 Mariia V. Maksimova1
Mariia V. Maksimova1 Ghislaine J. M. W. van Thiel1Yke Tromp1Rosan Lechner2Johannes J. M. van Delden1
Ghislaine J. M. W. van Thiel1Yke Tromp1Rosan Lechner2Johannes J. M. van Delden1 Lourens T. Bloem3*
Lourens T. Bloem3*- 1Department of Bioethics and Health Humanities, Julius Center for Health Sciences and Primary Care, University Medical Center Utrecht, Utrecht, Netherlands
- 2Department of Clinical Genetics, Erasmus Medical Center, Rotterdam, Netherlands
- 3Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Utrecht, Netherlands
The European Medicines Agency’s conditional marketing authorization (CMA) aims to expedite patient access to medicines for unmet medical needs by shifting a part of the drug development process post-authorization. We highlight ethical issues surrounding CMA, comprising (i) the complexity of defining unmet medical need; (ii) poor understanding of CMA and its impact on informed consent; (iii) hope versus unrealistic optimism; (iv) implications of prolonged post-authorization studies and potential patient harm; (v) rights and duties of patients surrounding participation in post-authorization studies; (vi) access to previously authorized CMA medicines; and (vii) the “benefit slippage” phenomenon, defined as the gradual shift of strict criteria to less strict criteria. We propose a comprehensive research agenda to address these ethical issues, and stress the need for multi-stakeholder engagement to ensure patient-centered use of CMA.
1 Introduction
To enable early access to promising medicines for patients with unmet medical needs–such as rare diseases or poorly treatable cancers–regulatory authorities have implemented expedited pathways that allow marketing authorization of medicines based on preliminary (“non-comprehensive”) data. Expedited pathways are in place in for example the European Union (EU), the United States (US), Japan, Canada, and other countries (1). In the EU, the European Medicines Agency’s (EMA) conditional marketing authorization (CMA) has been in place since 2006 (Box 1). Until the end of 2023, 89 CMAs were granted, of which almost half were granted in the last four years (N = 42, 47%; Figure 1).
| BOX 1 Characteristics of the conditional marketing authorization in the European Union (2). |
| In the European Union, medicines can be granted conditional marketing authorization (CMA) based on non-comprehensive data when they are (i) intended for treatment, prevention or diagnosis of a seriously debilitating or life-threatening disease, (ii) designated an orphan medicine by the European Commission, or (iii) to be used in emergency situations, such as the coronavirus disease 2019 (COVID-19) pandemic. In addition, the following requirements should be met: |
| i. The benefit-risk balance of the medicine is considered positive based on the available evidence; |
| ii. It is likely that the developer can provide comprehensive data in a timely manner to resolve important uncertainties; |
| iii. An unmet medical need is expected to be fulfilled by the medicine; and |
| iv. The benefits to public health of the immediate availability of the medicine outweigh the risks inherent in the fact that additional data are still required. |
| To facilitate that comprehensive data become available and important remaining uncertainties about safety and efficacy are resolved, post-authorization studies called “specific obligations” are imposed as legally binding conditions of the CMA. Each year, the developer must submit a report describing the status of the specific obligations, after which the CMA can be renewed for another year. A continued positive benefit-risk balance is critical for maintaining the CMA. Upon completion of the specific obligations, the CMA can be converted to a standard marketing authorization. |
| The CMA is distinct from the authorization under exceptional circumstances for which comprehensive data are not expected to become available at all. |
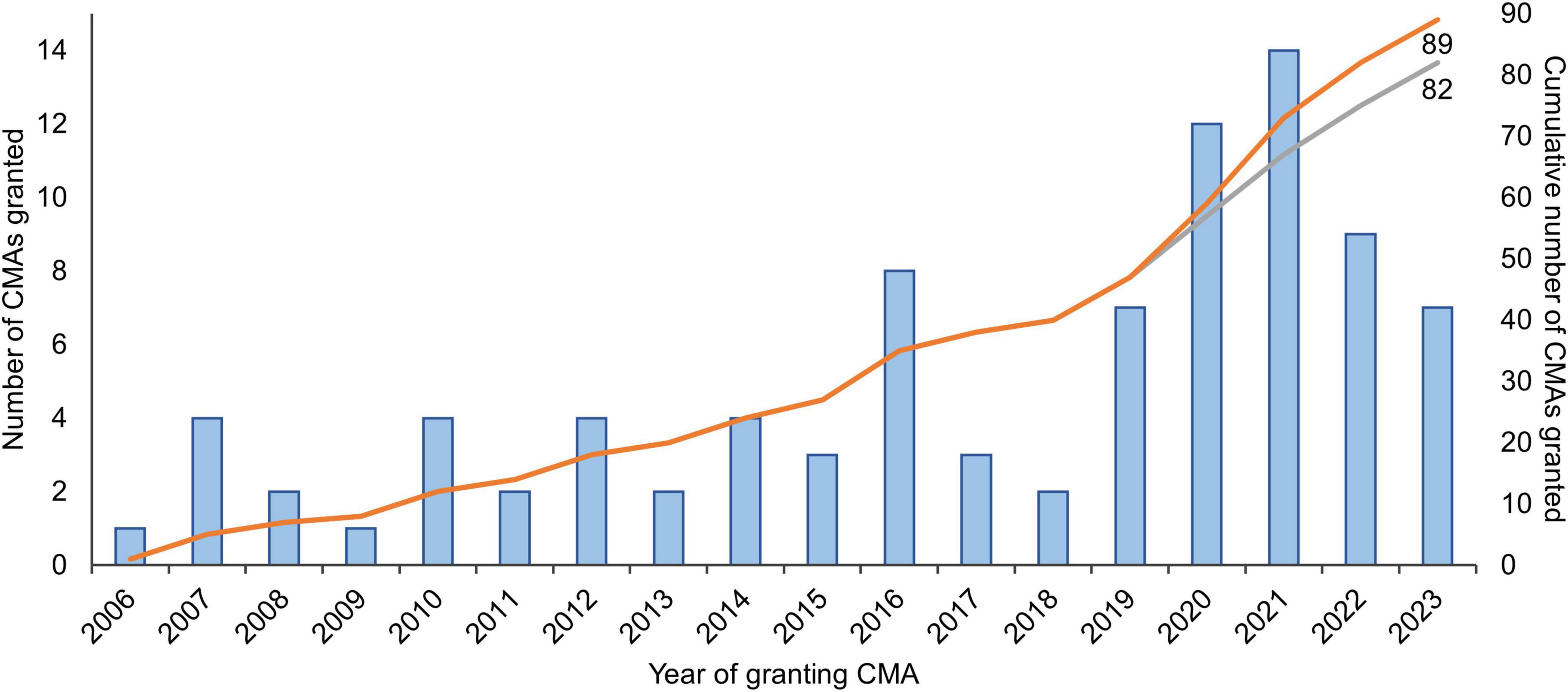
Figure 1. Number of medicines granted conditional marketing authorization in 2006-2023 in the European Union. The gray curve excludes the seven coronavirus disease 2019 (COVID-19) vaccines and treatments that were granted CMA (N = 2 in 2020; N = 4 in 2021; and N = 1 in 2022). Adapted from Bloem et al. (3) and updated with data from the European Commission’s Union Register of medicinal products (4). CMA, conditional marketing authorization.
CMA aims to provide patients with early access to promising medicines through regulatory flexibility. It can offer a valuable opportunity for patients who often have no other options to meet their medical need. On the other hand, CMA comes with heightened uncertainty about benefits and risks and impacts several aspects of the drug development process. Moreover, in Europe, actual patient access is often dependent on national pricing and reimbursement processes following authorization. In this paper, we present an overview of ethical issues relevant to CMA and propose a research agenda to help further the responsible introduction of new medicines granted CMA in clinical practice and the use of CMA as a regulatory tool.
2 From CMA requirements to ethical issues
The CMA pathway forms a unique context in which urgent patient needs must be balanced with ensuring the safety and efficacy of medicines. This context builds on three ethically relevant characteristics of CMA (Box 1). The first characteristic is unmet medical need. The CMA’s primary aim is to provide patients with unmet medical needs with early access to promising medicines. Therefore, the main ethical driver for the CMA is the ethical principle of beneficence, which entails the duty to promote the health and wellbeing of patients with unmet medical needs. The second characteristic is uncertainty about the benefit-risk balance. The CMA introduces risks inherent to the reliance on initial, non-comprehensive data about the safety and efficacy of a new medicine. The third characteristic comprises mandatory post-authorization studies to obtain comprehensive data and resolve important remaining uncertainties (hereafter CMA studies). During these studies, patients in clinical practice face increased uncertainty about benefits and risks compared to patients treated with medicines granted standard marketing authorization. The initial assessment of the benefit-risk balance is not always confirmed with the emerging data from CMA studies (3, 5). As a result, medicines’ indications may be restricted or entire marketing authorizations withdrawn, revoked or not renewed. For example, the third-line treatment indication for ovarian cancer was recently removed for rucaparib (Rubraca) (6, 7), as well as the treatment indication for RET mutation-negative medullary thyroid carcinoma for vandetanib (Caprelsa) (8). The renewal of the marketing authorization of ataluren (Translarna) for Duchenne muscular dystrophy is currently (June 2024) under discussion (9), while the marketing authorization of belantamab mafodotin (Blenrep) for multiple myeloma was previously not renewed (10, 11) and that of olaratumab (Lartruvo) for soft tissue sarcoma revoked (12, 13).
The combination of non-comprehensive evidence at initial marketing authorization and the subsequent mandatory CMA studies moves a part of the drug development process, traditionally performed in the highly controlled research setting, into regular clinical practice. Medicines that not (yet) meet the scientific standard of comprehensive evidence are nonetheless released in the clinical setting in which different actors make decisions and the ethico-legal regulations for treatment apply alongside those for post-authorization research. This results in a hybrid situation in which the medicine is still in the research stage as well as part of (standard) clinical care. Current ethical norms and regulations guiding the drug development process are not optimally suited for such hybrids.
3 Ethical issues related to CMA
Ethical issues related to CMA (Figure 2) are described below and derived from an in-depth analysis of the context of CMA and its implications for the ethical delivery of healthcare. The issues described in this paper are highlighted for their direct impact on CMA stakeholders and their potential to inform and refine ethical guidelines and policy development. Therefore, for each ethical issue, needs for future inquiry are discussed, establishing a comprehensive research agenda for CMA. Although the analysis focuses on ethical issues in the context of CMA, some may also apply to other contexts.
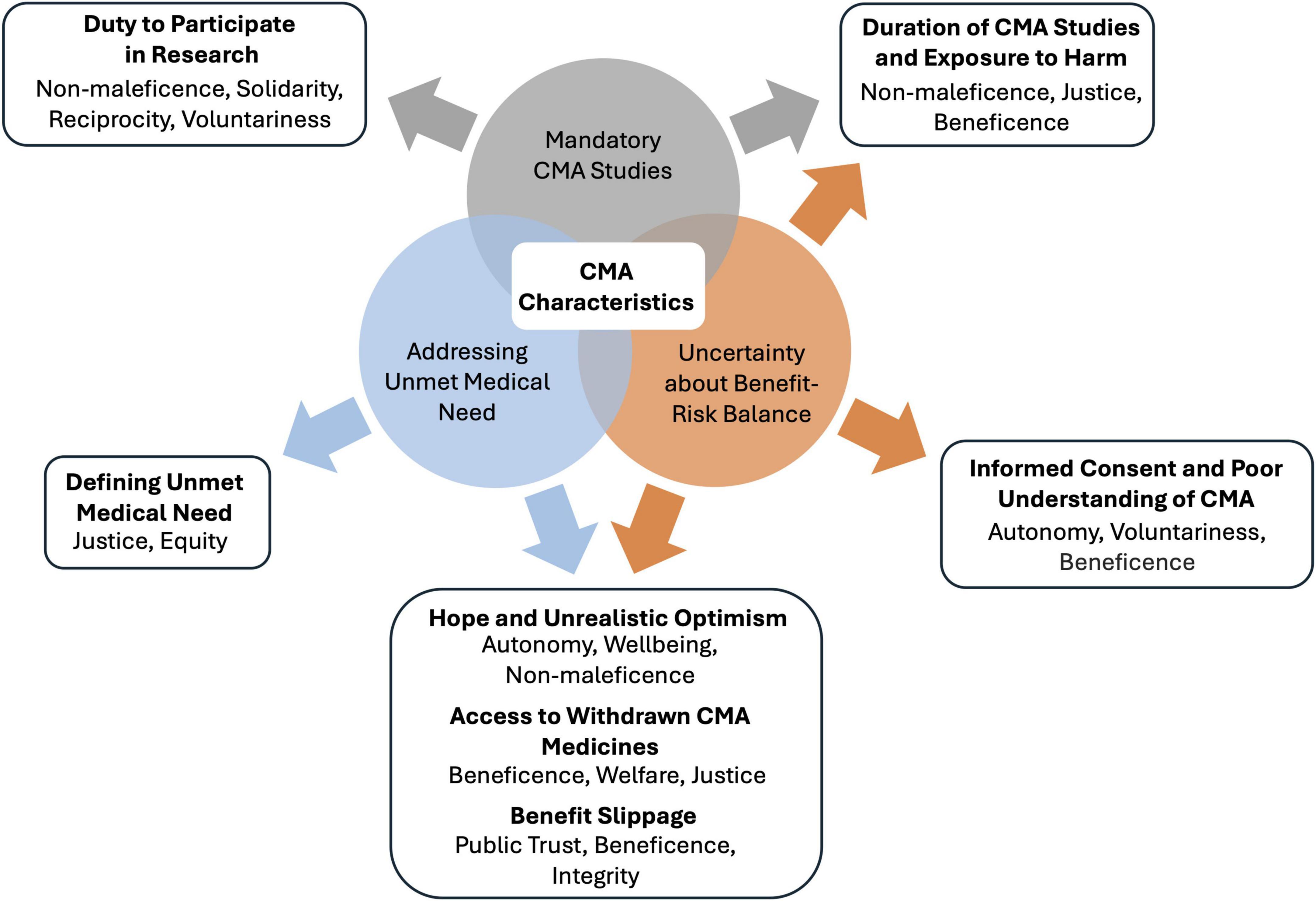
Figure 2. Relationships between characteristics of conditional marketing authorization (CMA) and ethical issues. CMA requirements result in three ethically relevant characteristics of CMA: addressing an unmet medical need, uncertainty about the benefit-risk balance, and mandatory CMA studies. These characteristics and their intersections provoke ethical issues, which are listed in bold. Below each ethical issue, the implicated ethical principles and values are shown. CMA, conditional marketing authorization.
3.1 Defining unmet medical need
Defining “unmet medical need” for CMA presents an ethical challenge as this definition impacts priority setting in diseases and patient groups for expedited regulatory pathways. The current EU regulatory framework identifies unmet medical need as “a condition for which there exists no satisfactory method of diagnosis, prevention or treatment authorized in the Union or, even if such a method exists, in relation to which the medicinal product concerned will be of major therapeutic advantage to those affected” (2). Yet, interpretations vary widely among stakeholders and countries. Vreman et al. (14) identified 16 distinct definitions. Of these, six definitions considered the severity or burden of the disease, and one considered the size of the patient population affected (14).
Various stakeholders have suggested changes to the definition of unmet medical need (15–17). In the light of the reform of the EU pharmaceutical legislation, a definition based on meaningful or substantial reduction in morbidity and mortality was recently proposed (18, 19). However, such proposals may introduce even more ambiguity because of the subjective nature of what is considered a “substantial reduction.” The current debate shows that the assessment of unmet medical need is highly dependent on the scope and the value framework of each stakeholder (14, 17).
Given the significant influence of the definition of unmet medical need on healthcare priorities and resource allocation, researching how to refine this definition is important. Such research should aim to include perspectives of different stakeholders. One way to achieve this could be through multi-criteria decision analyses (20). Additionally, comparative studies across different healthcare systems could provide insights into how varying definitions of unmet medical need impact healthcare delivery and innovation.
3.2 Informed consent and poor understanding of CMA
Voluntary informed consent is a fundamental ethical requirement for clinical care as well as medical research. To fulfill this requirement, patients, physicians, and researchers need to understand the uncertainties surrounding the benefit-risk balance of medicines granted CMA. Studies show that patients and healthcare professionals often misunderstand the disparities in data that may underlie new medicines. For example, Schumacher et al. (21) found that in the context of experimental cancer treatment trials, around 80% of patients did not understand that the benefits of treatment were uncertain and that participation was associated with additional risks. Similarly, Woloshin and Schwartz (22) noted widespread misconceptions among the public about approvals by the US Food and Drug Administration (FDA), with many believing that such approvals guarantee efficacy or the absence of severe side effects.
Physicians also need to have sufficient understanding to communicate effectively with their patients (23, 24). Research shows, however, that physicians often misinterpret the robustness of evidence used for regulatory decision-making by the FDA, leading to overestimated benefits and underestimated risks (23, 25–27). Moreover, uncertainties about the benefit-risk balance are seldom communicated by regulators to physicians, patients, or the public (28).
This jeopardizes the physician’s duty to adequately explain information and subsequently impacts voluntariness and the quality of the patient’s informed consent (29). Research should investigate the understanding of CMA among European patients and physicians, including the broader perceptions of EMA authorizations. In addition, it is important to further study and implement existing strategies to improve the informed consent process (30). Furthermore, there has been a call for attention to specific groups, particularly children, as specific guidelines in this area are currently lacking (31).
3.3 Hope and unrealistic optimism
A combination of dire need and the glimpse of a solution may foster unrealistic expectations of medicines granted CMA among patients and families. Scholars have debated the role of hope, optimism, and realism in access to investigational drugs (32, 33).
False or unrealistic optimism is a cognitive bias where individuals believe they are less likely to experience adverse outcomes and more likely to experience positive outcomes. Unrealistic optimism is distinct from being misinformed (as discussed in relation to informed consent) since it operates as an internal bias undermining the accurate appreciation of risks and benefits of a treatment. This can adversely affect patients’ health and wellbeing (34–36). In ethical terms, unrealistic optimism may compromise autonomy in decision-making which is characterized by intentionality and freedom from controlling influences. For instance, unrealistic optimism shapes how patients decide not only about treatment but also about other life plans, leading them to make choices they would not have considered if they had more realistic expectations (37).
On the other hand, some scholars suggest that unrealistic optimism is not as harmful as is often thought. They argue that unless such optimism leads to choices that are clearly misaligned with patients’ values and goals, it may not be detrimental to autonomy (38–41). Unrealistic optimism, experienced as hope, can have positive effects in healthcare, such as resilience and helping to endure adversity. Hope as resilience was a positive factor associated with recovery, and empowerment (42).
The distinction between beneficial and detrimental expectations is crucial, and conflating them overlooks the possible harms of false hope or optimism (43). For medicines granted CMA, this distinction is essential given their provisional benefit-risk balance, which may foster unrealistic patient expectations. To address this issue, further empirical research could serve as a starting point to gain more insight in relevant factors. As expectations, understanding, and hope are interlinked in medical decision-making, this research should be conducted alongside research on enhancing informed consent processes, as discussed in the previous section.
3.4 Duration of CMA studies and exposure to harm
The CMA of ataluren is currently under discussion after 10 years of CMA status and the indication of vandetanib was restricted after 11 years (8, 9). These cases highlight ethical concerns about the potential harms of prolonged use under CMA. These harms go beyond direct effects of the medicines (44). They also include the missed opportunity for alternative treatments, and the societal harm of inefficient resource allocation. In ethical terms, the timelines until the potential non-renewal and restriction of the CMAs for ataluren and vandetanib raise the question: Was the duty to minimize risk and avoid harm safeguarded sufficiently?
In contrast, the CMAs for olaratumab and belantamab mafodotin were revoked and not renewed after less than three and four years on the market, respectively (10–13). This variance highlights the challenges in determining the feasibility and acceptable duration of CMA studies. However, addressing these problems in the conduct of CMA studies requires a tailored approach. For instance, in rare diseases, patient recruitment challenges can cause delays. Adding pressing time frames could result in effective treatments being restricted, withdrawn, revoked or not renewed prematurely or put undue pressure on patients. It could also increase the uncertainty, when, for example, reliance on surrogate endpoints in clinical research would increase to shorten the time to study completion.
Further research should identify the ethical and social factors that define the optimal duration of CMA studies. This exploration should aim to inform a discussion on acceptable timeframes for the completion of CMA studies and the subsequent decision-making process regarding the conversion or withdrawal, revocation or non-renewal of CMAs, thereby prioritizing the perspectives of patients, physicians, and society.
3.5 Duty to participate in research
The need for efficient CMA studies to resolve uncertainties about safety and efficacy raises another ethical question: Should access to a medicine that is granted CMA come with a duty to participate in CMA studies? Advocates for such a duty argue that individuals who enjoy the benefits of the healthcare system have a moral responsibility to contribute to the medical knowledge that supports it (45–47). They posit that participation in biomedical research is a form of reciprocity rooted in justice and fairness, especially when it can prevent harm to others. Specifically, for medicines granted CMA, participation in research by patients who receive these medicines can accelerate the collection of comprehensive data, limit the use of ineffective or unsafe medicines and thereby safeguard the interests of other patients and optimize public resource allocation.
However, this stance encounters several ethical challenges. First, it may interfere with the principle of voluntariness of research participation—a cornerstone of research ethics underscored in international guidelines (48, 49). The question of voluntariness becomes particularly complex among vulnerable groups who may feel compelled to participate due to unmet medical needs. Framing this duty as contributing to the common good and as inherently beneficial could inadvertently boost hope and potentially unrealistic expectations. Second, CMA studies may involve additional medical procedures, which could be invasive or risky. It is unclear which level of risk or burden would be acceptable under a duty to participate.
Investigating how to shape a duty to participate in a way that avoids these ethical pitfalls could be the focus of further research. For instance, conceptualizing this duty as an act of solidarity in advancing medical practice rather than a forced obligation could shift perspectives. Solidarity, as suggested by Hollestelle et al. (50), is fostered not through imposition but by empowering individuals. This raises a compelling question for future research: How can we encourage patient participation in post-authorization studies in general and CMA studies in particular in an empowering way?
3.6 Access to previously authorized CMA medicines
The experience with restricting indications and withdrawing, revoking or not renewing CMAs due to unmet specific obligations is limited. Meanwhile, losing access to a medicine poses an under-discussed ethical issue in the conceivable scenario when individual patients demonstrate benefit despite a lack of evidence of a favorable benefit-risk balance at the population level.
This scenario shares similarities with post-trial access, as delineated in the Declaration of Helsinki’s Article 34 (49). The principles underpinning post-trial access—emphasizing individual welfare—offer a precedent for interpreting the right to access treatments proven beneficial on a personal level (51). One can suggest that, by analogy, there is prima facie right to individual arrangements after withdrawn, revoked or not renewed CMAs in case of proven personal benefit. However, this interpretation must be approached with caution, recognizing that individual patient improvements might be overestimated or caused by additional factors (52–54). Notably, regulating access to previously authorized CMA medicines may be difficult if guidelines continue to recommend this use while the medicine remains authorized for another (part of the) indication (55). Given the above, there is a need for research on ethically justified and reasonable arrangements for previously authorized CMA medicines, such as a tailored fade-out process.
3.7 Benefit slippage
The aim of CMA to help patients with unmet medical needs may result in so-called “benefit slippage.” Juth (56) identifies benefit slippage as the gradual shift of strict criteria for medical interventions to less strict criteria. He describes this phenomenon observed in neonatal screening programs, where the bar for including conditions lowers over time. This trend occurs because new conditions are added to the screening based on their similarity to previously included ones rather than direct health benefits (56). A similar phenomenon could arise in the context of CMA, where the drive to address unmet medical needs may lead to the acceptance of medicines with a higher level of uncertainty regarding their benefit-risk balance. This may erode original stringent criteria for authorization and undermine the primary aim of the regulatory framework: protecting public health. A previous study observed that applications for CMA in oncology have changed from the use of CMA as a “rescue option” to its proactive use by pharmaceutical companies, suggesting that these companies have learned what types of data are considered acceptable for CMA (3). The increasing use of single-arm trials and surrogate endpoints to support CMA has been a cause of concern, and their use is not limited to CMA (5).
Concerns about benefit slippage have also been expressed by, for example, Swedish governmental organizations. They fear that the European Commission’s proposals for increased use of expedited regulatory pathways will lower evidence standards (57). Future research should examine the phenomenon of benefit slippage in the context of CMA, including whether and how evidence standards may shift over time and the implications for patient care and policy-making.
4 Discussion
Our study of the ethical issues related to CMA reveals the merging of research and clinical practices as a root cause of ethical tension. Historically, these two domains have been distinct (54), but within the realm of CMA, they increasingly overlap, as CMA moves a part of the drug development process into clinical care. CMA is not alone in this trend; several other hybrid practices have emerged, such as highly individualized therapies or N-of-1 clinical strategies, highlighting a broader shift toward integrating research and clinical care (54, 58, 59). This overlap indicates the need for a joint ethical framework to navigate the complexities in such a hybrid healthcare setting, including ethical norms, guidelines, and regulations.
To further improve the regulatory and clinical practices of CMA, it is crucial to actively involve patients, clinicians, and ethicists. Their collaboration is essential in areas like defining unmet medical need, ensuring informed consent, and managing patient expectations. Empirical research can provide insights relevant to specific ethical complexities in the context of CMA. Optimal use of existing best practices (i.e., for informed consent) can also help diminish ethical tension. Addressing the complexities in optimizing CMA study durations, potential restriction, withdrawal, revocation or non-renewal of CMA, and the phenomenon of benefit slippage requires a collaborative ecosystem that comprises all stakeholders, including pharmaceutical companies and policymakers.
In conclusion, the research agenda proposed in this paper provides the starting point for fostering an ethically grounded and patient-centered approach to using CMA, prioritizing welfare and minimizing the risk of harm in the introduction and use of promising new medicines.
Author contributions
MM: Conceptualization, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. GT: Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing. YT: Formal analysis, Investigation, Writing – original draft, Writing – review & editing. RL: Formal analysis, Writing – review & editing. JD: Supervision, Writing – review & editing. LB: Conceptualization, Formal analysis, Investigation, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. The work for this article was in part funded by ZonMw (The Netherlands Organisation for Health Research and Development), through the TAILORED project: Toward a human iPSC neuronal platform for neurodevelopmental disorder therapeutic discovery, project number 10250022110002 (2021-2026). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Centre for Innovation in Regulatory Science.R&D Briefing 81: New drug approvals in six major authorities 2011–2020: Focus on facilitated regulatory pathways and worksharing. London: Centre for Innovation in Regulatory Science (2020).
2. European Medicines Agency.CHMP guideline on the scientific application and the practical arrangements necessary to implement commission regulation (EC) No 507/2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of regulation (EC) No 726/2004 (EMA/CHMP/509951/2006, Rev. 1). Amsterdam: European Medicines Agency (2016).
3. Bloem L, Schelhaas J, Lopez-Anglada L, Herberts C, van Hennik P, Tenhunen O. European conditional marketing authorization in a rapidly evolving treatment landscape: A comprehensive study of anticancer medicinal products in 2006-2020. Clin Pharmacol Ther. (2023) 114:148–60. doi: 10.1002/cpt.2906
5. Koole S, Huisman A, Timmers L, Westgeest H, van Breugel E, Sonke G, et al. Lessons learned from postmarketing withdrawals of expedited approvals for oncology drug indications. Lancet Oncol. (2024) 25:e126–35. doi: 10.1016/s1470-2045(23)00592-2
6. European Medicines Agency.EMA recommends restricting use of cancer medicine Rubraca. Amsterdam: European Medicines Agency (2022).
7. European Medicines Agency.CHMP assessment report for Rubraca (EMA/CHMP/238139/2018). Amsterdam: European Medicines Agency (2022).
8. European Medicines Agency.CHMP assessment report for Caprelsa (EMA/CHMP/30610/2023). Amsterdam: European Medicines Agency (2022).
9. European Medicines Agency.EMA confirms recommendation for non-renewal of authorisation of Duchenne muscular dystrophy medicine Translarna. Amsterdam: European Medicines Agency (2024).
10. European Medicines Agency.EMA confirms recommendation for non-renewal of authorisation of multiple myeloma medicine Blenrep. Amsterdam: European Medicines Agency (2023).
11. European Medicines Agency.CHMP assessment report for Blenrep (EMA/72045/2024). Amsterdam: European Medicines Agency (2023).
12. European Medicines Agency.CHMP assessment report for Lartruvo (EMA/254126/2019). Amsterdam: European Medicines Agency (2019).
13. European Medicines Agency.EMA recommends withdrawal of marketing authorisation for cancer medicine Lartruvo. Amsterdam: European Medicines Agency (2019).
14. Vreman R, Heikkinen I, Schuurman A, Sapede C, Garcia J, Hedberg N, et al. Unmet Medical Need: An Introduction to Definitions and Stakeholder Perceptions. Value Health. (2019) 22:1275–82. doi: 10.1016/j.jval.2019.07.007
15. European Medicines Agency.European medicines agency – payer community meeting. Amsterdam: European Medicines Agency (2017).
16. European Medicines Agency.European medicines agency and European union payer community meeting. Amsterdam: European Medicines Agency (2019).
17. Zhang K, Kumar G, Skedgel C. Towards a new understanding of unmet medical need. Appl Health Econ Health Policy. (2021) 19:785–8. doi: 10.1007/s40258-021-00655-3
18. Wölken T. DRAFT REPORT on the proposal for a regulation of the European Parliament and of the Council laying down Union procedures for the authorisation and supervision of medicinal products for human use and establishing rules governing the European Medicines Agency. Strasbourg: European Parliament (2023).
19. European Commission.Proposal for a REGULATION OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL laying down Union procedures for the authorisation and supervision of medicinal products for human use and establishing rules governing the European medicines agency, amending regulation (EC) No 1394/2007 and regulation (EU) No 536/2014 and repealing Regulation (EC) No 726/2004, Regulation (EC) No 141/2000 and regulation (EC) No 1901/2006. Brussels: European Commission (2023).
20. Cleemput I, Devriese S, Kohn L, Westhovens R. A multi-criteria decision approach for ranking unmet needs in healthcare. Health Policy. (2018) 122:878–84. doi: 10.1016/j.healthpol.2018.06.010
21. Schumacher A, Sikov W, Quesenberry M, Safran H, Khurshid H, Mitchell K, et al. Informed consent in oncology clinical trials: A Brown University Oncology Research Group prospective cross-sectional pilot study. PLoS One. (2017) 12:e0172957. doi: 10.1371/journal.pone.0172957
22. Woloshin S, Schwartz L. Communicating data about the benefits and harms of treatment: A randomized trial. Ann Intern Med. (2011) 155:87–96. doi: 10.7326/0003-4819-155-2-201107190-00004
23. Dhruva S, Kesselheim A, Woloshin S, Ji R, Lu Z, Darrow J, et al. Physicians’ perspectives On FDA regulation of drugs and medical devices: A national survey. Health Aff (Millwood). (2024) 43:27–35. doi: 10.1377/hlthaff.2023.00466
24. Medendorp N, Stiggelbout A, Aalfs C, Han P, Smets E, Hillen MA. A scoping review of practice recommendations for clinicians’ communication of uncertainty. Health Expect. (2021) 24:1025–43. doi: 10.1111/hex.13255
25. Kesselheim A, Woloshin S, Eddings W, Franklin J, Ross K, Schwartz L. Physicians’ knowledge about FDA approval standards and perceptions of the “breakthrough therapy” designation. JAMA. (2016) 315:1516–8. doi: 10.1001/jama.2015.16984
26. Kesselheim A, Woloshin S, Lu Z, Tessema F, Ross K, Schwartz L. Physicians’ perspectives on FDA approval standards and off-label drug marketing. JAMA Intern Med. (2019) 179:707–9. doi: 10.1001/jamainternmed.2018.8121
27. Paquin R, Boudewyns V, O’Donoghue A, Aikin K. Physician perceptions of the FDA’s breakthrough therapy designation: An update. Oncologist. (2022) 27:e85–8. doi: 10.1093/oncolo/oyab021
28. Davis C, Wagner A, Salcher-Konrad M, Scowcroft H, Mintzes B, Pokorny A, et al. Communication of anticancer drug benefits and related uncertainties to patients and clinicians: Document analysis of regulated information on prescription drugs in Europe. BMJ. (2023) 380:e073711. doi: 10.1136/bmj-2022-073711
29. Nelson R, Merz J. Voluntariness of consent for research: An empirical and conceptual review. Med Care. (2002) 40:V69–80. doi: 10.1097/01.Mlr.0000023958.28108.9c
30. i-Consent Consortium.Guidelines for tailoring the informed consent process in clinical studies. (2021). Available online at: https://elsi.health-ri.nl/sites/elsi/files/2021-04/iCONSENT-Guidelines.pdf (accessed March 11, 2024).
31. Lepola P, Kindred M, Giannuzzi V, Glosli H, Dehlinger-Kremer M, Dalrymple H, et al. Informed consent and assent guide for paediatric clinical trials in Europe. Arch Dis Child. (2022) 107:582–90. doi: 10.1136/archdischild-2021-322798
32. Hordijk M, Vermeulen S, Bunnik E. The ‘false hope’ argument in discussions on expanded access to investigational drugs: A critical assessment. Med Health Care Philos. (2022) 25:693–701. doi: 10.1007/s11019-022-10106-y
33. Bunnik E, Aarts N, van de Vathorst S. The changing landscape of expanded access to investigational drugs for patients with unmet medical needs: Ethical implications. J Pharm Policy Pract. (2017) 10:10. doi: 10.1186/s40545-017-0100-3
35. Jansen L. Two concepts of therapeutic optimism. J Med Ethics. (2011) 37:563–6. doi: 10.1136/jme.2010.038943
36. Jansen L. Informed consent, therapeutic misconception, and unrealistic optimism. Perspect Biol Med. (2020) 63:359–73. doi: 10.1353/pbm.2020.0024
37. Delaney J. Does unrealistic optimism undermine patient autonomy? (2023) 26:100859. doi: 10.1016/j.jemep.2022.100859
38. Blumenthal-Barby J, Ubel P. In defense of “Denial”: Difficulty knowing when beliefs are unrealistic and whether unrealistic beliefs are bad. Am J Bioeth. (2018) 18:4–15. doi: 10.1080/15265161.2018.1498934
39. Majumder M, Scott C. Off-target effects of a defense of denial. Am J Bioeth. (2018) 18:22–4. doi: 10.1080/15265161.2018.1498944
40. Weinfurt K. Propositions and pragmatics. Am J Bioeth. (2018) 18:18–20. doi: 10.1080/15265161.2018.1498943
41. Jefferson A, Bortolotti L. Why (Some) unrealistic optimism is permissible in patient decision making. Am J Bioeth. (2018) 18:27–9. doi: 10.1080/15265161.2018.1498940
42. Olsman E. Hope in health care: A synthesis of review studies. In: S van den Heuvel editor. Historical and multidisciplinary perspectives on hope. Cham: Springer International Publishing (2020). p. 197–214.
43. Eijkholt M. Medicine’s collision with false hope: The false hope harms (FHH) argument. Bioethics. (2020) 34:703–11. doi: 10.1111/bioe.12731
44. Pace J, Ghinea N, Kerridge I, Lipworth W. An ethical framework for the creation, governance and evaluation of accelerated access programs. Health Policy (2018) 122:984–90. doi: 10.1016/j.healthpol.2018.07.014
45. Harris J. Scientific research is a moral duty. J Med Ethics. (2005) 31:242–8. doi: 10.1136/jme.2005.011973
46. Schaefer G, Emanuel E, Wertheimer A. The obligation to participate in biomedical research. JAMA. (2009) 302:67–72. doi: 10.1001/jama.2009.931
47. London A. For the common good: Philosophical foundations of research ethics. Oxford: Oxford University Press (2021).
48. Cioms.International ethical guidelines for health-related research involving humans. Geneva: CIOMS (2017).
49. World Medical Association. World medical association declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA. (2013) 310:2191–4. doi: 10.1001/jama.2013.281053
50. Hollestelle M, van der Graaf R, Sturkenboom M, van Delden J. Stimulating solidarity to improve knowledge on medications used during pregnancy: A contribution from the ConcePTION project. BMC Med Ethics. (2023) 24:44. doi: 10.1186/s12910-023-00924-x
51. Mastroleo I. Post-trial obligations in the declaration of Helsinki 2013: Classification, reconstruction and interpretation. Dev World Bioeth. (2016) 16:80–90. doi: 10.1111/dewb.12099
52. Thieffry S, Klein P, Baulac M, Plumb J, Pelgrims B, Steeves S, et al. Understanding the challenge of comparative effectiveness research in focal epilepsy: A review of network meta-analyses and real-world evidence on antiepileptic drugs. Epilepsia. (2020) 61:595–609. doi: 10.1111/epi.16476
53. Raman G, Balk E, Lai L, Shi J, Chan J, Lutz J, et al. Evaluation of person-level heterogeneity of treatment effects in published multiperson N-of-1 studies: Systematic review and reanalysis. BMJ Open. (2018) 8:e017641. doi: 10.1136/bmjopen-2017-017641
54. Defelippe V, van Thiel G, Otte W, Schutgens R, Stunnenberg B, Cross H, et al. Toward responsible clinical n-of-1 strategies for rare diseases. Drug Discov Today. (2023) 28:103688. doi: 10.1016/j.drudis.2023.103688
55. Gyawali B, Rome B, Kesselheim A. Regulatory and clinical consequences of negative confirmatory trials of accelerated approval cancer drugs: Retrospective observational study. BMJ. (2021) 374:n1959. doi: 10.1136/bmj.n1959
56. Juth N. Justifying the expansion of neonatal screening: Two cases. Public Health Ethics. (2019) 12:250–60. doi: 10.1093/phe/phz013
57. Kleja M. Swedish stakeholders: New approval procedures for medicines risk weakening evidence standards: Euractiv. (2022). Available online at: https://www.euractiv.com/section/health-consumers/news/swedish-stakeholders-new-approval-procedures-for-medicines-risk-weakening-evidence-standards/ (accessed March 11, 2024).
58. Lee S, Caruncho M, Chung W, Johnston J, Tabb K, Appelbaum P. Individualized interventions for rare genetic conditions and the research-treatment spectrum: Stakeholder perspectives. Genet Med. (2023) 25:100832. doi: 10.1016/j.gim.2023.100832
Keywords: conditional marketing authorization, research ethics, clinical ethics, expedited regulatory pathways, unmet medical need, uncertainty, informed consent, European Medicines Agency
Citation: Maksimova MV, van Thiel GJMW, Tromp Y, Lechner R, van Delden JJM and Bloem LT (2024) Balancing ethical norms and duties for the introduction of new medicines through conditional marketing authorization: a research agenda. Front. Med. 11:1408553. doi: 10.3389/fmed.2024.1408553
Received: 28 March 2024; Accepted: 13 June 2024;
Published: 24 June 2024.
Edited by:
Carla Matos Torre, University of Lisbon, PortugalReviewed by:
Cristina Sampaio, CHDI Foundation, United StatesViviana Giannuzzi, Fondazione per la Ricerca Farmacologica Gianni Benzi Onlus, Italy
Copyright © 2024 Maksimova, van Thiel, Tromp, Lechner, van Delden and Bloem. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lourens T. Bloem, bC50LmJsb2VtQHV1Lm5s