 Yanjiang Liu
Yanjiang Liu Tao Zhang2
Tao Zhang2 He Wei
He Wei- 1School of Basic Medical Sciences, Chengdu Medical College, Chengdu, China
- 2School of Bioscience and Technology, Chengdu Medical College, Chengdu, China
Intestinal fibrosis is a common complication of chronic intestinal diseases with the characteristics of fibroblast proliferation and extracellular matrix deposition after chronic inflammation, leading to lumen narrowing, structural and functional damage to the intestines, and life inconvenience for the patients. However, anti-inflammatory drugs are currently generally not effective in overcoming intestinal fibrosis making surgery the main treatment method. The development of intestinal fibrosis is a slow process and its onset may be the result of the combined action of inflammatory cells, local cytokines, and intestinal stromal cells. The aim of this study is to elucidate the pathogenesis [e.g., extracellular matrix (ECM), cytokines and chemokines, epithelial-mesenchymal transition (EMT), differentiation of fibroblast to myofibroblast and intestinal microbiota] underlying the development of intestinal fibrosis and to explore therapeutic advances (such as regulating ECM, cytokines, chemokines, EMT, differentiation of fibroblast to myofibroblast and targeting TGF-β) based on the pathogenesis in order to gain new insights into the prevention and treatment of intestinal fibrosis.
1 Introduction
Fibrosis is a pathological physiological process in which the body responds to damage caused by harmful substances, such as physical, chemical, and mechanical injuries, infections, and autoimmune reactions, which can occur in various types of tissue injuries, particularly in chronic inflammatory diseases. The formation of fibrosis is due to the accumulation of extracellular matrix components, e.g., collagen and fibronectin, and is also an important stage in the repair of organs and tissues (1, 2). However, if the stimulus of fibrogenesis persists or manifests itself abnormally, tissue fibrosis and scarring may occur, which may even lead to organ dysfunction (3).
It is well-known that the formation of intestinal fibrosis is associated with inflammatory bowel disease (IBD), collagenous colitis, radiation enteropathy, eosinophilic enteropathy, cystic fibrosis, and other diseases (4–6). IBD is a chronic inflammatory condition with multiple potential causes, whereas radiation enteropathy is a specific type of intestinal damage caused by radiation therapy, eosinophilic enteropathy involves eosinophilic infiltration causing inflammation in the gastrointestinal tract and cystic fibrosis is a genetic disorder affecting both the respiratory and digestive systems due to mutations in the CFTR gene. Among these diseases, IBD, including Crohn’s disease (CD) and ulcerative colitis (UC), which is characterized by relapsing and chronic inflammation, and is associated with a 1.4 to 2.2-fold increased risk of colorectal cancer, is the main cause of intestinal fibrosis (7–10). Intestinal fibrosis is one of the most threatening complications of CD, affecting more than 50% of patients, and can progress from fibrotic strictures to penetrating or stenotic lesions. UC has long been considered a mucosal disease with no or minimal fibrosis. However, recent studies have shown that fibrosis can also be detectable in acute and chronic UC and in all sections of the colon, which correlates with the severity of the inflammation (11).
The imbalance of fibrotic regulation caused by the continuous irritation of intestinal inflammation is one of the typical features of intestinal fibrosis. In this context, the lumen diameter gradually decreases, resulting in intestinal stenosis, which can easily cause intestinal obstruction and even endanger the patient’s life (10, 12). Although the treatment of various gastrointestinal diseases has improved significantly due to the rapid development of medical technology and diagnostics, the formation of intestinal fibrosis is still high. In addition, pathological mechanisms play a crucial role in understanding and treating intestinal fibrosis. In this context, this study reviewed the progress of recent development of strategies for the treatment of intestinal fibrosis on the basis of elucidating the pathogenesis of intestinal fibrosis (Figure 1).
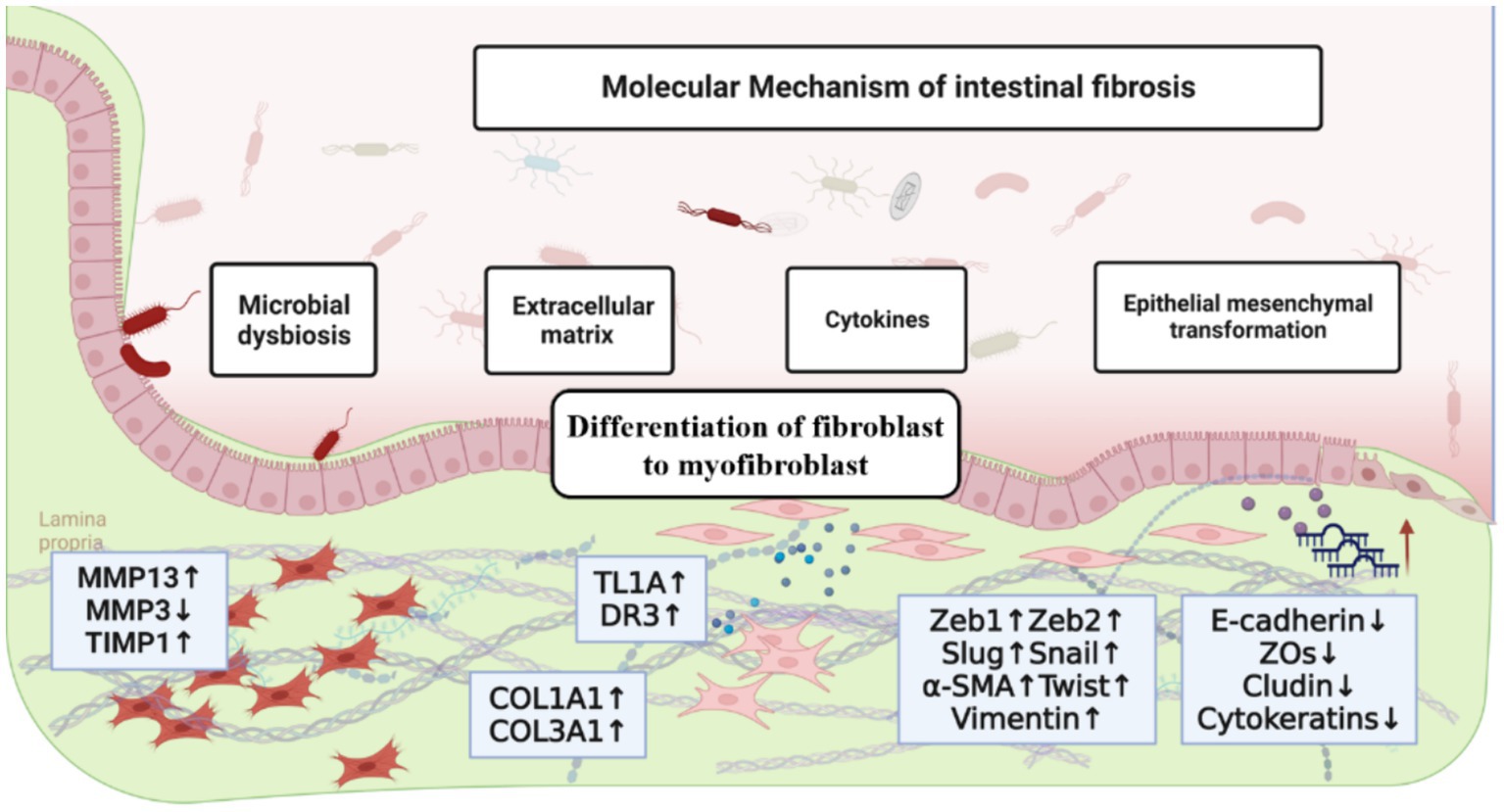
Figure 1. The molecular mechanism of intestinal fibrosis, including the influence of extracellular matrix, cytokines and chemokines, epithelial-mesenchymal transition, differentiation of fibroblast to myofibroblast and intestinal microbiota.
2 The pathogenesis of intestinal fibrosis
2.1 Extracellular matrix
The development of organ fibrosis is closely associated to the formation and activation of fibroblasts and the resulting deposition of extracellular matrix (ECM). The ECM is a complex network of proteins, elastin, hyaluronic acid, collagen and polysaccharides that surrounds and supports cells within tissues and organs and plays a crucial role in maintaining tissue integrity, providing mechanical support, and enabling various cellular functions (13, 14). Intestinal fibrosis occurs when there is an imbalance between the production and degradation of ECM components (15). It is reported that hyaluronic acid, one of the high molecular weight polymers under normal conditions, could be cleaved into low molecular weight fragments during excessive inflammatory actions, which promotes fibroblast proliferation and myofibroblast differentiation, thereby accelerating the fibrosis process (16). In addition, the low molecular weight fragments of hyaluronic acid help recruit immune cells to the site of inflammation, leading to the release of various inflammatory mediators and growth factors that further trigger fibrosis.
During the fibrosis formation process, antifibrotic and various profibrotic factors play a regulatory role, with the regulation of tissue inhibitors of metalloproteinases (TIMPs) and matrix metalloproteinases (MMPs) being of great importance. MMPs can degrade most of the extracellular matrix, such as MMP-1, MMP-9, MMP13, etc., while TIMPs can regulate the activity of MMPs. An imbalance in the MMPs/TIMPs system can lead to abnormal ECM deposition, contributing to the development of intestinal fibrosis (17, 18). It was found that MMP3 expression decreased in the fibrotic strictures of the intestinal mucosa in patients with CD, while TIMP1 expression was increased (19). Furthermore, researchers have observed significant upregulation of MMP13 in colon biopsies from the constricted areas compared to non-constricted areas in CD patients (20).
2.2 Cytokines and chemokines
Cytokines and chemokines secreted by immune cells and nonimmune cells act as coordinators of persistent inflammatory microenvironments, including interleukins (ILs), transforming growth factor-alpha (TNF-α), TGF-β, platelet-derived growth factor, insulin-like growth factor-1 and epidermal growth factor (21). The profibrotic effects result from the proliferation of fibroblasts caused by various growth factors and cytokines and provide a plausible avenue for new therapeutic targets in fibrotic diseases. Among these, ILs, TNF-α and TGF-β were the mostly widely studied. ILs involved in intestinal fibrosis generally include IL-11, IL-33, IL-34, IL-17, IL-36, etc., in which IL-11, a member of the IL-6 family, is considered a profibrotic cytokine that secreted by stromal cells and epithelial cells during tissue injury. Schäfer et al. found that transgenic expression of fibroblast-specific IL-11 or IL-11 injection in mice leads to cardiac and renal fibrosis and organ failure, while genetic deletion of IL-11RA can prevent the disease. Their experimental data illustrate that IL11 is a promising therapeutic target for IBD, particularly in the context of resistance to TNF therapy (22). Milara et al. reported that IL-11 and IL-11Rα are overexpressed in the pulmonary arteries of patients with idiopathic pulmonary fibrosis, which contributes to pulmonary artery remodeling and regulation of pulmonary arterial hypertension. Their work elucidates the role of IL-11 and IL-11Rα as driving factors in pulmonary arterial hypertension, suggesting their involvement in the process of pulmonary arterial remodeling (23). IL-33, a member of the IL-1 family that passively released upon cell damage and necrosis, is also involved in the fibrosis formation process. It was reported that IL-33 is closely associated with fibrotic progression in pediatric CD of the ileum due to increased epithelial expression (24). Remarkably, a recent study demonstrated a novel association between the IL-33/ST2 signaling pathway and intestinal dysbiosis in intestinal fibrosis. The authors observed that adherent invasive Escherichia coli (AIEC) colonization induced ST2 expression in the intestinal epithelium, thereby enhancing the IL-33/ST2 signaling pathway and ultimately promoting intestinal fibrosis. Their findings contribute to the expanding array of research investigating the underlying processes of fibrostenosis in CD, suggesting that eosinophils and mediators originating from eosinophils could serve as promising therapeutic targets for CD-associated strictures (25). Furthermore, IL-34 shows significantly increased expression in the damaged intestine of patients with CD and UC, which plays a crucial role in mediating crosstalk between immune cells and stromal cells and influences the production of fibrotic molecules by promoting signaling pathway activation (26). Another study reported that IL-34 stimulates the expression and secretion of COL1A1 and COL3A1 collagen by fibroblasts via a p38 MAPK-dependent mechanism, thereby promoting fibrogenesis (27).
Tumor necrosis factor-like ligand 1A (TL1A), a member of the TNF family, interacts with death receptor-3 (DR3) to form the TL1A/DR3 costimulatory system, while abnormal TL1A/DR3 signaling is involved in chronic inflammation and fibrogenesis formation process, and may contribute to the development of colitis-associated inflammation and fibrosis through the regulation of immune responses or inflammatory factors (28, 29). TGF-β has three subtypes, namely TGF-β1, TGF-β2, and TGF-β3, which play a crucial role in cellular development, proliferation and differentiation. They are the main regulatory factors driving organ fibrosis and play an important role in tissue fibrosis processes (30, 31). Smad proteins, the downstream proteins and signaling transducers in the TGF-β pathway, can form the TGF-β/Smad signaling pathway with TGF-β. Phosphorylated type II TGF-β receptor and TGF-β ligand combine to form heterodimeric complexes, which can recruit and activate type I receptors, promoting the activation and phosphorylation of downstream Smad2/Smad3 proteins. Smad4 can bind to P-Smad2/3 to form a complex, which then enters the cell nucleus to regulate relevant target genes (32, 33). It was reported that normal intestinal myofibroblasts primarily release TGF-β3, while myofibroblasts in ulcerative colitis express both TGF-β1 and TGF-β3. In fibroblast cultures derived from CD fibrotic tissues, the expression of TGF-β3 is significantly reduced, but the release of TGF-β2 is enhanced (32, 34). Furthermore, it was also discovered that TGF-β1 selectively enhances the production of collagen protein in human intestinal smooth muscle cells in vitro and plays an important role as an inflammatory mediator in the pathogenesis of intestinal fibrosis (35). Berger et al. showed that abrogation of TGF-β expression in T cells was completely able to disrupt T/B cell homeostasis and induce T cell-mediated inflammatory lesions in various organs, including the intestine (36). If the termination of wound healing due to various factors is delayed, TGF-β will persist in its overexpression, further fueling tissue fibrosis. Additionally, TGF-β plays a role in converting fibroblasts into myofibroblasts, which actively contribute to the deposition of ECM (37). Moreover, TGF-β has the capability to stimulate fibroblasts to produce more TGF-β internally, thereby establishing an autocrine loop that significantly contributes to the progression of fibrosis (38).
2.3 Epithelial-mesenchymal transition
EMT is a biological process in which epithelial cells undergo a transformation into mesenchymal cells, which, on the other hand, are more migratory and have the ability to differentiate into various cell types such as fibroblasts, smooth muscle cells, and immune cells (39). In addition, the loss of cell polarity, the enhanced invasion abilities, the downregulation of epithelial markers such as adhesion molecules, tight junction proteins (E-cadherin, ZOs and claudin), and cytokeratins, and upregulation of mesenchymal markers, including α-smooth muscle actin (α-SMA), vimentin, fibronectin, collagen and the EMT-associated transcription factors (Twist, Snail, Slug and Zeb1/2) are also features of the mesenchymal phenotype (40). Notably, a mounting body of evidence has emerged in recent years, underscoring a profound association between EMT and the development of intestinal fibrosis. EMT emerges as a pivotal indicator of intestinal fibrosis, characterized by the loss of epithelial polarity and phenotype, accompanied by a consequential metamorphosis into mesenchymal cells. Noteworthy, abnormal expression of EMT-associated molecules has been observed within fibrotic lesions of patients afflicted with CD affecting the intestines (41). It was also reported that EMT is involved in fistula formation, an abnormal channel between two epithelial cells that is associated with fibrosis (42).
The process of EMT in intestinal fibrosis is regulated by various signaling pathways and other mediators. Wang et al. demonstrate that miR-21 is significantly upregulated in intestinal tissue of CD patients with fibrotic strictures, followed by reduced PTEN expression, increased expression of EMT markers and mTOR, and imbalance in MMP9 /TIMP1. Knocking down miR-21 can reduce EMT in vitro and inhibit TNBS-induced intestinal fibrosis in vivo by inhibiting EMT and balancing MMPs/TIMPs. Their findings indicate that inhibiting miR-21 with antagonists could be a potential new therapeutic target for the treatment of CD fibrosis complications (43). Ortiz-Masi et al. elucidated the connection between the increased recognition of EMT and the WNT2b/FZD4 interaction in the intestinal tissue of CD patients. WNT2b stimulation elicits in vitro EMT by instigating the induction of FZD4, thereby potentially orchestrating the transmural engagement that characterizes penetrating CD. A comprehensive comprehension of FZD4’s involvement in this intricate mechanism may pave the way for novel therapeutic strategies aimed at averting complications arising from CD based on their findings (44). Certain pro-inflammatory cytokines, e.g., IL-17A, have been shown to involve fibrotic properties and are associated with fibrosis in multiple organs, including the intestine. The findings presented by Zhang et al. underscore the involvement of the EMT pathway as a key mechanism employed by IL-17A to drive intestinal fibrosis in both clinical CD patients and in a model of TNBS-induced chronic colitis. These results shed light on the potential of targeting EMT as a promising strategy for the development of anti-fibrotic therapeutics, with the prospect of mitigating IL-17A-mediated effects (45). To assess the functional state of TGF-β1 signaling within inflamed tissues of individuals with inflammatory bowel disease (IBD) and investigate the potential amelioration of impaired epithelial adhere and junction in IBD through the inhibition of TGF-β1 signaling using small molecules, Ghorbaninejad et al. directed their attention toward the modulation of EMT pathways. Their findings revealed the activation of both canonical and non-canonical TGF-β1 signaling cascades in inflamed intestinal epithelial cells, leading to the induction of EMT and disruption of epithelial tight junctions, which serve as distinguishing features of IBD (46). In addition, mediators such as Toll-like receptor 4 are also involved in inducing EMT and promoting intestinal fibrosis (43, 47). In addition to EMT, endothelial-mesenchymal transition (EndoMT) has also been reported as a novel mechanism of fibrosis (48).
2.4 Differentiation of fibroblast to myofibroblast
It is worth noting that the differentiation of fibroblast to myofibroblast also plays a crucial role in the development of intestinal fibrosis as myofibroblasts contribute to tissue repair and wound healing by secreting collagen and other matrix proteins (49, 50). In pathological conditions, persistent micro-stimuli activation of epithelial cells leads to the production of numerous cytokines at the site of injury. Cytokines like transforming growth factor TGF-β and tumor necrosis factor TNF-α trigger fibroblast proliferation, migration, accumulation, and differentiation into myofibroblasts, forming fibroblastic foci at sites of progressive intestinal injury and repair (51). The appearance of fibroblastic foci is a key feature in intestinal pathogenesis, indicating fibrotic areas during active progression and serving as a histopathological marker for irreversible fibrosis. These foci are primarily composed of myofibroblasts expressing α-SMA (52, 53). Myofibroblasts differ in ultrastructure and function from fibroblasts and smooth muscle cells, secreting an abundance of matrix metalloproteinases that disrupt the tissue matrix balance. Fibroblasts can also synthesize gelatinase to degrade the basement membrane during migration, hampering tissue regeneration. Myofibroblasts release angiotensinogen, promoting lung epithelial cell apoptosis and affecting tissue repair and remodeling. The activation of fibroblasts into myofibroblasts by injured intestinal epithelial cells perpetuates lung epithelial cell damage, creating a harmful cycle that contributes to the development of intestinal fibrosis.
2.5 Intestinal microbiota
Intestinal microbiota, also known as gut microbiota, refers to the community of microorganisms that inhabit the human gastrointestinal tract and form a relatively stable ecosystem through interactions with the host. It is estimated that there are 1010–1014 bacteria in the human gut, consisting of approximately 500–1,500 different bacterial species. As the largest and most complex ecosystem in the human body, the gut microbiota and its thousands of metabolic products have a profound impact on various aspects of host physiological activities (32). There seems to be increasing evidence that the gut microbiota influences intestinal fibrosis. Numerous investigations have demonstrated that chronic infection with Salmonella leads to the manifestation of profound and enduring intestinal fibrosis in mice, concomitant with the elicitation of protease expression in macrophages and epithelial cells (54). Furthermore, it has been observed that fibrosis can be directly instigated by microbial derivatives, particularly constituents of the cellular wall. Moreover, the existence of fecal matter or anaerobic bacteria in the gastrointestinal tract has been identified as a potential trigger for intestinal fibrosis (55).
In a study conducted by Jacob et al., the investigation focused on elucidating the impact of diverse microbial communities and their interactions with TL1A on the development of fibrosis. Remarkably, the study revealed the complete elimination of the pro-fibrotic and inflammatory phenotype induced by TL1A overexpression in the absence of a resident microbiota. To further unravel the intricate dynamics, the authors orally administered fecal samples obtained from specific pathogen-free (SPF) mice and healthy human donors to germ-free (GF) wild-type and TL1A-transgenic (Tl1a-Tg) mice. The outcomes unequivocally demonstrated that colonization with SPF microbiota resulted in escalated collagen deposition and activation of fibroblasts within the intestines of TL1A-Tg mice. Conversely, under GF conditions, fibroblast migration and activation were notably diminished, underscoring the indispensable role of specific microbial populations in TL1A-mediated intestinal fibrosis and fibroblast activation (56). In another investigation by Zhao et al., it was discovered that pretreatment with antibiotics augmented the ability of mice to restore their intestinal microbiota subsequent to radiation exposure. Intriguingly, this pretreatment effectively curtailed lipopolysaccharide (LPS) levels and modulated the polarization of ileal macrophages by inhibiting the TLR4/MyD88/NF-κB signaling pathway. Moreover, downregulation of TGF-β1, phosphorylated Smad-3, and SMA protein levels, coupled with upregulation of E-cadherin protein expression, was observed. Consequently, these alterations culminated in a reduction of inflammation and prevention of intestinal fibrosis. Significantly, these beneficial effects translated into improved post-radiotherapy survival rates and ameliorated intestinal damage (57). In summary, the involvement of the gut microbiota in the pathogenesis of intestinal fibrosis can manifest through direct mechanisms or via inflammation-mediated processes. Consequently, modulation of the gut microbiota may represent a promising therapeutic avenue for the treatment of intestinal fibrosis.
3 Research progress in the treatment of intestinal fibrosis
As the understanding of the pathogenesis of intestinal fibrosis deepens, new research into appropriate treatments has also emerged. Although traditional treatments, e.g., antibiotics and surgery, are used for intestinal fibrosis, there are currently no clinically approved targeted antifibrotic drugs for it. The development of therapeutic drugs primarily focuses on the underlying mechanisms of intestinal fibrosis with the aim of improving patient outcomes. Some recent promising therapies on intestinal fibrosis were summarized in Table 1.
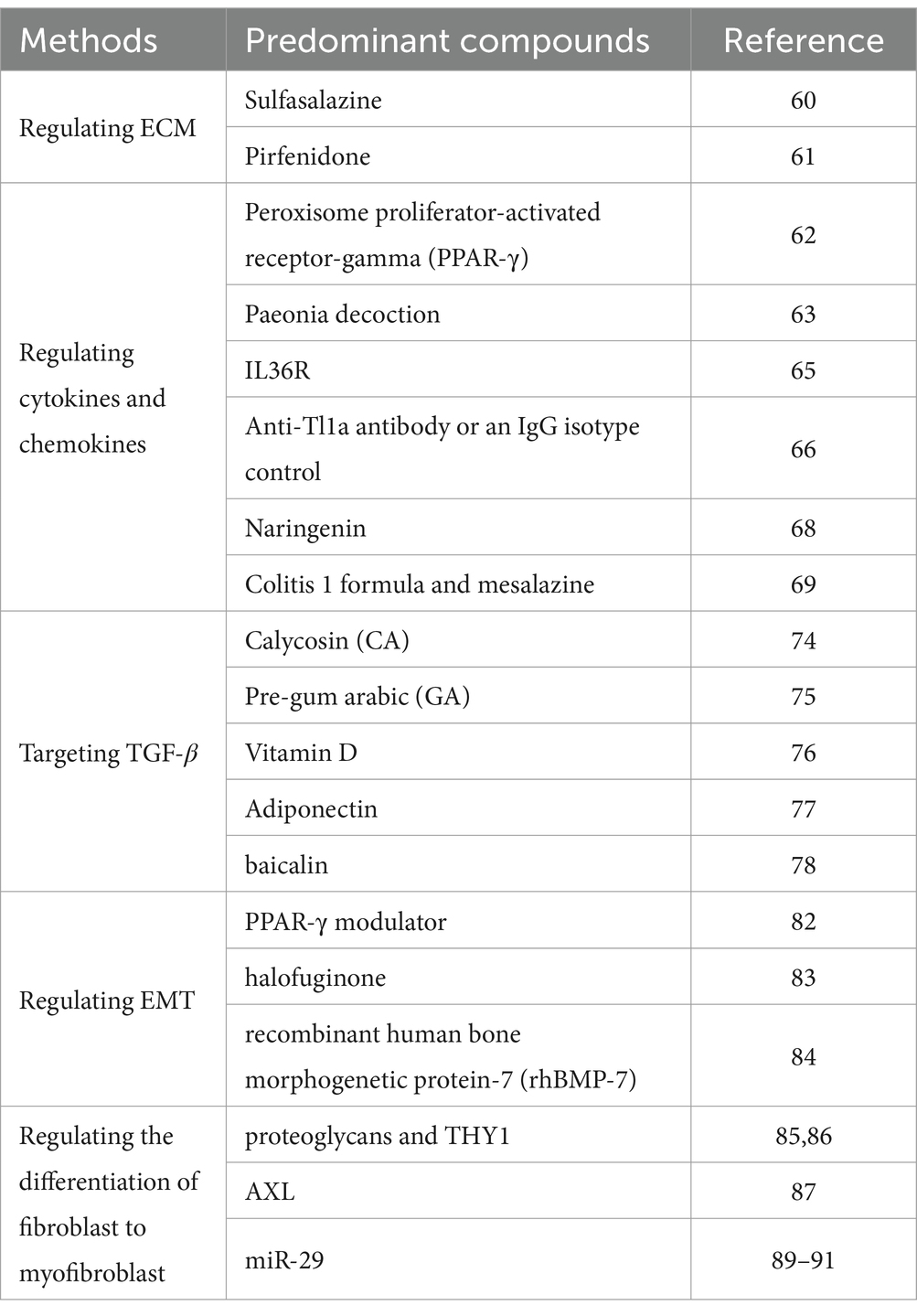
Table 1. Some recent promising therapies on intestinal fibrosis.
3.1 Regulating ECM
As mentioned above, intestinal fibrosis is primarily caused by an imbalance in ECM deposition and degradation, regulated by MMPs and TIMPs. Therefore, targeting ECM regulation with drugs can achieve certain therapeutic goals. It is reported that fibroblast activation protein (FAP) is involved in fibrosis by regulating ECM deposition. The inhibition of FAP could reduce the production of type I collagen and TIMP1, suggesting that targeting FAP can restore ECM homeostasis in fibrotic strictures of patients with CD (58). Furthermore, in a mouse model of chronic intestinal inflammation, increased transcription of plasminogen activator inhibitor-1 (PAI-1) was observed in active fibrotic lesions, promoting TNBS-induced intestinal fibrosis. Oral administration of a PAI-1 inhibitor in this mouse model upregulates MMP9 protein, reduces collagen accumulation, and inhibits fibrogenesis. Some drugs are already available for ECM modulation. Sulfasalazine, a molecule with anti-inflammatory activity, protects the colonic mucosa from TNBS-induced damage and inhibits intestinal fibrosis by regulating the balance of TIMP/MMP proteins and ECM degradation (59). Pirfenidone is an antifibrotic drug that treats intestinal fibrosis by inhibiting the proliferation of intestinal fibroblasts and suppressing collagen production (60).
3.2 Regulating cytokines and chemokines
Cytokines and chemokines play a crucial role in the development of intestinal fibrosis and are effective targets for the treatment of intestinal fibrosis. It has been found that peroxisome proliferator-activated receptor-gamma (PPAR-γ) has anti-inflammatory and antifibrotic effects in multiple organs and can downregulate the production of pro-inflammatory cytokines interleukins. In preliminary clinical trials and experimental models of intestinal fibrosis, PPAR-γ has shown improvement in the fibrotic formation process. Therefore, studying new molecules that can improve inflammation and fibrotic lesions as PPAR-γ agonists may be an effective treatment for preventing and treating IBD and intestinal fibrosis (61). It is also reported that paeonia decoction (a traditional Chinese herbal formula) can improve radiation-induced enteritis in C57BL/6 mice and its potential protective mechanism may be associated with reduced expression of interleukins, thereby alleviating inflammation and improving intestinal fibrosis (62). Additionally, IL-36 promotes the secretion of pro-fibrotic mediators, and the expression of MMP13 is regulated by IL36R signaling, while MMP13-deficient mice exhibit less fibrosis in a chronic dextran sodium sulfate (DSS) model, with reduced numbers of αSMA+ fibroblasts. Therefore, the inhibition of IL-36R signaling has been proposed as a potential therapeutic strategy for fibrotic diseases (20). In mice with DSS or TNBS-induced colitis, IL-36 induces the expression of fibrosis-related genes in regulatory fibroblasts. Remarkably, the reduction of chronic colitis and intestinal fibrosis could be achieved by inhibiting or knocking out the IL36R gene in mice and the fibrosis phenomenon could also be significantly reduced by injecting of antibodies against IL-36R (63).
It is believed that blocking TL1A may have potential therapeutic effects in intestinal fibrosis. Li et al. reported that a chronic colitis model was established by injecting CD4+CD45RBhigh naive T cells obtained from either C57BL/6 wild type mice or LCK-CD2-Tl1a-GFP transgenic mice into recombinase activating gene-1-deficient mice via intraperitoneal injection. The colitis model mice were subjected to either prophylactic or therapeutic treatment using anti-Tl1a antibody or an IgG isotype control. Histopathological changes in colonic tissue were assessed using Hematoxylin and eosin staining, Masson’s trichrome staining, and sirius red staining. Additionally, immunohistochemical staining was performed to evaluate the expression levels of collagen I, collagen III, TIMP1, vimentin, α-SMA, and TGF-β1/Smad3. The findings revealed that inhibiting the activation of intestinal fibroblasts and reducing collagen synthesis through the use of anti-Tl1a antibody contributed to the alleviation of intestinal inflammation and fibrosis in the chronic colitis model induced by T cell transfer. This effect may be attributed to the inhibition of the TGF-β1/Smad3 signaling pathway (64).
Recent studies also indicate that naringenin can improve DSS-induced colitis and colonic fibrosis in mice. After intervention with naringenin, UC mice showed a significant increase in body weight, a significant decrease in the ratio of colon weight to length, and a significant reduction in levels of TNF-α, IL-1β, IL-6, and the degree of colonic fibrosis. This demonstrates that naringenin can alleviate intestinal inflammation, improve intestinal barrier function, and inhibit DSS-induced colitis fibrosis (65, 66). The combination of colitis 1 formula and mesalazine has been found to be more effective in treating ulcerative colitis than mesalazine alone. This combined treatment reduces levels of TNF-α, IL-6, and IL-8, regulates fibrosis-related growth factors, and lowers inflammatory factor levels to promote ulcer healing (67).
3.3 Targeting TGF-β
Targeted therapy against TGF-β is the most promising anti-fibrotic treatment approach, as it is the major molecular mediator of fibrosis, with TGF-β1 being the most important among them. It was reported that local activation of angiotensin II strongly stimulates the production of TGF-β1. Angiotensin II is the main effector of the renin-angiotensin system, and its activity is elevated in the colonic mucosa of patients with CD. Therefore, it is believed that angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor antagonists may play a role in the process of intestinal fibrosis formation, for example, captopril (68–70). The cholesterol-lowering drug simvastatin can also regulate TGF-β and exert its effects. Additionally, direct targeting of drugs has significant effects (71).
In a study conducted by Liu et al., the potential antifibrotic properties of calycosin (CA) were examined using CCD-18Co cells, which are human intestinal fibroblasts induced by TGF-β1. The MTT method was employed to determine the optimal concentration of CA. Furthermore, the expression levels of α-smooth muscle actin (α-SMA), collagen I, and the TGF-β/Smad pathway were assessed using real-time polymerase chain reaction and western blot analysis. The experimental findings indicated that CA exhibited concentrations of 12.5, 25, and 50 μmol/L. Notably, CA displayed inhibitory effects on the expression of α-SMA and collagen I. Furthermore, CA demonstrated the ability to modulate the TGF-β/Smad signaling pathway. Specifically, CA hindered the expression of p-Smad2, p-Smad3, Smad4, and TGF-β1, while enhancing the expression of Smad7, thereby suppressing the TGF-β/Smad pathway. Consequently, CA exhibited promise in mitigating intestinal fibrosis through the inhibition of the TGF-β/Smad pathway (72).
Al-Araimi et al. reported that colitis was induced in C57BL/6 mice using DSS, followed by their transition to normal drinking water to monitor the recovery process. The mice were divided into two groups: the pre-gum arabic (GA) group received 140 g/L GA prior to colitis induction, while the post-GA group received GA after colitis induction. Body weight changes, disease activity index (DAI), and histological assessment were used to evaluate disease activity and recovery. The expression levels of proinflammatory, anti-inflammatory, and fibrotic markers were measured in the colonic tissues. During the recovery phase, the pre-GA group exhibited an increase in body weight without significant differences in DAI scores. Moreover, this group demonstrated lower histological colitis scores compared to the post-GA group, which exhibited higher DAI and histological scores during recovery. Notably, the pre-GA group displayed increased expression of proinflammatory markers during the recovery phase, while the expression of fibrotic markers, including TGF-β1 and procollagen I, was reduced. The reduction in fibrotic marker expression correlated with decreased collagen staining and increased proliferation of epithelial cells. The administration of GA exhibited protective and ameliorative effects on the severity of DSS-induced colitis, reducing colonic fibrosis and suppressing TGF-β1 expression (73).
Tao et al. reported that mice deficient in vitamin D were randomly assigned to two groups at the weaning stage (week 4) and provided either a vitamin D-deficient or vitamin D-sufficient diet. Intestinal fibrosis was induced by administering 2,4,6-trinitrobenzene sulfonic acid every 6 weeks, starting from week 8. At week 14, the levels of extracellular matrix (ECM) production and total collagen were assessed in the colon. Additionally, the TGF-β1/Smad3 signaling pathway was investigated in isolated colonic subepithelial myofibroblasts (SEMF). The expression of vitamin D receptor (VDR), α-SMA, and Collagen I was evaluated in both normal SEMF and VDR-null SEMF following exposure to TGF-β1 and/or 1,25(OH)2D3. The findings indicate that vitamin D has a preventive effect on intestinal fibrosis in mice with chronic colitis and vitamin D deficiency, and its mechanism may be related to the inhibition of the TGF-β1/Smad3 pathway through VDR induction in SEMF (74). Furthermore, the treatment of intestinal fibrosis with adiponectin, baicalin, and the spleen-strengthening medicine are also related TGF-β (75–77).
3.4 Regulating EMT
Targeting EMT is another treatment for intestinal fibrosis and Atractylenolide III (ATL-III) may inhibit TGF-β1-induced EMT in IEC-6 cells by activating the AMPK signaling pathway, thereby inhibiting intestinal fibrosis (78). PPAR-γ is a member of the nuclear hormone receptor superfamily and acts as a ligand-activated transcription factor with various roles in lipid metabolism, inflammation, cell proliferation, and fibrosis. Significant impairment of PPAR-γ expression has been observed in colonic epithelial cells of IBD patients, suggesting that disruption of PPAR-γ signaling may be a key step in the pathogenesis of IBD. PPAR-γ overexpression can prevent tissue fibrosis, while its deficiency can increase fibrosis (79). It was reported that a novel PPAR-γ modulator, GED-0507-34 Levo, can improve DSS-induced intestinal fibrosis by regulating EMT mediators and pro-fibrotic molecules (80). Recently, halofuginone was used to inhibit TGF-β1-induced EMT in IPEC-J2 cells through the eIF2α/SMAD signaling pathway, making them a potential anti-EMT drug for the treatment of intestinal fibrosis (81). Furthermore, there is also evidence that recombinant human bone morphogenetic protein-7 (rhBMP-7) has anti-fibrotic effects, as it can inhibit TGF-β1-induced EMT associated with intestinal fibrosis (82).
3.5 Regulating the differentiation of fibroblast to myofibroblast
Myofibroblasts primarily stem from resident fibroblasts within the tissue, but can also arise from epithelial and endothelial cells, as well as other mesenchymal precursors. Various factors shape their differentiation, including cytokines, growth factors, the composition and rigidity of the extracellular matrix, and cell surface molecules like proteoglycans and THY1, which is also the key point in the treatment of intestinal fibrosis by regulating the differentiation of fibroblast to myofibroblast (51, 83). Steiner et al. found that activation of the AXL pathway occurs in models of intestinal fibrosis. Blocking AXL signaling using the compound BGB324 effectively prevents matrix stiffness and TGF-β1-induced fibrosis in human colonic myofibroblasts. Moreover, suppressing AXL with BGB324 enhances the vulnerability of myofibroblasts to apoptosis. Additionally, treatment with BGB324 inhibits the expression of fibrotic genes and proteins induced by TGF-β1 in human intestinal organoids (84).
Numerous studies have highlighted the dual role of miR-29 in tumor progression, particularly emphasizing its significant inhibitory impact on fibrosis (85). For instance, Yuan R et al. demonstrated that miR-29a contained in exosomes derived from human adipose-derived mesenchymal stem cells (hADSCs) directly interacts with TGF-β2, effectively suppressing the TGF-β/Smad signaling pathway and consequently impeding the development of pathological scars (86). Moreover, lncRNA H19 has been identified as a regulator targeting miR-29b to modulate the behavior of oral mucosal fibroblasts (87). In a different study, Yang J et al. established an in vitro model of silicosis cells with manipulated levels of miR-29c and observed that miR-29c successfully impedes the SiO2-induced trans-differentiation of lung fibroblasts in vitro (88). The competitive binding of lncRNA TUG1 with miR-29c enhances the hypoxia-induced fibroblast-to-myofibroblast transition (FMT) in cardiac fibroblasts (89). Consequently, circHIPK3 and the miR-29 family present themselves as promising therapeutic targets for fibrosis that merit additional investigation.
4 Future perspective
Currently, our understanding of the pathological mechanisms of intestinal fibrosis is still limited. Further research is needed to investigate the pathogenesis of intestinal fibrosis, such as inflammation-mediated fibrosis, extracellular matrix remodeling, and cellular activation, in order to reveal its development and progression mechanisms. With a deeper understanding of the mechanisms underlying intestinal fibrosis, we can explore new therapeutic targets. Factors such as cytokines, signaling pathways, and cell types that play a role in the fibrosis process are worth further investigation (90). Exploring these targets may lead to new treatment strategies. Intestinal fibrosis may be driven by multiple different mechanisms, thus individual variations may exist. Developing personalized treatment strategies and selecting the optimal treatment methods based on the specific pathological mechanisms of each patient can help improve treatment outcomes and prognosis. Due to the complexity of intestinal fibrosis, a single treatment may not be sufficient to completely reverse fibrosis (91). Therefore, combining multiple treatment modalities such as medication, surgical intervention, and physical therapy may produce better treatment outcomes.
5 Conclusion
In summary, the ECM, cytokines, and epithelial-mesenchymal transition play a crucial role in the occurrence, development, and treatment of intestinal fibrosis. In recent years, significant progress has been made in understanding the mechanisms and transformations of intestinal fibrosis, providing a comprehensive understanding of its pathogenesis and new strategies for clinical treatment. To some extent, the symptoms of intestinal fibrosis can be controlled, and long-term remission is possible. Although research on the cellular and molecular mechanisms underlying the pathogenesis of intestinal fibrosis is still in its early stages, anti-fibrotic therapy remains a challenging clinical need. However, as our understanding of the pathogenesis of intestinal fibrosis continues to grow, the emergence of new anti-fibrotic treatments holds great promise in effectively controlling disease progression and alleviating patient suffering.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
YL: Writing – original draft. TZ: Investigation, Supervision, Writing – review & editing. KP: Writing – review & editing. HW: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by a grant from the Science & Technology Department of Sichuan Province (23NSFSC1222).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Henderson, NC, Rieder, F, and Wynn, TA. Fibrosis: from mechanisms to medicines. Nature. (2020) 587:555–66. doi: 10.1038/s41586-020-2938-9
2. Bamias, G, Pizarro, TT, and Cominelli, F. Immunological regulation of intestinal fibrosis in inflammatory bowel disease. Inflamm Bowel Dis. (2022) 28:337–49. doi: 10.1093/ibd/izab251
3. Zhao, X, Kwan, JYY, Yip, K, Liu, PP, and Liu, FF. Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Discov. (2020) 19:57–75. doi: 10.1038/s41573-019-0040-5
4. Wiciński, M, Gębalski, J, Mazurek, E, Podhorecka, M, Śniegocki, M, Szychta, P, et al. The influence of polyphenol compounds on human gastrointestinal tract microbiota. Nutrients. (2020) 12:350. doi: 10.3390/nu12020350
5. Mathur, R, Alam, MM, Zhao, XF, Liao, Y, Shen, J, Morgan, S, et al. Induction of autophagy in Cx3crl+ mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis. Mucosal Immunol. (2019) 12:612–23. doi: 10.1038/s41385-019-0146-4
6. Yang, Y, Zeng, QS, Zou, M, Zeng, J, Nie, J, Chen, DF, et al. Targeting gremlin 1 prevents intestinal fibrosis progression by inhibiting the fatty acid oxidation of fibroblast cells. Front Pharmacol. (2021) 12:663774. doi: 10.3389/fphar.2021.663774
7. Wijnands, AM, Mahmoud, R, Lutgens, MWMD, and Oldenburg, B. Surveillance and management of colorectal dysplasia and cancer in inflammatory bowel disease: current practice and future perspectives. Eur J Intern Med. (2021) 93:35–41. doi: 10.1016/j.ejim.2021.08.010
8. Hayashi, Y, and Nakase, H. The molecular mechanisms of intestinal inflammation and fibrosis in Crohn's disease. Front Physiol. (2022) 13:845078. doi: 10.3389/fphys.2022.845078
9. Watanabe, D, and Kamada, N. Contribution of the gut microbiota to intestinal fibrosis in Crohn's disease. Front Med. (2022) 9:1826240. doi: 10.3389/fmed.2022.826240
10. Wang, R, Wang, DJ, Wang, HW, Wang, TY, Weng, YJ, and Zhang, YL. Therapeutic targeting of Nrf2 signaling by maggot extracts ameliorates inflammation-associated intestinal fibrosis in chronic DSS-induced colitis. Front Immunol. (2022) 12:67059. doi: 10.3389/fimmu.2021.670159
11. Hai, SS, Liu, WX, Zhang, HL, Yuan, LL, Dong, L, and Peng, N. Progress of NRF2 in inflammatory bowel disease-associated intestinal fibrosis. Chin J Gastroenterol Hepatol. (2023) 32:1190–4. doi: 10.3969/j.issn.1006-5709.2023.10.023
12. Garg, M, Royce, SG, Tikellis, C, Shalluel, C, Batu, D, Velkoskaet, E, et al. Imbalance of the renin-angiotensin system may contribute to inflammation and fibrosis in IBD: a novel therapeutic target? Gut. (2020) 69:841–51. doi: 10.1136/gutjnl-2019-318512
13. Moretti, L, Stalfort, J, Barker, TH, and Abebayehu, D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J Biol Chem. (2022) 298:101530. doi: 10.1016/j.jbc.2021.101530
14. Deng, Z, Fear, MW, Suk-Choi, Y, Wood, FM, Allahham, A, Mutsaers, SE, et al. The extracellular matrix and mechanotransduction in pulmonary fibrosis. Int J Biochem Cell Biol. (2020) 126:105802. doi: 10.1016/j.biocel.2020.105802
15. Speca, S, Giusti, I, Rieder, F, and Latella, G. Cellular and molecular mechanisms of intestinal fibrosis. World J Gastroenterol. (2012) 18:3635–61. doi: 10.3748/wjg.v18.i28.3635
16. Wu, X, Lin, X, Tan, J, Liu, Z, He, J, Hu, F, et al. Cellular and molecular mechanisms of intestinal fibrosis. Gut Liver. (2023) 17:360–74. doi: 10.5009/gnl220045
17. Zhao, X, Chen, J, Sun, H, Zhang, Y, and Zou, D. New insights into fibrosis from the ECM degradation perspective: the macrophage-MMP-ECM interaction. Cell Biosci. (2022) 12:117. doi: 10.1186/s13578-022-00856-w
18. Medina, C, and Radomski, MW. Role of matrix metalloproteinases in intestinal inflammation. J Pharmacol Exp Ther. (2006) 318:933–8. doi: 10.1124/jpet.106.103465
19. de Bruyn, JR, van den Brink, GR, Steenkamer, J, Buskens, CJ, Bemelman, WA, Meisner, S, et al. Fibrostenotic phenotype of myofibroblasts in Crohn's disease is dependent on tissue stiffness and reversed by LOX inhibition. J Crohns Colitis. (2018) 12:849–59. doi: 10.1093/ecco-jcc/jjy036
20. Koop, K, Enderle, K, Hillmann, M, Ruspeckhofer, L, Vieth, M, Sturm, G, et al. Interleukin 36 receptor-inducible matrix metalloproteinase 13 mediates intestinal fibrosis. Front Immunol. (2023) 14:1163198. doi: 10.3389/fimmu.2023.1163198
21. Park, JM, Kim, J, Lee, YJ, Bae, SU, and Lee, HW. Inflammatory bowel disease-associated intestinal fibrosis. J Pathol Transl Med. (2023) 57:60–6. doi: 10.4132/jptm.2022.11.02
22. Lim, WW, Ng, B, Widjaja, A, Xie, C, Su, L, Ko, N, et al. Transgenic interleukin 11 expression causes cross-tissue fibro-inflammation and an inflammatory bowel phenotype in mice. PLoS One. (2020) 15:e0227505. doi: 10.1371/journal.pone.0227505
23. Milara, J, Roger, I, Montero, P, Artigues, E, Escrivá, J, and Cortijo, J. IL-11 system participates in pulmonary artery remodeling and hypertension in pulmonary fibrosis. Respir Res. (2022) 23:313. doi: 10.1186/s12931-022-02241-0
24. Masterson, JC, Capocelli, KE, Hosford, L, Biette, K, McNamee, EN, de Zoeten, EF, et al. Eosinophils and IL-33 perpetuate chronic inflammation and fibrosis in a pediatric population with stricturing Crohn's ileitis. Inflamm Bowel Dis. (2015) 21:2429–40. doi: 10.1097/MIB.0000000000000512
25. Imai, J, Kitamoto, S, Sugihara, K, Nagao-Kitamoto, H, Hayashi, A, Morhardt, TL, et al. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. (2019) 12:632–43. doi: 10.1038/s41385-019-0138-4
26. Monteleone, G, Franzè, E, Troncone, E, Maresca, C, and Marafini, I. Interleukin-34 mediates cross-talk between stromal cells and immune cells in the gut. Front Immunol. (2022) 13:873332. doi: 10.3389/fimmu.2022.873332
27. Franzè, E, Dinallo, V, Laudisi, F, Di Grazia, A, Di Fusco, D, Colantoni, A, et al. Interleukin-34 stimulates gut fibroblasts to produce collagen synthesis. J Crohns Colitis. (2020) 14:1436–45. doi: 10.1093/ecco-jcc/jjaa073
28. Jia, WX, Yang, MY, Han, F, Luo, YX, Wu, MY, Li, CY, et al. Effect and mechanism of TL1A expression on epithelial-mesenchymal transition during chronic colitis-related intestinal fibrosis. Mediat Inflamm. (2021) 2021:5927064. doi: 10.1155/2021/5927064
29. Jacob, N, Kumagai, K, Abraham, JP, Shimodaira, Y, Ye, Y, Luu, J, et al. Direct signaling of TL1A-DR3 on fibroblasts induces intestinal fibrosis in vivo. Sci Rep. (2020) 10:18189. doi: 10.1038/s41598-020-75168-5
30. Soufla, G, Porichis, F, Sourvinos, G, Vassilaros, S, and Spandidos, DA. Transcriptional deregulation of VEGF, FGF2, TGF-beta1, 2, 3 and cognate receptors in breast tumorigenesis. Cancer Lett. (2006) 235:100–13. doi: 10.1016/j.canlet.2005.04.022
31. Yun, SM, Kim, SH, and Kim, EH. The molecular mechanism of transforming growth factor-β signalinging for intestinal fibrosis: a mini-review. Front Pharmacol. (2019) 10:162. doi: 10.3389/fphar.2019.00162
32. Hotta, Y, Uchiyama, K, Takagi, T, Kashiwagi, S, Nakano, T, Mukai, R, et al. Transforming growth factor β1-induced collagen production in myofibroblasts is mediated by reactive oxygen species derived from NADPH oxidase 4. Biochem Biophys Res Commun. (2018) 506:557–62. doi: 10.1016/j.bbrc.2018.10.116
33. McKaig, BC, Hughes, K, Tighe, PJ, and Mahida, YR. Differential expression of TGF-beta isoforms by normal and inflammatory bowel disease intestinal myofibroblasts. Am J Phys Cell Phys. (2002) 282:C172–82. doi: 10.1152/ajpcell.00048.2001
34. Naguib, R, and El-Shikh, WM. Clinical significance of hepatocyte growth factor and transforming growth factor-beta-1 levels in assessing disease activity in inflammatory bowel disease. Can J Gastroenterol Hepatol. (2020) 2020:2104314. doi: 10.1155/2020/2104314
35. Graham, MF, Bryson, GR, and Diegelmann, RF. Transforming growth factor beta 1 selectively augments collagen synthesis by human intestinal smooth muscle cells. Gastroenterology. (1990) 99:447–53. doi: 10.1016/0016-5085(90)91028-5
36. Berger, JR, Miller, CS, and Danaher, RJ. Distribution and quantity of sites of John Cunningham virus persistence in immunologically healthy patients: correlation with John Cunningham virus antibody and urine John Cunningham virus DNA. JAMA Neurol. (2017) 74:437–44. doi: 10.1001/jamaneurol.2016.5537
37. Zhu, H, Luo, H, Shen, Z, Hu, X, Sun, L, and Zhu, X. Transforming growth factor-β1 in carcinogenesis, progression, and therapy in cervical cancer. Tumour Biol. (2016) 37:7075–83. doi: 10.1007/s13277-016-5028-8
38. Travis, MA, and Sheppard, D. TGF-β activation and function in immunity. Annu. Rev. Immunol. (2014) 32:51–82. doi: 10.1146/annurev-immunol-032713-120257
39. Lu, W, and Kang, Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev Cell. (2019) 49:361–74. doi: 10.1016/j.devcel.2019.04.010
40. Brabletz, S, Schuhwerk, H, Brabletz, T, and Stemmler, MP. Dynamic EMT: a multi-tool for tumor progression. EMBO J. (2021) 40:e108647. doi: 10.15252/embj.2021108647
41. Wang, Z, Zhou, H, Cheng, F, Zhang, Z, and Long, S. MiR-21 regulates epithelial-mesenchymal transition in intestinal fibrosis of Crohn's disease by targeting PTEN/mTOR. Dig Liver Dis. (2022) 54:1358–66. doi: 10.1016/j.dld.2022.04.007
42. Ortiz-Masiá, D, Gisbert-Ferrándiz, L, Bauset, C, Coll, S, and Mamie, C. Succinate activates EMT in intestinal epithelial cells through SUCNR1: a novel protagonist in fistula development. Cells. (2020) 9:1104–11. doi: 10.3390/cells9051104
43. Ortiz-Masià, D, Salvador, P, Macias-Ceja, DC, Gisbert-Ferrándiz, L, Esplugues, JV, Manyé, J, et al. WNT2b activates epithelial-mesenchymal transition through FZD4: relevance in penetrating Crohn’s disease. J Crohns Colitis. (2020) 14:230–9. doi: 10.1093/ecco-jcc/jjz134
44. Zhang, HJ, Zhang, YN, Zhou, H, Guan, L, Li, Y, and Sun, MJ. IL-17A promotes initiation and development of intestinal fibrosis through EMT. Dig Dis Sci. (2018) 63:2898–909. doi: 10.1007/s10620-018-5234-x
45. Ghorbaninejad, M, Abdollahpour-Alitappeh, M, Shahrokh, S, Fayazzadeh, S, Asadzadeh-Aghdaei, H, and Meyfour, A. TGF-β receptor I inhibitor may restrict the induction of EMT in inflamed intestinal epithelial cells. Exp Biol Med (Maywood). (2023) 8:665–76. doi: 10.1177/153537022311519
46. Jun, YK, Kwon, SH, Yoon, HT, Park, H, Soh, H, Lee, HJ, et al. Toll-like receptor 4 regulates intestinal fibrosis via cytokine expression and epithelial-mesenchymal transition. Sci Rep. (2020) 10:19867. doi: 10.1038/s41598-020-76880-y
47. Wang, Y, Huang, B, Jin, T, Ocansey, DKW, Jiang, J, and Mao, F. Intestinal fibrosis in inflammatory bowel disease and the prospects of mesenchymal stem cell therapy. Front Immunol. (2022) 13:835005. doi: 10.3389/fimmu.2022.835005
48. Yang, X, Guo, Y, Chen, C, Shao, B, Zhao, L, Zhou, Q, et al. Interaction between intestinal microbiota and tumor immunity in the tumor microenvironment. Immunology. (2021) 164:476–93. doi: 10.1111/imm.13397
49. Marconi, GD, Fonticoli, L, Rajan, TS, Lanuti, P, Della, RY, Pierdomenico, SD, et al. Transforming growth factor-beta1 and human gingival fibroblast-to-myofibroblast differentiation: molecular and morphological modifications. Front Physiol. (2021) 12:676512. doi: 10.3389/fphys.2021.676512
50. Kis, K, Liu, X, and Hagood, JS. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev Mol Med. (2011) 13:e27. doi: 10.1017/S1462399411001967
51. Zhang, H, Zhou, Y, Wen, D, and Wang, J. Noncoding RNAs: master regulator of fibroblast to myofibroblast transition in fibrosis. Int J Mol Sci. (2023) 24:1801. doi: 10.3390/ijms24021801
52. Willis, BC, duBois, RM, and Borok, Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. (2006) 3:377–82. doi: 10.1513/pats.200601-004TK
53. Hashimoto, S, Gon, Y, and Takeshita, I. Transforming growth factor-β1 induces phenotypic modulation of human lung fibroblasts to myofibroblasts through a c-Jun-NH2-terminal kinase-dependent pathway. Am J Respir Crit Care Med. (2001) 163:152–7. doi: 10.1164/ajrccm.163.1.2005069
54. Ehrhardt, K, Steck, N, Kappelhoff, R, Stein, S, Rieder, F, Gordon, IO, et al. Persistent salmonella enterica serovar typhimurium infection induces protease expression during intestinal fibrosis. Inflamm Bowel Dis. (2019) 25:1629–43. doi: 10.1093/ibd/izz070
55. Rieder, F . The gut microbiome in intestinal fibrosis: environmental protector or provocateur? Sci Transl Med. (2013) 5:190. doi: 10.1126/scitranslmed.300473
56. Jacob, N, Jacobs, JP, Kumagai, K, Ha, CWY, Kanazawa, Y, Lagishetty, V, et al. Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal Immunol. (2018) 11:1466–76. doi: 10.1038/s41385-018-0055-y
57. Zhao, Z, Cheng, W, Qu, W, Shao, G, and Liu, S. Antibiotic alleviates radiation-induced intestinal injury by remodeling microbiota, reducing inflammation, and inhibiting fibrosis. ACS Omega. (2020) 5:2967–77. doi: 10.1021/acsomega.9b03906
58. Truffi, M, Sorrentino, L, Monieri, M, Fociani, P, Mazzucchelli, S, Bonzini, M, et al. Inhibition of fibroblast activation protein restores a balanced extracellular matrix and reduces fibrosis in Crohn's disease strictures ex vivo. Inflamm Bowel Dis. (2018) 24:332–45. doi: 10.1093/ibd/izx0008
59. Chen, H, Xu, H, Luo, L, Qiao, L, Wang, Y, Xu, M, et al. Thalidomide prevented and ameliorated pathogenesis of Crohn's disease in mice via regulation of inflammatory response and fibrosis. Front Pharmacol. (2019) 10:1486. doi: 10.3389/fphar.2019.01486
60. Cui, Y, Zhang, M, Leng, C, Blokzijl, T, Jansen, BH, Dijkstra, G, et al. Pirfenidone inhibits cell proliferation and collagen I production of primary human intestinal fibroblasts. Cells. (2020) 9:775. doi: 10.3390/cells9030775
61. Vetuschi, A, Pompili, S, Gaudio, E, Latella, G, and Sferra, R. PPAR-γ with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Eur Rev Med Pharmacol Sci. (2018) 22:8839–48. doi: 10.26355/eurrev_201812_16652
62. Li, Z, Gao, Y, Du, L, Yuan, Y, Huang, W, Fu, X, et al. Anti-inflammatory and anti-apoptotic effects of Shaoyao decoction on X-ray radiation-induced enteritis of C57BL/6 mice. J Ethnopharmacol. (2022) 292:115158. doi: 10.1016/j.jep.2022.115158
63. Scheibe, K, Kersten, C, Schmied, A, Vieth, M, Primbs, T, Carlé, B, et al. Inhibiting interleukin 36 receptor signaling reduces fibrosis in mice with chronic intestinal inflammation. Gastroenterology. (2019) 156:1082–1097.e11. doi: 10.1053/j.gastro.2018.11.029
64. Li, H, Song, J, Niu, G, Zhang, H, Guo, J, Shih, DQ, et al. TL1A blocking ameliorates intestinal fibrosis in the T cell transfer model of chronic colitis in mice. Pathol Res Pract. (2018) 214:217–27. doi: 10.1016/j.prp.2017.11.017
65. Danese, S, Klopocka, M, Scherl, EJ, Romatowski, J, Allegretti, JR, Peeva, E, et al. Anti-TL1A antibody PF-06480605 safety and efficacy for ulcerative colitis: a phase 2a single-arm study. Clin Gastroenterol Hepatol. (2021) 19:2324–2332.e6. doi: 10.1016/j.cgh.2021.06.011
66. Wand, KY, Huang, CX, Liu, JY, Qin, XH, Liu, ZQ, and Song, JL. Study on the improvement effect of naringenin on colonic fibrosis in DSS-induced colitis mic. China Food Sci Technol Soc. (2022) 2:220–1. doi: 10.26914/c.cnkihy.2022.050972
67. Wang, FX . Study on the therapeutic effect of colitis 1 formula combined with mesalazine in the treatment of ulcerative colitis. Chinese Medical Innovation. (2022) 19:87–90. doi: 10.26914/c.cnkihy.2022.050843
68. Wolf, G, Zahner, G, Schroeder, R, and Stahl, RA. Transforming growth factor beta mediates the angiotensin-II-induced stimulation of collagen type IV synthesis in cultured murine proximal tubular cells. Nephrol Dial Transplant. (1996) 11:263–9. doi: 10.1093/ndt/11.2.263
69. Hume, GE, Fowler, EV, Lincoln, D, Eri, R, Templeton, D, Florin, TH, et al. Angiotensinogen and transforming growth factor beta1: novel genes in the pathogenesis of Crohn's disease. J Med Genet. (2006) 43:e51. doi: 10.1136/jmg.2005.040477
70. Wengrower, D, Zanninelli, G, Pappo, O, Latella, G, Sestieri, M, Villanova, A, et al. Prevention of fibrosis in experimental colitis by captopril: the role of tgf-beta1. Inflamm Bowel Dis. (2004) 10:536–45. doi: 10.1097/00054725-200409000-00007
71. Burke, JP, Watson, RW, Murphy, M, Docherty, NG, Coffey, JC, and O'Connell, PR. Simvastatin impairs smad-3 phosphorylation and modulates transforming growth factor beta1-mediated activation of intestinal fibroblasts. Br J Surg. (2009) 96:541–51. doi: 10.1002/bjs.6577
72. Liu, J, Deng, T, Wang, Y, Zhang, M, Zhu, G, Fang, H, et al. Calycosin inhibits intestinal fibrosis on CCD-18Co cells via modulating transforming growth factor-β/smad signaling pathway. Pharmacology. (2019) 104:81–9. doi: 10.1159/000500186
73. Al-Araimi, A, Al Kindi, IA, Bani Oraba, A, AlKharusi, A, Ali, BH, Zadjali, R, et al. Gum arabic supplementation suppresses colonic fibrosis after acute colitis by reducing transforming growth factor β1 expression. J Med Food. (2021) 24:1255–63. doi: 10.1089/jmf.2021.0007
74. Tao, Q, Wang, B, Zheng, Y, Jiang, X, Pan, Z, and Ren, J. Vitamin D prevents the intestinal fibrosis via induction of vitamin D receptor and inhibition of transforming growth factor-beta1/Smad3 pathway. Dig Dis Sci. (2015) 60:868–75. doi: 10.1007/s10620-014-3398-6
75. Xie, M, Xiong, Z, Yin, S, Xiong, J, Li, X, Jin, L, et al. Adiponectin alleviates intestinal fibrosis by enhancing AMP-activated protein kinase phosphorylation. Dig Dis Sci. (2022) 67:2232–43. doi: 10.1007/s10620-021-07015-0
76. Xu, HJ, Zhu, Q, Ding, K, and Tan, YY. Research progress on the mechanism of baicalin in treating inflammatory bowel disease. Modern drugs clin prac. (2023) 38:219–23. doi: 10.3892/mmr.2023.13526
77. Zhen, L, Wen, XL, and Duan, SL. Mechanism of jianpi 1ingchang tongluo decoction on intestinal fibrosis induced by sodium trinitrobenzene sulfonate in mice with severe inflammatory bowel disease. JETCM. (2022) 31:1362–6. doi: 10.3969/j.issn.1004-745X.2022.09.011
78. Huang, M, Jiang, W, Luo, C, Yang, M, and Ren, Y. Atractylenolide III inhibits epithelial mesenchymal transition in small intestine epithelial cells by activating the AMPK signalinging pathway. Mol Med Rep. (2022) 25:98. doi: 10.3892/mmr.2022.12614
79. Latella, G, Di Gregorio, J, Flati, V, Rieder, F, and Lawrance, IC. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand J Gastroenterol. (2015) 50:53–65. doi: 10.3109/00365521.2014.968863
80. Di Gregorio, J, Sferra, R, Speca, S, Vetuschi, A, Dubuquoy, C, Desreumaux, P, et al. Role of glycogen synthase kinase-3β and PPAR-γ on epithelial-to-mesenchymal transition in DSS-induced colorectal fibrosis. PLoS One. (2017) 12:e0171093. doi: 10.1371/journal.pone.0171093
81. Duan, M, Wei, X, Cheng, Z, Liu, D, Fotina, H, Xia, X, et al. Involvement of eIF2α in halofuginone-driven inhibition of TGF-β1-induced EMT. J Biosci. (2020) 45:71. doi: 10.1007/s12038-020-00042-5
82. Santacroce, G, Lenti, MV, and Di Sabatino, A. Therapeutic targeting of intestinal fibrosis in Crohn's disease. Cells. (2022) 11:429. doi: 10.3390/cells11030429
83. Jalili Angourani, K, Mazhari, S, Farivar, S, Salman Mahini, D, Rouintan, A, and Baghaei, K. Fibroblast-myofibroblast crosstalk after exposure to mesenchymal stem cells secretome. Gastroenterol Hepatol Bed Bench. (2018) 11:S73–9. doi: 10.22037/GHFBB.V0I0.1539
84. Steiner, CA, Rodansky, ES, Johnson, LA, Berinstein, JA, Cushing, KC, Huang, S, et al. AXL is a potential target for the treatment of intestinal fibrosis. Inflamm Bowel Dis. (2021) 27:303–16. doi: 10.1093/ibd/izaa169
85. Kwon, JJ, Factora, TD, Dey, S, and Kota, J. 1 a systematic review of miR-29 in cancer. Mol Ther Oncolytics. (2019) 12:173–94. doi: 10.1016/j.omto.2018.12.011
86. Yuan, R, Dai, X, Li, Y, Li, C, and Liu, L. Exosomes from miR-29a-modified adipose-derived mesenchymal stem cells reduce excessive scar formation by inhibiting TGF-beta 2/Smad3 signaling. Mol Med Rep. (2021) 24:12398. doi: 10.3892/MMR.2021.12398
87. Yu, CC, Liao, YW, Hsieh, PL, and Chang, YC. Targeting lncRNA H19/miR-29b/COL1A1 axis impedes myofibroblast activities of precancerous oral submucous fibrosis. Int J Mol Sci. (2021) 22:2216. doi: 10.3390/ijms22042216
88. Yang, J, Zhang, Y, Huang, L, Lan, YJ, and Zhang, Q. The inhibition effect of miR-29c on lung fibroblasts trans-differentiation induced by SiO2. Chin J Ind Hyg Occup Dis. (2019) 37:321–6. doi: 10.3760/cma.j.issn.1001-9391.2019.05.001
89. Zhu, Y, Feng, Z, Jian, Z, and Xiao, Y. Long noncoding RNA TUG1 promotes cardiac fibroblast transformation to myofibroblasts via miR-29c in chronic hypoxia. Mol Med Rep. (2018) 18:3451–60. doi: 10.3892/mmr.2018.9327
90. Sabbagh, C, Mauvais, F, Tuech, JJ, Tresallet, C, Ortega-Debalon, P, Mathonnet, M, et al. Impact of a procalcitonin-based algorithm on the quality of management of patients with uncomplicated adhesion-related small bowel obstruction assessed by a textbook outcome: a multicenter cluster-randomized open-label controlled trial. BMC Gastroenterol. (2022) 22:90. doi: 10.1186/s12876-022-02144-w
Keywords: intestinal fibrosis, pathological mechanisms, therapeutic research, epithelial-mesenchymal transition, cytokines and chemokines
Citation: Liu Y, Zhang T, Pan K and Wei H (2024) Mechanisms and therapeutic research progress in intestinal fibrosis. Front. Med. 11:1368977. doi: 10.3389/fmed.2024.1368977
Edited by:
Pradeep Kumar Shukla, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Rakesh Kumar, Washington University in St. Louis, United StatesShirin Fatma, University of Illinois at Urbana-Champaign, United States
Copyright © 2024 Liu, Zhang, Pan and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kejian Pan, cGFua2VqaWFuMjAwNUBjbWMuZWR1LmNu; He Wei, d2VpaGVAY21jLmVkdS5jbg==