBP-β and metabolic health. Nat Commun. (2013) 4:1528. doi: 10.1038/ncomms2537
27. Reda D, Elshopakey GE, Albukhari TA, Almehmadi SJ, Refaat B, Risha EF, et al. Vitamin D3 alleviates nonalcoholic fatty liver disease in rats by inhibiting hepatic oxidative stress and inflammation via the SREBP-1-c/ PPARα-NF-κB/IR-S2 signaling pathway. Front Pharmacol. (2023) 14:1164512. doi: 10.3389/fphar.2023.1164512
28. Luukkonen PK, Sädevirta S, Zhou Y, Kayser B, Ali A, Ahonen L, et al. Saturated fat is more metabolically harmful for the human liver than unsaturated fat or simple sugars. Diabetes Care. (2018) 41:1732–9. doi: 10.2337/dc18-0071
29. Leung KC, Doyle N, Ballesteros M, Waters MJ, Ho KK. Insulin regulation of human hepatic growth hormone receptors: divergent effects on biosynthesis and surface translocation. Clin Endocrinol Metab. (2000) 85:4712–20. doi: 10.1210/jcem.85.12.7017
30. Pang Y, Liu Z, Han H, Wang B, Li W, Mao C, et al. Peptide SMIM30 promotes HCC development by inducing SRC/YES1 membrane anchoring and MAPK pathway activation. J Hepatol. (2020) 73:1155–69. doi: 10.1016/j.jhep.2020.05.028
31. Arab JP, Arrese M, Trauner M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu Rev Pathol. (2018) 13:321–50. doi: 10.1146/annurev-pathol-020117-043617
32. Hammerich L, Tacke F. Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatol. (2023) 20:633–46. doi: 10.1038/s41575-023-00807-x
33. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol. (2018) 15:738–52. doi: 10.1038/s41575-018-0065-y
34. Azzu V, Vacca M, Virtue S, Allison M, Vidal-Puig A. Adipose tissue-liver cross talk in the control of whole-body metabolism: implications in nonalcoholic fatty liver disease. Gastroenterology. (2020) 158:1899–912. doi: 10.1053/j.gastro.2019.12.054
35. Ringelhan M, Pfister D, O'Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. (2018) 19:222–32. doi: 10.1038/s41590-018-0044-z
36. Martín-Fernández M, Arroyo V, Carnicero C, Sigüenza R, Busta R, Mora N, et al. Role of oxidative stress and lipid peroxidation in the pathophysiology of NAFLD. Antioxidants. (2022) 11:2217. doi: 10.3390/antiox11112217
37. Polimeni L. Oxidative stress: new insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World J Hepatol. (2015) 7:1325. doi: 10.4254/wjh.v7.i10.1325
38. Koek GH, Liedorp PR, Bast A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin Chim Acta. (2011) 412:1297–305. doi: 10.1016/j.cca.2011.04.013
39. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. (2014) 10:9–17. doi: 10.1038/nchembio.1416
40. Yu Y-C, Luu HN, Wang R, Thomas CE, Glynn NW, Youk AO, et al. Serum biomarkers of iron status and risk of hepatocellular carcinoma development in patients with nonalcoholic fatty liver disease. Cancer Epidemiol Biomarkers Prev. (2022) 31:230–5. doi: 10.1158/1055-9965.EPI-21-0754
41. Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS biology. (2018) 16:e2006203. doi: 10.1371/journal.pbio.2006203
42. Sun Y, Zheng C, Li T, He X, Yang F, Guo W, et al. GB1a activates SIRT6 to regulate lipid metabolism in mouse primary hepatocytes. Int J Mol Sci. (2023) 24:9540. doi: 10.3390/ijms24119540
43. Pradere JP, Troeger J, Dapito D, Mencin AA, Schwabe RF. Toll-like receptor 4 and hepatic fibrogenesis. Semin Liver Dis. (2010) 30:232–44. doi: 10.1055/s-0030-1255353
44. Balmer ML, Slack E, de Gottardi A, Lawson MAE, Hapfelmeier S, Miele L, et al. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci Transl Med. (2014) 6:237ra66. doi: 10.1126/scitranslmed.3008618
45. Venetsanaki V, Karabouta Z, Polyzos SA. Farnesoid X nuclear receptor agonists for the treatment of nonalcoholic steatohepatitis. Eur J Pharmacol. (2019) 863:172661. doi: 10.1016/j.ejphar.2019.172661
46. Fuchs CD, Claudel T, Scharnagl H, Stojakovic T, Trauner M. FXR controls CHOP expression in steatohepatitis. FEBS Lett. (2017) 591:3360–8. doi: 10.1002/1873-3468.12845
47. Takahashi S, Luo Y, Ranjit S, Xie C, Libby AE, Orlicky DJ, et al. Bile acid sequestration reverses liver injury and prevents progression of nonalcoholic steatohepatitis in western diet–fed mice. J Biol Chem. (2020) 295:4733–47. doi: 10.1074/jbc.RA119.011913
48. Zhang X, Coker OO, Chu ES, Fu K, Lau HCH, Wang Y-X, et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. (2021) 70:761–74. doi: 10.1136/gutjnl-2019-319664
49. Pan Y, Zhang X. Diet and gut microbiome in fatty liver and its associated liver cancer. J Gastroenterol Hepatol. (2022) 37:7–14. doi: 10.1111/jgh.15713
50. Behary J, Amorim N, Jiang X-T, Raposo A, Gong L, McGovern E, et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun. (2021) 12:187. doi: 10.1038/s41467-020-20422-7
51. Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y, et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov. (2017) 7:522–38. doi: 10.1158/2159-8290.CD-16-0932
52. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science. (2018) 360:eaan5931. doi: 10.1126/science.aan5931
53. Longo M, Meroni M, Paolini E, Erconi V, Carli F, Fortunato F, et al. TM6SF2/PNPLA3/MBOAT7 loss-of-function genetic variants impact on NAFLD development and progression both in patients and in in vitro models. Cell Mol Gastroenterol Hepatol. (2022) 13:759–88. doi: 10.1016/j.jcmgh.2021.11.007
54. Zhao Z, Meng J, Su R, Zhang J, Chen J, Ma X, et al. Epitranscriptomics in liver disease: basic concepts and therapeutic potential. J Hepatol. (2020) 73:664–79. doi: 10.1016/j.jhep.2020.04.009
55. Yang Y, Cai J, Yang X, Wang K, Sun K, Yang Z, et al. Dysregulated m6A modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Molecular Therapy. (2022) 30:2342–53. doi: 10.1016/j.ymthe.2022.02.021
56. Soronen J, Yki-Järvinen H, Zhou Y, Sädevirta S, Sarin AP, Leivonen M, et al. Novel hepatic microRNAs upregulated in human nonalcoholic fatty liver disease. Physiol Rep. (2016) 4:e12661. doi: 10.14814/phy2.12661
57. Latorre J, Moreno-Navarrete JM, Mercader JM, Sabater M, Rovira Ò, Gironès J, et al. Decreased lipid metabolism but increased FA biosynthesis are coupled with changes in liver microRNAs in obese subjects with NAFLD. Int J Obes. (2017) 41:620–30. doi: 10.1038/ijo.2017.21
58. C. Gordon S, Fraysse J, Li S, Ozbay AB, Wong RJ. Disease severity is associated with higher healthcare utilization in nonalcoholic steatohepatitis medicare patients. Am J Gastroenterol. (2020) 115:562–74. doi: 10.14309/ajg.0000000000000484
59. Ntandja Wandji LC, Ningarhari M, Lassailly G, Dharancy S, Boleslawski E, Mathurin P, et al. Liver transplantation in alcohol-related liver disease and alcohol-related hepatitis. J Clin Exp Hepatol. (2023) 13:127–38. doi: 10.1016/j.jceh.2022.06.013
60. Albhaisi S, Chowdhury A, Sanyal AJ. Non-alcoholic fatty liver disease in lean individuals. JHEP Reports. (2019) 1:329–41. doi: 10.1016/j.jhepr.2019.08.002
61. Samoylova ML, Mehta N, Roberts JP, Yao FY. Predictors of ultrasound failure to detect hepatocellular carcinoma. Liver Transplantation. (2018) 24:1171–7. doi: 10.1002/lt.25202
62. Loomba R, Lim JK, Patton H, El-Serag HB. AGA clinical practice update on screening and surveillance for hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: expert review. Gastroenterology. (2020) 158:1822–30. doi: 10.1053/j.gastro.2019.12.053
63. Park HJ, Jang HY, Kim SY, Lee SJ, Won HJ, Byun JH, et al. Non-enhanced magnetic resonance imaging as a surveillance tool for hepatocellular carcinoma: comparison with ultrasound. J Hepatol. (2020) 72:718–24. doi: 10.1016/j.jhep.2019.12.001
64. Kogiso T, Tokushige K. The current view of nonalcoholic fatty liver disease-related hepatocellular carcinoma. Cancers. (2021) 13:516. doi: 10.3390/cancers13030516
65. Department of Medical Administration. National health and health commission of the People's Republic of China. Guidelines for diagnosis and treatment of primary liver cancer in China (2024 edition). Chin J Gen Surg. (2024) 33:475–530. doi: 10.12025/j.issn.1008-6358.2024.20240065
66. Younossi ZM, Loomba R, Anstee QM, Rinella ME, Bugianesi E, Marchesini G, et al. Diagnostic modalities for nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, and associated fibrosis. Hepatology. (2018) 68:349–60. doi: 10.1002/hep.29721
67. Vilar-Gomez E, Chalasani N. Non-invasive assessment of non-alcoholic fatty liver disease: clinical prediction rules and blood-based biomarkers. J Hepatol. (2018) 68:305–15. doi: 10.1016/j.jhep.2017.11.013
68. Castera L, Friedrich-Rust M, Loomba R. Noninvasive assessment of liver disease in patients with nonalcoholic fatty liver disease. Gastroenterology. (2019) 156:1264–81.e4. doi: 10.1053/j.gastro.2018.12.036
69. Ito T, Ishigami M, Zou B, Tanaka T, Takahashi H, Kurosaki M, et al. The epidemiology of NAFLD and lean NAFLD in japan: a meta-analysis with individual and forecasting analysis, 1995-2040. Hepatol Int. (2021) 15:366–79. doi: 10.1007/s12072-021-10143-4
70. Younossi Z, Aggarwal P, Shrestha I, Fernandes J, Johansen P, Augusto M, et al. The burden of non-alcoholic steatohepatitis: a systematic review of health-related quality of life and patient-reported outcomes. JHEP Rep. (2022) 4:100525. doi: 10.1016/j.jhepr.2022.100525
71. Toyoda H, Johnson PJ. The ALBI score: from liver function in patients with HCC to a general measure of liver function. JHEP Rep. (2022) 4:100557. doi: 10.1016/j.jhepr.2022.100557
72. Zhou JH, Cai JJ, She ZG, Li HL. Noninvasive evaluation of nonalcoholic fatty liver disease: current evidence and practice. World J Gastroenterol. (2019) 25:1307–26. doi: 10.3748/wjg.v25.i11.1307
73. Huang DQ, Fowler KJ, Liau J, Cunha GM, Louie AL, An JY, et al. Comparative efficacy of an optimal exam between ultrasound versus abbreviated MRI for HCC screening in NAFLD cirrhosis: a prospective study. Aliment Pharmacol Ther. (2022) 55:820–7. doi: 10.1111/apt.16844
74. Kjaergaard M, Lindvig KP, Thorhauge KH, Andersen P, Hansen JK, Kastrup N, et al. Using the ELF test, FIB-4 and NAFLD fibrosis score to screen the population for liver disease. J Hepatol. (2023) 79:277–86. doi: 10.1016/j.jhep.2023.04.002
75. Sanyal AJ, Shankar SS, Yates KP, Bolognese J, Daly E, Dehn CA, et al. Diagnostic performance of circulating biomarkers for non-alcoholic steatohepatitis. Nat Med. (2023) 29:2656–64. doi: 10.1038/s41591-023-02539-6
76. Vali Y, Lee J, Boursier J, Petta S, Wonders K, Tiniakos D, et al. Biomarkers for staging fibrosis and non-alcoholic steatohepatitis in non-alcoholic fatty liver disease (the LITMUS project): a comparative diagnostic accuracy study. Lancet Gastroenterol Hepatol. (2023) 8:714–25. doi: 10.1016/S2468-1253(23)00017-1
77. Newsome PN, Sasso M, Deeks JJ, Paredes A, Boursier J, Chan WK, et al. FibroScan-AST (FAST) score for the non-invasive identification of patients with non-alcoholic steatohepatitis with significant activity and fibrosis: a prospective derivation and global validation study. Lancet Gastroenterol Hepatol. (2020) 5:362–73. doi: 10.1016/S2468-1253(19)30383-8
78. Noureddin M, Truong E, Gornbein JA, Saouaf R, Guindi M, Todo T, et al. MRI-based (MAST) score accurately identifies patients with NASH and significant fibrosis. J Hepatol. (2022) 76:781–7. doi: 10.1016/j.jhep.2021.11.012
79. Best J, Bilgi H, Heider D, Schotten C, Manka P, Bedreli S, et al. The GALAD scoring algorithm based on AFP, AFP-L3, and DCP significantly improves detection of BCLC early stage hepatocellular carcinoma. Z Gastroenterol. (2016) 54:1296–305. doi: 10.1055/s-0042-119529
80. Li D, Mallory T, Satomura S. AFP-L3: a new generation of tumor marker for hepatocellular carcinoma. Clin Chim Acta. (2001) 313:15–9. doi: 10.1016/S0009-8981(01)00644-1
81. Sun B, Huang Z, Wang B, Yu Y, Lin S, Luo L, et al. Significance of glypican-3 (GPC3) expression in hepatocellular cancer diagnosis. Med Sci Monit Int Med J Exp Clin Res. (2017) 23:850–5. doi: 10.12659/MSM.899198
82. Mossad NA, Mahmoud EH, Osman EA, Mahmoud SH, Shousha HI. Evaluation of squamous cell carcinoma antigen-immunoglobulin M complex (SCCA-IGM) and alpha-L-fucosidase (AFU) as novel diagnostic biomarkers for hepatocellular carcinoma. Tumour Biol. (2014) 35:11559–64. doi: 10.1007/s13277-014-2467-y
83. Hou S-C, Xiao M-B, Ni R-Z, Ni W-K, Jiang F, Li X-Y, et al. Serum GP73 is complementary to AFP and GGT-II for the diagnosis of hepatocellular carcinoma. Oncol Lett. (2013) 6:1152–8. doi: 10.3892/ol.2013.1522
84. Galle PR, Forner A, Llovet JM, Mazzaferro V, Piscaglia F, Raoul J-L, et al. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. (2018) 69:182–236. doi: 10.1016/j.jhep.2018.03.019
85. Karim G, Bansal MB. Resmetirom: an orally administered, smallmolecule, liver-directed, β-selective THR agonist for the treatment of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. TouchREV Endocrinol. (2023) 19:60–70. doi: 10.17925/EE.2023.19.1.60
86. Luo X, Li N, Zhao X, Liao C, Ye R, Cheng C, et al. DHRS2 mediates cell growth inhibition induced by trichothecin in nasopharyngeal carcinoma. J Exp Clin Cancer Res. (2019) 38:300. doi: 10.1186/s13046-019-1301-1
87. Zhang Y, Jia J, Jin W, Cao J, Fu T, Ma D, et al. Lidocaine inhibits the proliferation and invasion of hepatocellular carcinoma by downregulating USP14 induced PI3K/akt pathway. Pathol Res Pract. (2020) 216:152963. doi: 10.1016/j.prp.2020.152963
88. Gamede M, Mabuza L, Ngubane P, Khathi A. Plant-derived oleanolic acid ameliorates markers associated with non-alcoholic fatty liver disease in a diet-induced pre-diabetes rat model. Diabetes Metab Syndr Obes. (2019) 12:1953–62. doi: 10.2147/DMSO.S218626
89. Zhang H, Yang L, Wang Y, Huang W, Li Y, Chen S, et al. Oxymatrine alleviated hepatic lipid metabolism via regulating miR-182 in non-alcoholic fatty liver disease. Life Sci. (2020) 257:118090. doi: 10.1016/j.lfs.2020.118090
90. Xu G, Huang K, Zhou J. Hepatic AMP kinase as a potential target for treating nonalcoholic fatty liver disease: evidence from studies of natural products. Curr Med Chem. (2018) 25:889–907. doi: 10.2174/0929867324666170404142450
91. Ma T, Tian X, Zhang B, Li M, Wang Y, Yang C, et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature. (2022) 603:159–65. doi: 10.1038/s41586-022-04431-8
92. Kim DS, Jeong SK, Kim HR, Kim DS, Chae SW, Chae HJ. Metformin regulates palmitate-induced apoptosis and ER stress response in HepG2 liver cells. Immunopharmacol Immunotoxicol. (2010) 32:251–7. doi: 10.3109/08923970903252220
93. Friedman SL, Pinzani M. Hepatic fibrosis 2022: unmet needs and a blueprint for the future. Hepatology. (2022) 75:473–88. doi: 10.1002/hep.32285
94. Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. (2013) 381:468–75. doi: 10.1016/S0140-6736(12)61425-1
95. Mabire M, Hegde P, Hammoutene A, Wan J, Caër C, Sayegh RA, et al. MAIT cell inhibition promotes liver fibrosis regression via macrophage phenotype reprogramming. Nat Commun. (2023) 14:1830. doi: 10.1038/s41467-023-37453-5
96. Rius B, Duran-Güell M, Flores-Costa R, López-Vicario C, Lopategi A, Alcaraz-Quiles J, et al. The specialized proresolving lipid mediator maresin 1 protects hepatocytes from lipotoxic and hypoxia-induced endoplasmic reticulum stress. FASEB J. (2017) 31:5384–98. doi: 10.1096/fj.201700394R
97. Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J. (2017) 31:1273–88. doi: 10.1096/fj.201601222R
98. Liu C, Schönke M, Spoorenberg B, Lambooij JM, van der Zande HJP, Zhou E, et al. FGF21 protects against hepatic lipotoxicity and macrophage activation to attenuate fibrogenesis in nonalcoholic steatohepatitis. Elife. (2023) 12:e83075. doi: 10.7554/eLife.83075
99. Yuan L, Han X, Li W, Ren D, Yang X. Isoorientin prevents hyperlipidemia and liver injury by regulating lipid metabolism, antioxidant capability, and inflammatory cytokine release in high-fructose-fed mice. J Agric Food Chem. (2016) 64:2682–9. doi: 10.1021/acs.jafc.6b00290
100. Singal AG, Kanwal F, Llovet JM. Global trends in hepatocellular carcinoma epidemiology: implications for screening, prevention and therapy. Nat Rev Clin Oncol. (2023) 20:864–84. doi: 10.1038/s41571-023-00825-3
101. Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J Hepatol. (2020) 72:816–27. doi: 10.1016/j.jhep.2019.11.024
102. Ratziu V, Sanyal A, Harrison SA, Wong VW-S, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology. (2020) 72:892–905. doi: 10.1002/hep.31108
103. Yu LX, Schwabe RF. The gut microbiome and liver cancer: mechanisms and clinical translation. Nat Rev Gastroenterol Hepatol. (2017) 14:527–39. doi: 10.1038/nrgastro.2017.72
104. Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, Wargo JA. The microbiome, cancer, and cancer therapy. Nat Med. (2019) 25:377–88. doi: 10.1038/s41591-019-0377-7
105. Depommier C, Everard A, Druart C, Plovier H, Van Hul M, Vieira-Silva S, et al. Supplementation with akkermansia muciniphila in overweight and obese human volunteers: a proof-of-concept exploratory study. Nat Med. (2019) 25:1096–103. doi: 10.1038/s41591-019-0495-2
106. Greten TF, Lai CW Li G, Staveley-O'Carroll KF. Targeted and immune-based therapies for hepatocellular carcinoma. Gastroenterology. (2019) 156:510–24. doi: 10.1053/j.gastro.2018.09.051
107. Jarvis H, Craig D, Barker R, Spiers G, Stow D, Anstee QM, et al. Metabolic risk factors and incident advanced liver disease in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of population-based observational studies. PLoS Med. (2020) 17:e1003100. doi: 10.1371/journal.pmed.1003100
108. Engström A, Wintzell V, Melbye M, Svanström H, Svanström B, Gudbjörnsdottir S, et al. Association of glucagon-like peptide-1 receptor agonists with serious liver events among patients with type 2 diabetes: a Scandinavian Cohort Study. Hepatology. (2024) 79:1401–11. doi: 10.1097/HEP.0000000000000712
109. Wang P, Kang D, Cao W, Wang Y, Liu Z. Diabetes mellitus and risk of hepatocellular carcinoma: a systematic review and meta-analysis. Diabetes Metab Res Rev. (2012) 28:109–22. doi: 10.1002/dmrr.1291
110. Hsu W-H, Sue S-P, Liang H-L, Tseng C-W, Lin H-C, Wen W-L, et al. Dipeptidyl peptidase 4 inhibitors decrease the risk of hepatocellular carcinoma in patients with chronic hepatitis C infection and type 2 diabetes mellitus: a nationwide study in taiwan. Front Public Health. (2021) 9:711723. doi: 10.3389/fpubh.2021.711723
111. Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. (2019) 392:2705–17. doi: 10.1016/S0140-6736(18)31785-9
112. Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. (2018) 391:1174–85. doi: 10.1016/S0140-6736(18)30474-4
113. Galle PR, Dufour JF, Peck-Radosavljevic M, Trojan J, Vogel A. Systemic therapy of advanced hepatocellular carcinoma. Future Oncol. (2021) 17:1237–51. doi: 10.2217/fon-2020-0758
114. Fernández M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. (2009) 50:604–20. doi: 10.1016/j.jhep.2008.12.011
115. Takayasu K, Shima Y, Muramatsu Y, Moriyama N, Yamada T, Makuuchi M, et al. Hepatocellular carcinoma: treatment with intraarterial iodized oil with and without chemotherapeutic agents. Radiology. (1987) 163:345–51. doi: 10.1148/radiology.163.2.3031724
116. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages? J. Hepatol. (2020) 73:757–70. doi: 10.1016/j.jhep.2020.04.025
117. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. New Engl J Med. (2021) 385:1547–58. doi: 10.1056/NEJMoa2036205
118. Gao Y, Zhang W, Zeng L-Q, Bai H, Li J, Zhou J, et al. Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol. (2020) 36:101635. doi: 10.1016/j.redox.2020.101635
119. D'Abbondanza M, Ministrini S, Pucci G, Migliola EN, Martorelli E-E, Gandolfo V, et al. Very low-carbohydrate ketogenic diet for the treatment of severe obesity and associated non-alcoholic fatty liver disease: the role of sex differences. Nutrients. (2020) 12:2748. doi: 10.3390/nu12092748
120. Noakes TD, Windt J. Evidence that supports the prescription of low-carbohydrate high-fat diets: a narrative review. Br J Sports Med. (2017) 51:133–9. doi: 10.1136/bjsports-2016-096491
121. Holmer M, Lindqvist C, Petersson S, Moshtaghi-Svensson J, Tillander V, Brismar TB, et al. Treatment of NAFLD with intermittent calorie restriction or low-carb high-fat diet - a randomised controlled trial. JHEP Rep. (2021) 3:100256. doi: 10.1016/j.jhepr.2021.100256
122. Longo M, Meroni M, Paolini E, Macchi C, Dongiovanni P. Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): new perspectives for a fairy-tale ending? Metabolism. (2021) 117:154708. doi: 10.1016/j.metabol.2021.154708
123. Baumeister SE, Schlesinger S, Aleksandrova K, Jochem C, Jenab M, Gunter MJ, et al. Association between physical activity and risk of hepatobiliary cancers: a multinational cohort study. J Hepatol. (2019) 70:885–92. doi: 10.1016/j.jhep.2018.12.014
124. Baumeister SE, Leitzmann MF, Linseisen J, Schlesinger S. Physical activity and the risk of liver cancer: a systematic review and meta-analysis of prospective studies and a bias analysis. JNCI: J Natl Cancer Inst. (2019) 111:1142–51. doi: 10.1093/jnci/djz111
125. Farzanegi P, Dana A, Ebrahimpoor Z, Asadi M, Azarbayjani MA. Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): roles of oxidative stress and inflammation. Eur J Sport Sci. (2019) 19:994–1003. doi: 10.1080/17461391.2019.1571114
126. Simon TG, Ma Y, Ludvigsson JF, Chong DQ, Giovannucci EL, Fuchs CS, et al. Association between aspirin use and risk of hepatocellular carcinoma. JAMA Oncol. (2018) 4:1683. doi: 10.1001/jamaoncol.2018.4154
127. Simon TG, Duberg AS, Aleman S, Chung RT, Chan AT, Ludvigsson JF. Association of aspirin with hepatocellular carcinoma and liver-related mortality. N Engl J Med. (2020) 382:1018–28. doi: 10.1056/NEJMoa1912035
128. Abdelmalak J, Tan N, Con D, Eslick G, Majeed A, Kemp W, et al. The effect of aspirin use on incident hepatocellular carcinoma-an updated systematic review and meta-analysis. Cancers. (2023) 15:3518. doi: 10.3390/cancers15133518
129. Hassan MM, Curley SA, Li D, Kaseb A, Davila M, Abdalla EK, et al. Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer. (2010) 116:1938–46. doi: 10.1002/cncr.24982
130. Fröhlich E, Wahl R. Chemotherapy and chemoprevention by thiazolidinediones. Biom Res Int. (2015) 2015:845340. doi: 10.1155/2015/845340
131. Yang Y, Zhao L, Huang B, Wang RY, Yuan SX, Tao QF, et al. Pioglitazone, a PPARγ agonist, inhibits growth and invasion of human hepatocellular carcinoma via blockade of the rage signaling. Mol Carcinog. (2015) 54:1584–95. doi: 10.1002/mc.22231
132. Facciorusso A, Aziz MAAE, Singh S, Pusceddu S, Milione M, Giacomelli L, et al. Statin use decreases the incidence of hepatocellular carcinoma: an updated meta-analysis. Cancers. (2020) 12:874. doi: 10.3390/cancers12040874
133. Li S, Saviano A, Erstad DJ, Hoshida Y, Fuchs BC, Baumert T, et al. Risk factors, pathogenesis, and strategies for hepatocellular carcinoma prevention: emphasis on secondary prevention and its translational challenges. J Clin Med. (2020) 9:3817. doi: 10.3390/jcm9123817
Keywords: metabolic dysfunction-associated steatotic liver disease, hepatocellular carcinoma, inflammatory response, oxidative stress, the intestinal microbiome
Citation: Ma Y, Wang J, Xiao W and Fan X (2024) A review of MASLD-related hepatocellular carcinoma: progress in pathogenesis, early detection, and therapeutic interventions. Front. Med. 11:1410668. doi: 10.3389/fmed.2024.1410668
Received: 07 April 2024; Accepted: 20 May 2024;
Published: 04 June 2024.
Edited by:
Gian Paolo Caviglia, University of Turin, ItalyReviewed by:
Kyle Poulsen, University of Texas Health Science Center at Houston, United StatesAaron Balasingam Koenig, INOVA Health System, United States
Antonella Mosca, Bambino Gesù Children's Hospital (IRCCS), Italy
Miriam Longo, IRCCS Ca 'Granda Foundation Maggiore Policlinico Hospital, Italy
Copyright © 2024 Ma, Wang, Xiao and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoming Fan, ZmFueGlhb20xOTg3QDE2My5jb20=
†These authors have contributed equally to this work