 Simona Granata
Simona Granata
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 08 November 2023
Sec. Nephrology
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1303110
This article is part of the Research Topic The Kidney in Auto-Immune and Auto-Inflammatory Processes: Definitions, Mechanisms, and Biomarkers - Volume II View all 4 articles
Kidney transplantation is the best available renal replacement therapy for patients with end-stage kidney disease and is associated with better quality of life and patient survival compared with dialysis. However, despite the significant technical and pharmaceutical advances in this field, kidney transplant recipients are still characterized by reduced long-term graft survival. In fact, almost half of the patients lose their allograft after 15–20 years. Most of the conditions leading to graft loss are triggered by the activation of a large immune-inflammatory machinery. In this context, several inflammatory markers have been identified, and the deregulation of the inflammasome (NLRP3, NLRP1, NLRC4, AIM2), a multiprotein complex activated by either whole pathogens (including fungi, bacteria, and viruses) or host-derived molecules, seems to play a pivotal pathogenetic role. However, the biological mechanisms leading to inflammasome activation in patients developing post-transplant complications (including, ischemia-reperfusion injury, rejections, infections) are still largely unrecognized, and only a few research reports, reviewed in this manuscript, have addressed the association between abnormal activation of this pathway and the onset/development of major clinical effects. Finally, the regulation of the inflammasome machinery could represent in future a valuable therapeutic target in kidney transplantation.
Kidney transplantation is the best treatment for patients with end-stage kidney disease (ESKD), a clinical condition characterized by severe alterations in body homeostasis that require the initiation of renal replacement therapy to ensure patient survival. It is associated with a better quality of life and survival compared with dialysis treatment (1).
In addition, during the last two decades, the significant advancement of surgical and organ preservation techniques and the optimization of immunosuppressive drug management has led to an important improvement in short-term graft survival (more than 90% after 1 year) (2). However, despite this clinical success, the long-term allograft outcome remains unsatisfactory. The 10-year graft survival rate is 65.5% among living donor transplant recipients and 49.5% among kidney transplant recipients from deceased donors (3).
Several demographic and clinical factors have been considered responsible for this condition (e.g., donor/recipient age, marginality of the kidney, delayed graft function (DGF), acute rejection, infections, and glomerulonephritis recurrence), but the entire biological machinery involved in graft loss is still only partially defined.
Numerous biological elements have been associated with pathogenetic alterations leading to post-transplant complications, but inflammation seems to play a pivotal role in almost all clinical allograft alterations. In addition, several reports have recently suggested that the inflammasome, a multiprotein complex activated by either whole pathogens (including fungi, bacteria, and viruses) or host-derived molecules as in chronic kidney disease (4–6), could be involved in these post-transplant complications (7).
In this review, which focuses only on kidney transplantation, we provide a brief overview of the potential role of inflammasomes in the pathogenesis of major acute and chronic allograft complications (including ischemia/reperfusion injury, graft rejections, post-transplant infections and recurrent glomerulonephritis) (Figure 1).
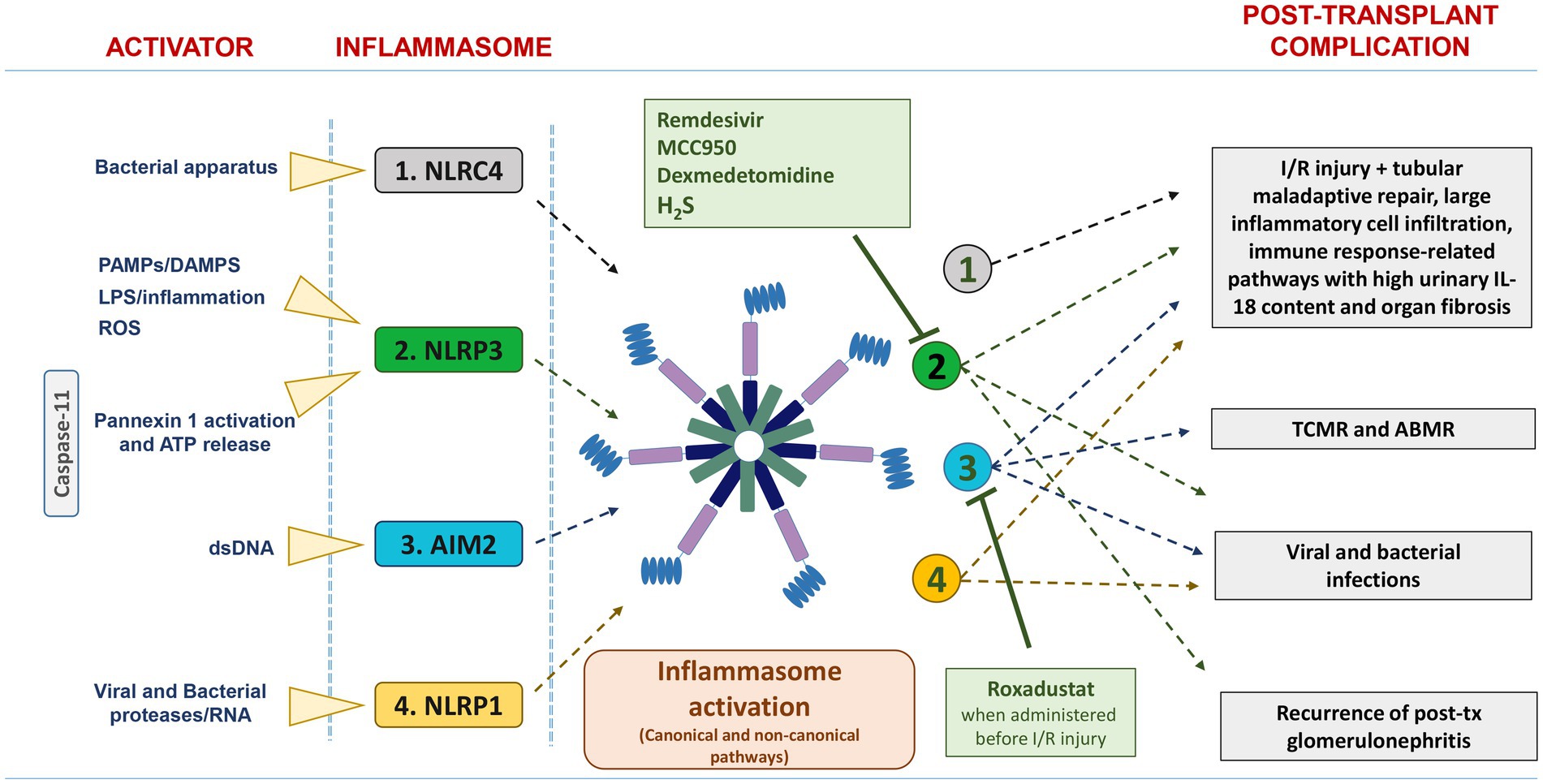
Figure 1. List of the main activators and inflammasomes involved in post-transplant complications. NLRC4, NLRP3, and AIM2 are activated in IRI/DGF, and their persistent over-expression was associated with tubular maladaptive repair, large inflammatory cell infiltration, and organ fibrosis. Up-regulation of AIM2 inflammasome has been found in acute rejection, particularly TCMR. NLRP3, AIM2, and NLRP1 are involved in viral and bacterial infection. The pharmacological inhibitors Remdesivir, MCC950, dexmedetomidine, and H2S suppress NLRP3 inflammasome activation, whereas Roxadustat downregulates AIM2 inflammasome activation.
The inflammasome is a multiprotein complex comprising three classes of molecules: receptor/sensor, adaptor, and effector. Inflammasome sensors (or pattern recognition receptors, PRR) belong to two classes: nucleotide oligomerization domain (NOD)-like receptors (NLRs), and absent in melanoma-2 (AIM2)-like receptors (ALRs) (8). The adaptor molecule used in most inflammasomes is apoptosis-associated speck-like protein (ASC) that enables the recruitment of pro-caspase 1, an effector molecule that mediates maturation of pro-interleukin (IL)-1β and pro-IL-18 to fully active interleukins that are crucial for host defense responses to infection and injury (9). In addition, inflammasomes trigger the pyroptosis (10) a programmed pro-inflammatory cell death mediated by gasdermin D (GSDMD), which forms pores in the plasma membrane, inducing the release of IL-1β and IL-18, water influx, cell swelling, and consequent osmotic lysis (11–14).
The assembly of inflammasomes begins with the activation of PRRs (15, 16). These molecules are capable of recognizing various pathogen-associated molecular patterns (PAMPs) (such as lipopolysaccharide (LPS) and flagellin) and danger-associated molecular patterns (DAMPs) such as uric acid, ATP, DNA, and High Mobility Group 1 (HMGB1) which arise during microbial infections and sterile injury, respectively (15).
The human genome encodes for 23 NLRs; however, only a few NLR proteins, such as NLRP1, NLRP3, NLRP6, NLRP7, NLRP9, NLRP12, and NLRC4, have been found to form inflammasomes and activate caspase-1 (8). These NLRs share some similar structures: C-terminal leucine-rich repeat (LRR) domains, which are responsible for ligand sensing, and central nucleotide binding domains (NACHT), which elicit ATP-induced oligomerization. The N-terminal region contains the pyrin domain (PYD) in NLRP and one or more caspase recruitment (CARD) domains in NLRC proteins (17, 18).
The AIM2 protein consists of two domains: (1) a C-terminal HIN domain, which allows for the detection of double-stranded DNA in the cytoplasm, and (2) a PYD domain for the recruitment of ASCs and formation of the inflammasome complex (19, 20).
NLRP1, NLRP3, NLRP6, NLRP7, NLRP9, NLRP12, and NLRC4 belong to the group of “canonical inflammasomes” that induce the activation of IL-1β and IL-18 through caspase-1, whereas caspase-11 (in mouse) and caspase-4 and -5 (in humans) are defined “non-canonical” (21). These non-canonical inflammasomes consist of three main domains: an N-terminal CARD, a p20, and a C-terminal p10 (22). Although activated by different types of PAMPs and DAMPs, they have similar downstream effector functions, including the activation of GSDMD-mediated pyroptosis and the activation of caspase-1, leading to subsequent proteolytic maturation of IL-1β and IL-18 and their secretion through GSDMD-mediated pores (23–26).
Among the canonical inflammasomes, NLRP3 is the most studied, being largely involved in both innate and adaptive immunity after activation by either whole pathogens, including fungi, bacteria, and viruses, or by host-derived molecules, such as fibrillar amyloid-β (Aβ) peptide, as well as extracellular ATP and glucose (27–31).
The NLRP3 inflammasome may be activated by both canonical and noncanonical pathways.
This pathway consists of two steps: priming and activation. Priming signals include microbial components or endogenous cytokines that bind to the Toll-like receptor (TLR), FAS-associated death domain protein (FADD) or IL-1R ligands (32–34) and, through the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (35), may induce the expression of genes that encode pro-IL-1β, pro-IL-18, and NLRP3 (36, 37).
After this phase, in the activation step, a second signal, such as ATP, ion fluxes (K+, Ca++, Cl−) (28), β-amyloids, particles (such as uric acid, silica, aluminum), reactive oxygen species (ROS), and dysfunctional organelles, is required to form and assembly the inflammasome (10, 38–40).
Inflammasome activation may be regulated by post-translational modifications. For example, ATP and TLR-4 may activate the deubiquitination of NLRP3 in macrophages (41) by the enzyme BRCA1/BRCA2-containing complex subunit 3 (BRCC3) (42) and c-Jun N-terminal kinase (JNK1)-mediated NLRP3 S194 phosphorylation may induce NLRP3 deubiquitination with a subsequent inflammasome assembly (43).
Direct and indirect mechanisms of epigenetic regulation may activate the inflammasome. Wang et al. reported that overexpression of miR-377, through activation of the p38 MAPK/TXNIP/NLRP3 inflammasome pathway, was able to promote oxidative stress in diabetic nephropathy and prevent a severe glomerular podocyte inflammation (44). Other authors, reported that miR-711 suppressed NLRP3 expression by inhibiting FADD and NFkB (45), whereas miR-22, miR-30e and miR-223 by targeting directly the 3’UTR of NLRP3 gene (46–48).
Wu et al., showed that the NLRP3 inflammasome enhanced the chronic deleterious effect of hypoxia on proximal tubular cells and that miR-155, a positive-regulator of NLRP3 signals by inhibiting the targeted FOXO3a gene, could facilitate this biological effect (49).
These findings confirm previous results which showed that in tubular renal cells, the overexpression of miR-155 promotes upregulation of caspase-1, IL-1β, and IL-18, whereas knockdown of miR-155 attenuated the inflammatory cell death, suggested a potential use of the anti-miR-155 as preventive strategy against pyroptosis (50).
However, all these findings should be further studied and validated before being translated in the clinic.
The non-canonical activation of the NLRP3 inflammasome is caspase-1 independent and is mediated by caspase-4 and caspase-5 in humans and caspase-11 in mice (24).
These non-canonical caspases interact directly with LPS (51) and can activate both the NLRP3 inflammasome for cytokine production and GSDMD to mediate pyroptosis (11, 52–55).
Caspase-4, like caspase-1, may cleave GSDMD and pro-IL-18, but it seems not able to cleave IL-1β. Instead, caspase-5 cleaves GSDMD, but it has a very weak pro-cytokine activity (56), and caspase-11 has a proteolytic activity limited to GSDMD only (57).
In addition, caspase-8 may activate both canonical and non-canonical NLRP3 inflammasome pathways (33), but this effect needs to be better elucidated.
Ischemia–reperfusion injury (IRI) is an almost inevitable process occurring after kidney transplantation, which includes a wide range of biological insults caused by the initial transient surgical warm ischemia followed by the cold ischemic period due to hypothermal maintenance/preservation of the organ (58).
In many recipients (approximately 15–25%), this process may lead to delayed graft function (DGF), a condition of no post-transplant recovery of renal function that requires the maintenance of dialysis treatment to ensure patient survival. Moreover, IRI activates a complex and multi-factorial biological network that may trigger acute allograft rejection and the early onset of interstitial fibrosis and tubular atrophy (IFTA). All these events may accelerate graft loss (58, 59).
In this process, activation of the inflammatory and innate immune system, including the inflammasome pathway, has a primary role (60). NLRP1, caspase-1, and NLRP3 expression appeared significantly up-regulated in renal tubular epithelial cells after IRI (61–63) and their persistent over-expression was associated with tubular maladaptive repair, large inflammatory cell infiltration, and organ fibrosis (64). Moreover, NLRP3−/− mice but not Asc−/− or caspase-1−/− mice were protected from IRI, suggesting a direct effect of NLRP3 on tubular epithelial cells leading to IRI, but independent of inflammasome formation (65). However, during IRI, other pathways may be involved in this large biological networking (63). Recently, Lavallard et al. clearly demonstrated a direct activation of the NLRP3 inflammasome by ROS in rat islets undergoing IRI and that this effect was not prevented by N-acetyl-L-cysteine or caspase-1 inhibitors (66).
RNA-sequencing analysis in human kidney tissues obtained before ischemia (considered as a normal condition), 15 min after hypoxia (ischemia), and 10 min of reperfusion revealed significant up-regulation of the inflammasome (NLRC4) and immune response-related pathways after reperfusion (67, 68) with high urinary IL-18 content (69, 70).
IRI may be aggravated by a long cold storage time and may have a further negative impact on “fragile” organs from Extended Criteria Donors (ECD), which include donors over the age of 60 years or donors over the age of 50 years with 2 of the following 3 items: (1) history of high blood pressure, (2) serum creatinine ≥1.5 mg/dL, and (3) death due to stroke, with increased risk for allograft outcome (71, 72). These marginal organs, under specific stimulations, are often prone to release higher levels of pro-inflammatory cytokines and DAMPS compared to those from the Standard Criteria Donor (SDC). This may accelerate inflammatory-related allograft alterations (73).
In rat model of kidney transplantation, prolonged cold storage (18 h) induced a higher decline in renal function and a significant increment of IL-1β and NLRP3 protein levels compared with 2 h cold storage (this effect was similar in both deceased cardiac death (DCD) and living donor models) (74).
Moreover, the expression of several genes involved in the TLR-4 and inflammasome pathways was significantly upregulated in pre-implantation biopsies of ECDs compared with SDC, suggesting their potential role as therapeutic targets for new pharmacological treatments finalized to slow-down the progression of chronic allograft damage in marginal organs (75).
A promising therapeutic strategy to minimize IRI in kidney transplantation involves multipotent adult progenitor cells (MAPC). The addition of these cells to the normothermic machine perfusion solution significantly improved urine output, decreased the expression of NGAL (a main injury biomarker) and IL-1β, and upregulated anti-inflammatory and pro-tolerogenic cytokines (including IL-10 and Indolamine-2,3-dioxygenase) compared with untreated organs (76).
Activation of the NLRP3 inflammasome in IRI is also mediated by the non-canonical pathway. IRI may induce a marked increase in the expression of caspase-11 in renal tissue with subsequent pannexin 1 (panx1) activation by catalytic cleavage. This may facilitate ATP release and, through stimulation/activation of the NLRP3 inflammasome, promote kidney injury (77, 78).
Therefore, pharmacological inhibition of the NLRP3 inflammasome may represent a valuable therapeutic target to antagonize IRI-induced allograft damage.
Early post-transplant administration of MCC950 (50 mg/kg, i.p), a specific NLRP3 inhibitor that blocks inflammasome oligomerization, reduced the activation of caspase-1 and the release of IL-1β in allograft of mice that underwent IRI and ameliorated the post-transplant functional recovery of these organs (79).
Remdesivir, a widely employed antiviral nucleotide prodrug against COVID-19 (80), appears to antagonize acute kidney injury (AKI) by inhibiting NLRP3 inflammasome activation via the repression of NFkB and MAPK pathways in LPS-activated macrophages (81).
Roxadustat, a hypoxia-inducible factor (HIF) prolyl-hydroxylase inhibitor, when administered to mice before IRI (at a dose of 10 mg/kg for 5 days) alleviated renal damage by down-regulating AIM2 inflammasome with a consequent enhancement of HIF stabilization and activation of CD73/adenosine signaling (82).
A protective effect was also demonstrated for hydrogen sulfide (H2S), produced by the kidney under physiological conditions to promote kidney excretion, regulate renin release, and increase ATP production. H2S appears to have a protective role in kidney diseases by regulating oxidative stress, inflammation, and the renin–angiotensin–aldosterone system (83). In an animal model of IRI, the administration of H2S prior to surgery exerted this positive effect, at least partially, by Nrf2-mediated NLRP3 inflammasome inhibition (62).
Another drug with an inhibitory effect on the NLRP3 inflammasome is dexmedetomidine (DEX), a selective a2-adrenoreceptor agonist that regulates hemodynamics by reducing sympathetic tone, decreasing inflammatory response, inhibiting renin release, increasing glomerular filtration rate, and increasing secretion of sodium and water by the kidneys (84). Several animal studies have reported antioxidant, anti-apoptosis, and anti-inflammatory effects of DEX (85, 86).
Administration of DEX (30 μg/kg) 30 min before intraperitoneal injection of LPS in a mouse model of AKI attenuated renal injury by enhancing autophagy via the alpha2-adrenoreceptor/AMPK/mTOR pathway, which inhibits the NLRP3 inflammasome (87). This protective effect could explain the reduced incidence of DGF found in patients who received perioperative DEX during kidney transplantation (88).
GSK1070806, a humanized IgG1 antibody that binds and neutralizes the function of mature IL-18, has also been tested for the prevention of DGF in patients with DCD transplantation. However, administration of a single dose of this agent (3 mg/kg) just before kidney allograft reperfusion did not prevent this complication (89).
Acute rejection is a major complication of the allograft that could also occur very early post-transplantation and have a significant negative impact on graft survival. This condition, which could be triggered by humoral or T cellular immune dysfunctional regulation, has a complex and multi-factorial pathogenesis and a large involvement of the immune system.
Based on a few literature reports, inflammasomes may be involved in the onset and development of T-cell-mediated rejection (TCMR) and antibody-mediated rejection (ABMR).
Tejada et al., using a transcriptomics-based approach, revealed a significant enhancement of the AIM2 inflammasome in both T-cell- and antibody-mediated rejected allografts, even if the association was stronger for the former (90). These data were in line with those published by Venner et al. (91), which demonstrated only a small association between the AIM2 transcript and ABMR in a large biopsy transcriptomic analysis (703 biopsies) (92).
Asgari et al. showed that inflammasome activation could be induced by complement pathway mediators over-expressed during the rejection episode. In particular, C3a generated during inflammation engaged C3aR, increasing extracellular ATP, which, in turn, through P2X7 receptor signaling, activated inflammasome and IL-1β production by monocytes (93).
Moreover, donor-derived cell-free DNA (dd-cfDNA), deoxyribonucleic acid fragments that are released into the blood primarily during allograft rejection (94), could activate the inflammasome pathway (95). The binding of cfDNA to DNA sensor cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS) induces the synthesis of 2′3′ cyclic GMP–AMP (cGAMP), which activates stimulator of interferon genes protein (STING) and, in turn, the transcription of inflammatory genes via phosphorylation and activation of IRF3 (96). The cyclic dinucleotides produced by cGAS can also activate the NLRP3 inflammasome (97), and dd-cfDNA may directly activate the AIM2 inflammasome by binding to the HIN domain. However, additional studies are required to assess this important biological association.
Dessing et al., then, analyzing several NLRP3 SNPs in >1,200 matched donors and recipients, revealed that the NLRP3 gain-of-function SNP rs35829419 in donors was associated with an increased risk of acute rejection, whereas the NLRP3 loss-of-function SNP rs6672995 in recipients was associated with a reduced risk of rejection, particularly within the first year after transplantation (98). Unfortunately, the authors did not measure the specific production of inflammasome-related cytokines and did not clearly discriminate between cellular and humoral allograft rejection.
Finally, the administration of MCC950 and DEX, as in IRI, also demonstrated a significant protective effect against acute allograft rejection (79, 88), but with no real impact on short- and long-term allograft function (88). Nevertheless, the absence of large prospective studies and clinical trials on kidney transplantation makes it difficult to draw definitive conclusions.
Infections are frequent comorbidities in kidney transplant recipients that can directly or indirectly impact graft and patient survival. Although characterized by specific pathogenetic and biological mechanisms, these clinical complications may activate inflammation and, in some cases, the inflammasome pathway.
Epstein–Barr virus (EBV): it belongs to the human gamma herpesvirus family, with a seroprevalence of more than 90% in adults, and mainly infects B lymphocytes, epithelial cells, T lymphocytes, and NK cells (99).
In kidney transplant recipients, acute and reactivation of latent EBV infection may induce severe clinical complications, including the onset of a post-transplantation lymphoproliferative disorder (PTLD) (100–102), which may dramatically impact graft and patient survival.
Interestingly, the inflammasome pathway may play a role in maintaining the virus in a latent form. In fact, in EBV latent-containing B cells, Interferon Gamma Inducible Protein 16 (IFI16) recognizes the viral genome and, after recruitment of ASC and procaspase-1, forms an inflammasome complex, which moves into the cytoplasm, activates caspase-1, and cleaves pro-IL-1β, pro-IL-18, and pro-IL-33 into their mature forms (103). IFI16 and cytokines are subsequently sorted and released outside the cells via exosomes, representing a potential strategy to facilitate EBV persistence (103).
Another mechanism that continues viral latency in B lymphocytes is mediated by a physical interaction between IFI16 and the core constitutive heterochromatin machinery (KAP1 and SZF1), which silences the key EBV lytic switch protein (104).
Nevertheless, some authors have recently suggested that inflammasome pathways may also be involved in the EBV primary infection-related biological network. Lytic triggers, such as Histone deacetylase inhibitor (HDACi), DNA methyltransferase (DNMT) inhibitors, and Ig cross-linking, may induce caspase-1 activation through the thioredoxin-interacting protein (TXNIP)-NLRP3 inflammasome pathway, leading to partial loss of KAP1/TRIM28, a barrier to EBV lytic cycle entry (105, 106). However, all these observations should be confirmed in larger research and clinical studies.
The Human Herpesvirus-8 (HHV-8), also called Kaposi sarcoma Herpesvirus (KSHV), belongs to the family of DNA viruses Herpeseviridae and may be responsible for the onset/development of post-transplant cancer with single or multiple lesions on mucosal surfaces, including the skin, lungs, gastrointestinal tract, and lymphoid tissue (107). It can infect several different cell types, including endothelial cells, B cells, epithelial cells, dendritic cells, monocytes, and fibroblasts (108, 109).
The virus binds to several host cell surface receptors, such as integrins (including α3β1, αVβ5, and αVβ3), the cystine–glutamate transporter xCT, heparan sulfate, and the tyrosine protein kinase receptor EPHA2, and induces a signal transduction cascade, which results in cellular changes that allow the virus to enter the cell and traffic within the cytoplasm (110, 111).
Viral entry results in the delivery of the virion capsid into the cytoplasm, followed by its uncoating and the delivery of the HHV-8 genome into the nucleus, where it remains as an episome after circularization. Subsequently, the virus becomes latent or undergoes sporadic bouts of lytic reactivation during its lifecycle (108, 109).
Cellular entry or reactivation of virus activates an immune response via TLRs (TLR-3, TLR-4, and TLR-9) (112–114), retinoic acid-inducible gene I protein (RIG-I)-like receptors (RLRs), NLRs (NLRP1 and NLRP3) (115), ALRs and cyclic GMP-AMP synthase (cGAS)–STING (116).
The activation of these biological elements leads to the induction of type I interferon and NLR-dependent inflammasome pathways. However, Orf63, an HHV-8 tegument protein, with high sequence similarity to NLRP1, inhibits NLRP1 and NLRP3 inflammasomes by disrupting the association of NLRP1 and NLRP3 with ASC or procaspase-1 (115). Because Orf63 is a component of the tegument, it is released into the cytoplasm at the onset of primary infection, inhibiting host immunity.
Likewise, SOX protein, encoded by HHV-8 ORF37, with endo- and exo-nuclease activity for degrading cellular mRNA and processing the viral DNA genome, suppresses AIM2 inflammasome activation during the HHV-8 lytic cycle by disrupting AIM2: dsDNA polymerization and ASC recruitment and oligomerization (117).
Thus, targeting viral inhibitors of inflammasomes represents a therapeutic strategy for the treatment of viral infection.
BK polyomavirus (BKV): is a double-stranded DNA virus belonging to Polyomaviridae family and is highly prevalent in humans with an incidence of more than 80% in the general population (118). In immunocompetent individuals, primary BK virus infection is generally asymptomatic or results in mild symptoms, but in kidney transplantation, its reactivation in the tubular renal epithelial cells may induce cytotoxicity and allograft dysfunction (119).
Ribeiro et al., found that the inflammasome activator TLR-3 was upregulated in the tubule-interstitial allograft compartment of patients with polyomavirus-associated nephropathy (PVAN) along with IL-1β and IL-18 compared with acute rejection and pre-transplant donor biopsies (controls) (120, 121). However, surprisingly, the in vitro part of the study did not confirm the activation of the NLRP3 or AIM2 inflammasome pathways (120).
Severe acute respiratory syndrome (SARS) coronavirus 2 (SARS-CoV-2): is a positive-sense single-stranded RNA virus belonging to the coronavirus family responsible for coronavirus disease 2019 (COVID-19). This virus, which activates the NLRP3 inflammasome through the accessory protein viroporin E (122), could exacerbate the inflammatory machinery in infected kidney transplant recipients, leading to severe clinical complications and partly justify the positive effects of anti-inflammatory therapies in these fragile patients (123, 124).
These results, even if they did not allow to draw definitive conclusions, underline the necessity to study these pathways in kidney transplant patients.
Although inflammation plays a major role in urinary tract infections, only one study has clearly described the impact of the inflammasome in these common post-transplant complications.
It has been demonstrated that recombinant purified E. coli α-hemolysin caused deubiquitination, oligomerization, and activation of the NLRP3 inflammasome in response to K+ concentration perturbations in THP-1 macrophages. This led to mitochondrial dysfunction, and activation of the immune response, resulting in cell death (125). Additional studies are required to better address this important topic.
Glomerulonephritis (GNs), the fourth most common cause of allograft loss, are major clinical problems occurring in the post-transplant period. Most patients affected by these conditions may experience a significant reduction in allograft survival and develop severe systemic alterations (nephrotic and nephritic syndrome, acute allograft dysfunction, accelerated development of chronic allograft nephropathy) (126).
However, although no specific studies have been published regarding the impact of inflammasomes on the pathogenesis of recurrent glomerulonephritis (rGNs) after kidney transplantation, we can postulate their involvement in these disorders (127). In particular, hyperactivation of the NLRP3 inflammasome in infiltrating macrophages and podocytes (128, 129) and in serum of IgAN nephropathy (as demonstrated by high circulating level of IL-18 and IL-1β) is associated with a fast progression of this disease (127, 130).
In patients with focal segmental glomerulosclerosis (FSGS), necroptosis, a pro-inflammatory lytic form of programmed cell death activated in podocytes, activates the NLRP3 inflammasome pathway. As reported by Hu et al., the inhibition of necroptosis in adriamycin (ADR)-induced nephropathy, through the inhibition of this inflammasome in podocytes attenuates proteinuria levels and kidney histological damage (131).
In patients with ANCA-associated vasculitis (AAV), the serum level of IL-18 was elevated (132) and the tissue protein content of NOD2, NLRP3, and NLRC5 was higher than that in healthy controls, demonstrating the role of the inflammasome in these disorders. In addition, kidney tissue expression levels of NOD2 and NLRC5 were significantly correlated with the severity of renal lesions in AAV (133).
Therefore, despite the role of the inflammasomes in the pathogenesis of GNs in the native kidney, the impact of these biological pathways in the post-transplant recurrence should be addressed with specific research projects.
Our paper reveals the great interest of the transplant community in understanding the pathophysiological role of inflammasomes in kidney transplantation. Although the role of these biological pathways has been well elucidated in several native kidney disorders (134), few data regarding their direct contribution to allograft and systemic post-transplant complications are available (Table 1). Additionally, it underlines that a deep comprehension of the contribution of the inflammasomes in the biological machinery associated with acute and chronic allograft alterations may facilitate the discovery of novel previously unrecognized therapeutic targets, the identification of early diagnostic and prognostic biomarkers, and accelerate the initiation of clinical studies/trials involving inflammasome regulators (e.g., MCC950) finalized to slow down the progression of chronic allograft alterations and allograft functional failure. In this context, innovative biomolecular research strategies (including the high-throughput omics technologies) and an integrated/multi-disciplinary collaborative approach, which facilitate the interaction between different professionals (including molecular biologists, clinicians, bioinformaticians), could help accelerate this discovery process.
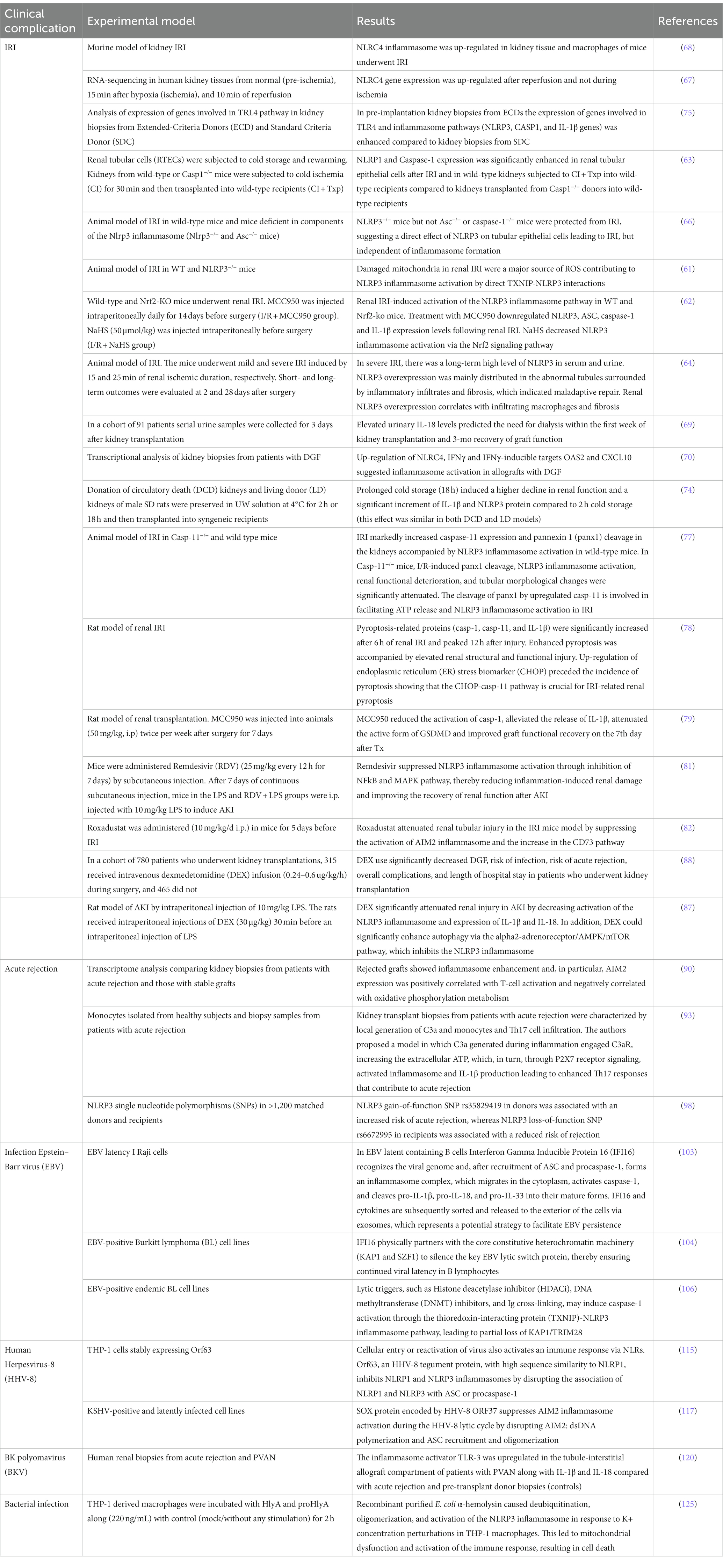
Table 1. Main studies focusing on the role of inflammasome pathways in clinical complications after kidney transplantation.
SG: Writing – original draft. DL: Writing – original draft. GS: Writing – review & editing. AP: Writing – review & editing. GZ: Conceptualization, Funding acquisition, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the European Union - Next Generation EU - NRRP M6C2 - Investment 2.1 Enhancement and strengthening of biomedical research in the NHS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Poggio, ED, Augustine, JJ, Arrigain, S, Brennan, DC, and Schold, JD. Long-term kidney transplant graft survival-making progress when most needed. Am J Transplant. (2021) 21:2824–32. doi: 10.1111/ajt.16463
2. Meier-Kriesche, HU, Schold, JD, and Kaplan, B. Long-term renal allograft survival: have we made significant progress or is it time to rethink our analytic and therapeutic strategies? Am J Transplant. (2004) 4:1289–95. doi: 10.1111/j.1600-6143.2004.00515.x
3. United States Renal Data System. 2021 USRDS annual data report: Epidemiology of kidney disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (2021).
4. Huang, G, Zhang, Y, Zhang, Y, and Ma, Y. Chronic kidney disease and NLRP3 inflammasome: pathogenesis, development and targeted therapeutic strategies. Biochem Biophys Rep. (2022) 33:101417. doi: 10.1016/j.bbrep.2022.101417
5. Lucafò, M, Granata, S, Bonten, EJ, McCorkle, R, Stocco, G, Caletti, C, et al. Hypomethylation of NLRP3 gene promoter discriminates glucocorticoid-resistant from glucocorticoid-sensitive idiopathic nephrotic syndrome patients. Clin Transl Sci. (2021) 14:964–75. doi: 10.1111/cts.12961
6. Granata, S, Masola, V, Zoratti, E, Scupoli, MT, Baruzzi, A, Messa, M, et al. NLRP3 inflammasome activation in dialyzed chronic kidney disease patients. PLoS One. (2015) 10:e0122272. doi: 10.1371/journal.pone.0122272
7. Burke, RM, Dale, BL, and Dholakia, S. The NLRP3 Inflammasome: relevance in solid organ transplantation. Int J Mol Sci. (2021) 22:10721. doi: 10.3390/ijms221910721
8. Sharma, D, and Kanneganti, TD. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. (2016) 213:617–29. doi: 10.1083/jcb.201602089
9. Broz, P, and Dixit, VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
10. Zheng, D, Liwinski, T, and Elinav, E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. (2020) 6:36. doi: 10.1038/s41421-020-0167-x
11. Liu, X, Zhang, Z, Ruan, J, Pan, Y, Magupalli, VG, Wu, H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
12. Sborgi, L, Rühl, S, Mulvihill, E, Pipercevic, J, Heilig, R, Stahlberg, H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. (2016) 35:1766–78. doi: 10.15252/embj.201694696
13. Fink, SL, and Cookson, BT. Pillars article: Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. (2006) 8:1812–25. doi: 10.1111/j.1462-5822.2006.00751.x
14. Yu, P, Zhang, X, Liu, N, Tang, L, Peng, C, and Chen, X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. (2021) 6:128. doi: 10.1038/s41392-021-00507-5
15. Broz, P, and Monack, DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol. (2013) 13:551–65. doi: 10.1038/nri3479
16. Platnich, JM, and Muruve, DA. NOD-like receptors and inflammasomes: a review of their canonical and non-canonical signaling pathways. Arch Biochem Biophys. (2019) 670:4–14. doi: 10.1016/j.abb.2019.02.008
17. Schroder, K, and Tschopp, J. The inflammasomes. Cells. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
18. Fu, J, and Wu, H. Structural mechanisms of NLRP3 Inflammasome assembly and activation. Annu Rev Immunol. (2023) 41:301–16. doi: 10.1146/annurev-immunol-081022-021207
19. Fernandes-Alnemri, T, Yu, JW, Datta, P, Wu, J, and Alnemri, ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. (2009) 458:509–13. doi: 10.1038/nature07710
20. Hornung, V, Ablasser, A, Charrel-Dennis, M, Bauernfeind, F, Horvath, G, Caffrey, DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. (2009) 458:514–8. doi: 10.1038/nature07725
21. Ding, J, and Shao, F. SnapShot: the noncanonical Inflammasome. Cells. (2017) 168:544–544.e1. doi: 10.1016/j.cell.2017.01.008
22. Yi, YS. Functional crosstalk between non-canonical caspase-11 and canonical NLRP3 inflammasomes during infection-mediated inflammation. Immunology. (2020) 159:142–55. doi: 10.1111/imm.13134
23. Hagar, JA, Powell, DA, Aachoui, Y, Ernst, RK, and Miao, EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. (2013) 341:1250–3. doi: 10.1126/science.1240988
24. Kayagaki, N, Warming, S, Lamkanfi, M, Vande Walle, L, Louie, S, Dong, J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. (2011) 479:117–21. doi: 10.1038/nature10558
25. Matikainen, S, Nyman, TA, and Cypryk, W. Function and regulation of noncanonical Caspase-4/5/11 Inflammasome. J Immunol. (2020) 204:3063–9. doi: 10.4049/jimmunol.2000373
26. Downs, KP, Nguyen, H, Dorfleutner, A, and Stehlik, C. An overview of the non-canonical inflammasome. Mol Asp Med. (2020) 76:100924. doi: 10.1016/j.mam.2020.100924
27. Gross, O, Poeck, H, Bscheider, M, Dostert, C, Hannesschläger, N, Endres, S, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. (2009) 459:433–6. doi: 10.1038/nature07965
28. Mariathasan, S, Weiss, DS, Newton, K, McBride, J, O'Rourke, K, Roose-Girma, M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. (2006) 440:228–32. doi: 10.1038/nature04515
29. Kanneganti, TD, Body-Malapel, M, Amer, A, Park, JH, Whitfield, J, Franchi, L, et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem. (2006) 281:36560–8. doi: 10.1074/jbc.M607594200
30. Muruve, DA, Pétrilli, V, Zaiss, AK, White, LR, Clark, SA, Ross, PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. (2008) 452:103–7. doi: 10.1038/nature06664
31. Halle, A, Hornung, V, Petzold, GC, Stewart, CR, Monks, BG, Reinheckel, T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. (2008) 9:857–65. doi: 10.1038/ni.1636
32. Allam, R, Lawlor, KE, Yu, EC, Mildenhall, AL, Moujalled, DM, Lewis, RS, et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. (2014) 15:982–90. doi: 10.15252/embr.201438463
33. Gurung, P, Anand, PK, Malireddi, RK, Vande Walle, L, Van Opdenbosch, N, Dillon, CP, et al. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol. (2014) 192:1835–46. doi: 10.4049/jimmunol.1302839
34. McKee, CM, and Coll, RC. NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. J Leukoc Biol. (2020) 108:937–52. doi: 10.1002/JLB.3MR0720-513R
35. Kelley, N, Jeltema, D, Duan, Y, and He, Y. The NLRP3 Inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:3328. doi: 10.3390/ijms20133328
36. Bauernfeind, FG, Horvath, G, Stutz, A, Alnemri, ES, MacDonald, K, Speert, D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
37. Qiao, Y, Wang, P, Qi, J, Zhang, L, and Gao, C. TLR-induced NF-κB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. (2012) 586:1022–6. doi: 10.1016/j.febslet.2012.02.045
38. Martinon, F, Pétrilli, V, Mayor, A, Tardivel, A, and Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. (2006) 440:237–41. doi: 10.1038/nature04516
39. Hornung, V, Bauernfeind, F, Halle, A, Samstad, EO, Kono, H, Rock, KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. (2008) 9:847–56. doi: 10.1038/ni.1631
40. Dostert, C, Pétrilli, V, Van Bruggen, R, Steele, C, Mossman, BT, and Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. (2008) 320:674–7. doi: 10.1126/science.1156995
41. Juliana, C, Fernandes-Alnemri, T, Kang, S, Farias, A, Qin, F, and Alnemri, ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. (2012) 287:36617–22. doi: 10.1074/jbc.M112.407130
42. Py, BF, Kim, MS, Vakifahmetoglu-Norberg, H, and Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. (2013) 49:331–8. doi: 10.1016/j.molcel.2012.11.009
43. Song, N, Liu, ZS, Xue, W, Bai, ZF, Wang, QY, Dai, J, et al. NLRP3 phosphorylation is an essential priming event for Inflammasome activation. Mol Cell. (2017) 68:185–197.e6. doi: 10.1016/j.molcel.2017.08.017
44. Wang, W, Ding, XQ, Gu, TT, Song, L, Li, JM, Xue, QC, et al. Pterostilbene and allopurinol reduce fructose-induced podocyte oxidative stress and inflammation via microRNA-377. Free Radic Biol Med. (2015) 83:214–26. doi: 10.1016/j.freeradbiomed.2015.02.029
45. Boursereau, R, Abou-Samra, M, Lecompte, S, Noel, L, and Brichard, SM. Downregulation of the NLRP3 inflammasome by adiponectin rescues Duchenne muscular dystrophy. BMC Biol. (2018) 16:33. doi: 10.1186/s12915-018-0501-z
46. Feng, X, Luo, Q, Wang, H, Zhang, H, and Chen, F. MicroRNA-22 suppresses cell proliferation, migration and invasion in oral squamous cell carcinoma by targeting NLRP3. J Cell Physiol. (2018) 233:6705–13. doi: 10.1002/jcp.26331
47. Haneklaus, M, Gerlic, M, Kurowska-Stolarska, M, Rainey, AA, Pich, D, McInnes, IB, et al. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1β production. J Immunol. (2012) 189:3795–9. doi: 10.4049/jimmunol.1200312
48. Li, D, Yang, H, Ma, J, Luo, S, Chen, S, and Gu, Q. MicroRNA-30e regulates neuroinflammation in MPTP model of Parkinson's disease by targeting Nlrp3. Hum Cell. (2018) 31:106–15. doi: 10.1007/s13577-017-0187-5
49. Wu, X, Chang, SC, Jin, J, Gu, W, and Li, S. NLRP3 inflammasome mediates chronic intermittent hypoxia-induced renal injury implication of the microRNA-155/FOXO3a signaling pathway. J Cell Physiol. (2018) 233:9404–15. doi: 10.1002/jcp.26784
50. Wu, H, Huang, T, Ying, L, Han, C, Li, D, Xu, Y, et al. MiR-155 is involved in renal ischemia-reperfusion injury via direct targeting of FoxO3a and regulating renal tubular cell Pyroptosis. Cell Physiol Biochem. (2016) 40:1692–705. doi: 10.1159/000453218
51. Shi, J, Zhao, Y, Wang, Y, Gao, W, Ding, J, Li, P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. (2014) 514:187–92. doi: 10.1038/nature13683
52. Casson, CN, Yu, J, Reyes, VM, Taschuk, FO, Yadav, A, Copenhaver, AM, et al. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc Natl Acad Sci U S A. (2015) 112:6688–93. doi: 10.1073/pnas.1421699112
53. Baker, PJ, Boucher, D, Bierschenk, D, Tebartz, C, Whitney, PG, D'Silva, DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol. (2015) 45:2918–26. doi: 10.1002/eji.201545655
54. Viganò, E, Diamond, CE, Spreafico, R, Balachander, A, Sobota, RM, and Mortellaro, A. Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun. (2015) 6:8761. doi: 10.1038/ncomms9761
55. Mulvihill, E, Sborgi, L, Mari, SA, Pfreundschuh, M, Hiller, S, and Müller, DJ. Mechanism of membrane pore formation by human gasdermin-D. EMBO J. (2018) 37:e98321. doi: 10.15252/embj.201798321
56. Bibo-Verdugo, B, Snipas, SJ, Kolt, S, Poreba, M, and Salvesen, GS. Extended subsite profiling of the pyroptosis effector protein gasdermin D reveals a region recognized by inflammatory caspase-11. J Biol Chem. (2020) 295:11292–302. doi: 10.1074/jbc.RA120.014259
57. Ramirez, MLG, Poreba, M, Snipas, SJ, Groborz, K, Drag, M, and Salvesen, GS. Extensive peptide and natural protein substrate screens reveal that mouse caspase-11 has much narrower substrate specificity than caspase-1. J Biol Chem. (2018) 293:7058–67. doi: 10.1074/jbc.RA117.001329
58. Zhao, H, Alam, A, Soo, AP, George, AJT, and Ma, D. Ischemia-reperfusion injury reduces long term renal graft survival: mechanism and beyond. EBioMedicine. (2018) 28:31–42. doi: 10.1016/j.ebiom.2018.01.025
59. Situmorang, GR, and Sheerin, NS. Ischaemia reperfusion injury: mechanisms of progression to chronic graft dysfunction. Pediatr Nephrol. (2019) 34:951–63. doi: 10.1007/s00467-018-3940-4
60. Su, X, Liu, B, Wang, S, Wang, Y, Zhang, Z, Zhou, H, et al. NLRP3 inflammasome: a potential therapeutic target to minimize renal ischemia/reperfusion injury during transplantation. Transpl Immunol. (2022) 75:101718. doi: 10.1016/j.trim.2022.101718
61. Wen, Y, Liu, YR, Tang, TT, Pan, MM, Xu, SC, Ma, KL, et al. mROS-TXNIP axis activates NLRP3 inflammasome to mediate renal injury during ischemic AKI. Int J Biochem Cell Biol. (2018) 98:43–53. doi: 10.1016/j.biocel.2018.02.015
62. Su, Y, Wang, Y, Liu, M, and Chen, H. Hydrogen sulfide attenuates renal I/R-induced activation of the inflammatory response and apoptosis via regulating Nrf2-mediated NLRP3 signaling pathway inhibition. Mol Med Rep. (2021) 24:518. doi: 10.3892/mmr.2021.12157
63. Jain, S, Plenter, R, Jeremy, R, Nydam, T, Gill, RG, and Jani, A. The impact of Caspase-1 deletion on apoptosis and acute kidney injury in a murine transplant model. Cell Signal. (2021) 85:110039. doi: 10.1016/j.cellsig.2021.110039
64. Zheng, Z, Xu, K, Li, C, Qi, C, Fang, Y, Zhu, N, et al. NLRP3 associated with chronic kidney disease progression after ischemia/reperfusion-induced acute kidney injury. Cell Death Discov. (2021) 7:324. doi: 10.1038/s41420-021-00719-2
65. Lavallard, V, Cottet-Dumoulin, D, Wassmer, CH, Rouget, C, Parnaud, G, Brioudes, E, et al. NLRP3 Inflammasome is activated in rat pancreatic islets by transplantation and hypoxia. Sci Rep. (2020) 10:7011. doi: 10.1038/s41598-020-64054-9
66. Shigeoka, AA, Mueller, JL, Kambo, A, Mathison, JC, King, AJ, Hall, WF, et al. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J Immunol. (2010) 185:6277–85. doi: 10.4049/jimmunol.1002330
67. Park, M, Kwon, CH, Ha, HK, Han, M, and Song, SH. RNA-Seq identifies condition-specific biological signatures of ischemia-reperfusion injury in the human kidney. BMC Nephrol. (2020) 21:398. doi: 10.1186/s12882-020-02025-y
68. Guo, Y, Zhang, J, Lai, X, Chen, M, and Guo, Y. Tim-3 exacerbates kidney ischaemia/reperfusion injury through the TLR-4/NF-κB signalling pathway and an NLR-C4 inflammasome activation. Clin Exp Immunol. (2018) 193:113–29. doi: 10.1111/cei.13126
69. Hall, IE, Yarlagadda, SG, Coca, SG, Wang, Z, Doshi, M, Devarajan, P, et al. IL-18 and urinary NGAL predict dialysis and graft recovery after kidney transplantation. J Am Soc Nephrol. (2010) 21:189–97. doi: 10.1681/ASN.2009030264
70. McGuinness, D, Mohammed, S, Monaghan, L, Wilson, PA, Kingsmore, DB, Shapter, O, et al. A molecular signature for delayed graft function. Aging Cell. (2018) 17:e12825. doi: 10.1111/acel.12825
71. Port, FK, Bragg-Gresham, JL, Metzger, RA, Dykstra, DM, Gillespie, BW, Young, EW, et al. Donor characteristics associated with reduced graft survival: an approach to expanding the pool of kidney donors. Transplantation. (2002) 74:1281–6. doi: 10.1097/00007890-200211150-00014
72. Merion, RM, Ashby, VB, Wolfe, RA, Distant, DA, Hulbert-Shearon, TE, Metzger, RA, et al. Deceased-donor characteristics and the survival benefit of kidney transplantation. JAMA. (2005) 294:2726–33. doi: 10.1001/jama.294.21.2726
73. Mazeti-Felicio, CM, Caldas, HC, Fernandes-Charpiot, IMM, Dezotti, CZ, Baptista, MASF, and Abbud-Filho, M. Preimplantation kidney biopsies of extended criteria donors have a heavier inflammatory burden than kidneys from standard criteria donors. Transplant Direct. (2017) 3:e180. doi: 10.1097/TXD.0000000000000671
74. Wang, XW, Guo, RD, Ma, JG, Wang, YW, and Zou, XF. Effects of prolonged cold ischemia on the DCD kidney function and Inflammasome expression in rat kidney transplants. Transpl Immunol. (2022) 74:101511. doi: 10.1016/j.trim.2021.101511
75. Florim, GMS, Caldas, HC, Gonçalves, NN, Bueno, GOBE, Baptista, MASF, Fernandes-Charpiot, IMM, et al. Activation of HMGB1-TLR4 pathway and Inflammasome contribute to enhanced inflammatory response in extended criteria and kidneys with KDPI ≥85. Transplantation. (2020) 104:724–30. doi: 10.1097/TP.0000000000003048
76. Thompson, ER, Bates, L, Ibrahim, IK, Sewpaul, A, Stenberg, B, McNeill, A, et al. Novel delivery of cellular therapy to reduce ischemia reperfusion injury in kidney transplantation. Am J Transplant. (2021) 21:1402–14. doi: 10.1111/ajt.16100
77. Yin, F, Zheng, PQ, Zhao, LQ, Wang, YZ, Miao, NJ, Zhou, ZL, et al. Caspase-11 promotes NLRP3 inflammasome activation via the cleavage of pannexin1 in acute kidney disease. Acta Pharmacol Sin. (2022) 43:86–95. doi: 10.1038/s41401-021-00619-2
78. Yang, JR, Yao, FH, Zhang, JG, Ji, ZY, Li, KL, Zhan, J, et al. Ischemia-reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am J Physiol Renal Physiol. (2014) 306:F75–84. doi: 10.1152/ajprenal.00117.2013
79. Zou, XF, Gu, JH, Duan, JH, Hu, ZD, and Cui, ZL. The NLRP3 inhibitor Mcc950 attenuates acute allograft damage in rat kidney transplants. Transpl Immunol. (2020) 61:101293. doi: 10.1016/j.trim.2020.101293
80. Eastman, RT, Roth, JS, Brimacombe, KR, Simeonov, A, Shen, M, Patnaik, S, et al. Remdesivir: a review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent Sci. (2020) 6:672–83. doi: 10.1021/acscentsci.0c00489
81. Yin, L, Zhao, H, Zhang, H, Li, Y, Dong, Y, Ju, H, et al. Remdesivir alleviates acute kidney injury by inhibiting the activation of NLRP3 Inflammasome. Front Immunol. (2021) 12:652446. doi: 10.3389/fimmu.2021.652446
82. Yang, H, Wu, Y, Cheng, M, Zhang, M, Qiu, X, Liu, S, et al. Roxadustat (FG-4592) protects against ischaemia-induced acute kidney injury via improving CD73 and decreasing AIM2 inflammasome activation. Nephrol Dial Transplant. (2023) 38:858–75. doi: 10.1093/ndt/gfac308
83. Feng, J, Lu, X, Li, H, and Wang, S. The roles of hydrogen sulfide in renal physiology and disease states. Ren Fail. (2022) 44:1289–308. doi: 10.1080/0886022X.2022.2107936
84. Shen, M, Wang, S, Wen, X, Han, XR, Wang, YJ, Zhou, XM, et al. Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed Pharmacother. (2017) 95:885–93. doi: 10.1016/j.biopha.2017.08.125
85. Kutanis, D, Erturk, E, Besir, A, Demirci, Y, Kayir, S, Akdogan, A, et al. Dexmedetomidine acts as an oxidative damage prophylactic in rats exposed to ionizing radiation. J Clin Anesth. (2016) 34:577–85. doi: 10.1016/j.jclinane.2016.06.031
86. Wang, Y, Mao, X, Chen, H, Feng, J, Yan, M, Wang, Y, et al. Dexmedetomidine alleviates LPS-induced apoptosis and inflammation in macrophages by eliminating damaged mitochondria via PINK1 mediated mitophagy. Int Immunopharmacol. (2019) 73:471–81. doi: 10.1016/j.intimp.2019.05.027
87. Yang, T, Feng, X, Zhao, Y, Zhang, H, Cui, H, Wei, M, et al. Dexmedetomidine enhances autophagy via α2-AR/AMPK/mTOR pathway to inhibit the activation of NLRP3 Inflammasome and subsequently alleviates lipopolysaccharide-induced acute kidney injury. Front Pharmacol. (2020) 11:790. doi: 10.3389/fphar.2020.00790
88. Chen, J, Perez, R, de Mattos, AM, Wang, C, Li, Z, Applegate, RL 2nd, et al. Perioperative Dexmedetomidine improves outcomes of kidney transplant. Clin Transl Sci. (2020) 13:1279–87. doi: 10.1111/cts.12826
89. Wlodek, E, Kirkpatrick, RB, Andrews, S, Noble, R, Schroyer, R, Scott, J, et al. A pilot study evaluating GSK1070806 inhibition of interleukin-18 in renal transplant delayed graft function. PLoS One. (2021) 16:e0247972. doi: 10.1371/journal.pone.0247972
90. Franchon Marques Tejada, N, Ziroldo Lopes, JV, Duarte Gonçalves, LE, Andrade, MC, da Conceição, I, Franco, GR, et al. AIM2 as a putative target in acute kidney graft rejection. Front Immunol. (2022) 13:839359. doi: 10.3389/fimmu.2022.839359
91. Venner, JM, Hidalgo, LG, Famulski, KS, Chang, J, and Halloran, PF. The molecular landscape of antibody-mediated kidney transplant rejection: evidence for NK involvement through CD16a fc receptors. Am J Transplant. (2015) 15:1336–48. doi: 10.1111/ajt.13115
92. Venner, JM, Famulski, KS, Badr, D, Hidalgo, LG, Chang, J, and Halloran, PF. Molecular landscape of T cell-mediated rejection in human kidney transplants: prominence of CTLA4 and PD ligands. Am J Transplant. (2014) 14:2565–76. doi: 10.1111/ajt.12946
93. Asgari, E, Le Friec, G, Yamamoto, H, Perucha, E, Sacks, SS, Köhl, J, et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. (2013) 122:3473–81. doi: 10.1182/blood-2013-05-502229
94. Knight, SR, Thorne, A, and Lo Faro, ML. Donor-specific cell-free DNA as a biomarker in solid organ transplantation. A systematic review. Transplantation. (2019) 103:273–83. doi: 10.1097/TP.0000000000002482
95. Dholakia, S, De Vlaminck, I, and Khush, KK. Adding insult on injury: immunogenic role for donor-derived cell-free DNA? Transplantation. (2020) 104:2266–71. doi: 10.1097/TP.0000000000003240
96. Decout, A, Katz, JD, Venkatraman, S, and Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. (2021) 21:548–69. doi: 10.1038/s41577-021-00524-z
97. Abdul-Sater, AA, Tattoli, I, Jin, L, Grajkowski, A, Levi, A, Koller, BH, et al. Cyclic-di-GMP and cyclic-di-AMP activate the NLRP3 inflammasome. EMBO Rep. (2013) 14:900–6. doi: 10.1038/embor.2013.132
98. Dessing, MC, Kers, J, Damman, J, Navis, GJ, Florquin, S, and Leemans, JC. Donor and recipient genetic variants in NLRP3 associate with early acute rejection following kidney transplantation. Sci Rep. (2016) 6:36315. doi: 10.1038/srep36315
99. Smatti, MK, Al-Sadeq, DW, Ali, NH, Pintus, G, Abou-Saleh, H, and Nasrallah, GK. Epstein-Barr virus epidemiology, serology, and genetic variability of LMP-1 oncogene among healthy population: an update. Front Oncol. (2018) 8:211. doi: 10.3389/fonc.2018.00211
100. Caillard, S, Lelong, C, Pessione, F, and Moulin, B, French PTLD Working Group. Post-transplant lymphoproliferative disorders occurring after renal transplantation in adults: report of 230 cases from the French registry. Am J Transplant. (2006) 6:2735–42. doi: 10.1111/j.1600-6143.2006.01540.x
101. Starzl, TE, Nalesnik, MA, Porter, KA, Ho, M, Iwatsuki, S, Griffith, BP, et al. Reversibility of lymphomas and lymphoproliferative lesions developing under cyclosporin-steroid therapy. Lancet. (1984) 1:583–7. doi: 10.1016/S0140-6736(84)90994-2
102. Le, J, Durand, CM, Agha, I, and Brennan, DC. Epstein-Barr virus and renal transplantation. Transplant Rev (Orlando). (2017) 31:55–60. doi: 10.1016/j.trre.2016.12.001
103. Ansari, MA, Singh, VV, Dutta, S, Veettil, MV, Dutta, D, Chikoti, L, et al. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J Virol. (2013) 87:8606–23. doi: 10.1128/JVI.00805-13
104. Xu, H, Li, X, Rousseau, BA, Akinyemi, IA, Frey, TR, Zhou, K, et al. IFI16 partners with KAP1 to maintain Epstein-Barr virus latency. J Virol. (2022) 96:e0102822. doi: 10.1128/jvi.01028-22
105. Li, X, Burton, EM, Koganti, S, Zhi, J, Doyle, F, Tenenbaum, SA, et al. KRAB-ZFP repressors enforce quiescence of oncogenic human herpesviruses. J Virol. (2018) 92:e00298–18. doi: 10.1128/JVI.00298-18
106. Burton, EM, Goldbach-Mansky, R, and Bhaduri-McIntosh, S. A promiscuous inflammasome sparks replication of a common tumor virus. Proc Natl Acad Sci U S A. (2020) 117:1722–30. doi: 10.1073/pnas.1919133117
107. Campistol, JM, and Schena, FP. Kaposi's sarcoma in renal transplant recipients--the impact of proliferation signal inhibitors. Nephrol Dial Transplant. (2007) 22:i17–22. doi: 10.1093/ndt/gfm089
108. Bechtel, JT, Liang, Y, Hvidding, J, and Ganem, D. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J Virol. (2003) 77:6474–81. doi: 10.1128/JVI.77.11.6474-6481.2003
109. Cesarman, E, Damania, B, Krown, SE, Martin, J, Bower, M, and Whitby, D. Kaposi sarcoma. Nat Rev Dis Primers. (2019) 5:9. doi: 10.1038/s41572-019-0060-9
110. Kumar, B, Roy, A, Veettil, MV, and Chandran, B. Insight into the roles of E3 ubiquitin ligase c-Cbl, ESCRT machinery, and host cell signaling in Kaposi's sarcoma-associated herpesvirus entry and trafficking. J Virol. (2018) 92:e01376–17. doi: 10.1128/JVI.01376-17
111. Kumar, B, and Chandran, B. KSHV entry and trafficking in target cells-hijacking of cell signal pathways, actin and membrane dynamics. Viruses. (2016) 8:305. doi: 10.3390/v8110305
112. West, J, and Damania, B. Upregulation of the TLR3 pathway by Kaposi's sarcoma-associated herpesvirus during primary infection. J Virol. (2008) 82:5440–9. doi: 10.1128/JVI.02590-07
113. Lagos, D, Vart, RJ, Gratrix, F, Westrop, SJ, Emuss, V, Wong, PP, et al. Toll-like receptor 4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell Host Microbe. (2008) 4:470–83. doi: 10.1016/j.chom.2008.09.012
114. West, JA, Gregory, SM, Sivaraman, V, Su, L, and Damania, B. Activation of plasmacytoid dendritic cells by Kaposi's sarcoma-associated herpesvirus. J Virol. (2011) 85:895–904. doi: 10.1128/JVI.01007-10
115. Gregory, SM, Davis, BK, West, JA, Taxman, DJ, Matsuzawa, S, Reed, JC, et al. Discovery of a viral NLR homolog that inhibits the inflammasome. Science. (2011) 331:330–4. doi: 10.1126/science.1199478
116. Hopcraft, SE, and Damania, B. Tumour viruses and innate immunity. Philos Trans R Soc Lond Ser B Biol Sci. (2017) 372:20160267. doi: 10.1098/rstb.2016.0267
117. Zhang, X, Lan, Q, Zhang, M, Wang, F, Shi, K, Li, X, et al. Inhibition of AIM2 inflammasome activation by SOX/ORF37 promotes lytic replication of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. (2023) 120:e2300204120. doi: 10.1073/pnas.2300204120
118. Knowles, WA, Pipkin, P, Andrews, N, Vyse, A, Minor, P, Brown, DW, et al. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol. (2003) 71:115–23. doi: 10.1002/jmv.10450
119. Shen, CL, Wu, BS, Lien, TJ, Yang, AH, and Yang, CY. BK polyomavirus nephropathy in kidney transplantation: balancing rejection and infection. Viruses. (2021) 13:487. doi: 10.3390/v13030487
120. Ribeiro, A, Wörnle, M, Motamedi, N, Anders, HJ, Gröne, EF, Nitschko, H, et al. Activation of innate immune defense mechanisms contributes to polyomavirus BK-associated nephropathy. Kidney Int. (2012) 81:100–11. doi: 10.1038/ki.2011.311
121. Stokman, G, Kers, J, Yapici, Ü, Hoelbeek, JJ, Claessen, N, de Boer, OJ, et al. Predominant tubular Interleukin-18 expression in polyomavirus-associated nephropathy. Transplantation. (2016) 100:e88–95. doi: 10.1097/TP.0000000000001086
122. Guarnieri, JW, Angelin, A, Murdock, DG, Schaefer, P, Portluri, P, Lie, T, et al. SARS-COV-2 viroporins activate the NLRP3-inflammasome by the mitochondrial permeability transition pore. Front Immunol. (2023) 14:1064293. doi: 10.3389/fimmu.2023.1064293
123. Gandolfini, I, Delsante, M, Fiaccadori, E, Zaza, G, Manenti, L, Degli Antoni, A, et al. COVID-19 in kidney transplant recipients. Am J Transplant. (2020) 20:1941–3. doi: 10.1111/ajt.15891
124. Fernández-Ruiz, M, and Aguado, JM. Immunomodulatory therapies for COVID-19 in solid organ transplant recipients. Curr Transplant Rep. (2020) 7:379–89. doi: 10.1007/s40472-020-00306-x
125. Verma, V, Kumar, P, Gupta, S, Yadav, S, Dhanda, RS, Thorlacius, H, et al. α-Hemolysin of uropathogenic E. coli regulates NLRP3 inflammasome activation and mitochondrial dysfunction in THP-1 macrophages. Sci Rep. (2020) 10:12653. doi: 10.1038/s41598-020-69501-1
126. Allen, PJ, Chadban, SJ, Craig, JC, Lim, WH, Allen, RDM, Clayton, PA, et al. Recurrent glomerulonephritis after kidney transplantation: risk factors and allograft outcomes. Kidney Int. (2017) 92:461–9. doi: 10.1016/j.kint.2017.03.015
127. Wu, X, Zhao, L, Li, K, and Yang, J. The role of NLRP3 Inflammasome in IgA nephropathy. Medicina (Kaunas). (2022) 59:82. doi: 10.3390/medicina59010082
128. Peng, W, Pei, GQ, Tang, Y, Tan, L, and Qin, W. IgA1 deposition may induce NLRP3 expression and macrophage transdifferentiation of podocyte in IgA nephropathy. J Transl Med. (2019) 17:406. doi: 10.1186/s12967-019-02157-2
129. Zhang, L, Wang, XZ, Li, YS, Zhang, L, and Hao, LR. Icariin ameliorates IgA nephropathy by inhibition of nuclear factor kappa b/Nlrp3 pathway. FEBS Open Bio. (2016) 7:54–63. doi: 10.1002/2211-5463.12161
130. Hua, KF, Yang, SM, Kao, TY, Chang, JM, Chen, HL, Tsai, YJ, et al. Osthole mitigates progressive IgA nephropathy by inhibiting reactive oxygen species generation and NF-κB/NLRP3 pathway. PLoS One. (2013) 8:e77794. doi: 10.1371/journal.pone.0077794
131. Hu, H, Li, M, Chen, B, Guo, C, and Yang, N. Activation of necroptosis pathway in podocyte contributes to the pathogenesis of focal segmental glomerular sclerosis. Clin Exp Nephrol. (2022) 26:1055–66. doi: 10.1007/s10157-022-02258-1
132. Hultgren, O, Andersson, B, Hahn-Zoric, M, and Almroth, G. Serum concentration of interleukin-18 is up-regulated in patients with ANCA-associated vasculitis. Autoimmunity. (2007) 40:529–31. doi: 10.1080/08916930701622783
133. Wang, LY, Sun, XJ, Chen, M, and Zhao, MH. The expression of NOD2, NLRP3 and NLRC5 and renal injury in anti-neutrophil cytoplasmic antibody-associated vasculitis. J Transl Med. (2019) 17:197. doi: 10.1186/s12967-019-1949-5
Keywords: inflammasome, kidney transplantation, NLRP3, post-transplant complications, immune response
Citation: Granata S, La Russa D, Stallone G, Perri A and Zaza G (2023) Inflammasome pathway in kidney transplantation. Front. Med. 10:1303110. doi: 10.3389/fmed.2023.1303110
Edited by:
Gian Marco Ghiggeri, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Pasquale Esposito, University of Genoa, ItalyCopyright © 2023 Granata, La Russa, Stallone, Perri and Zaza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gianluigi Zaza, gianluigi.zaza@unifg.it
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.