 Philomène Lavis
Philomène Lavis Ani Garabet3
Ani Garabet3 Alessandra Kupper Cardozo
Alessandra Kupper Cardozo Benjamin Bondue
Benjamin Bondue
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 19 September 2024
Sec. Pulmonary Medicine
Volume 11 - 2024 | https://doi.org/10.3389/fmed.2024.1393778
This article is part of the Research TopicBiomarkers for Progressive Pulmonary FibrosisView all 8 articles
Idiopathic pulmonary fibrosis (IPF) is a rare, chronic, and progressive interstitial lung disease with an average survival of approximately 3 years. The evolution of IPF is unpredictable, with some patients presenting a relatively stable condition with limited progression over time, whereas others deteriorate rapidly. In addition to IPF, other interstitial lung diseases can lead to pulmonary fibrosis, and up to a third have a progressive phenotype with the same prognosis as IPF. Clinical, biological, and radiological risk factors of progression were identified, but no specific biomarkers of fibrogenesis are currently available. A recent interest in the fibroblast activation protein alpha (FAPα) has emerged. FAPα is a transmembrane serine protease with extracellular activity. It can also be found in a soluble form, also named anti-plasmin cleaving enzyme (APCE). FAPα is specifically expressed by activated fibroblasts, and quinoline-based specific inhibitors (FAPI) were developed, allowing us to visualize its distribution in vivo by imaging techniques. In this review, we discuss the use of FAPα as a useful biomarker for the progression of lung fibrosis, by both its assessment in human fluids and/or its detection by imaging techniques and immunohistochemistry.
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive interstitial lung disease (ILD) with an average survival of approximately 3 years (1). The only drugs available, nintedanib and pirfenidone, slow the progression of the disease, but the patient will still progress to respiratory failure. Lung transplantation is the only possible cure for IPF patients (2). In addition to IPF, there are many other diseases associated with the development of pulmonary fibrosis, such as hypersensitivity pneumonitis, occupational pneumonitis, connective tissue diseases, or idiopathic non-specific interstitial pneumonia. A progressive phenotype can also be present in these conditions, and accordingly, it has been shown that 18–32% of non-IPF ILD progressed despite an appropriate treatment (3–6) with a similar prognosis as IPF patients and a median survival of 3.7 years (7–9). Progressive pulmonary fibrosis (PPF) is defined by the presence of two of the three following criteria: (1) an absolute decline of more than 5% of forced vital capacity (FVC) or 10% absolute decline of the diffusion capacity of the lungs for carbon monoxide (DLCO) in 12 months; (2) worsening of the symptoms; (3) worsening of the fibrotic features on high-resolution computed tomography (HRCT) (10). PPF patients treated with nintedanib had a reduced decline of FVC compared to patients receiving placebo (9), indicating the necessity to identify markers of disease progression to better guide doctors in the choice of treatment.
Hypersensitivity and occupational pneumonitis, stage IV sarcoidosis, and ILD related to rheumatoid arthritis and systemic sclerosis are the main causes of PPF with an identified etiology (10, 11). In addition to the etiology of ILD, other risk factors were identified, particularly the presence of a usual interstitial pneumonia pattern (UIP) on the HRCT, the severity score of HRCT, and lower basal values of DLCO, FVC, or saturation of peripheral oxygen (5, 12–15). In progressor patients, survival is also influenced by age and sex, with older men being at greater risk of death (8). Moreover, each disease has associated risk factors for progression. For example, patients with rheumatoid arthritis are more likely to develop an ILD if they are obese, active smokers, or have an active disease (16). Regarding patients with ILD associated with systemic sclerosis, black ethnicity and the presence of anti-topoisomerase I (SCL-70) antibodies are risk factors of severity (17, 18).
In addition to those risk factors which are population-based factors, in clinical practice, there is an urgent need to find reliable patient-specific markers of fibrogenesis progression in order to guide clinicians in their choice of treatment, to determine the individual prognosis of each patient and the disease activity at a specific time (personalized medicine). Different blood biomarkers have already been studied in patients with IPF to assess their risk of progression or their survival (Table 1), but none is currently recommended for daily clinical practice, and even less so for PPF (2, 10). In the present article, we review different studies that reveal fibroblast activation protein α (FAPα) as a potential novel marker of fibrogenesis progression in fibrotic lung diseases.
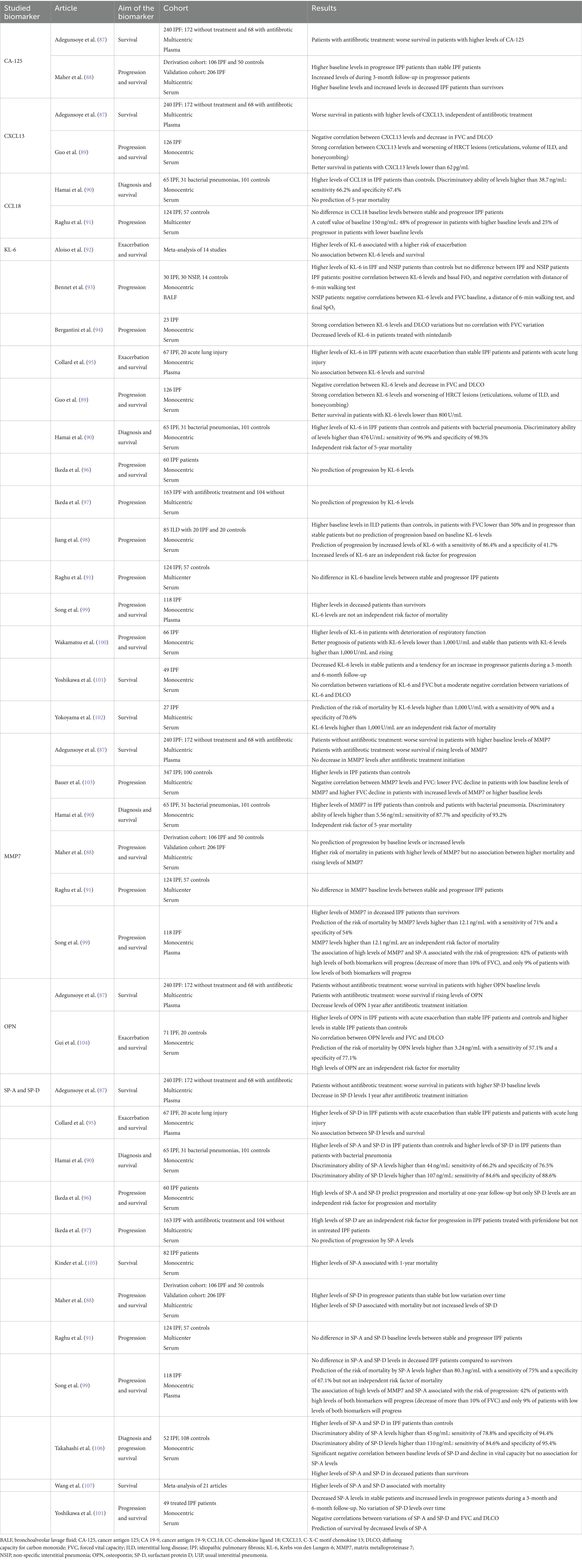
Table 1. Summary of the potential blood biomarkers evaluated in the literature regarding progression and survival in patients with idiopathic pulmonary fibrosis (IPF).
FAPα is a 170 kDa transmembrane serine protease with extracellular activity. It is a member of the prolyl peptidases, along with dipeptidyl peptidase IV (DPPIV), with whom it shares 70% of amino acid sequence homology (19, 20). In addition to a dipeptidyl peptidase enzymatic activity, FAPα has its own endopeptidase enzymatic activity, also known as gelatinase (21, 22). Based on the enzymatic homology between FAPα and DPPIV, it was demonstrated that neuropeptide Y, brain natriuretic peptides, substance P, and peptide YY were FAPα substrates. However, it displayed no effect on chemokines cleaved by DPPIV (23). The dipeptidyl peptidase enzymatic activity of FAPα also inactivates fibroblast growth factor 21, through a cleavage of its C-terminal extremity (24, 25). Regarding its endopeptidase activity, FAPα has an antifibrotic activity as it complements matrix metalloproteinase 1 (MMP1) to cleave collagens I and III (Figure 1) but it is not able to cleave them alone. Of note, no enzymatic activity on collagen IV was detected (20, 26). Moreover, a soluble form of FAPα was identified, also named anti-plasmin cleaving enzyme (APCE) (27). APCE acts as FAPα and complements MMP1 to cleave collagens (26). In addition, APCE cleaves the N-terminal extremity of α2-anti-plasmin, generating a substrate that reduces blood clot degradation and delays fibrinolysis (28). Of note, human and murine FAPα share 89% amino acid sequence and have similar enzymatic activities (29).
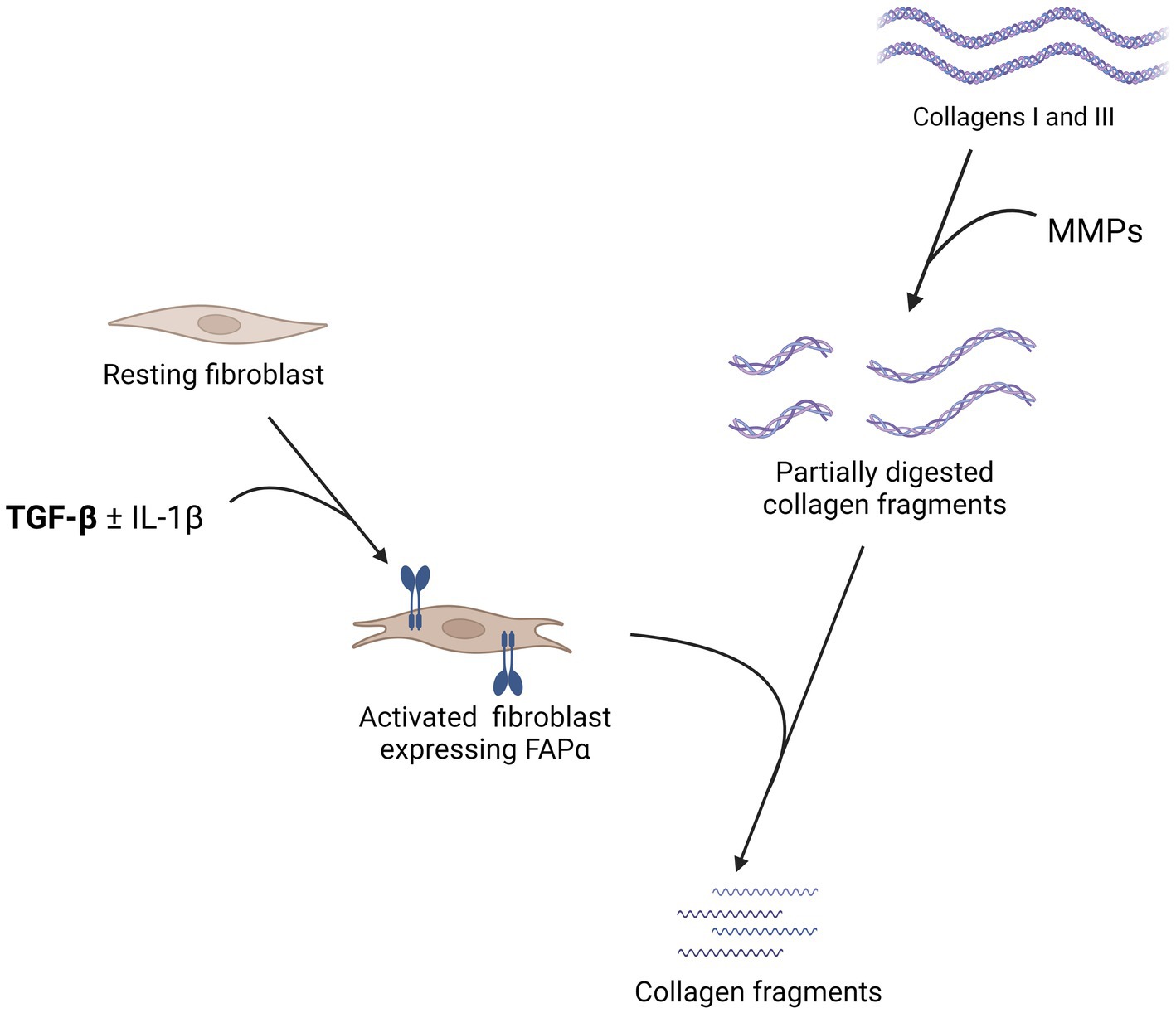
Figure 1. Summary of the pathophysiology of the fibroblast activation protein α (FAPα) in lung fibrosis. The expression of FAPα by fibroblasts is stimulated by transforming growth factor β (TGF-β) with a synergistic effect of interleukin 1β (IL-1β). FAPα is not able to cleave directly collagens I and III but complements matrix metalloproteinases (MMP) by cleaving partially digested collagen fragments. In the absence of FAPα, these partially digested collagen fragments accumulate in the lungs and more extensive fibrosis is observed. Created with Biorender.com.
FAPα is transiently expressed in certain fetal mesenchymal tissues but not in healthy adult tissues (30). Its expression is mainly found on fibroblasts from scar tissues, in pathological conditions of organ fibrosis and in the tumor microenvironment (30–34). FAPα contributes to tumor growth and metastasis through extracellular matrix degradation. Its expression is stimulated by transforming growth factor-β (TGF-β) with a synergistic effect of interleukin 1β (IL-1β) (35). In melanocytes and primary melanoma cells, FAPα expression was stimulated by ultraviolet radiation (36), whereas in an ovarian cancer cell line, FAPα induction was dependent on collagen I exposure (37). FAPα-expressing fibroblasts are different from myofibroblasts (expressing α-smooth muscle actin—αSMA) in their function; thus, FAPα-expressing fibroblasts tended to have a proteolytic profile, while αSMA-myofibroblasts were less involved in extracellular matrix remodeling but had contractibility properties (38).
It was previously demonstrated that FAPα was expressed in lesions of lung fibrosis, particularly in fibrotic interstitium and in fibroblast foci of IPF patients (39). Moreover, Wenlong et al. (40) were able to visualize lesions of fibrosis in vivo in mice by coupling FAPα with luciferase. They confirmed that FAPα was expressed by fibroblasts. However, its role in lung fibrogenesis is controversial; some studies show a pro-fibrotic role, whereas others show an antifibrotic one. Fan et al. (41) used two models to study lung fibrosis in FAPα-knock-out (FAPKO) mice: the bleomycin model and the thoracic irradiation model. The bleomycin model is the most widely used animal model to study lung fibrosis as it induces significant fibrosis in 3 weeks. During the first week, a strong immune response is observed, and lesions of fibrosis subsequently develop to reach a maximum at 21 days after the initiation of the model. However, after 28 days, the fibrosis tends to resolve (42–44). The thoracic irradiation model also induces a strong lung fibrosis that is similar to what is observed in humans after thoracic radiotherapy. The fibrosis takes, however, more time to develop between 24 and 30 weeks (43). In both models, Fan et al. (41) observed that wild-type (WT) mice had a better survival and developed less fibrosis than FAPKO mice. In the basal state, FAPKO mice had already more hydroxyproline lung content than WT mice. Moreover, they observed that isolated lung fibroblasts from FAPKO mice exposed to TGF-β differentiated more into myofibroblasts. Finally, they observed that the FAPKO mice presented an accumulation of collagen fragments in the lungs, confirming that FAPα cleaves products of MMP rather than complete collagen fibers (26). This antifibrotic role of FAPα was also observed by Kimura et al. (45) using the chronic bleomycin mouse model in two types of FAPα-deficient mice: FAPKO mice and mice depleted of FAP+ cells. The chronic bleomycin model has the advantage of inducing an irreversible lung fibrosis, and the histological lesions are more similar to what is observed in IPF patients (46). These two mouse lines showed exacerbated lung fibrosis, and the FAPKO mice a higher lung infiltration of immune cells (45). However, they did not observe any significant difference regarding fibrosis between transgenic and WT mice when lung fibrosis was induced by constitutive expression of active TGF-β. They hypothesized that the diverse fibrosis stimulus can trigger different subtypes of fibroblasts and therefore modulate the fibrotic response. Indeed, bleomycin could activate fibroblasts in FAP+ cells that present a proteolytic profile, whereas TGF-β would be more involved in fibroblast-to-myofibroblast differentiation and thus in tissue contraction (38, 45). On the contrary, Egger et al. (47) observed pro-fibrotic properties of FAPα. They induced fibrosis in WT mice with the chronic bleomycin model and treated them with an inhibitor of FAPα, namely, PT100. PT100-treated mice had a better survival and presented fewer areas of pulmonary fibrosis. It is to be noted that PT100 is an inhibitor of all the DPP family and not only FAPα (48). The contradictory results could therefore be explained by this lack of specificity, which is not found in mouse models using FAPKO mice or mice depleted of FAP+ cells (41, 45).
Other inhibitors of FAPα were also studied in fibrosis affecting other organs. Yang et al. (49) performed a model of liver fibrosis and assessed an antifibrotic treatment with another quinoline-based FAPα inhibitor. Indeed, FAPα is expressed by hepatic stellate cells and activated fibroblasts in cirrhotic liver but not in healthy liver (31). The inhibitor of FAPα led to a lower mononuclear immune infiltrate, reduced collagen levels, and less fibrosis. As the proliferation of hepatocytes was higher, they hypothesized that the inhibitor also promoted regeneration of hepatocytes (49). Finally, Dienus et al. (32) studied fibroblasts from keloid scars and observed a higher expression of FAPα. The use of a FAPα inhibitor on isolated keloid fibroblasts reduced their invasion but did not influence procollagen I and fibronectin synthesis.
Overall, these studies showed contradictory results, with mice inactivated for FAPα appearing to be more susceptible to fibrosis (41, 45), whereas pharmacological inhibitors of FAPα seem to reduce the development of fibrosis in various mouse models (32, 47, 49). Therefore, more studies need to be performed to better understand FAPα pathophysiology as well as the precise mechanism of action of these inhibitors before considering it as a potential therapeutic target.
Currently, there are no available biomarkers to identify fibrosis activity, particularly lung fibrogenesis. Various proteins were identified to be associated with a worse prognosis of IPF and therefore associated with the severity of the disease as cancer antigen 125, cancer antigen 19-9, or MMP7, but direct biomarkers of fibrogenesis are still missing (50). Moreover, the use of these biomarkers is not validated yet in daily clinical practice (10). FAPα is specifically expressed in fibroblast foci that are lesions of active fibrosis at the interface between healthy lung tissue and already fibrosed lung. Moreover, a soluble form of FAPα could be identified in biological liquids, rendering this protein a promising candidate as a biomarker for progressive lung fibrosis (21, 39). In the following paragraphs, we will discuss different studies that provide substantial evidence to support this assertion.
Our group showed first that bleomycin-treated mice presented higher concentrations of FAPα in bronchoalveolar lavage fluid (BALF) as compared to control mice during the active phase of fibrogenesis. Importantly, a significant dose-response effect was observed. The levels of FAPα in BALF were correlated with weight loss and quantity of fibrosis. Moreover, a significant decrease in FAPα concentrations in BALF was observed in nintedanib-treated mice. Accordingly, IPF patients also showed higher levels of FAPα in BALF as compared to healthy controls. In addition, when IPF patients were classified into stable or progressors according to the 2022 ATS/ERS/JRS/ALAT Clinical Practice Guidelines (10), patients with a progressive disease had higher FAPα levels than controls and stable patients (Figure 2) (51). Finally, FAPα BALF levels higher than 192.5 pg/mL could predict the risk of progression, transplantation, or death with a sensitivity of 100% and a specificity of 90% (51). Of note, no association between FAPα concentrations with disease progression nor survival was found in a study analyzing the serum of 149 IPF patients (52), indicating local and not serum FAPα as a candidate biomarker for lung fibrosis. No comparison with control patients was performed (52).

Figure 2. Assessment of FAPα levels in bronchoalveolar lavage fluid (BALF) from control subjects (CTRL) and patients with idiopathic pulmonary fibrosis (IPF) with either a stable or a progressive phenotype according to the 2022 ATS/ERS/JRS/ALAT Clinical Practice Guidelines (10). Data are reproduced from Lavis et al. (51).
Although promising, the data on BALF come from a single center and more studies using independent cohorts of patients should be analyzed to affirm FAPα as a biomarker of fibrotic lung diseases. Interestingly, other studies have also evaluated its use in other fibrotic diseases. Particularly, Uitte de Willige et al. (53) observed that FAPα plasma levels decreased in patients after transplantation for severe liver damage following hepatitis C infection. Lower plasma FAPα concentrations were associated with a lower fibrotic score in patients with non-alcoholic fatty liver disease and could also exclude severe fibrosis in patients with metabolic syndrome (54). Keane et al. (55) also showed that the activity of soluble FAPα was higher in cirrhotic patients. On the contrary, no significant difference was observed between patients with various atherosclerotic diseases and healthy controls. However, they observed that FAPα concentrations were higher in men than women and were associated with hyperlipidemia and body weight (56). Moreover, two independent groups did not highlight significant differences in blood FAPα levels in patients with systemic sclerosis, a disease characterized by a certain degree of skin fibrosis and involvement of other organs such as the lungs (57, 58).
In conclusion, currently, there are few studies evaluating the association of FAPα plasma or serum levels with fibrosis and the data are still controversial. On the contrary, preliminary data on the use of FAPα BALF concentrations as biomarker of lung fibrosis seem encouraging. As mentioned earlier, these results were obtained on a small cohort of IPF patients and multicenter studies should be performed to validate FAPα BALF levels as a biomarker of IPF and possibly as other progressive ILD.
As previously described, FAPα is a marker of activated fibroblasts and cancer-associated fibroblasts. Based on specific enzymatic inhibitors of FAPα (FAPI), radiotracers were thus recently developed to identify in vivo the tumoral microenvironment. They are considered as pan-cancer radiotracers as PET/CT imaging of FAPI labeled with gallium-68 (68Ga) can specifically identify various prevalent cancers and their metastasis as breast, lung, colorectal, and prostate carcinoma (59). In addition to identifying cancer-associated fibroblasts, FAPI imaging seems also useful for non-oncological conditions, particularly some auto-immune diseases such as rheumatoid arthritis and Crohn’s disease but also fibrotic lesions in the heart and lungs (60–63). Particularly, 68Ga-FAPI was first tested in patients with lung cancer and lesions of lung fibrosis. Whereas the uptake of the radiotracer was identical between the tumor and the lesions of fibrosis, an early uptake of 68Ga-FAPI was observed in fibrotic lesions as compared to malignant ones (63). The uptake of 68Ga-FAPI in fibrotic lesions correlated with lung density (63) and was higher in IPF patients than controls (64). Bergmann et al. (65) evaluated the uptake of 68Ga-FAPI-04 in patients with systemic sclerosis-associated ILD and observed a heterogeneous uptake in the fibrotic lung, significantly higher than what was observed in controls. In this study, the highest uptake was observed in patients with extended ILD disease, greater impairment of respiratory tests, history of progression or progressive disease, or higher activity score of the disease. Moreover, they found a cutoff value of 68Ga-FAPI-04 uptake allowing them to predict the risk of progression. Finally, they noticed an association between the radiotracer uptake and the response to nintedanib, with an increased uptake in a patient with a worsening of its respiratory function, a status quo in two stable patients and a decreased uptake in two patients with an improvement of their respiratory function. The use of 68Ga-FAPI-04 was beneficial to evaluate myocardial fibrosis in patients with systemic sclerosis as the uptake was higher than patients without myocardial fibrosis and was associated with the risk of arrhythmia and heart failure (66). 68Ga-FAPI-46 was also able to identify lesions of pulmonary fibrosis following COVID-19 that were not detected by fluorodeoxyglucose coupled with fluor-18 (18F-FDG) (67). One study evaluated FAPI-74 coupled with 18F in IPF patients, where they observed that the uptake of the radiotracer was higher in IPF patients than the controls, and a strong correlation was observed between lung density and 18F-FAPI-74 uptake (64). Liu et al. (68) used single-photon emission computed tomography (SPECT) to detect FAPI by coupling it with 99Technetium. In agreement with the studies described above, a significantly higher uptake of FAPI was observed in the lower lobes of IPF patients than controls. SPECT imaging has advantage of being cheaper and delivering less radiation than PET/CT, therefore being a good alternative.
Preclinical murine models of lung fibrosis were also established to evaluate various FAPI-based radiotracers. Rosenkrans et al. (69) assessed 68Ga-FAPI-46 in the bleomycin lung model on days 7 and 14 after the instillation. They observed a higher and significant lung uptake of the radiotracer in bleomycin-treated mice than controls at these two timepoints, in contrast to lung density that was significantly different only at day 14. Compared with the uptake of 18F-FDG, less background in the heart and the brain were observed. Our group obtained similar results with 18F-FAPI-74; thus, a higher and significant uptake of the tracer was observed in bleomycin-treated mice than controls at days 10 and 16 after the instillation. We did not observe any significant difference at day 3, during the inflammatory phase nor at day 28 when the fibrosis is already established. Our results showed that 18F-FAPI-74 is a specific marker of fibrogenesis as lung uptake was strongly correlated with the development of fibrosis, assessed by lung density, lung content of hydroxyproline, and the Ashcroft modified scale. However, we could not observe significant differences in 18F-FAPI-74 uptake in mice treated with different doses of bleomycin that caused different levels of fibrosis (51).
FAPI uptake specificity for fibrogenesis was also observed in a model of tendinopathy with a high uptake of the tracer at day 7 after the injury and no significant difference after 4 weeks (70) and also allowed to discriminate inflammatory and fibrotic lesions in IgG4-related disease (71).
Song et al. (72) used a model of lung fibrosis induced by paraquat poisoning and observed a higher uptake of Al18F-NODA-FAPI-04 in poisoned mice than controls. Two patients after paraquat poisoning were also included, and a diffuse uptake in the lung bases was observed.
In summary, various studies showed that FAPI radiotracer uptake was higher in patients with lung fibrosis (IPF and non-IPF) as compared to controls, without being able to discriminate different etiologies of fibrosis. Compared to FDG, FAPI seems to be a promising specific marker of fibrogenesis that could help to differentiate active from inactive fibrotic lesions (Figure 3). Another limitation of 18F-FDG is that the intensity of the uptake was partly related to lung density, and this limitation is not found for FAPI radiotracers (73). Moreover, the response to antifibrotic treatments could not be assessed by 18F-FDG uptake (74), whereas encouraging preliminary results showed an association between the response to nintedanib and the uptake of FAPI in ILD patients with associated systemic sclerosis (65). 68Ga-DOTATATE, an analog of the somatostatin receptor was also previously assessed as a biomarker of lung fibrosis (75). However, contrary to FAPI, it is not entirely specific to fibroblasts and the detection of the somatostatin receptor 2a was found in alveolar macrophages, smooth muscle cells, epithelial bronchial cells, focally on endothelial cells and alveolar type 2 cells in bleomycin-treated mice (76). Moreover, a comparison between 18F-FDG uptake and 68Ga-DOTATATE was performed and showed that both presented the same distribution and uptake intensity (75). The assessment of 129Xenon red blood cell uptake by magnetic resonance enables pulmonary diffusion to be evaluated and thus reflects changes in lung microstructures (77, 78). However, it is not a direct marker of fibrogenesis. Recently, it was demonstrated that over a 12-month follow-up period, the tracer uptake was modified despite the absence of any significant decline in the pulmonary function tests. However, their cohort was quite heterogeneous with untreated patients and patients already receiving an antifibrotic treatment, making it impossible to assess whether 129Xenon uptake predicts response to antifibrotic treatment (78). As this imaging technique also seems promising, a study to compare FAPI and 129Xenon uptake to assess ILD progression and response to antifibrotic treatment could be performed.
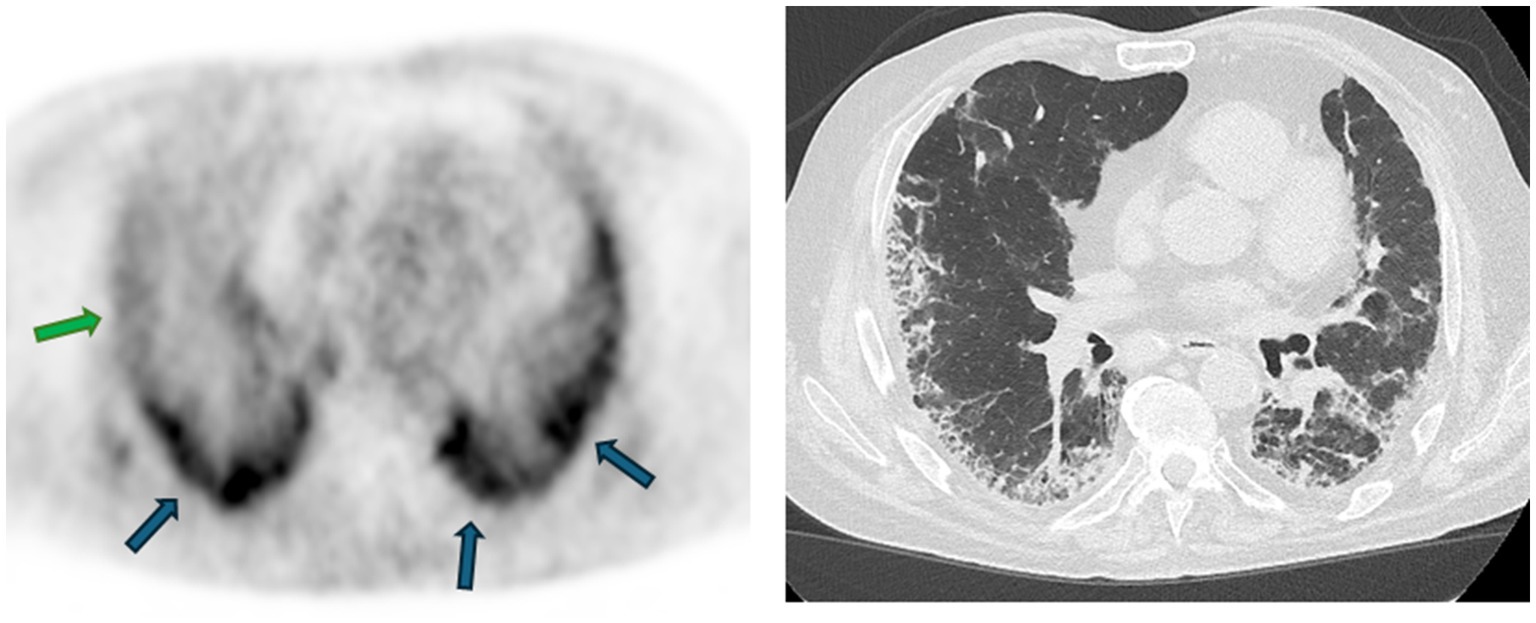
Figure 3. 68Ga-FAPI uptake assessed by PET/CT in a patient with idiopathic pulmonary fibrosis and the corresponding high-resolution CT. An intense uptake of the radiotracer is observed in some subpleural lesions (blue arrows), whereas some present a weak uptake (green arrow), suggesting a lower fibrogenesis activity kindly provided by the Department of Nuclear Medicine of the Hôpital Universitaire de Bruxelles (HUB), Brussels, Belgium.
Currently, FAPI coupled with 68Ga is the mostly used radiotracer (33, 63, 65), but some studies evaluating FAPI coupled with 18F or 99Tc have also demonstrated their feasibility and their ability to identify fibrotic lesions (64, 68). Further studies should be carried out to identify which pairing (subtype of FAPI and labeling) gives the best results in identifying lesions of fibrogenesis, with the lowest background and unspecific uptake. Overall, FAPI imaging is a promising tool to help clinicians determine the best timing to start antifibrotic treatments in non-IPF pulmonary fibrosis or evaluate the impact of other therapeutical interventions. We should keep in mind that even if the results are encouraging, more studies should be carried out to exactly determine how FAPI PET/CT imaging could be used as part of patient management.
The identification of fibroblast foci on lung slides from patients with lung fibrosis is a hallmark of UIP (the pattern observed in IPF). In addition to UIP, some fibroblast foci can be observed in other conditions, such as non-specific interstitial pneumonia (NSIP). Fibroblast foci are found at the interface between healthy and fibrotic lung tissue, thus representing the active pulmonary fibrotic lesion (79). However, the origin of fibroblasts present in fibroblast foci is a matter of debate. One hypothesis is that they originated from alveolar epithelial cells undergoing epithelial-to-mesenchymal transition (EMT) under the influence of TGF-β (80). Another research group suggested that type 2 alveolar epithelial cells that had transited to a mesenchymal phenotype were not directly the main source of fibroblasts and myofibroblasts but promoted a pro-fibrotic environment leading to more fibroblast-to-myofibroblast differentiation (81).
The majority of the studies tend to show that the number and size of fibroblast foci are associated with poorer survival in patients with lung fibrosis (82, 83). Accordingly, the number of fibroblast foci was higher in patients with UIP than patients with NSIP in which the prognosis is better than in UIP (82). Collard et al. (84) observed a correlation between the number of fibroblast foci and a decrease in FVC and worsening of dyspnea at 6-month follow-up.
FAPα was shown to be a sensitive and specific marker of activated fibroblasts, with an expression found in fibroblast foci and fibrotic interstitium of UIP lesions but not on lung slides from patients with emphysema or healthy subjects (33, 39). Acharya et al. (39) also observed that FAPα expression on lung slides allowed the identification of some fibroblast foci that were not visible on the hematoxylin-eosin slide. They also showed that FAPα is more specific to fibroblast foci, as αSMA, a marker of transition of fibroblast to myofibroblasts, is also expressed by smooth muscle cells and therefore detected in vascular interstitium. In line with these studies, we also observed a clear staining of FAPα in fibroblast foci of patients with IPF (Figure 4). As the number of fibroblast foci seems to be associated with the prognosis of patients with lung fibrosis, FAPα immunohistochemistry staining could help clarifying the prognosis of patients for whom a cryobiopsy or a surgical lung biopsy is necessary for diagnosis. We have recently observed immunohistochemistry staining of FAPα in some hyperplastic alveolar epithelial cells that may be undergoing EMT, a hypothesis that needs to be tested (51).
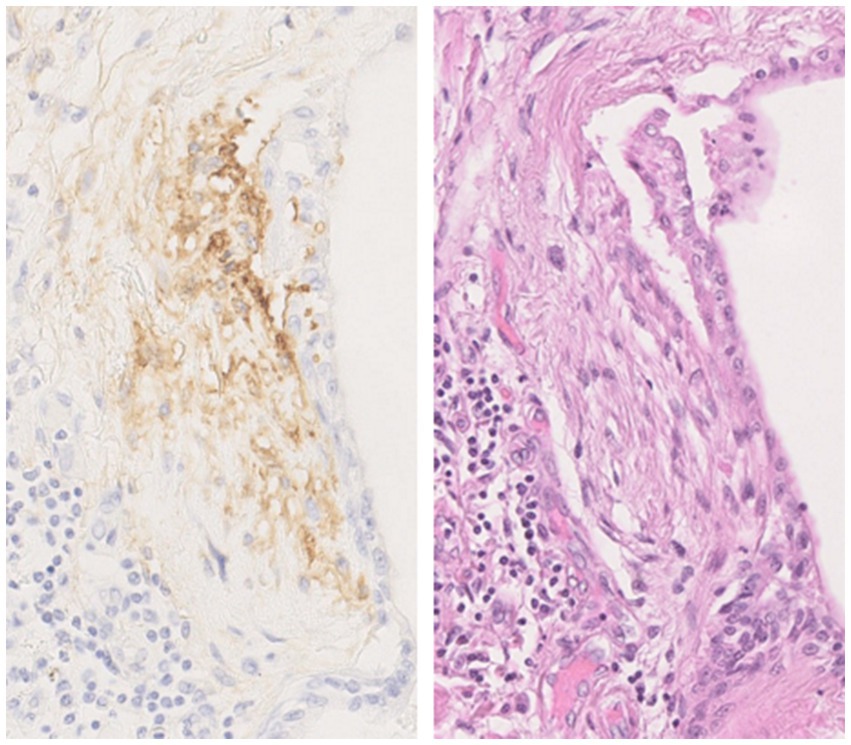
Figure 4. Fibroblast activation protein α (FAPα) immunostaining in a fibroblast foci from a lung explant of a patient with idiopathic pulmonary fibrosis with the corresponding hematoxylin-eosin staining. The use of FAPα immunostaining enables rapid identification of fibroblast foci and a more accurate assessment of their number. Magnification fold 200×. The images come from the Biobank of Pneumology—HUB. The protocol for FAPα immunostaining was detailed in Lavis et al. (51).
Overall, FAPα staining appears to be a sensible marker of fibroblast foci that could be implemented in routine practice to help identifying fibroblast foci and therefore precise patients’ prognosis. Some studies could also be conducted to confirm the expression of FAPα by alveolar epithelial cells and verify whether it can be a novel marker of cells undergoing EMT in lung fibrosis.
Specific biomarkers allowing to identify IPF patients at risk of rapid progression at diagnosis or during the course of the disease are urgently needed. Moreover, a significant number of other lung fibrosis can evolve despite adequate treatment, and these biomarkers would be extremely helpful for these conditions. In this context, interest in FAPα has grown strongly in recent years, in particular due to its specificity for activated fibroblasts and the development of specific quinoline-based inhibitors that are able to target this protein and therefore visualize it by imaging techniques. In addition, the presence of a soluble form of FAPα, enabling the measurement of it in biological fluids, makes it a potential biomarker for the evaluation and follow-up of lung fibrosis. The perspectives regarding the use of FAPα as a biomarker of lung fibrogenesis are summarized in Figure 5.
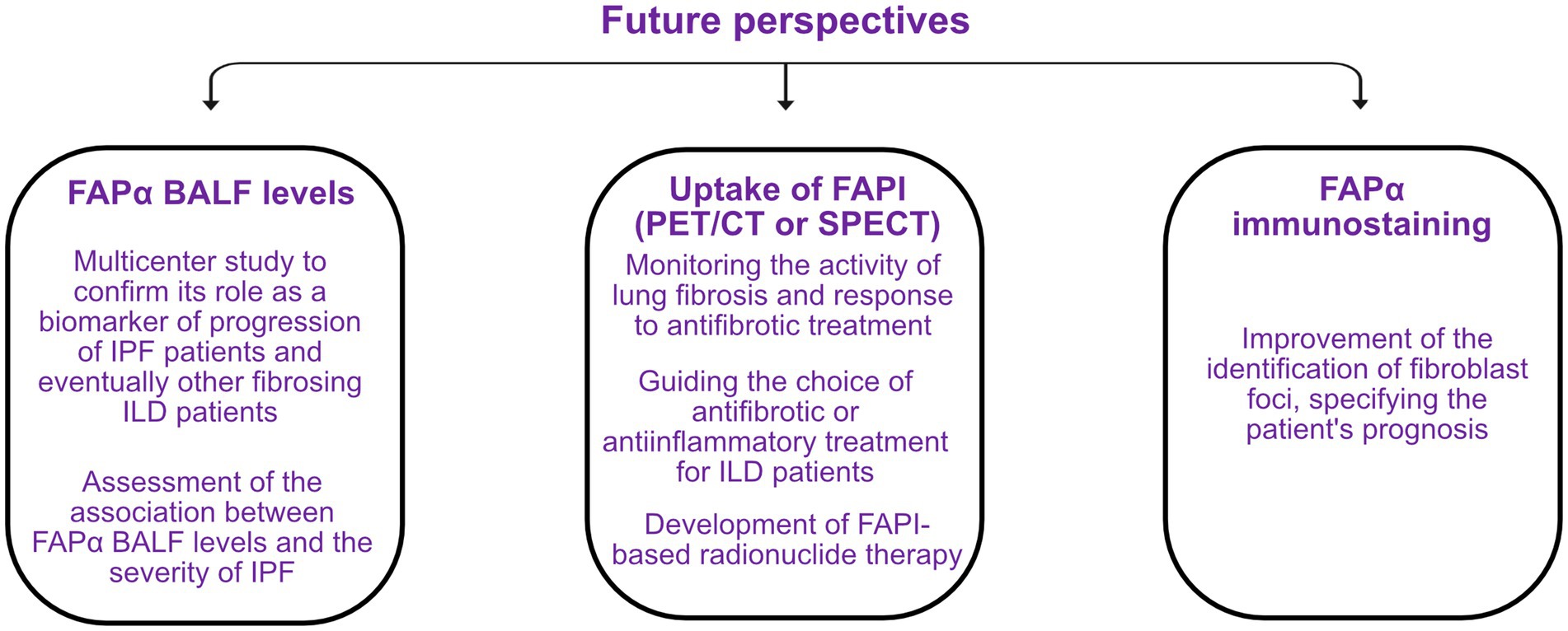
Figure 5. Perspectives on the use of the fibroblast activation protein α (FAPα) as a future biomarker of fibrogenesis in idiopathic pulmonary fibrosis (IPF) and other fibrosing interstitial lung diseases (ILD). BALF, bronchoalveolar lavage fluid; FAPI, FAPα inhibitor; PET/CT, positron emission tomography/computed tomography; SPECT, single-photon emission computed tomography.
Among fluids of interest, the assessment of FAPα in BALF seems particularly promising. Indeed, FAPα BALF levels are higher in IPF patients than controls and are associated with the progression of the disease (51). Currently, a BALF is performed in the majority of patients with a suspected ILD (2) and FAPα measurement could therefore be easily implemented as one of the parameters to be analyzed. Our study involved a limited number of IPF patients, and these results should be validated in larger multicenter studies but could eventually provide an assessment of a patient’s risk of progression at diagnosis. FAPα concentrations should also be measured on BALF from non-IPF ILD patients and could allow the identification of PPF patients in this group. The association between FAPα BALF levels and the severity of the disease could also be assessed.
For some ILD patients, the diagnosis of the etiology of lung fibrosis is difficult, and then, a surgical lung biopsy or a cryobiopsy is recommended (2). As FAPα helps identifying more precisely fibroblast foci and their number and size seem to be associated with the prognosis of the patient, this staining could be performed on lung samples and define the exact number of active lesions of the patient (39).
The detection of FAPI through the use of PET/CT or SPECT appears to be a powerful tool to identify active fibrogenesis, independent of the etiology of lung fibrosis. This could help monitor the activity of the disease, allowing to adapt drug doses to the patient and also assessing the response to antifibrotic treatment, as previously shown by Bergmann et al. (65). Compared to 18F-FDG, it is more specific for fibrogenesis, not influenced by lung density, and shows less background in brain and heart. Moreover, the uptake seems to be linked to active fibrotic processes, not inflammation (51, 69, 71). In certain ILD patients, this could help determine whether their pulmonary disease is predominantly inflammatory or fibrosing and thus help guide the choice of treatment. A future perspective on the use of FAPI is radionuclide therapy, directly targeting FAPα+ fibroblasts. Preliminary studies already assessed its efficacy in various advanced and progressive cancers, as FAPα is expressed by cancer-associated fibroblasts, and promising results were obtained (85). The treatment also seemed to be well tolerated (85, 86). Such studies on ILD patients could be considered given the specificity of FAPI for activated fibroblasts, mediating the progression of IPF and other PPF.
In brief, the identification of FAPα, whether in BALF, by nuclear imaging, or on lung sections, appears promising for assessing the risk of progression in fibrotic patients, response to treatment, and survival.
PL: Conceptualization, Writing – original draft, Writing – review & editing. AG: Writing – original draft, Writing – review & editing. AC: Funding acquisition, Writing – original draft, Writing – review & editing. BB: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. PL has a PhD scholarship from the Belgian National Fund for Scientific Research (FNRS-FC 43697) and is a grant holder of Fonds Erasme. BB received grants from Fonds Erasme and FNRS (CDR-J.0017.24).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ley, B, Collard, HR, and King, TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2011) 183:431–40. doi: 10.1164/rccm.201006-0894CI
2. Raghu, G, Remy-Jardin, M, Myers, JL, Richeldi, L, Ryerson, CJ, Lederer, DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2018) 198:e44–68. doi: 10.1164/rccm.201807-1255ST
3. Guler, SA, Winstone, TA, Murphy, D, Hague, C, Soon, J, Sulaiman, N, et al. Does systemic sclerosis-associated interstitial lung disease burn out? Specific phenotypes of disease progression. Ann Am Thorac Soc. (2018) 15:1427–33. doi: 10.1513/AnnalsATS.201806-362OC
4. Reiseter, S, Gunnarsson, R, Mogens Aaløkken, T, Lund, MB, Mynarek, G, Corander, J, et al. Progression and mortality of interstitial lung disease in mixed connective tissue disease: a long-term observational nationwide cohort study. Rheumatology. (2018) 57:255–62. doi: 10.1093/rheumatology/kex077
5. Zamora-Legoff, JA, Krause, ML, Crowson, CS, Ryu, JH, and Matteson, EL. Progressive decline of lung function in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheumatol. (2017) 69:542–9. doi: 10.1002/art.39971
6. Nasser, M, Larrieu, S, Si-Mohamed, S, Ahmad, K, Boussel, L, Brevet, M, et al. Progressive fibrosing interstitial lung disease: a clinical cohort (the PROGRESS study). Eur Respir J. (2021) 57:2002718. doi: 10.1183/13993003.02718-2020
7. Brown, KK, Martinez, FJ, Walsh, SLF, Thannickal, VJ, Prasse, A, Schlenker-Herceg, R, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J. (2020) 55:2000085. doi: 10.1183/13993003.00085-2020
8. Nasser, M, Larrieu, S, Boussel, L, Si-Mohamed, S, Bazin, F, Marque, S, et al. Estimates of epidemiology, mortality and disease burden associated with progressive fibrosing interstitial lung disease in France (the PROGRESS study). Respir Res. (2021) 22:162. doi: 10.1186/s12931-021-01749-1
9. Flaherty, KR, Wells, AU, Cottin, V, Devaraj, A, Walsh, SLF, Inoue, Y, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. (2019) 381:1718–27. doi: 10.1056/NEJMoa1908681
10. Raghu, G, Remy-Jardin, M, Richeldi, L, Thomson, CC, Inoue, Y, Johkoh, T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2022) 205:e18–47. doi: 10.1164/rccm.202202-0399ST
11. Cottin, V, Hirani, NA, Hotchkin, DL, Nambiar, AM, Ogura, T, Otaola, M, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. (2018) 27:180076. doi: 10.1183/16000617.0076-2018
12. Rajan, SK, Cottin, V, Dhar, R, Danoff, S, Flaherty, KR, Brown, KK, et al. Progressive pulmonary fibrosis: an expert group consensus statement. Eur Respir J. (2023) 61:2103187. doi: 10.1183/13993003.03187-2021
13. Alfieri, V, Crisafulli, E, Visca, D, Chong, WH, Stock, C, Mori, L, et al. Physiological predictors of exertional oxygen desaturation in patients with fibrotic interstitial lung disease. Eur Respir J. (2020) 55:1901681. doi: 10.1183/13993003.01681-2019
14. Gimenez, A, Storrer, K, Kuranishi, L, Soares, MR, Ferreira, RG, and Pereira, CAC. Change in FVC and survival in chronic fibrotic hypersensitivity pneumonitis. Thorax. (2018) 73:391–2. doi: 10.1136/thoraxjnl-2017-210035
15. Fan, J-J, Gu, J-M, Xiao, S-Y, Jia, M-Y, and Han, G-L. Risk factors for progression of pulmonary fibrosis: a single-centered, retrospective study. Front Med. (2024) 11:1335758. doi: 10.3389/fmed.2024.1335758
16. Kronzer, VL, Huang, W, Dellaripa, PF, Huang, S, Feathers, V, Lu, B, et al. Lifestyle and clinical risk factors for incident rheumatoid arthritis-associated interstitial lung disease. J Rheumatol. (2021) 48:656–63. doi: 10.3899/jrheum.200863
17. Assassi, S, Sharif, R, Lasky, RE, McNearney, TA, Estrada-Y-Martin, RM, Draeger, H, et al. Predictors of interstitial lung disease in early systemic sclerosis: a prospective longitudinal study of the GENISOS cohort. Arthritis Res Ther. (2010) 12:R166. doi: 10.1186/ar3125
18. Greidinger, EL, Flaherty, KT, White, B, Rosen, A, Wigley, FM, and Wise, RA. African-American race and antibodies to topoisomerase I are associated with increased severity of scleroderma lung disease. Chest. (1998) 114:801–7. doi: 10.1378/chest.114.3.801
19. Goldstein, LA, Ghersi, G, Piñeiro-Sánchez, ML, Salamone, M, Yeh, Y, Flessate, D, et al. Molecular cloning of seprase: a serine integral membrane protease from human melanoma. Biochim Biophys Acta. (1997) 1361:11–9. doi: 10.1016/s0925-4439(97)00032-x
20. Piñeiro-Sánchez, ML, Goldstein, LA, Dodt, J, Howard, L, Yeh, Y, Tran, H, et al. Identification of the 170-kDa melanoma membrane-bound gelatinase (seprase) as a serine integral membrane protease. J Biol Chem. (1997) 272:7595–601. doi: 10.1074/jbc.272.12.7595
21. Collins, PJ, McMahon, G, O’Brien, P, and O’Connor, B. Purification, identification and characterisation of seprase from bovine serum. Int J Biochem Cell Biol. (2004) 36:2320–33. doi: 10.1016/j.biocel.2004.05.006
22. Sun, S, Albright, CF, Fish, BH, George, HJ, Selling, BH, Hollis, GF, et al. Expression, purification, and kinetic characterization of full-length human fibroblast activation protein. Protein Expr Purif. (2002) 24:274–81. doi: 10.1006/prep.2001.1572
23. Keane, FM, Nadvi, NA, Yao, T-W, and Gorrell, MD. Neuropeptide Y, B-type natriuretic peptide, substance P and peptide YY are novel substrates of fibroblast activation protein-α. FEBS J. (2011) 278:1316–32. doi: 10.1111/j.1742-4658.2011.08051.x
24. Dunshee, DR, Bainbridge, TW, Kljavin, NM, Zavala-Solorio, J, Schroeder, AC, Chan, R, et al. Fibroblast activation protein cleaves and inactivates fibroblast growth factor 21. J Biol Chem. (2016) 291:5986–96. doi: 10.1074/jbc.M115.710582
25. Zhen, EY, Jin, Z, Ackermann, BL, Thomas, MK, and Gutierrez, JA. Circulating FGF21 proteolytic processing mediated by fibroblast activation protein. Biochem J. (2016) 473:605–14. doi: 10.1042/BJ20151085
26. Christiansen, VJ, Jackson, KW, Lee, KN, and McKee, PA. Effect of fibroblast activation protein and alpha2-antiplasmin cleaving enzyme on collagen types I, III, and IV. Arch Biochem Biophys. (2007) 457:177–86. doi: 10.1016/j.abb.2006.11.006
27. Lee, KN, Jackson, KW, Christiansen, VJ, Lee, CS, Chun, J-G, and McKee, PA. Antiplasmin-cleaving enzyme is a soluble form of fibroblast activation protein. Blood. (2006) 107:1397–404. doi: 10.1182/blood-2005-08-3452
28. Lee, KN, Jackson, KW, Christiansen, VJ, Chung, KH, and McKee, PA. A novel plasma proteinase potentiates alpha2-antiplasmin inhibition of fibrin digestion. Blood. (2004) 103:3783–8. doi: 10.1182/blood-2003-12-4240
29. Niedermeyer, J, Enenkel, B, Park, JE, Lenter, M, Rettig, WJ, Damm, K, et al. Mouse fibroblast-activation protein--conserved Fap gene organization and biochemical function as a serine protease. Eur J Biochem. (1998) 254:650–4. doi: 10.1046/j.1432-1327.1998.2540650.x
30. Rettig, WJ, Su, SL, Fortunato, SR, Scanlan, MJ, Raj, BK, Garin-Chesa, P, et al. Fibroblast activation protein: purification, epitope mapping and induction by growth factors. Int J Cancer. (1994) 58:385–92. doi: 10.1002/ijc.2910580314
31. Levy, MT, McCaughan, GW, Abbott, CA, Park, JE, Cunningham, AM, Müller, E, et al. Fibroblast activation protein: a cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology. (1999) 29:1768–78. doi: 10.1002/hep.510290631
32. Dienus, K, Bayat, A, Gilmore, BF, and Seifert, O. Increased expression of fibroblast activation protein-alpha in keloid fibroblasts: implications for development of a novel treatment option. Arch Dermatol Res. (2010) 302:725–31. doi: 10.1007/s00403-010-1084-x
33. Yang, P, Luo, Q, Wang, X, Fang, Q, Fu, Z, Li, J, et al. Comprehensive analysis of fibroblast activation protein expression in interstitial lung diseases. Am J Respir Crit Care Med. (2023) 207:160–72. doi: 10.1164/rccm.202110-2414OC
34. Li, M, Deng, Z, Xie, C, Chen, J, Yuan, Z, Rahhal, O, et al. Fibroblast activating protein promotes the proliferation, migration, and activation of fibroblasts in oral submucous fibrosis. Oral Dis. (2023) 30:1252–63. doi: 10.1111/odi.14602
35. Chen, H, Yang, W-W, Wen, Q-T, Xu, L, and Chen, M. TGF-beta induces fibroblast activation protein expression; fibroblast activation protein expression increases the proliferation, adhesion, and migration of HO-8910PM. Exp Mol Pathol. (2009) 87:189–94. doi: 10.1016/j.yexmp.2009.09.001
36. Wäster, P, Rosdahl, I, Gilmore, BF, and Seifert, O. Ultraviolet exposure of melanoma cells induces fibroblast activation protein-α in fibroblasts: implications for melanoma invasion. Int J Oncol. (2011) 39:193–202. doi: 10.3892/ijo.2011.1002
37. Kennedy, A, Dong, H, Chen, D, and Chen, W-T. Elevation of seprase expression and promotion of an invasive phenotype by collagenous matrices in ovarian tumor cells. Int J Cancer. (2009) 124:27–35. doi: 10.1002/ijc.23871
38. Avery, D, Govindaraju, P, Jacob, M, Todd, L, Monslow, J, and Puré, E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. (2018) 67:90–106. doi: 10.1016/j.matbio.2017.12.003
39. Acharya, PS, Zukas, A, Chandan, V, Katzenstein, A-LA, and Puré, E. Fibroblast activation protein: a serine protease expressed at the remodeling interface in idiopathic pulmonary fibrosis. Hum Pathol. (2006) 37:352–60. doi: 10.1016/j.humpath.2005.11.020
40. Wenlong, L, Leilei, Y, Wei, F, Yi, C, Jing, T, Lanzhi, M, et al. Luciferase expression is driven by the promoter of fibroblast activation protein-α in murine pulmonary fibrosis. Biotechnol Lett. (2015) 37:1757–63. doi: 10.1007/s10529-015-1855-8
41. Fan, M-H, Zhu, Q, Li, H-H, Ra, H-J, Majumdar, S, Gulick, DL, et al. Fibroblast activation protein (FAP) accelerates collagen degradation and clearance from lungs in mice. J Biol Chem. (2016) 291:8070–89. doi: 10.1074/jbc.M115.701433
42. Chaudhary, NI, Schnapp, A, and Park, JE. Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am J Respir Crit Care Med. (2006) 173:769–76. doi: 10.1164/rccm.200505-717OC
43. Moore, BB, and Hogaboam, CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L152–60. doi: 10.1152/ajplung.00313.2007
44. Liu, T, De Los Santos, FG, and Phan, SH. The bleomycin model of pulmonary fibrosis. Methods Mol Biol. (2017) 1627:27–42. doi: 10.1007/978-1-4939-7113-8_2
45. Kimura, T, Monslow, J, Klampatsa, A, Leibowitz, M, Sun, J, Liousia, M, et al. Loss of cells expressing fibroblast activation protein has variable effects in models of TGF-β and chronic bleomycin-induced fibrosis. Am J Physiol Lung Cell Mol Physiol. (2019) 317:L271–82. doi: 10.1152/ajplung.00071.2019
46. Degryse, AL, Tanjore, H, Xu, XC, Polosukhin, VV, Jones, BR, McMahon, FB, et al. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2010) 299:L442–52. doi: 10.1152/ajplung.00026.2010
47. Egger, C, Cannet, C, Gérard, C, Suply, T, Ksiazek, I, Jarman, E, et al. Effects of the fibroblast activation protein inhibitor, PT100, in a murine model of pulmonary fibrosis. Eur J Pharmacol. (2017) 809:64–72. doi: 10.1016/j.ejphar.2017.05.022
48. Cunningham, CC. Talabostat. Expert Opin Investig Drugs. (2007) 16:1459–65. doi: 10.1517/13543784.16.9.1459
49. Yang, A-T, Kim, Y-O, Yan, X-Z, Abe, H, Aslam, M, Park, K-S, et al. Fibroblast activation protein activates macrophages and promotes parenchymal liver inflammation and fibrosis. Cell Mol Gastroenterol Hepatol. (2023) 15:841–67. doi: 10.1016/j.jcmgh.2022.12.005
50. Karampitsakos, T, Juan-Guardela, BM, Tzouvelekis, A, and Herazo-Maya, JD. Precision medicine advances in idiopathic pulmonary fibrosis. EBioMedicine. (2023) 95:104766. doi: 10.1016/j.ebiom.2023.104766
51. Lavis, P, Pingitore, J, Doumont, G, Garabet, A, Van Simaeys, G, Lacroix, S, et al. Usefulness of FAPα assessment in bronchoalveolar lavage as a marker of fibrogenesis: results of a preclinical study and first report in patients with idiopathic pulmonary fibrosis. Respir Res. (2023) 24:254. doi: 10.1186/s12931-023-02556-6
52. Prior, TS, Hoyer, N, Davidsen, JR, Shaker, SB, Deleuran, BW, Bendstrup, E, et al. Fibroblast activation protein and disease severity, progression and survival in IPF. Eur Respir J. (2022):60. doi: 10.1183/13993003.congress-2022.510
53. Uitte de Willige, S, Keane, FM, Bowen, DG, Malfliet, JJMC, Zhang, HE, Maneck, B, et al. Circulating fibroblast activation protein activity and antigen levels correlate strongly when measured in liver disease and coronary heart disease. PLoS One. (2017) 12:e0178987. doi: 10.1371/journal.pone.0178987
54. Williams, KH, Viera de Ribeiro, AJ, Prakoso, E, Veillard, AS, Shackel, NA, Bu, Y, et al. Lower serum fibroblast activation protein shows promise in the exclusion of clinically significant liver fibrosis due to non-alcoholic fatty liver disease in diabetes and obesity. Diabetes Res Clin Pract. (2015) 108:466–72. doi: 10.1016/j.diabres.2015.02.024
55. Keane, FM, Yao, T-W, Seelk, S, Gall, MG, Chowdhury, S, Poplawski, SE, et al. Quantitation of fibroblast activation protein (FAP)-specific protease activity in mouse, baboon and human fluids and organs. FEBS Open Bio. (2013) 4:43–54. doi: 10.1016/j.fob.2013.12.001
56. Uitte de Willige, S, Malfliet, JJMC, Deckers, JW, Dippel, DWJ, Leebeek, FWG, and Rijken, DC. Plasma levels of soluble fibroblast activation protein in arterial thrombosis: determinants and cleavage of its substrate alpha-2-antiplasmin. Int J Cardiol. (2015) 178:105–10. doi: 10.1016/j.ijcard.2014.10.091
57. Sinnathurai, P, Lau, W, Vieira de Ribeiro, AJ, Bachovchin, WW, Englert, H, Howe, G, et al. Circulating fibroblast activation protein and dipeptidyl peptidase 4 in rheumatoid arthritis and systemic sclerosis. Int J Rheum Dis. (2018) 21:1915–23. doi: 10.1111/1756-185X.13031
58. Kosałka-Węgiel, J, Lichołai, S, Pacholczak-Madej, R, Dziedzina, S, Milewski, M, Kuszmiersz, P, et al. Serum IL-17 and TNFα as prognostic biomarkers in systemic sclerosis patients: a prospective study. Rheumatol Int. (2023) 44:119–28. doi: 10.1007/s00296-023-05499-9
59. Kratochwil, C, Flechsig, P, Lindner, T, Abderrahim, L, Altmann, A, Mier, W, et al. 68Ga-FAPI PET/CT: tracer uptake in 28 different kinds of cancer. J Nucl Med. (2019) 60:801–5. doi: 10.2967/jnumed.119.227967
60. Chen, L, Zhong, X, Li, L, Li, X, Liu, Y, Guo, C, et al. [68Ga]Ga-FAPI-04 PET/CT on assessing Crohn’s disease intestinal lesions. Eur J Nucl Med Mol Imaging. (2023) 50:1360–70. doi: 10.1007/s00259-023-06107-5
61. Luo, Y, Pan, Q, Zhou, Z, Li, M, Wei, Y, Jiang, X, et al. 68Ga-FAPI PET/CT for rheumatoid arthritis: a prospective study. Radiology. (2023) 307:e222052. doi: 10.1148/radiol.222052
62. Mayola, MF, and Thackeray, JT. The potential of fibroblast activation protein-targeted imaging as a biomarker of cardiac remodeling and injury. Curr Cardiol Rep. (2023) 25:515–23. doi: 10.1007/s11886-023-01869-8
63. Röhrich, M, Leitz, D, Glatting, FM, Wefers, AK, Weinheimer, O, Flechsig, P, et al. Fibroblast activation protein-specific PET/CT imaging in fibrotic interstitial lung diseases and lung cancer: a translational exploratory study. J Nucl Med. (2022) 63:127–33. doi: 10.2967/jnumed.121.261925
64. Mori, Y, Kramer, V, Novruzov, E, Mamlins, E, Röhrich, M, Fernández, R, et al. Initial results with [18F]FAPI-74 PET/CT in idiopathic pulmonary fibrosis. Eur J Nucl Med Mol Imaging. (2023) 51:1605–11. doi: 10.1007/s00259-023-06564-y
65. Bergmann, C, Distler, JHW, Treutlein, C, Tascilar, K, Müller, A-T, Atzinger, A, et al. 68Ga-FAPI-04 PET-CT for molecular assessment of fibroblast activation and risk evaluation in systemic sclerosis-associated interstitial lung disease: a single-centre, pilot study. Lancet Rheumatol. (2021) 3:e185–94. doi: 10.1016/S2665-9913(20)30421-5
66. Treutlein, C, Distler, JHW, Tascilar, K, Fakhouri, SC, Györfi, A-H, Atzinger, A, et al. Assessment of myocardial fibrosis in patients with systemic sclerosis using [68Ga]Ga-FAPI-04-PET-CT. Eur J Nucl Med Mol Imaging. (2023) 50:1629–35. doi: 10.1007/s00259-022-06081-4
67. Sviridenko, A, Boehm, A, di Santo, G, Uprimny, C, Nilica, B, Fritz, J, et al. Enhancing clinical diagnosis for patients with persistent pulmonary abnormalities after COVID-19 infection: the potential benefit of 68 Ga-FAPI PET/CT. Clin Nucl Med. (2022) 47:1026–9. doi: 10.1097/RLU.0000000000004437
68. Liu, Y, Zhang, Q, Zhang, Y, Wang, J, Wu, Y, Yang, G, et al. 99mTc-labeled FAPI SPECT imaging in idiopathic pulmonary fibrosis: preliminary results. Pharmaceuticals. (2023) 16:1434. doi: 10.3390/ph16101434
69. Rosenkrans, ZT, Massey, CF, Bernau, K, Ferreira, CA, Jeffery, JJ, Schulte, JJ, et al. [68Ga]Ga-FAPI-46 PET for non-invasive detection of pulmonary fibrosis disease activity. Eur J Nucl Med Mol Imaging. (2022) 49:3705–16. doi: 10.1007/s00259-022-05814-9
70. Liu, Z, Zhou, H, Li, P, Wang, Z, Tu, T, Ezzi, SHA, et al. Fibroblast activation protein-targeted PET/CT with Al18F-NODA-FAPI-04 for in vivo imaging of tendon healing in rat achilles tendon injury models. Am J Sports Med. (2023) 51:3790–801. doi: 10.1177/03635465231208843
71. Schmidkonz, C, Rauber, S, Atzinger, A, Agarwal, R, Götz, TI, Soare, A, et al. Disentangling inflammatory from fibrotic disease activity by fibroblast activation protein imaging. Ann Rheum Dis. (2020) 79:1485–91. doi: 10.1136/annrheumdis-2020-217408
72. Song, C-Y, Liu, Z-F, Wang, P, Su, X-H, and Lu, Y-Q. Assessment of pulmonary fibrosis induced by paraquat using Al18F-NODA-FAPI-04 PET/CT. Intern Emerg Med. (2023) 18:1673–9. doi: 10.1007/s11739-023-03327-3
73. Castiaux, A, Van Simaeys, G, Goldman, S, and Bondue, B. Assessment of 18F-FDG uptake in idiopathic pulmonary fibrosis: influence of lung density changes. Eur J Hybrid Imaging. (2018) 2:27. doi: 10.1186/s41824-018-0045-z
74. Bondue, B, Castiaux, A, Van Simaeys, G, Mathey, C, Sherer, F, Egrise, D, et al. Absence of early metabolic response assessed by 18F-FDG PET/CT after initiation of antifibrotic drugs in IPF patients. Respir Res. (2019) 20:10. doi: 10.1186/s12931-019-0974-5
75. Win, T, Screaton, NJ, Porter, J, Endozo, R, Wild, D, Kayani, I, et al. Novel positron emission tomography/computed tomography of diffuse parenchymal lung disease combining a labeled somatostatin receptor analogue and 2-deoxy-2[18F]fluoro-D-glucose. Mol Imaging. (2012) 11:91–8. doi: 10.2310/7290.2011.00030
76. Borie, R, Fabre, A, Prost, F, Marchal-Somme, J, Lebtahi, R, Marchand-Adam, S, et al. Activation of somatostatin receptors attenuates pulmonary fibrosis. Thorax. (2008) 63:251–8. doi: 10.1136/thx.2007.078006
77. Weatherley, ND, Stewart, NJ, Chan, H-F, Austin, M, Smith, LJ, Collier, G, et al. Hyperpolarised xenon magnetic resonance spectroscopy for the longitudinal assessment of changes in gas diffusion in IPF. Thorax. (2019) 74:500–2. doi: 10.1136/thoraxjnl-2018-211851
78. Eaden, JA, Weatherley, ND, Chan, H-F, Collier, G, Norquay, G, Swift, AJ, et al. Hyperpolarised xenon-129 diffusion-weighted magnetic resonance imaging for assessing lung microstructure in idiopathic pulmonary fibrosis. ERJ Open Res. (2023) 9:00048-2023. doi: 10.1183/23120541.00048-2023
79. Smith, ML. The histologic diagnosis of usual interstitial pneumonia of idiopathic pulmonary fibrosis. Where we are and where we need to go. Mod Pathol. (2022) 35:8–14. doi: 10.1038/s41379-021-00889-5
80. Yamaguchi, M, Hirai, S, Tanaka, Y, Sumi, T, Miyajima, M, Mishina, T, et al. Fibroblastic foci, covered with alveolar epithelia exhibiting epithelial-mesenchymal transition, destroy alveolar septa by disrupting blood flow in idiopathic pulmonary fibrosis. Lab Investig. (2017) 97:232–42. doi: 10.1038/labinvest.2016.135
81. Hill, C, Jones, MG, Davies, DE, and Wang, Y. Epithelial-mesenchymal transition contributes to pulmonary fibrosis via aberrant epithelial/fibroblastic cross-talk. J Lung Health Dis. (2019) 3:31–5. doi: 10.29245/2689-999X/2019/2.1149
82. Harada, T, Watanabe, K, Nabeshima, K, Hamasaki, M, and Iwasaki, H. Prognostic significance of fibroblastic foci in usual interstitial pneumonia and non-specific interstitial pneumonia. Respirology. (2013) 18:278–83. doi: 10.1111/j.1440-1843.2012.02272.x
83. Mäkelä, K, Mäyränpää, MI, Sihvo, H-K, Bergman, P, Sutinen, E, Ollila, H, et al. Artificial intelligence identifies inflammation and confirms fibroblast foci as prognostic tissue biomarkers in idiopathic pulmonary fibrosis. Hum Pathol. (2021) 107:58–68. doi: 10.1016/j.humpath.2020.10.008
84. Collard, HR, Cool, CD, Leslie, KO, Curran-Everett, D, Groshong, S, and Brown, KK. Organizing pneumonia and lymphoplasmacytic inflammation predict treatment response in idiopathic pulmonary fibrosis. Histopathology. (2007) 50:258–65. doi: 10.1111/j.1365-2559.2006.02554.x
85. Fendler, WP, Pabst, KM, Kessler, L, Fragoso Costa, P, Ferdinandus, J, Weber, M, et al. Safety and efficacy of 90Y-FAPI-46 radioligand therapy in patients with advanced sarcoma and other cancer entities. Clin Cancer Res. (2022) 28:4346–53. doi: 10.1158/1078-0432.CCR-22-1432
86. Assadi, M, Rekabpour, SJ, Jafari, E, Divband, G, Nikkholgh, B, Amini, H, et al. Feasibility and therapeutic potential of 177Lu-fibroblast activation protein Inhibitor-46 for patients with relapsed or refractory cancers: a preliminary study. Clin Nucl Med. (2021) 46:e523–30. doi: 10.1097/RLU.0000000000003810
87. Adegunsoye, A, Alqalyoobi, S, Linderholm, A, Bowman, WS, Lee, CT, Pugashetti, JV, et al. Circulating plasma biomarkers of survival in antifibrotic-treated patients with idiopathic pulmonary fibrosis. Chest. (2020) 158:1526–34. doi: 10.1016/j.chest.2020.04.066
88. Maher, TM, Oballa, E, Simpson, JK, Porte, J, Habgood, A, Fahy, WA, et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med. (2017) 5:946–55. doi: 10.1016/S2213-2600(17)30430-7
89. Guo, L, Yang, Y, Liu, F, Jiang, C, Yang, Y, Pu, H, et al. Clinical research on prognostic evaluation of subjects with IPF by peripheral blood biomarkers, quantitative imaging characteristics and pulmonary function parameters. Arch Bronconeumol. (2020) 56:365–72. doi: 10.1016/j.arbres.2019.08.020
90. Hamai, K, Iwamoto, H, Ishikawa, N, Horimasu, Y, Masuda, T, Miyamoto, S, et al. Comparative study of circulating MMP-7, CCL18, KL-6, SP-A, and SP-D as disease markers of idiopathic pulmonary fibrosis. Dis Markers. (2016) 2016:4759040–8. doi: 10.1155/2016/4759040
91. Raghu, G, Richeldi, L, Jagerschmidt, A, Martin, V, Subramaniam, A, Ozoux, M-L, et al. Idiopathic pulmonary fibrosis: prospective, case-controlled study of natural history and circulating biomarkers. Chest. (2018) 154:1359–70. doi: 10.1016/j.chest.2018.08.1083
92. Aloisio, E, Braga, F, Puricelli, C, and Panteghini, M. Prognostic role of Krebs von den Lungen-6 (KL-6) measurement in idiopathic pulmonary fibrosis: a systematic review and meta-analysis. Clin Chem Lab Med. (2021) 59:1400–8. doi: 10.1515/cclm-2021-0199
93. Bennett, D, Salvini, M, Fui, A, Cillis, G, Cameli, P, Mazzei, MA, et al. Calgranulin B and KL-6 in bronchoalveolar lavage of patients with IPF and NSIP. Inflammation. (2019) 42:463–70. doi: 10.1007/s10753-018-00955-2
94. Bergantini, L, Bargagli, E, Cameli, P, Cekorja, B, Lanzarone, N, Pianigiani, L, et al. Serial KL-6 analysis in patients with idiopathic pulmonary fibrosis treated with nintedanib. Respir Investig. (2019) 57:290–1. doi: 10.1016/j.resinv.2019.02.001
95. Collard, HR, Calfee, CS, Wolters, PJ, Song, JW, Hong, S-B, Brady, S, et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2010) 299:L3–7. doi: 10.1152/ajplung.90637.2008
96. Ikeda, K, Shiratori, M, Chiba, H, Nishikiori, H, Yokoo, K, Saito, A, et al. Serum surfactant protein D predicts the outcome of patients with idiopathic pulmonary fibrosis treated with pirfenidone. Respir Med. (2017) 131:184–91. doi: 10.1016/j.rmed.2017.08.021
97. Ikeda, K, Chiba, H, Nishikiori, H, Azuma, A, Kondoh, Y, Ogura, T, et al. Serum surfactant protein D as a predictive biomarker for the efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis: a post-hoc analysis of the phase 3 trial in Japan. Respir Res. (2020) 21:316. doi: 10.1186/s12931-020-01582-y
98. Jiang, Y, Luo, Q, Han, Q, Huang, J, Ou, Y, Chen, M, et al. Sequential changes of serum KL-6 predict the progression of interstitial lung disease. J Thorac Dis. (2018) 10:4705–14. doi: 10.21037/jtd.2018.07.76
99. Song, JW, Do, KH, Jang, SJ, Colby, TV, Han, S, and Kim, DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest. (2013) 143:1422–9. doi: 10.1378/chest.11-2735
100. Wakamatsu, K, Nagata, N, Kumazoe, H, Oda, K, Ishimoto, H, Yoshimi, M, et al. Prognostic value of serial serum KL-6 measurements in patients with idiopathic pulmonary fibrosis. Respir Investig. (2017) 55:16–23. doi: 10.1016/j.resinv.2016.09.003
101. Yoshikawa, T, Otsuka, M, Chiba, H, Ikeda, K, Mori, Y, Umeda, Y, et al. Surfactant protein A as a biomarker of outcomes of anti-fibrotic drug therapy in patients with idiopathic pulmonary fibrosis. BMC Pulm Med. (2020) 20:27. doi: 10.1186/s12890-020-1060-y
102. Yokoyama, A, Kondo, K, Nakajima, M, Matsushima, T, Takahashi, T, Nishimura, M, et al. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology. (2006) 11:164–8. doi: 10.1111/j.1440-1843.2006.00834.x
103. Bauer, Y, White, ES, de Bernard, S, Cornelisse, P, Leconte, I, Morganti, A, et al. MMP-7 is a predictive biomarker of disease progression in patients with idiopathic pulmonary fibrosis. ERJ Open Res. (2017) 3:00074-2016. doi: 10.1183/23120541.00074-2016
104. Gui, X, Qiu, X, Xie, M, Tian, Y, Min, C, Huang, M, et al. Prognostic value of serum Osteopontin in acute exacerbation of idiopathic pulmonary fibrosis. Biomed Res Int. (2020) 2020:3424208–10. doi: 10.1155/2020/3424208
105. Kinder, BW, Brown, KK, McCormack, FX, Ix, JH, Kervitsky, A, Schwarz, MI, et al. Serum surfactant protein-A is a strong predictor of early mortality in idiopathic pulmonary fibrosis. Chest. (2009) 135:1557–63. doi: 10.1378/chest.08-2209
106. Takahashi, H, Fujishima, T, Koba, H, Murakami, S, Kurokawa, K, Shibuya, Y, et al. Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med. (2000) 162:1109–14. doi: 10.1164/ajrccm.162.3.9910080
Keywords: FAP, fibroblast activation protein, FAPI, PPF, IPF, progression, fibrosis, biomarker
Citation: Lavis P, Garabet A, Cardozo AK and Bondue B (2024) The fibroblast activation protein alpha as a biomarker of pulmonary fibrosis. Front. Med. 11:1393778. doi: 10.3389/fmed.2024.1393778
Edited by:
Rosane Duarte Achcar, National Jewish Health (United States), United StatesReviewed by:
Ahmed Fahim, Royal Wolverhampton Hospitals NHS Trust, United KingdomCopyright © 2024 Lavis, Garabet, Cardozo and Bondue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin Bondue, YmVuamFtaW4uYm9uZHVlQHVsYi5iZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.