 Patrik Krumpolec1*
Patrik Krumpolec1* Dominik Kodada1,2Dominik Hadžega1Oliver Petrovič1Klaudia Babišová1Erik Dosedla3
Dominik Kodada1,2Dominik Hadžega1Oliver Petrovič1Klaudia Babišová1Erik Dosedla3 Zuzana Turcsányiová3
Zuzana Turcsányiová3 Gabriel Minárik1
Gabriel Minárik1- 1Medirex Group Academy n.o., Nitra, Slovakia
- 2Department of Clinical Biology, Genetics and Clinical Genetics, Faculty of Medicine, Comenius University in Bratislava, Bratislava, Slovakia
- 3Department of Gynaecology and Obstetrics, Faculty of Medicine, Pavol Jozef Šafárik University in Košice, Košice, Slovakia
Background: The mode of delivery represents an epigenetic factor with potential to affect further development of the individual by multiple mechanisms. DNA methylation may be one of them, representing a major epigenetic mechanism involving direct chemical modification of the individual’s DNA. This pilot study aims to examine whether a specific mode of delivery induces changes of DNA methylation by comparing the umbilical cord blood and peripheral blood of the newborns.
Methods: Blood samples from infants born by vaginal delivery and caesarean section were analysed to prepare the Methylseq library according to NEBNext enzymatic Methyl-seq Methylation Library Preparation Kit with further generation of target-enriched DNA libraries using the Twist Human Methylome Panel. DNA methylation status was determined using Illumina next-generation sequencing (NGS).
Results: We identified 168 differentially methylated regions in umbilical cord blood samples and 157 regions in peripheral blood samples. These were associated with 59 common biological, metabolic and signalling pathways for umbilical cord and peripheral blood samples.
Conclusion: Caesarean section is likely to represent an important epigenetic factor with the potential to induce changes in the genome that could play an important role in development of a broad spectrum of disorders. Our results could contribute to the elucidation of how epigenetic factors, such as a specific mode of delivery, could have adverse impact on health of an individual later in their life.
Introduction
In the most recent three decades, a growing number of deliveries by caesarean section (CS) has been observed as the worldwide rate has risen from 7% in 1990 to about 21% at present (1, 2). In countries such as the Dominican Republic, Brazil, Cyprus, Egypt and Turkey, CSs even outnumber vaginal deliveries (VDs). Moreover, this number continues to increase as about 29% of all births are likely to be performed using caesarean section by 2030 (2).
Along with the rising rate of CSs, there has also been a trend of an increased share of CS due to non-clinical reasons (maternal preference or other social factors). The excessive use of CS has raised public health concerns (3) as an increasing volume of evidence indicates long-term impact of CS on the maternal and also the child’s state of health. The molecular mechanisms involved in outcomes of CS are not entirely explained (4, 5), but an important role could play an infant stress during labour. Although it represents an advantage in preparation for extrauterine life, the stress experienced by infants varies vastly between delivery modes (6). There is a flush of stress hormones in the infant during VD. Especially hormones like catecholamines and cortisol are responsible for readiness of the respiratory system for gas exchange, increased blood flow, activation of the CNS and mobilization of fuel processing (7, 8). However, this high expression of stress hormones also triggers a cascade of hormones and cytokines involved in inflammatory defence pathways (7, 9). Compared to CS, infants born via VD have higher levels of such hormone release (7, 8, 10, 11), greater complexity of intestinal flora (12) and higher immune response (13). It is therefore not surprising that epidemiological studies showed that children delivered by CS are more likely to develop respiratory disorders, neurological diseases (e.g., autism spectrum disorders, schizophrenia) (14, 15) and immune-related diseases (e.g., asthma, skin atopy, coeliac disease and juvenile arthritis, type 1 diabetes or obesity) (1, 16–22).
Although the involved biological mechanisms are not yet completely understood, recent evidence suggests that epigenetic alterations (e.g., changes in DNA methylation, histone modifications and non-coding RNA) play a certain role.
DNA methylation is an epigenetic process in which non-sequence-based regulatory information can be mitotically transferred between the mother and daughter cells. In this process, a methyl group is added at the 5-position of a cytosine-guanine (CpG) dinucleotide. Methylated cytosines can be distributed within the genome on CpG islands, shores, shelves and in the so-called open sea regions (23). Shores are regions within 2 kb from CpG islands, shelves are regions 2–4 kb from CpG islands and open sea regions are isolated CpG sites in the genome without a specific designation or without any kind of CpG content enrichment (referred in the literature also as other regions) (24).
DNA methylation can be associated with inhibition of gene expression by interference with transcription binding proteins, which influences chromatin remodelling (25). About 90% of all methylated CpG sites are localized in transposable, repetitive elements, while others occur in gene promoters. During early mammalian embryogenesis, DNA methylation patterns are completely reset with further de novo methylation and demethylation events allowing tissue differentiation (26). Almost all CpG sites are de novo methylated except for the sites labelled as CpG islands that are protected by virtue of cis-acting regulatory sequences (27). To a certain extent, all further changes in DNA methylation are conserved and gene-specific. For example, pluripotency genes become de novo methylated at the time of gastrulation, while tissue-specific regulatory regions undergo demethylation during embryonic development or during adult stem cell differentiation (28–31). It was believed that DNA methylation patterns created during development do not undergo further modifications, however, recent studies showed that methylation could occur also postnatally in terminally differentiated tissues. This can be induced by internal signalization [e.g., DNA methylation changes in the liver induced by testosterone (32–34)] as well as by effect of environment or due to specific behaviors which represent epigenetic factors (35). Moreover, after onset, such changes can persist for extended time periods (27).
It appears that the mode of delivery represents one of the crucial epigenetic events associated with changes in DNA methylation that could have profound impact on human health in later stages of life.
This study provides evaluation how the mode of delivery may induce changes in newborn’s genome that can potentially lead to a broad spectrum of health-related complications in the future. We have also identified biological and metabolic pathways that could be affected by differences in DNA methylation as the major driver of such delivery-dependent modifications in the genome.
To our knowledge, there are only a few works that analyze dynamics of changes in DNA methylation in early postnatal life. Our results indicate that changes in DNA methylation (i) occur not only at the time of birth, as was described in the literature so far, (ii) but also postnatally and (ii) there could be mechanism to adopt to such changes. Therefore our work has pointed out the need of continual study of changes in DNA methylation during the life to understand their mechanisms and potential consequences.
Methods
Demographic and clinical data
The study includes 12 healthy newborn infants from the Slovakian population. All of them were delivered in full term as singletons. Exclusion criteria were multiple pregnancies, preterm delivery (gestational age <37 weeks), pregnancy resulting from in vitro fertilisation, maternal diabetes, preeclampsia, congenital infection and chromosomal disorders. We compared two modes of delivery: vaginal delivery (VD; n = 7) and elective caesarean section (CS; n = 5). In two cases, vaginal delivery was induced.
Samples
From each study subject, 5 mL EDTA blood from the umbilical cord was sampled upon delivery and 2 mL EDTA blood from a peripheral vein on 2nd/3rd day of infants’ postnatal age. Sample analyses were performed separately as on the two sub-groups comprising umbilical cord and peripheral blood (as two separate single time-point analyses). In the next step, a comparison was performed on these two different datasets based on delivery mode (CS vs. VD). We evaluated also changes in DNA methylation in time, i.e., upon delivery and on 2nd/3rd day after delivery.
DNA preparation
Genomic DNA was extracted using DNeasy Blood & Tissue Kit (QIAGEN) following the manufacturer’s instructions. DNA quantification was performed with Qubit dsDNA Broad Range Quantitation Assay (Thermo Fisher Scientific).
Genomic DNA (with the volume of 100 μL) was sheared using a focused ultrasound on Covaris S220 system (sonication conditions were optimized, the applied settings are shown in the Table 1). Ultrasonication-based DNA fragmentation was selected to avoid the removal of methyl groups from DNA by enzymatic fragmentation. Fragmented genomic DNA was cleaned up using 1.2× sample volume of AMPure XP beads (Beckman Coulter, United States).
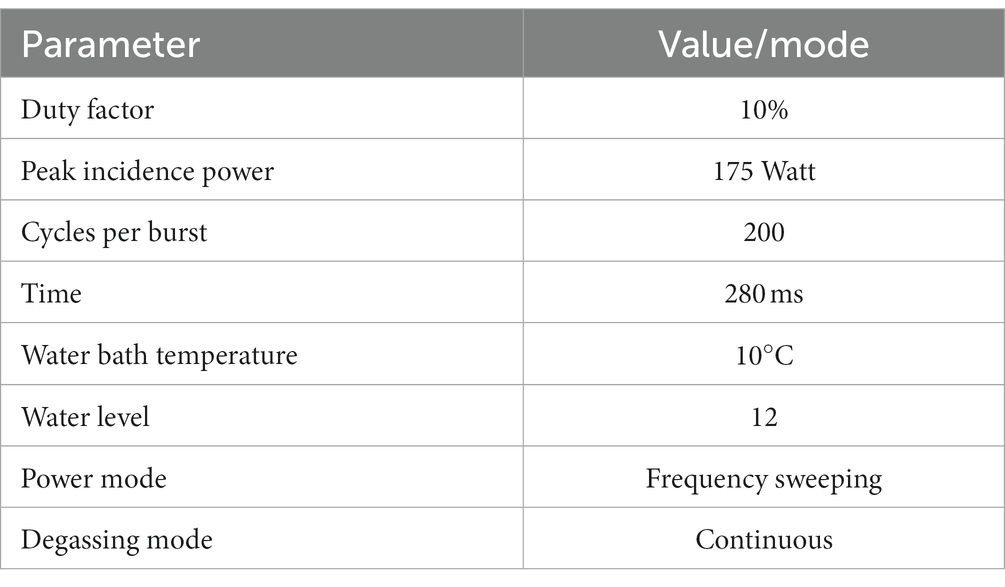
Table 1. Summary of Covaris operating conditions.
MethylSeq library preparations
Only the samples with suitable size (average of 240–290 bp) were allowed to proceed to preparation of the MethylSeq library pursuant to NEBNext enzymatic Methyl-seq Methylation Library Preparation Kit (Twist Bioscience). These were used to generate target-enriched DNA libraries by Twist Human Methylome Panel (Twist Bioscience).
Mechanically sheared DNA underwent end repair. After ligation of methylated adapters, 5-methylcytosines were oxidized by TET2 enzyme and in further APOBEC treatment, unmethylated cytosines were deaminated to uraciles. The converted library was then indexed by PCR amplification. After quality check, eight libraries were pooled in equimolar quantity with further overnight hybridization with a custom double-stranded DNA panel to target specific regions. Fragments of interest were captured with streptavidin binding beads. The pool of libraries was amplified by qPCR. Quantification and validation of enriched libraries was performed by Qubit dsDNA High Sensitivity Quantitation Assay (Thermo Fisher Scientific) and Agilent Bioanalyzer High Sensitivity DNA kit. Out of the 16 original pairs of samples, 12 pairs qualified for sequencing on Illumina next-generation sequencing (NGS) systems (NextSeq 500 and NextSeq 2000 by Illumina) using 2 × 75 or 2 × 100 paired-end sequencing kits and protocols, respectively.
MethylSeq data analysis
The entire analysis was performed using the BaseSpace Sequence Hub (BSSH) cloud service. In the first step, the BCL files were converted into FASTQ files. Next, FASTQ files were submitted as the input to the DRAGEN Methylation pipeline. A conversion of the bases from C-to-T, and G-to-A was then performed. The bisulfite converted reads were afterwards aligned to the reference genome, in our case it was the hg19 alt-aware UCSC reference genome already present in the BSSH. Targeted methylation calling was performed once the converted reads were aligned to the reference genome. The regions subjected to the analysis were provided in the BED file from Twist Bioscience. In the background, BSSH utilizes the Bismark Bisulfite Read Mapper to map the reads and to identify the local cytosine methylation status. As a result, we obtained the BAM files as well as the Cytosine Report TXT file, which then served as an input to the MethylKit application.
The MethylKit application within the BSSH represents the second phase of the computational analysis. Here, the input data representing the output from the previous phase were provided. At the same time, the conceptual division was determined between the samples considered experimental and the samples used as a reference set.
Methylation pathway analysis
Pathway analysis was based on genes where differentially methylated CpG sites were observed. The list of these genes was compared against the Kyoto Encyclopaedia of Genes and Genomes (KEGG) with the aim to identify biological, metabolic and signalling pathways that could be related to genes affected by changes in methylation.
Data analysis
Statistical analyses were performed using SAS Jump Statistics Software (United States) and MS Excel software. Data are presented as mean values ± SD. Paired t-test was used to assess the difference between the VD group and the CS group. Pearson correlation was used to determine the association state between variables. Statistical significance was considered at p < 0.05, and for trends, p < 0.10.
Results
Descriptive statistics
The study includes 12 healthy newborn infants delivered in full term as singletons. Detailed characteristics of the study groups are available in the Table 2. Seven newborns were delivered by VD, while five newborns by elective CS. In two cases, the vaginal delivery was induced. None of the CS represented an emergency. Maternal age ranged from 24 to 37 years, none of mothers have a history of smoking. Ten mothers were of Slovak ethnic origin, one was of Albanian and one of Polish ethnic origin. Two cases in the VD group and one infant in the CS group recquired artificial feeding without any other complications. In one case in the VD group, partial syndaktyl was diagnosed.

Table 2. Characteristics of the study groups.
We did not observe any significant differences between the VD group and the CS group, except for higher delivery weight in the CS group, which has a character of a trend (p = 0.078). The magnitude of correlation between observed parameters was low (data not shown).
Methylation distribution in umbilical cord blood samples
In the umbilical cord blood samples, the total of 3,595,242 CpG sites was compared for all analyzed samples. In the comparison of vaginal delivery and caesarean section, from the total number of 4,352 differentially methylated bases, 1,776 bases were hypermethylated, while 2,576 bases were hypomethylated. Three thousand seventeen bases (69.3%) were assigned as significantly differentially methylated. These were localized into 168 differentially methylated regions, whereas 78 regions were hypermethylated and 90 regions were hypomethylated. As shown in the Figure 1A, 13% of methylation CpG probe sites differences were found on the CpG islands, 17% on the shores and shelves and a predominant portion of CpG sites was in open sea regions. The highest distribution of differentially methylated CpG sites was observed within introns (46%) and in the intergenic regions (34%), while only 11 and 9% of CpG sites with different methylation were localized within promoters and exons, respectively (Figure 1B).
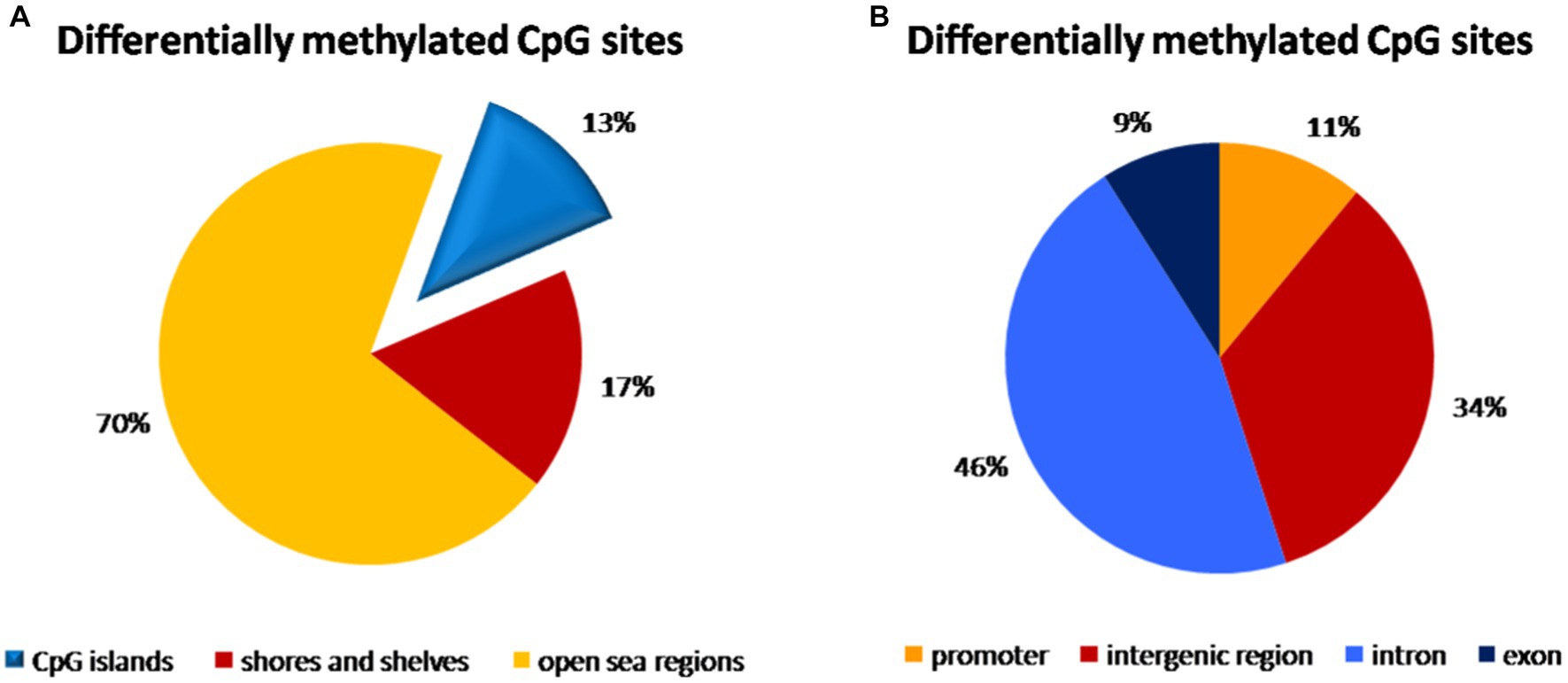
Figure 1. Distribution of differentially methylated CpG dinucleotide in the umbilical cord blood samples according to neighbourhood context (A) and functional genomic distribution (B). (A) A predominant portion of differentially methylated CpG probe sites (70%) was observed in open sea regions, 13% on the CpG islands and 17% on the shores and shelves. (B) The highest distribution of differentially methylated CpG probe sites was found within introns (46%) and in the intergenic regions (34%), while only 11% of CpG probe sites with different methylation were localized within promoters and 9% in exons.
Methylation distribution in peripheral blood samples
We compared 3,227,047 CpG sites common for all samples of peripheral blood. In the comparison of vaginal delivery and caesarean section, 3,853 bases were methylated differentially. Specifically, 1,873 bases were hypermethylated while 1,980 were hypomethylated. However, only 55.1% of them were assigned as significantly differentially methylated. We identified 79 hypermethylated and 78 hypomethylated regions, i.e., 157 regions showed differential methylation. Almost three quarters (73%) of all CpG sites with different methylation were identified in open sea regions of the genome, whereas 11% were localized on CpG islands and 16% were found on shores and shelves (Figure 2A). Introns accounted for 48% of differentially methylated CpG sites, 33% was in intergenic regions, while 10% in exons and only 9% in promoters (Figure 2B).
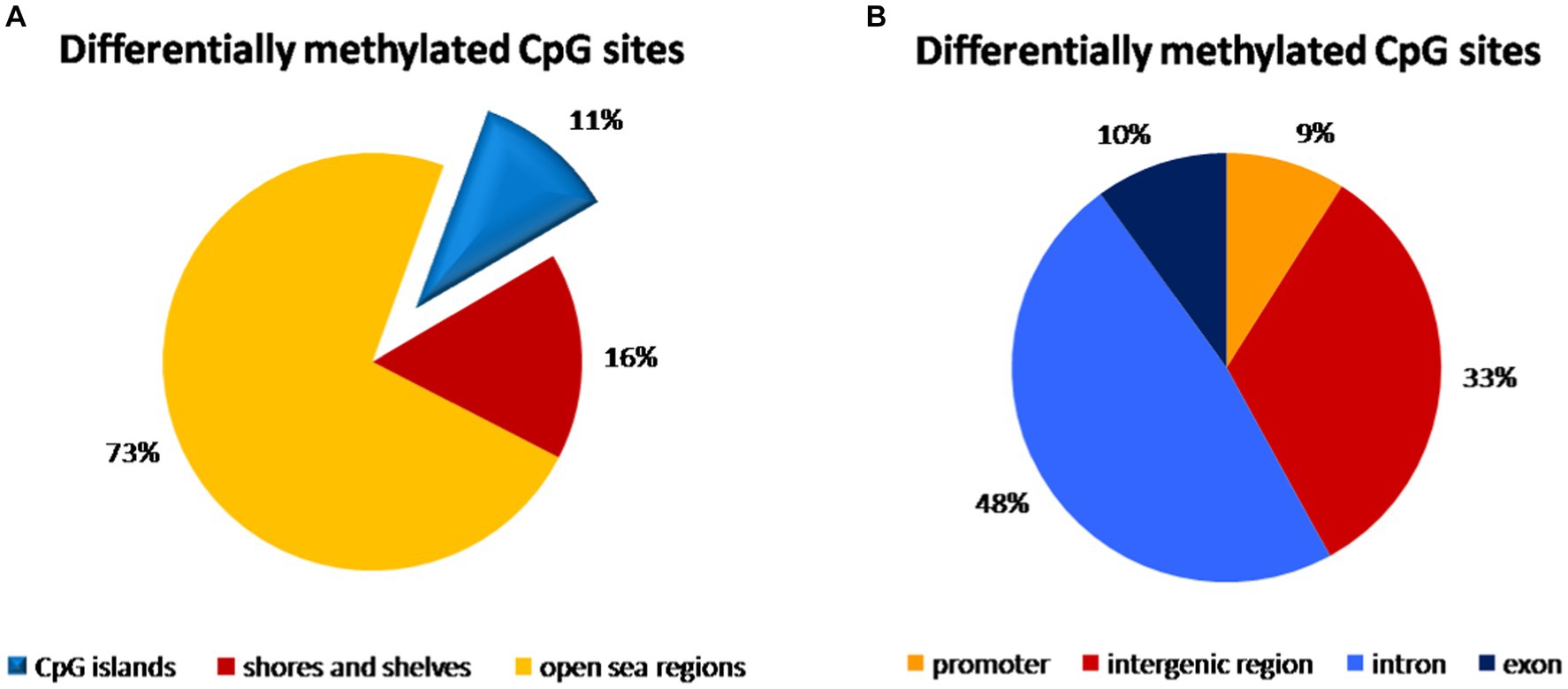
Figure 2. Distribution of differentially methylated CpG dinucleotide in the peripheral blood samples according to neighbourhood context (A) and functional genomic distribution (B). (A) Almost three quarters (73%) of all CpG probe sites with different methylation were identified in open sea regions of the genome, 11% were localized on CpG islands and 16% were found on shores and shelves. (B) The highest distribution of differentially methylated CpG probe sites was found within introns (48%) and in the intergenic regions (33%), while only 10% of CpG probe sites with different methylation were localized in exons and only 9% in promoters.
Chromosomal location of differentially methylated CpG sites
Although peripheral blood samples were obtained several days after delivery, the total number of all differentially methylated CpG sites is only about 10% lower than in umbilical cord blood samples. Final deviation between significantly differently methylated CpG sites across both types of samples reached more than 30%. Different methylation was approved in all somatic chromosomes and on X chromosome in peripheral blood samples. In umbilical cord blood samples, no significant changes in methylation of chromosome 18 and chromosome X were observed between vaginal delivery and caesarean section samples (Figure 3). The highest proportion of different methylation was identified on chromosomes 1, 2, 6 and 7 (about 7–8.5%). The lowest rate of differentially methylated CpG sites was on chromosomes 18 and 21 and on X chromosome (up to 1.8%).
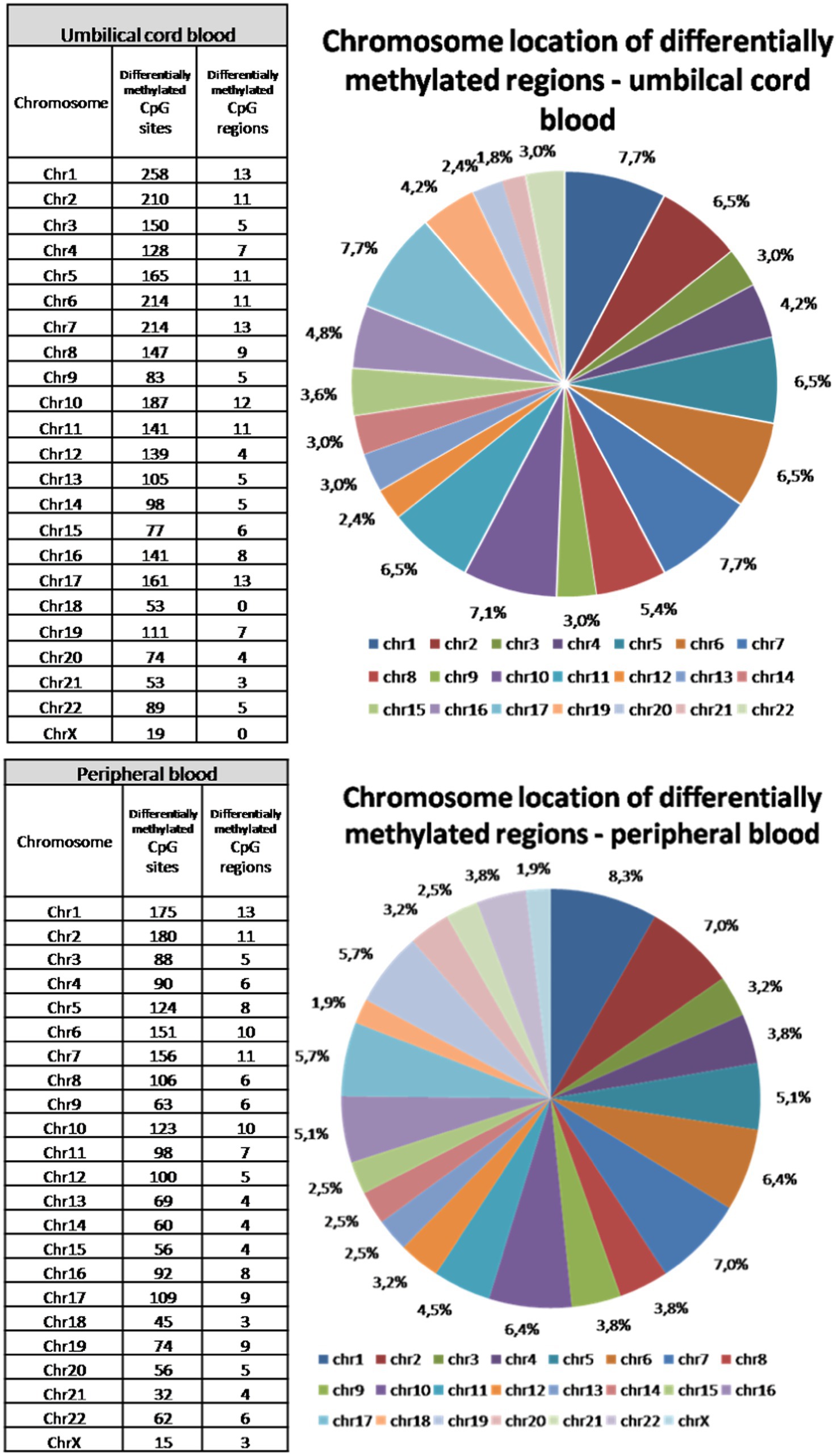
Figure 3. Chromosomal location of differentially methylated regions in the umbilical cord and peripheral blood samples. Differences in methylation was observed in all somatic chromosomes and also on X chromosome in peripheral blood samples, while there were no differences in methylation of chromosome 18 and chromosome X in umbilical cord blood samples. The highest proportion of different methylation was identified on chromosomes 1, 2, 6, and 7. The lowest rate of differentially methylated CpG sites was on chromosomes 18 and 21 and on X chromosome.
Methylation and pathway analysis
Based on genes affected by different methylation, we identified 59 common biological, metabolic and signalling pathways for umbilical cord and peripheral blood samples (Figure 4). In addition to the above, 20 pathways were assigned only to umbilical cord blood samples and 10 pathways only to peripheral blood samples (Table 3).
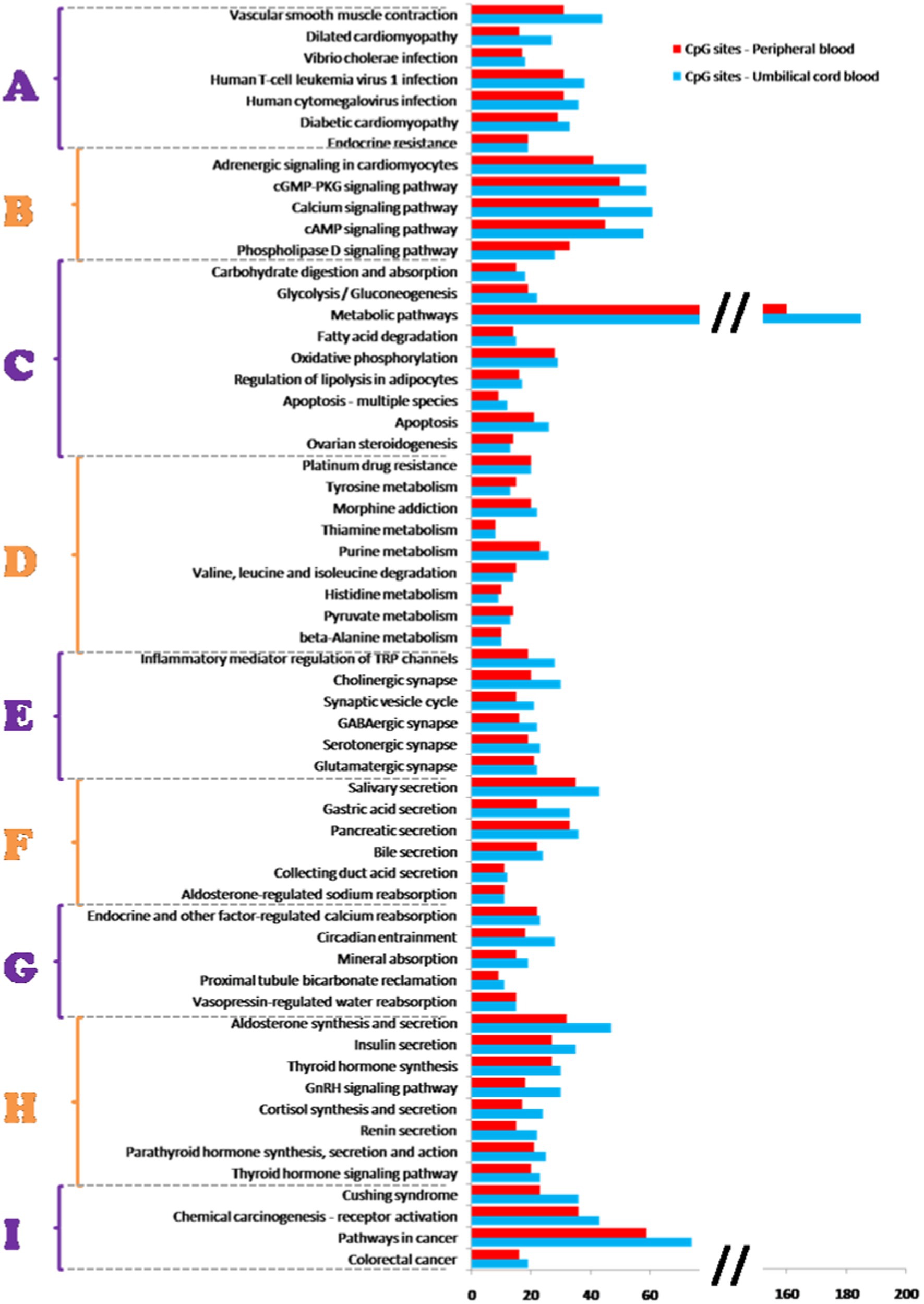
Figure 4. Methylation pathway analysis. Fifty-nine common biological, metabolic and signalling pathways for umbilical cord blood samples and peripheral blood samples identified based on a comparison of KEGG database with the analyzed genes affected by mode of delivery-related differences in DNA methylation. Pathways were grouped into 9 categories (A–I): A—diseases, B—signalling pathways, C—metabolic pathways, D—pathways of specific metabolites, E—synaptic pathways, F—synthesis and secretion of body fluids, G—mineral management, H—hormonal metabolism, I—carcinogenesis. Pathways are ordered with respect to their significance (from higher to lower). Significance based on g:SCS-adjusted p-value <0.05 (37).
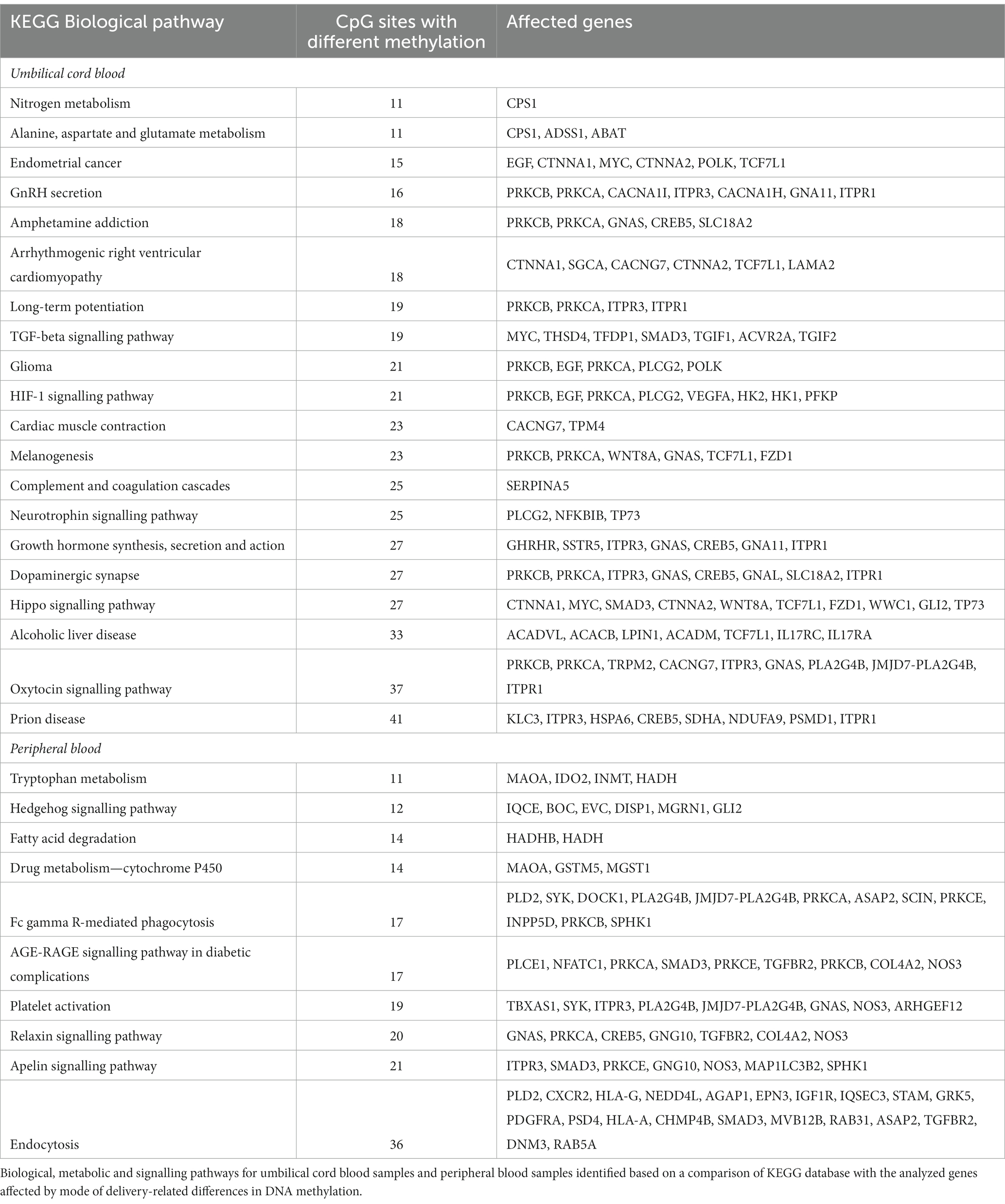
Table 3. Methylation pathway analysis.
Discussion
In our analyses comparing DNA methylation between samples from newborns delivered via conventional vaginal delivery and via caesarean section, we identified 168 regions in umbilical cord blood samples as well as 157 regions in peripheral blood samples with differential methylation. This indicates that mode of delivery could have profound impact on a wide range of genes and that caesarean section may represent an epigenetic factor with impact on DNA methylation state. Moreover, we also identified 59 biological pathways that are associated with genes from differentially methylated regions and not only at the time of birth but also later after the delivery. Such observation confirmed the ability of an epigenetic factor to change methylation status established during in utero development. Our observation that a considerable number of biological and signalling pathways are still changed even several days after delivery confirms the potential of persistence of induced changes. This process then creates space and time to induce further changes that could have adverse affect on the individual’s health status later in the life. Further research is needed to elucidate whether or not such changes persist for a longer time after delivery, or they are reversible.
Hypothesis of methylation state plasticity after delivery could be confirmed also by normalization of differences in methylation status in case of 21 biological pathways that disappeared after several days. On the other hand, we simultaneously identified other changes in DNA methylation state that could be associated with the need to amend the adverse state in newborns delivered by caesarean section (Table 3).
Identified pathways can be stratified into 9 categories based on affected processes and/or functions, as is shown in the Figure 4. Whereas almost none of the items in this categorization do not work as single separate units but are mutually interconnected, we try to discuss their role as components of several disorders and diseases which seem to be affected by changes in DNA methylation.
As shown in Figure 4, one of the pathways most affected by differential methylation is the Human cytomegalovirus (HCMV) infection pathway. HCMV infection is one of the most common infectious cause of congenital defects (38). About one fifth of children with congenital HCMV suffer from HCMV symptoms or long-term health problems, such as motor and intellectual disability, liver, lung and growth complication while hearing loss is the most common complication after congenital HCMV infection (39). In particularly severe cases, an HCMV infection can lead to pregnancy loss. It was observed that HCMV infection resulted in the re-localization of DNMT1 and DNMT3B from the nucleus in the cytoplasm. Such a draining of DNA methyltransferases from the nucleus may provide the opportunity to express viral proteins and replicate viral genome in a DNA methylation-free environment without formation of supressing complexes on viral promoters rather than by direct interaction with DNMTs (40). There could also be the possibility that HCMV could manipulate natural processes of the host cell designed to keep DNMTs outside of the nucleus because active retention of DNMT1 in the cytoplasm was also observed during early embryogenesis (41). Thus, our observation of potential relation between the changes in DNA methylation machinery and HCMV pathways suggests a potentially higher risk of developing symptoms or complications associated with HCMV infection in children delivered by caesarean section.
DNA methylation is considered as a host defence mechanism for inactivation of retrovirus expression (42). The first discovered human retrovirus was Human T-cell leukaemia virus type 1 (HTLV-1), which is associated with T-cell lymphoproliferative disorders (e.g., adult T-cell leukaemia/lymphoma, ATLL) and inflammatory diseases (e.g., HTLV-1-associated myelopathy, pneumonitis, uveitis, arthritis and myositis) (43). ATLL genomes are characterized by a prominent promoter-related CpG islands and DNA hypermethylation. Moreover, the level of hypermethylation is correlated with poor prognosis (44). Mutations in epigenetic regulators of DNA methylation TET methylcytosine dioxygenase 2 (TET2) and mixed-lineage leukaemia protein 3 (MLL3) were observed in ATLL patients, as well as CpG hypermethylation in gene encoding zinc finger transcription factors and major histocompatibility class I proteins (44, 45).
Nowadays, it is still unclear why ATLL takes decades to manifest itself, why only 5–10% of infected subjects develop symptoms and how HTLV-1-associated diseases can be prevented and treated. It is suggested that most individuals who develop ATLL in adult age were infected already as infants, probably via breast feeding (46). In the light of our observation of differences in DNA methylation between newborns delivered via VD and CS, we suggest that CS-related differences in DNA methylation could lead to higher susceptibility to development of ATLL symptoms and manifestations and that there is higher probability of HTLV-1 expression reactivation. Such a hypothesis requires further study whether or not CS could be associated with higher incidence of ATLL manifestation.
To date, multiple publications supported the importance of DNA methylation in a broad spectrum of cardiovascular diseases, including atherogenesis (47), coronary artery diseases (48, 49), dilated cardiomyopathy (50) and heart failure (51–53). In our analysis, we identified three pathways that could be potentially associated with different methylation (Figure 4). Vascular smooth muscle cells (VSMCs) are involved in maintaining the structural integrity and physiological function of blood vessels, regulating blood pressure and controlling vascular contraction and relaxation (54, 55). As individuals age, VSMCs suffer from mechanical stimulation, chronic inflammation, calcifications and other epigenetic events (56). The VSMCs proliferation is one of the primary features of vascular ageing-related diseases. Cell proliferation is tightly controlled by DNA methylation via regulation of gene transcription. DNMTs inhibit gene expression by promoting DNA methylation and regulating the proliferation of VSMCs. It has been observed that as DNA demethylation of PDGF increases PDGR mRNA and protein expression, while the hypomethylation of HIF-1α causes the proliferation and migration of VSMCs (57, 58). On the contrary, DNA hypermethylation of MFN2, PTEN and ER-α causes low expression of MFN2, PTEN and ER-α that leads also to the proliferation of VSMCs (59–61). Liu et al. (62) demonstrated that vascular injury and vascular disorders are associated with downregulation of expression of TET2, an enzyme with a key role in the DNA demethylation pathway. Abnormal DNA methylation and DNMTs suppression are also tightly linked to vascular diseases (63). DNA hypermethylation regulates cardio-metabolism by destroying nuclear respiratory factor 1-dependent oxidative metabolism (64). Moreover, in heart failure patients upregulation of DNMT3a and DNMT3b expression was observed (64). Additionally, in patients with dilated cardiomyopathies, association of aberrant DNA methylation was observed with significant differences in expression of ADORA2A and LY75 mRNA (50). Summarising the above, changes in DNA methylation potentially resulting from delivery by caesarean section associated with adverse changes in genome could increase the risk of developing cardiovascular complications later in the life.
Defects in biosynthesis and secretion of aldosterone, which represent another pathway affected by differentially methylated CpG islands in our analysis, can lead to sodium processing disorders manifested as hypotension, hyponatraemia, hyperkalaemia and acidosis. Aldosterone is important for regulation of sodium conservation in the kidney, colon, salivary and sweat glands. This adrenal steroid hormone acts via the mineralocorticoid receptor to promote active transport of sodium and excretion of potassium through activation of amiloride-sensitive sodium channels and a Na/K-ATP-ase pump (65). For example, patients with Addison’s disease are hyponatraemic and hyperkaliaemic because of potassium retention and excessive urine excretion of sodium (66). Thus, our observation of the association of different methylation with aldosterone-regulated sodium reabsorption is in compliance with reported sodium transport disorders. Gene expression of aldosterone synthase CYP11B2 is epigenetically controlled (67). Takeda et al. (68) show that infusion of angiotensin II in rats decreased the methylation ratio of the CYP11B2 gene and increased gene expression in the adrenal gland. They also observed that a low-sodium diet induced hypomethylation of CYP11B2 and increased its mRNA level parallel with synthesis of aldosterone. Moreover, aldosterone biosynthesis also includes the cAMP signalling pathway which is one of the pathways most affected by DNA methylation in our analysis (Figure 4).
As mentioned above, caesarean section is a potential epigenetic stress factor. In their work, Zhang et al. (69) tested hypothesis that chronic negative stress leads to alterations in DNA methylation of certain cardiac genes with further pathologic remodelling of the heart. The authors approved that altered methylation on specific genes is associated with the adrenergic signalling of cardiomyocyte pathways (e.g., via adrenergic receptor-α1) resulted in induced cardiac remodelling and arrhythmias. Also Xiao et al. (70) reported that abnormal DNA methylation within the CpG islands of the promoter region of voltage-gated potassium channel subfamily E genes has been linked to cardiac arrhythmias. We also found association of differentially methylated CpG sites with the pathway of adrenergic signalling in cardiomyocytes as well as with dilated cardiomyopathy pathway. Thus, changes in DNA methylation related to caesarean section can be tightly associated with cardiac dysfunction.
Another signalling pathway that could be influenced by modulation of DNA methylation state is cAMP signalling pathway. Adenosine 3′,5′-cyclic monophosphate (cAMP) is a highly regulated secondary messenger involved in numerous biological processes (71). Although it is known that chronic activation of the cAMP pathway results in cardiac hypertrophy and fibrosis, the underlying mechanisms linking elevated cAMP levels to cardiomyopathy are not fully elucidated. Fang et al. demonstrated that elevated level of cAMP analog N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate (DBcAMP) changed the expression of DNMTs and TETs as enzymes regulating genomic DNA methylation levels. Such a change induced increased global DNA methylation in HL-1 cardiomyocytes. Thus, DNA methylation mediates the upregulation of hypertrophy cardiomyopathy genes induced by cAMP. Possible explanation could involve physiological activation of the cAMP-dependent protein kinase (PKA) leading to phosphorylation of numerous transcription factors, including the cAMP response element-binding protein (CREB) (72). CREB then binds to cAMP response elements (CREs) on gene promoters and activates expression of involved genes. Similarly, during chronic activation of the cAMP pathway, increased cAMP induced hypermethylation in the promoter region with consequent activation of genes that are transcriptionally repressed under standard conditions (73).
Alterations in DNA methylation are well known in multiple oncological disorders. One of the leading causes of cancer-related mortality worldwide is colorectal cancer (CRC) (74), for which global DNA hypomethylation and depletion of 5-metylcytosine in CRC tissue was reported already in 1983 (75). Such DNA hypomethylation was accompanied by hypermethylation and transcriptional silencing of several tumour suppressor genes (76). Nowadays, it is known that hundreds of genes might be aberrantly methylated in the average CRC genome (77). It is important to keep in mind, that CRC is not a homogenous disease, but rather a heterogeneous disorder with different subtypes characterized by distinct genetic, cytogenetic and epigenetic alterations. Genomic instability is typical for CRC as a key a distinctive characteristic. Genome-scale analysis of aberrant DNA methylation identified four DNA methylation-based subgroups of CRC based on CpG island methylator phenotype (CIMP), specific subgroup of CRC that displays extensive levels of methylated genes (78). The first subgroup is a CIMP-high subgroup exhibiting a very high frequency of cancer-specific DNA hypermethylation, which is associated with MLH1 methylation and the BRAF V100E mutation. The second one is a CIMP-low subgroup associated with K-RAS mutations and characteristic for methylation of a subset of CIMP-high associated genes, rather than a unique group of CpG islands. The third one is a non-CIMP subgroup characterized by TP53 mutations and typical localization in the distal colon. Finally, the last one is a non-CIMP subgroup with low frequency of cancer-specific gene mutations and hypermethylation as well as with rectal localization (79).
To our knowledge, up to date, there has been published no work about the risk of CRC developing in the adulthood, which would be based on delivery mode history. Therefore, further research will be required to answer the question whether or not changes in DNA methylation occurring at the time of delivery could be associated with CRC onset in the adulthood.
There is rising evidence that changes in DNA methylation could be associated not only with physical functions but also with psychosocial functions and neuropsychological disorders. Similar to the above-mentioned hypothesis of stress-induced changes in DNA methylation state upon delivery by CS, several studies have reported that DNA methylation of NR3C1 gene could be affected by epigenetic factors such as quality of maternal care and experience of prenatal and childhood trauma (35, 80–83) which may lead to neuropsychiatric disorders, e.g., depression, anxiety and post-traumatic stress disorder. The mechanism behind these changes is not known but it may be associated with long-lasting dysregulation of the hypothalamus-pituitary-adrenal (84) axis. Hyperactivity of HPA axis induces an increase in secretion of cortisol, a predominant glucocorticoid in humans, which affects the central nervous system by binding to its two receptors encoded by the NR3C1 and NR3C2 genes. These receptors are expressed in the hippocampus, prefrontal cortex and the parvocellular nucleus of the hypothalamus (85). McGowan et al. (86) observed increased DNA methylation of the NR3C1 gene promoter in the hippocampus and prefrontal cortex in suicide victims with a history of childhood abuse. Thus, early-life disruption in the HPA-axis could be mediated by epigenetic mechanisms.
Chronic excess and attenuation of the endogenous diurnal variation in cortisol secretion leads to Cushing’s syndrome (87), which can be caused by an ACTH-producing pituitary adenoma (Cushing’s disease) or by a cortisol-producing adrenal adenoma. In our methylation pathway analysis, both cortisol synthesis and secretion as well as Cushing’s syndrome were identified as pathways with different DNA methylation between VD and CS. Lee et al. (88, 89) showed association of hypomethylation of FKBP5 gene in Cushing’s syndrome patients with anxiety-like behavior. Moreover, such methylation was reduced with glucocorticoid replacement therapy.
In concordance with observed associations between changes in DNA methylome and Cushing’s syndrome, we hypothesize that differences in DNA methylation could play a pathophysiological role in development of stress-related diseases.
Although there are several studies trying to shed more light on the relationship of changes in DNA methylation status and the mode of delivery, the results are inconsistent.
Franz et al. (90) observed hypermethylation of FOXP3, CD7, ELA2 and IRF1 genes for CS and VD samples and significantly higher methylation in ELA2 and IRF1 genes in the CS group compared to the VD group. However, global methylation did not differ significantly between the CS and VD groups. Virani et al. (91) observed lower level of global DNA methylation for total CS and planned CS compared to VD but such association disappeared after adjusting for confounding factors. Thus, they concluded that while mode of delivery may be associated with later health outcomes, it does not take effect via changes in global genomic methylation. Similarly, Franz et al. did not report any significant differences in global DNA methylation between CS and VD. However, the global analysis that was used in both papers, reported the average methylation levels in the interrogated restriction sites throughout the whole genome. Therefore, even if the changes were substantial, such method displays only small differences for similar amounts of hyper- and hypomethylation.
On the other hand, Almgren et al. (92), who analyzed CD34+ hematopoietic stem cells, found significant effect of mode of delivery on the global DNA methylation pattern. CD34+ cells from newborns delivered by CS were globally more DNA-methylated compared to DNA of infants delivered vaginally. They also showed in the locus-specific analysis that the functional relevance of differentially methylated loci involved processes such as immunoglobulin biosynthesis, regulation of glycolysis and ketone metabolism as well as regulation of food response. In another specific gene-targeted analysis, Chen et al. (4) identified 5 genes, whose gene expression level had profound implication for development of the immune system’s response in newborns. Such abnormality of the immune system could lead to chronic inflammation and autoimmune diseases.
Comparison of results across different studies is relatively complicated because of wide range of approaches used for detection of DNA methylation. In comparison with Chen et al., we did not observe any changes in methylation of immune system-related genes and also our methylation pathway analysis did not identify any pathway associated with the immune system. This may be explained by differences between Slovakian and Chinese population. Similarly to Almgren et al., we observed changes in DNA methylation associated with regulation of glucose metabolism, which may have implications for the state of health of the subjects in the future, e.g., because of reported higher risk of obesity and onset of diabetes in subjects with caesarean section in their medical history (93–95). Discrepancies in global DNA methylation between Almgren et al., who observed higher global DNA methylation and Franz et al. as well as Virani et al., who did not report any differences in global DNA methylation, could be explained by specific type of cells (CD34+ cells) used in the analysis by Almgren et al. and even here the observed higher level of methylation was relatively small (+2%). Comparison of our results with global DNA methylation could be based on the assumption that this approach delivers the average methylation levels in the interrogated restriction sites throughout the whole genome. Therefore, even if the changes were substantial, such method displays only small differences for similar amounts of hyper- and hypomethylation. Therefore, when Franz et al. evaluated also single gene methylation status, they observed hypermethylation for the group of genes comprising FOXP3, CD7, ELA2 and IRF1.
Omitting differences in reported DNA methylation at birth that could be affected by the method of evaluation as stated above, it will also be important to clarify whether the observed changes in DNA methylation could be long-lasting with potential of further implications in the future. In this paper, we outlined that some differences in DNA methylation may be amended while others may emerge shortly after delivery. Our results therefore represent a very good opportunity for further research focused on the consequences of differences in DNA methylation on the state of health in adolescence and adult age.
We are aware of some limitations of the present study. One of them is small number of involved subjects and intra-group diversity which could affect representativeness of the target population. Yet another limitation is the negative maternal history of tobacco use, which does not reflect natural distribution. It is also demanding to incorporate the entire range of epigenetic factors with the potential to induce changes in DNA methylation, e.g., medication during gravidity, mother-related health issues as well as complications during labour.
Conclusion
Our observation of differences in DNA methylation between blood samples from neonates delivered by conventional vaginal delivery and by caesarean section suggests that caesarean section probably represents an important epigenetic factor with the potential to induce changes in the genome. As we showed in our pathway analysis, these differences could play an important role in the development of various type of disorders. Our results could contribute to explaining how epigenetic factors such as mode of delivery could have an adverse impact on the individual’s health later in the life and which mechanisms could be crucial for development of such disorders.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/ena, PRJEB61787.
Ethics statement
The studies involving humans were approved by Ethics Committee of the Hospital Agel, Kosice-Saca, Slovakia (EK/2020-06). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
PK: Writing – original draft. DK: Writing – review & editing. DH: Writing – review & editing. OP: Writing – review & editing. KB: Writing – review & editing. ED: Writing – review & editing. ZT: Writing – review & editing. GM: Writing – review & editing.
Glossary
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This article was created with the support of the OP Integrated Infrastructure for the project: Center for Biomedical Research—BIOMEDIRES—II. phase, ITMS: 313011W428, co-financed by the European Regional Development Fund.
Acknowledgments
The authors thank all the participants of the present study and the workers in the Hospital Agel in Kosice-Saca.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2024.1291429/full#supplementary-material
References
1. Betrán, AP, Ye, J, Moller, AB, Zhang, J, Gülmezoglu, AM, and Torloni, MR. The increasing trend in caesarean section rates: global, regional and national estimates: 1990–2014. PLoS One. (2016) 11:e0148343. doi: 10.1371/journal.pone.0148343
2. Betran, AP, Ye, J, Moller, AB, Souza, JP, and Zhang, J. Trends and projections of caesarean section rates: global and regional estimates. BMJ Glob Health. (2021) 6:e005671. doi: 10.1136/bmjgh-2021-005671
3. Betran, AP, Torloni, MR, Zhang, JJ, and Gülmezoglu, AMThe WHO Working Group on Caesarean Section. WHO statement on caesarean section rates. BJOG. (2016) 123:667–70. doi: 10.1111/1471-0528.13526
4. Chen, Q, Ming, Y, Gan, Y, Huang, L, Zhao, Y, Wang, X, et al. The impact of cesarean delivery on infant DNA methylation. BMC Pregnancy Childbirth. (2021) 21:265. doi: 10.1186/s12884-021-03748-y
5. Lupu, VV, Miron, IC, Raileanu, AA, Starcea, IM, Lupu, A, Tarca, E, et al. Difficulties in adaptation of the mother and newborn via cesarean section versus natural birth-a narrative review. Life. (2023) 13:300. doi: 10.3390/life13020300
6. Chiș, A, Vulturar, R, Andreica, S, Prodan, A, and Miu, AC. Behavioral and cortisol responses to stress in newborn infants: effects of mode of delivery. Psychoneuroendocrinology. (2017) 86:203–8. doi: 10.1016/j.psyneuen.2017.09.024
7. Chen, DC, Nommsen-Rivers, L, Dewey, KG, and Lönnerdal, B. Stress during labor and delivery and early lactation performance. Am J Clin Nutr. (1998) 68:335–44. doi: 10.1093/ajcn/68.2.335
8. Miller, NM, Fisk, NM, Modi, N, and Glover, V. Stress responses at birth: determinants of cord arterial cortisol and links with cortisol response in infancy. BJOG. (2005) 112:921–6. doi: 10.1111/j.1471-0528.2005.00620.x
9. Malamitsi-Puchner, A, Protonotariou, E, Boutsikou, T, Makrakis, E, Sarandakou, A, and Creatsas, G. The influence of the mode of delivery on circulating cytokine concentrations in the perinatal period. Early Hum Dev. (2005) 81:387–92. doi: 10.1016/j.earlhumdev.2004.10.017
10. Heasman, L, Spencer, JA, and Symonds, ME. Plasma prolactin concentrations after caesarean section or vaginal delivery. Arch Dis Child Fetal Neonatal Ed. (1997) 77:F237–8. doi: 10.1136/fn.77.3.F237
11. Lubetzky, R, Ben-Shachar, S, Mimouni, F, and Dollberg, S. Mode of delivery and neonatal hematocrit. Am J Perinatol. (2000) 17:163–6. doi: 10.1055/s-2000-9291
12. Biasucci, G, Rubini, M, Riboni, S, Morelli, L, Bessi, E, and Retetangos, C. Mode of delivery affects the bacterial community in the newborn gut. Early Hum Dev. (2010) 86:13–5. doi: 10.1016/j.earlhumdev.2010.01.004
13. Yektaei-Karin, E, Moshfegh, A, Lundahl, J, Berggren, V, Hansson, LO, and Marchini, G. The stress of birth enhances in vitro spontaneous and IL-8-induced neutrophil chemotaxis in the human newborn. Pediatr Allergy Immunol. (2007) 18:643–51. doi: 10.1111/j.1399-3038.2007.00578.x
14. Curran, EA, Dalman, C, Kearney, PM, Kenny, LC, Cryan, JF, Dinan, TG, et al. Association between obstetric mode of delivery and autism spectrum disorder: a population-based sibling design study. JAMA Psychiatry. (2015) 72:935–42. doi: 10.1001/jamapsychiatry.2015.0846
15. O’Neill, SM, Curran, EA, Dalman, C, Kenny, LC, Kearney, PM, Clarke, G, et al. Birth by caesarean section and the risk of adult psychosis: a population-based cohort study. Schizophr Bull. (2016) 42:633–41. doi: 10.1093/schbul/sbv152
16. Hyde, MJ, and Modi, N. The long-term effects of birth by caesarean section: the case for a randomised controlled trial. Early Hum Dev. (2012) 88:943–9. doi: 10.1016/j.earlhumdev.2012.09.006
17. Thavagnanam, S, Fleming, J, Bromley, A, Shields, MD, and Cardwell, CR. A meta-analysis of the association between caesarean section and childhood asthma. Clin Exp Allergy. (2008) 38:629–33. doi: 10.1111/j.1365-2222.2007.02780.x
18. Dahlen, HG, Downe, S, Wright, ML, Kennedy, HP, and Taylor, JY. Childbirth and consequent atopic disease: emerging evidence on epigenetic effects based on the hygiene and EPIIC hypotheses. BMC Pregnancy Childbirth. (2016) 16:4. doi: 10.1186/s12884-015-0768-9
19. Decker, E, Engelmann, G, Findeisen, A, Gerner, P, Laaβ, M, Ney, D, et al. Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children. Pediatrics. (2010) 125:e1433–40. doi: 10.1542/peds.2009-2260
20. Cardwell, CR, Stene, LC, Joner, G, Cinek, O, Svensson, J, Goldacre, MJ, et al. Caesarean section is associated with an increased risk of childhood-onset type 1 diabetes mellitus: a meta-analysis of observational studies. Diabetologia. (2008) 51:726–35. doi: 10.1007/s00125-008-0941-z
21. Sevelsted, A, Stokholm, J, Bønnelykke, K, and Bisgaard, H. Cesarean section and chronic immune disorders. Pediatrics. (2015) 135:e92–8. doi: 10.1542/peds.2014-0596
22. Keag, OE, Norman, JE, and Stock, SJ. Long-term risks and benefits associated with cesarean delivery for mother, baby, and subsequent pregnancies: systematic review and meta-analysis. PLoS Med. (2018) 15:e1002494. doi: 10.1371/journal.pmed.1002494
23. Sandoval, J, Heyn, H, Moran, S, Serra-Musach, J, Pujana, MA, Bibikova, M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. (2011) 6:692–702. doi: 10.4161/epi.6.6.16196
24. Rechache, NS, Wang, Y, Stevenson, HS, Killian, JK, Edelman, DC, Merino, M, et al. DNA methylation profiling identifies global methylation differences and markers of adrenocortical tumors. J Clin Endocrinol Metab. (2012) 97:E1004–13. doi: 10.1210/jc.2011-3298
25. Jirtle, RL, and Skinner, MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. (2007) 8:253–62. doi: 10.1038/nrg2045
26. Smallwood, SA, and Kelsey, G. De novo DNA methylation: a germ cell perspective. Trends Genet. (2012) 28:33–42. doi: 10.1016/j.tig.2011.09.004
27. Cedar, H, and Bergman, Y. Programming of DNA methylation patterns. Annu Rev Biochem. (2012) 81:97–117. doi: 10.1146/annurev-biochem-052610-091920
28. Reizel, Y, Sabag, O, Skversky, Y, Spiro, A, Steinberg, B, Bernstein, D, et al. Postnatal DNA demethylation and its role in tissue maturation. Nat Commun. (2018) 9:2040. doi: 10.1038/s41467-018-04456-6
29. Ziller, MJ, Gu, H, Müller, F, Donaghey, J, Tsai, LTY, Kohlbacher, O, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. (2013) 500:477–81. doi: 10.1038/nature12433
30. Hon, GC, Rajagopal, N, Shen, Y, McCleary, DF, Yue, F, Dang, MD, et al. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet. (2013) 45:1198–206. doi: 10.1038/ng.2746
31. Sheaffer, KL, Kim, R, Aoki, R, Elliott, EN, Schug, J, Burger, L, et al. DNA methylation is required for the control of stem cell differentiation in the small intestine. Genes Dev. (2014) 28:652–64. doi: 10.1101/gad.230318.113
32. Cannon, MV, Pilarowski, G, Liu, X, and Serre, D. Extensive epigenetic changes accompany terminal differentiation of mouse hepatocytes after birth. G3. (2016) 6:3701–9. doi: 10.1534/g3.116.034785
33. Ehara, T, Kamei, Y, Yuan, X, Takahashi, M, Kanai, S, Tamura, E, et al. Ligand-activated PPARα-dependent DNA demethylation regulates the fatty acid beta-oxidation genes in the postnatal liver. Diabetes. (2015) 64:775–84. doi: 10.2337/db14-0158
34. Reizel, Y, Spiro, A, Sabag, O, Skversky, Y, Hecht, M, Keshet, I, et al. Gender-specific postnatal demethylation and establishment of epigenetic memory. Genes Dev. (2015) 29:923–33. doi: 10.1101/gad.259309.115
35. Weaver, IC, Cervoni, N, Champagne, FA, D’Alessio, AC, Sharma, S, Seckl, JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. (2004) 7:847–54. doi: 10.1038/nn1276
37. Reimand, J, Kull, M, Peterson, H, Hansen, J, and Vilo, J. G:profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. (2007) 35:W193–200. doi: 10.1093/nar/gkm226
38. Barton, M, Forrester, AM, and McDonald, J. Update on congenital cytomegalovirus infection: prenatal prevention, newborn diagnosis, and management. Paediatr Child Health. (2020) 25:395–6. doi: 10.1093/pch/pxaa083
39. Buxmann, H, Hamprecht, K, Meyer-Wittkopf, M, and Friese, K. Primary human cytomegalovirus (HCMV) infection in pregnancy. Dtsch Arztebl Int. (2017) 114:45–52. doi: 10.3238/arztebl.2017.0045
40. Esteki-Zadeh, A, Karimi, M, Strååt, K, Ammerpohl, O, Zeitelhofer, M, Jagodic, M, et al. Human cytomegalovirus infection is sensitive to the host cell DNA methylation state and alters global DNA methylation capacity. Epigenetics. (2012) 7:585–93. doi: 10.4161/epi.20075
41. Cardoso, MC, and Leonhardt, H. DNA methyltransferase is actively retained in the cytoplasm during early development. J Cell Biol. (1999) 147:25–32. doi: 10.1083/jcb.147.1.25
42. Verma, M. Viral genes and methylation. Ann N Y Acad Sci. (2003) 983:170–80. doi: 10.1111/j.1749-6632.2003.tb05972.x
43. Gessain, A, and Mahieux, R. Tropical spastic paraparesis and HTLV-1 associated myelopathy: clinical, epidemiological, virological and therapeutic aspects. Rev Neurol. (2012) 168:257–69. doi: 10.1016/j.neurol.2011.12.006
44. Kataoka, K, Shiraishi, Y, Takeda, Y, Sakata, S, Matsumoto, M, Nagano, S, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature. (2016) 534:402–6. doi: 10.1038/nature18294
45. Yeh, CH, Bai, XT, Moles, R, Ratner, L, Waldmann, TA, Watanabe, T, et al. Mutation of epigenetic regulators TET2 and MLL3 in patients with HTLV-I-induced acute adult T-cell leukemia. Mol Cancer. (2016) 15:15. doi: 10.1186/s12943-016-0500-z
46. Rosadas, C, and Taylor, GP. Mother-to-child HTLV-1 transmission: unmet research needs. Front Microbiol. (2019) 10:999. doi: 10.3389/fmicb.2019.00999
47. Ying, AK, Hassanain, HH, Roos, CM, Smiraglia, DJ, Issa, JJ, Michler, RE, et al. Methylation of the estrogen receptor-alpha gene promoter is selectively increased in proliferating human aortic smooth muscle cells. Cardiovasc Res. (2000) 46:172–9. doi: 10.1016/S0008-6363(00)00004-3
48. Sharma, P, Garg, G, Kumar, A, Mohammad, F, Kumar, SR, Tanwar, VS, et al. Genome wide DNA methylation profiling for epigenetic alteration in coronary artery disease patients. Gene. (2014) 541:31–40. doi: 10.1016/j.gene.2014.02.034
49. Sharma, P, Kumar, J, Garg, G, Kumar, A, Patowary, A, Karthikeyan, G, et al. Detection of altered global DNA methylation in coronary artery disease patients. DNA Cell Biol. (2008) 27:357–65. doi: 10.1089/dna.2007.0694
50. Haas, J, Frese, KS, Park, YJ, Keller, A, Vogel, B, Lindroth, AM, et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol Med. (2013) 5:413–29. doi: 10.1002/emmm.201201553
51. Koczor, CA, Lee, EK, Torres, RA, Boyd, A, Vega, JD, Uppal, K, et al. Detection of differentially methylated gene promoters in failing and nonfailing human left ventricle myocardium using computation analysis. Physiol Genomics. (2013) 45:597–605. doi: 10.1152/physiolgenomics.00013.2013
52. Movassagh, M, Choy, MK, Goddard, M, Bennett, MR, Down, TA, and Foo, RSY. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PLoS One. (2010) 5:e8564. doi: 10.1371/journal.pone.0008564
53. Movassagh, M, Choy, MK, Knowles, DA, Cordeddu, L, Haider, S, Down, T, et al. Distinct epigenomic features in end-stage failing human hearts. Circulation. (2011) 124:2411–22. doi: 10.1161/CIRCULATIONAHA.111.040071
54. Frismantiene, A, Philippova, M, Erne, P, and Resink, TJ. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal. (2018) 52:48–64. doi: 10.1016/j.cellsig.2018.08.019
55. Chi, C, Li, DJ, Jiang, YJ, Tong, J, Fu, H, Wu, YH, et al. Vascular smooth muscle cell senescence and age-related diseases: state of the art. Biochim Biophys Acta Mol basis Dis. (2019) 1865:1810–21. doi: 10.1016/j.bbadis.2018.08.015
56. Lacolley, P, Regnault, V, and Avolio, AP. Smooth muscle cell and arterial aging: basic and clinical aspects. Cardiovasc Res. (2018) 114:513–28. doi: 10.1093/cvr/cvy009
57. Zhang, D, Chen, Y, Xie, X, Liu, J, Wang, Q, Kong, W, et al. Homocysteine activates vascular smooth muscle cells by DNA demethylation of platelet-derived growth factor in endothelial cells. J Mol Cell Cardiol. (2012) 53:487–96. doi: 10.1016/j.yjmcc.2012.07.010
58. Wu, L, Pei, Y, Zhu, Y, Jiang, M, Wang, C, Cui, W, et al. Association of N6-methyladenine DNA with plaque progression in atherosclerosis via myocardial infarction-associated transcripts. Cell Death Dis. (2019) 10:909. doi: 10.1038/s41419-019-2152-6
59. Xu, L, Hao, H, Hao, Y, Wei, G, Li, G, Ma, P, et al. Aberrant MFN2 transcription facilitates homocysteine-induced VSMCs proliferation via the increased binding of c-Myc to DNMT1 in atherosclerosis. J Cell Mol Med. (2019) 23:4611–26. doi: 10.1111/jcmm.14341
60. Ma, SC, Zhang, HP, Jiao, Y, Wang, YH, Zhang, H, Yang, XL, et al. Homocysteine-induced proliferation of vascular smooth muscle cells occurs via PTEN hypermethylation and is mitigated by resveratrol. Mol Med Rep. (2018) 17:5312–9. doi: 10.3892/mmr.2018.8471
61. Min, J, Weitian, Z, Peng, C, Yan, P, Bo, Z, Yan, W, et al. Correlation between insulin-induced estrogen receptor methylation and atherosclerosis. Cardiovasc Diabetol. (2016) 15:156. doi: 10.1186/s12933-016-0471-9
62. Liu, R, Jin, Y, Tang, WH, Qin, L, Zhang, X, Tellides, G, et al. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. (2013) 128:2047–57. doi: 10.1161/CIRCULATIONAHA.113.002887
63. Hiltunen, MO, Turunen, MP, Häkkinen, TP, Rutanen, J, Hedman, M, Mäkinen, K, et al. DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc Med. (2002) 7:5–11. doi: 10.1191/1358863x02vm418oa
64. Pepin, ME, Drakos, S, Ha, CM, Tristani-Firouzi, M, Selzman, CH, Fang, JC, et al. DNA methylation reprograms cardiac metabolic gene expression in end-stage human heart failure. Am J Physiol Heart Circ Physiol. (2019) 317:H674–84. doi: 10.1152/ajpheart.00016.2019
65. Arai, K, Papadopoulou-Marketou, N, and Chrousos, GP. Aldosterone deficiency and resistance In: KR Feingold, B Anawalt, and MR Blackman, editors. Endotext. South Dartmouth, MA: Mdtext Com Inc. (2000)
66. White, PC. Disorders of aldosterone biosynthesis and action. N Engl J Med. (1994) 331:250–8. doi: 10.1056/NEJM199407283310408
67. Demura, M, and Bulun, SE. CpG dinucleotide methylation of the CYP19 I.3/II promoter modulates cAMP-stimulated aromatase activity. Mol Cell Endocrinol. (2008) 283:127–32. doi: 10.1016/j.mce.2007.12.003
68. Takeda, Y, Demura, M, Wang, F, Karashima, S, Yoneda, T, Kometani, M, et al. Epigenetic regulation of aldosterone synthase gene by sodium and angiotensin II. J Am Heart Assoc. (2018) 7:e008281. doi: 10.1161/JAHA.117.008281
69. Zhang, P, Li, T, Liu, YQ, Zhang, H, Xue, SM, Li, G, et al. Contribution of DNA methylation in chronic stress-induced cardiac remodeling and arrhythmias in mice. FASEB J. (2019) 33:12240–52. doi: 10.1096/fj.201900100R
70. Xiao, J, Sluijter, JP, Das, S, Yang, Y, and Shen, Z. A snapshot of genetic and epigenetic basis of arrhythmia and heart failure. Front Genet. (2015) 6:74. doi: 10.3389/fgene.2015.00074
71. Daniel, PB, Walker, WH, and Habener, JF. Cyclic AMP signaling and gene regulation. Annu Rev Nutr. (1998) 18:353–83. doi: 10.1146/annurev.nutr.18.1.353
72. Shaywitz, AJ, and Greenberg, ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. (1999) 68:821–61. doi: 10.1146/annurev.biochem.68.1.821
73. Kass, SU, Pruss, D, and Wolffe, AP. How does DNA methylation repress transcription? Trends Genet. (1997) 13:444–9. doi: 10.1016/S0168-9525(97)01268-7
74. Sung, H, Ferlay, J, Siegel, RL, Laversanne, M, Soerjomataram, I, Jemal, A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
75. Feinberg, AP, and Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. (1983) 301:89–92. doi: 10.1038/301089a0
76. Feinberg, AP, and Vogelstein, B. Hypomethylation of ras oncogenes in primary human cancers. Biochem Biophys Res Commun. (1983) 111:47–54. doi: 10.1016/S0006-291X(83)80115-6
77. Lao, VV, and Grady, WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. (2011) 8:686–700. doi: 10.1038/nrgastro.2011.173
78. Toyota, M, Ahuja, N, Ohe-Toyota, M, Herman, JG, Baylin, SB, and Issa, JPJ. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. (1999) 96:8681–6. doi: 10.1073/pnas.96.15.8681
79. Hinoue, T, Weisenberger, DJ, Lange, CPE, Shen, H, Byun, HM, van den Berg, D, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. (2012) 22:271–82. doi: 10.1101/gr.117523.110
80. Hompes, T, Izzi, B, Gellens, E, Morreels, M, Fieuws, S, Pexsters, A, et al. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. J Psychiatr Res. (2013) 47:880–91. doi: 10.1016/j.jpsychires.2013.03.009
81. Mulligan, CJ, D’Errico, N, Stees, J, and Hughes, D. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics. (2012) 7:853–7. doi: 10.4161/epi.21180
82. Oberlander, TF, Weinberg, J, Papsdorf, M, Grunau, R, Misri, S, and Devlin, AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. (2008) 3:97–106. doi: 10.4161/epi.3.2.6034
83. Romens, SE, McDonald, J, Svaren, J, and Pollak, SD. Associations between early life stress and gene methylation in children. Child Dev. (2015) 86:303–9. doi: 10.1111/cdev.12270
84. Mahajan, HB, Rashid, AS, Junnarkar, AA, Uke, N, Deshpande, SD, Futane, PR, et al. Integration of Healthcare 4.0 and blockchain into secure cloud-based electronic health records systems. Appl Nanosci. (2022) 13:2329–42. doi: 10.1007/s13204-021-02164-0
85. Reul, JM, and de Kloet, ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. (1985) 117:2505–11. doi: 10.1210/endo-117-6-2505
86. McGowan, PO, Sasaki, A, D’Alessio, AC, Dymov, S, Labonté, B, Szyf, M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. (2009) 12:342–8. doi: 10.1038/nn.2270
87. Newell-Price, J, Bertagna, X, Grossman, AB, and Nieman, LK. Cushing’s syndrome. Lancet. (2006) 367:1605–17. doi: 10.1016/S0140-6736(06)68699-6
88. Lee, RS, Tamashiro, KLK, Yang, X, Purcell, RH, Harvey, A, Willour, VL, et al. Chronic corticosterone exposure increases expression and decreases deoxyribonucleic acid methylation of Fkbp5 in mice. Endocrinology. (2010) 151:4332–43. doi: 10.1210/en.2010-0225
89. Lee, RS, Tamashiro, KLK, Yang, X, Purcell, RH, Huo, Y, Rongione, M, et al. A measure of glucocorticoid load provided by DNA methylation of Fkbp5 in mice. Psychopharmacology. (2011) 218:303–12. doi: 10.1007/s00213-011-2307-3
90. Franz, MB, Poterauer, M, Elhenicky, M, Stary, S, Birner, P, Vinatzer, U, et al. Global and single gene DNA methylation in umbilical cord blood cells after elective caesarean: a pilot study. Eur J Obstet Gynecol Reprod Biol. (2014) 179:121–4. doi: 10.1016/j.ejogrb.2014.05.038
91. Virani, S, Dolinoy, DC, Halubai, S, Jones, TR, Domino, SE, Rozek, LS, et al. Delivery type not associated with global methylation at birth. Clin Epigenetics. (2012) 4:8. doi: 10.1186/1868-7083-4-8
92. Almgren, M, Schlinzig, T, Gomez-Cabrero, D, Gunnar, A, Sundin, M, Johansson, S, et al. Cesarean delivery and hematopoietic stem cell epigenetics in the newborn infant: implications for future health? Am J Obstet Gynecol. (2014) 211:502.e1. doi: 10.1016/j.ajog.2014.05.014
93. Jensen, ET, Bertoni, AG, Crago, OL, Rotter, JI, Chen, YDI, Wood, A, et al. Cesarean delivery and insulin sensitivity in the older adult: the microbiome and insulin longitudinal evaluation study. J Endocr Soc. (2022) 6:bvac072. doi: 10.1210/jendso/bvac072
94. Chavarro, JE, Martín-Calvo, N, Yuan, C, Arvizu, M, Rich-Edwards, JW, Michels, KB, et al. Association of birth by cesarean delivery with obesity and type 2 diabetes among adult women. JAMA Netw Open. (2020) 3:e202605. doi: 10.1001/jamanetworkopen.2020.2605
Keywords: DNA methylation, 5-methyl cytosine, CpG sites, vaginal delivery, caesarean section
Citation: Krumpolec P, Kodada D, Hadžega D, Petrovič O, Babišová K, Dosedla E, Turcsányiová Z and Minárik G (2024) Changes in DNA methylation associated with a specific mode of delivery: a pilot study. Front. Med. 11:1291429. doi: 10.3389/fmed.2024.1291429
Edited by:
Yoshinori Okamoto, Meijo University, JapanReviewed by:
Aatish Mahajan, Translational Health Science and Technology Institute (THSTI), IndiaKamaldeen Olalekan Sanusi, Usmanu Danfodiyo University, Nigeria
Copyright © 2024 Krumpolec, Kodada, Hadžega, Petrovič, Babišová, Dosedla, Turcsányiová and Minárik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrik Krumpolec, cGF0cmlrLmtydW1wb2xlY0BtZWRpcmV4Z3JvdXBhY2FkZW15LnNr