 Toshihiko Kakiuchi
Toshihiko Kakiuchi Tetsuya Nosho1
Tetsuya Nosho1 Katsuya Tashiro
Katsuya Tashiro- 1Department of Pediatrics, Faculty of Medicine, Saga University, Saga, Japan
- 2Department of Pediatrics, Karatsu Red Cross Hospital, Karatsu, Japan
Carbamoyl-phosphate synthetase 1 (CPS1) deficiency is an autosomal recessive congenital urea cycle disorder (UCD) characterized by hyperammonemia. The recipients of liver transplantation (LT) for UCD are often children, and the potential donors are often the parents. Hereditary congenital diseases involving UCD entail the possibility of both parents being genetically heterozygous. Herein, we describe the case of a 12-year-old girl with CPS1 deficiency receiving a liver transplant (soon after birth) from her father, who had a heterozygous CPS1 mutation. She was referred to our hospital with respiratory distress after contracting two infections (respiratory syncytial virus and human metapneumovirus) within a short period, both of which presented with hyperammonemia. Medication for hyperammonemia quickly lowered the ammonia levels. The hyperammonemia was thought to be caused by the heterozygous mutation in the donor liver; moreover, it is likely that the low enzyme activity in the patient’s liver was increased due to the infections. This is the first study to report hyperammonemia in a CPS1 deficiency patient due to an infection after LT. Thus, patients with CPS1 deficiency should be aware of the development of hyperammonemia after LT.
1 Introduction
Carbamoyl-phosphate synthetase 1 (CPS1) deficiency is an autosomal recessive congenital urea cycle disorder (UCD) characterized by hyperammonemia, with a morbidity rate of 1/50,000 to 1/800,000 (1–3). The initial treatment for this CPS1 deficiency-based hyperammonemia involves protein restriction and supplementation with arginine, sodium benzoate, sodium phenylacetate/phenylbutyrate, L-citrulline, and carnitine (4). However, these treatments may not successfully avoid ammonia accumulation and recurrent hyperammonemia, resulting in neurologic sequelae that could lead to death (5).
Liver transplantation (LT) is the final treatment for CPS1 deficiency patients who have difficulty withdrawing the continuous plasmapheresis for hyperammonemia (6, 7). Currently, LT is the only curative treatment option until novel therapies become available (8). The recipients of LDLT for UCD are often children, and the potential donors are often the parents.
Herein, we report a case of hyperammonemia in a patient with CPS1 deficiency who received a liver donation from a heterozygous father.
2 Case report
A 12-year-old bedridden girl was referred to our hospital with respiratory distress. She was born healthy at 39 weeks gestation, but developed a fever from the second day after birth and was admitted to a secondary hospital, where she developed respiratory failure, jaundice, and convulsions, with ammonia levels > 400 μg/dL. The next day, she was referred to a tertiary care hospital, where continuous hemodiafiltration was immediately introduced. The ammonia level rose to a maximum of 2,700 μg/dL and declined rapidly, reaching near-normal levels after 2 days. Laboratory findings showed elevated plasma glutamine and alanine levels and decreased arginine and citrulline levels, with no increase in urinary orotic acid excretion; consequently, a biochemical diagnosis of CPS1 deficiency was reached. A genetic test showed a compound heterozygous pathogenic variant in the CPS1 gene (OMIM: 237300; c.1529del [p.Gly510AlafsTer5] in exon 34 and c.2339G>A [p.Arg780His] in exon 34; Table 1). Intensive care was provided, but the patient remained bedridden due to hyperammonitic encephalopathy. At 6 months of age, the patient underwent living-donor liver transplantation (LDLT) from her father. Her liver function was stabilized, and she was followed up at a secondary hospital with nutritional therapy for a long time.
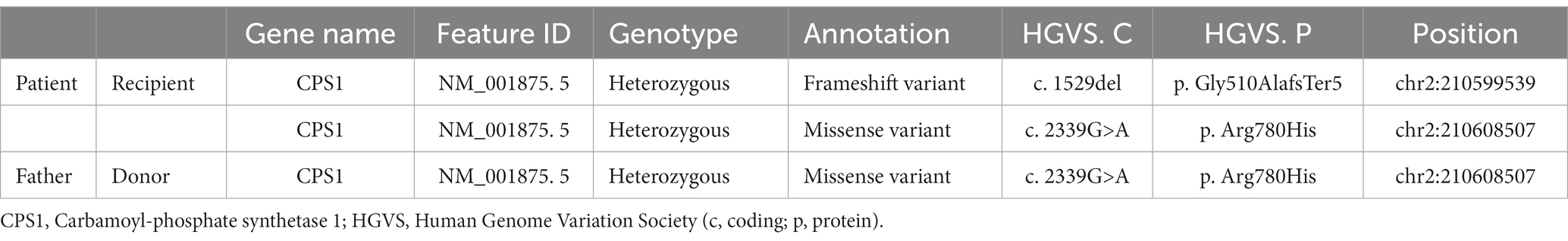
Table 1. The genetic test results of the patient (recipient) and her father (liver donor).
About 5 days ago, the patient was referred to our hospital with wheezing and retractions after contracting an infection with respiratory syncytial virus (RSV). Despite treatments at the referral hospital, the hypercapnia progressed. The laboratory data after admission to our hospital were as follows: ammonia, 324 μg/dL; pH, 7.170; pCO2, and 84.0 mmHg (Figure 1A). She was placed on mechanical ventilation, which rapidly resolved the hypercapnia. Administration of sodium phenylbutyrate (250 mg/kg), l-arginine hydrochloride (400 mg/kg), and sodium benzoate (250 mg/kg) was initiated, and the ammonia level was reduced to 130 μg/dL the next day. The respiratory condition remained stable thereafter. The ammonia level remained at the upper limit of the normal range, and the patient was transferred to her previous hospital on the 26th day of admission to our hospital.
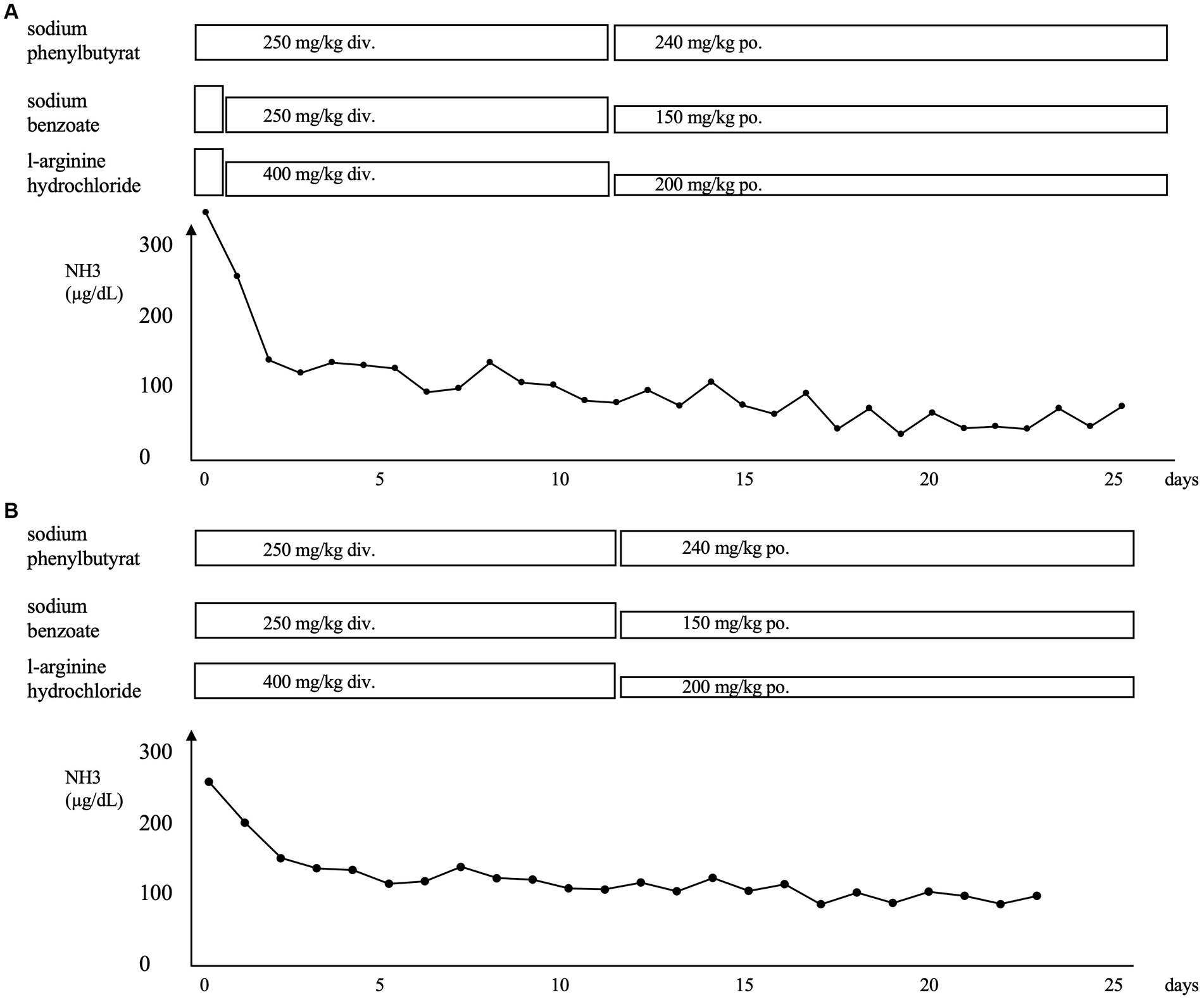
Figure 1. (A) Clinical course of the patient after the first admission to our hospital for respiratory syncytial virus infection. (B) Clinical course of the patient after admission to our hospital for human metapneumovirus infection. The administration of sodium phenylbutyrate, l-arginine hydrochloride, and sodium benzoate was initiated, and a rapid improvement in hyperammonemia was observed in both instances. NH3, ammonia; div., drip infusion into vein; po., per os.
Four months later, she developed another infection with human metapneumovirus with a worsening of the respiratory condition like the last time. Figure 1B shows the clinical course of the patient. The laboratory data were as follows: ammonia, 265 μg/dL; pH, 7.301; and pCO2, 103.0 mmHg. She was placed on mechanical ventilation, and the hyperammonemia was treated with the same medication as before via intravenous administration. The ammonia level dropped to 128 μg/dL the next day and gradually decreased until it returned to normal 2 weeks later. The patient was transferred to the previous hospital on the 23rd day after admission. Oral medication for high ammonia was continued. The patient was promptly discharged from the other hospital and has had no infections or hyperammonemia since then.
Genetic testing of the father showed a heterozygous pathogenic variant in the CPS1 gene (OMIM: 237300; c.2339G>A [p.Arg780His]) in exon 34 (Table 1), which matched that in the patient.
3 Discussion
The clinical course of the female patient in this study highlights that careful attention must be paid regarding the development of hyperammonemia after LTDT, particularly in cases where the donor is a relative with a heterozygous mutation for CPS1 deficiency.
LDLT for metabolic disorders is the key option in Japan. Most metabolic disorders have an autosomal recessive inheritance pattern. Parents who are obligate carriers of the recipient’s disorders become potential heterozygous donors (9). LDLT from heterozygous donors is a feasible option with a better quality of life in patients with CPS1 deficiency (10). On the other hand, in those with OTC deficiency, a transplant from an OTC deficiency heterozygous carrier should be avoided if another donor candidate is available due to the development of the potentially fatal hyperammonemia following LDLT (11). Wakiya et al. (12) proposed donor determination based on the ornithine transcarbamylase activity of the donor liver before LT.
To our knowledge, the present case is the first to report the development of hyperammonemia in a CPS1 deficiency patient due to an infection after LT. However, there are two unresolved issues in this case study. The first is that the causes of the two episodes of hyperammonemia were not wholly determined. The CPS1 enzyme activity in the donor liver was likely lower than normal, and the ability to process ammonia was decreased due to the infection. The father had never exhibited any symptoms suggesting hyperammonemia during infection. However, the enzyme activity levels in the donor livers were not measured in his life. Second, the types and severity of infections that cause hyperammonemia remain unclear. The patient experienced fever due to respiratory infections and aspiration pneumonia in the past but had never presented with hyperammonemia. This point is crucial as we continue to follow up on the patient; otherwise, the hyperammonemia may be overlooked, and medical overreach may result in frequent ammonia measurements.
In conclusion, as with OTC deficiency after LT, patients with CPS1 deficiency should be aware of the development of hyperammonemia at any time.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
TK: Conceptualization, Data curation, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing. TN: Data curation, Investigation, Writing – review & editing. MO: Data curation, Investigation, Writing – review & editing. KT: Conceptualization, Data curation, Investigation, Project administration, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors thank the patient’s family for providing consent and granting permission to draft and publish this case report.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Díez-Fernández, C, Gallego, J, Häberle, J, Cervera, J, and Rubio, V. The study of carbamoyl phosphate synthetase 1 deficiency sheds light on the mechanism for switching on/off the urea cycle. J Genet Genomics. (2015) 42:249–60. doi: 10.1016/j.jgg.2015.03.009
2. Summar, ML, Koelker, S, Freedenberg, D, le Mons, C, Haberle, J, Lee, HS, et al. The incidence of urea cycle disorders. Mol Genet Metab. (2013) 110:179–80. doi: 10.1016/j.ymgme.2013.07.008
3. Kurokawa, K, Yorifuji, T, Kawai, M, Momoi, T, Nagasaka, H, Takayanagi, M, et al. Molecular and clinical analyses of Japanese patients with carbamoylphosphate synthetase 1 (CPS1) deficiency. J Hum Genet. (2007) 52:349–54. doi: 10.1007/s10038-007-0122-9
4. Enns, GM, Berry, SA, Berry, GT, Rhead, WJ, Brusilow, SW, and Hamosh, A. Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N Engl J Med. (2007) 356:2282–92. doi: 10.1056/NEJMoa066596
5. McBride, KL, Miller, G, Carter, S, Karpen, S, Goss, J, and Lee, B. Developmental outcomes with early orthotopic liver transplantation for infants with neonatal-onset urea cycle defects and a female patient with late-onset ornithine transcarbamylase deficiency. Pediatrics. (2004) 114:e523–6. doi: 10.1542/peds.2004-0198
6. Häberle, J, Boddaert, N, Burlina, A, Chakrapani, A, Dixon, M, Huemer, M, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. (2012) 7:32. doi: 10.1186/1750-1172-7-32
7. Nakamura, K, Kido, J, Mitsubuchi, H, and Endo, F. Diagnosis and treatment of urea cycle disorder in Japan. Pediatr Int. (2014) 56:506–9. doi: 10.1111/ped.12439
8. Kido, J, Matsumoto, S, Häberle, J, Inomata, Y, Kasahara, M, Sakamoto, S, et al. Role of liver transplantation in urea cycle disorders: report from a nationwide study in Japan. J Inherit Metab Dis. (2021) 44:1311–22. doi: 10.1002/jimd.12415
9. Vimalesvaran, S, and Dhawan, A. Liver transplantation for pediatric inherited metabolic liver diseases. World J Hepatol. (2021) 13:1351–66. doi: 10.4254/wjh.v13.i10.1351
10. Kasahara, M, Sakamoto, S, Shigeta, T, Fukuda, A, Kosaki, R, Nakazawa, A, et al. Living-donor liver transplantation for carbamoyl phosphate synthetase 1 deficiency. Pediatr Transplant. (2010) 14:1036–40. doi: 10.1111/j.1399-3046.2010.01402.x
11. Rahayatri, TH, Uchida, H, Sasaki, K, Shigeta, T, Hirata, Y, Kanazawa, H, et al. Hyperammonemia in ornithine transcarbamylase-deficient recipients following living donor liver transplantation from heterozygous carrier donors. Pediatr Transplant. (2017) 21:e12848. doi: 10.1111/petr.12848
Keywords: carbamoyl-phosphate synthetase 1 deficiency disease, hyperammonemia, liver transplantation, ornithine transcarbamylase deficiency disease, heterozygous
Citation: Kakiuchi T, Nosho T, Oka M and Tashiro K (2024) Hyperammonemia in a carbamoyl-phosphate synthetase 1 deficiency recipient after living-donor liver transplantation from a carrier donor: a case report. Front. Med. 10:1327854. doi: 10.3389/fmed.2023.1327854
Edited by:
Marcos Mucenic, Santa Casa de Misericórdia de Porto Alegre, BrazilReviewed by:
Themis Silveira, Federal University of Rio Grande do Sul, BrazilGilda Porta, Hospital Sirio Libanes, Brazil
Copyright © 2024 Kakiuchi, Nosho, Oka and Tashiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshihiko Kakiuchi, a2FraXVjaHRAY2Muc2FnYS11LmFjLmpw