 Hasan Zulfiqar1†
Hasan Zulfiqar1† Zhiling Guo2†Ramala Masood Ahmad3
Zhiling Guo2†Ramala Masood Ahmad3 Zahoor Ahmed1
Zahoor Ahmed1 Peiling Cai4Xiang Chen1
Peiling Cai4Xiang Chen1 Yang Zhang5*
Yang Zhang5* Hao Lin1*
Hao Lin1* Zheng Shi6*
Zheng Shi6*- 1Yangtze Delta Region Institute (Huzhou), University of Electronic Science and Technology of China, Huzhou, Zhejiang, China
- 2Beidahuang Industry Group General Hospital, Harbin, China
- 3Department of Plant Breeding and Genetics, University of Agriculture Faisalabad, Faisalabad, Pakistan
- 4School of Basic Medical Sciences, Chengdu University, Chengdu, China
- 5Innovative Institute of Chinese Medicine and Pharmacy, Academy for Interdiscipline, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 6Clinical Genetics Laboratory, Clinical Medical College & Affiliated Hospital, Chengdu University, Chengdu, China
Snake venom contains many toxic proteins that can destroy the circulatory system or nervous system of prey. Studies have found that these snake venom proteins have the potential to treat cardiovascular and nervous system diseases. Therefore, the study of snake venom protein is conducive to the development of related drugs. The research technologies based on traditional biochemistry can accurately identify these proteins, but the experimental cost is high and the time is long. Artificial intelligence technology provides a new means and strategy for large-scale screening of snake venom proteins from the perspective of computing. In this paper, we developed a sequence-based computational method to recognize snake toxin proteins. Specially, we utilized three different feature descriptors, namely g-gap, natural vector and word 2 vector, to encode snake toxin protein sequences. The analysis of variance (ANOVA), gradient-boost decision tree algorithm (GBDT) combined with incremental feature selection (IFS) were used to optimize the features, and then the optimized features were input into the deep learning model for model training. The results show that our model can achieve a prediction performance with an accuracy of 82.00% in 10-fold cross-validation. The model is further verified on independent data, and the accuracy rate reaches to 81.14%, which demonstrated that our model has excellent prediction performance and robustness.
1 Introduction
Snake venom is a mixture of toxin proteins and other chemical molecules, which acts on the blood circulation system, nervous system or motion system of prey. It can make the prey lose resistance, and then achieve the purpose of predation. Many toxin enzymes have been isolated from snake venoms, such as serine proteinases, metalloproteinase and L-amino acid oxidases, which can interrupt the blood circulatory system, leading to blood clotting and heart failure. Moreover, the scientists found that the primary toxins of Pseudechis australis venom with antibacterial activity were phospholipases A2 and L-amino acid oxidases. The L-amino acid oxidase discovered in the venom of Crotalus adamanteus was the first pure toxin tested against bacteria. Since then, crude snake venom, portions of it, or refined components have all shown antibacterial activity. The mechanism of anti-microbial activity of snake toxin proteins is shown in Figure 1.
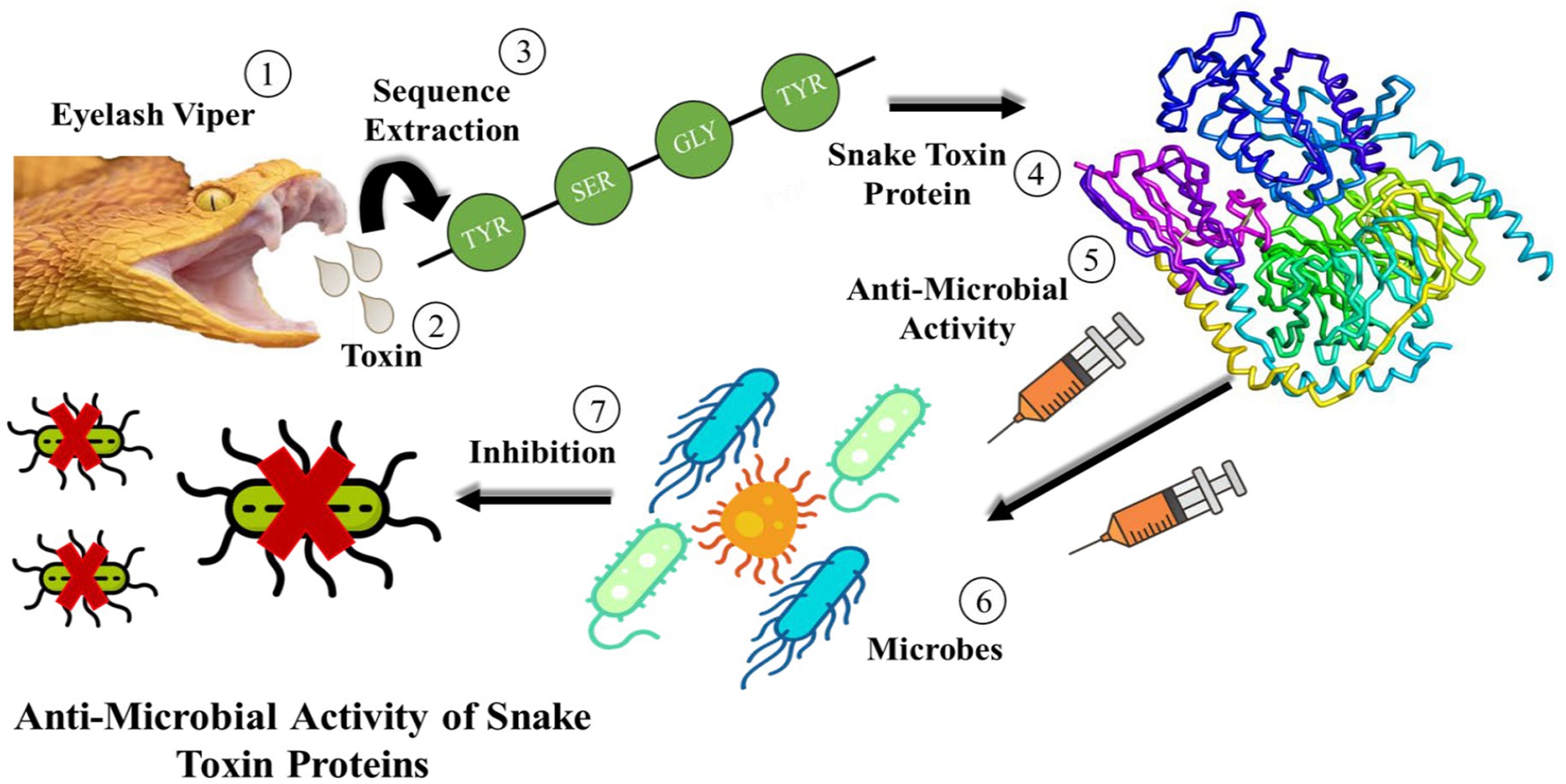
Figure 1. Schematic diagram of the anti-microbial activities of snake toxin proteins.
Many toxin proteins were found in snake venom, such as phospholipases A2, cysteine-rich secretory proteins (CRISP), α-dendrotoxins, β-dendrotoxins and γ-dendrotoxins which could interact with nervous system or molecules in nervous system (1, 2). Scientists have also obtained some venomous proteins, for example, three finger α-neurotoxins (α-3FNTx) and acetylcholine esterase proteins, which target motion system of prey and cause paralysis (3). Surprisingly, the components extracted from snakes can be used as drugs to cure various diseases (4). At present, scientists have extracted several drugs from snake toxin proteins for the treatment of heart related syndromes. For example, captopril is now used to treat hypertension and reduce the risk of heart failure after the heart attack (5). Therefore, the correct identification of snake venom protein is very important for the study of drug development based on snake venom. Biochemical technologies are complicated, tedious and expensive. Thus, there is an urgent need to develop bioinformatic tools that can precisely identify snake toxins in a short time. Current bioinformatic tools, such as FASTA (6), HAlign (7, 8) and BLAST (9) can search for similar sequences with the help of known protein databases. However, in the absence of homologous sequences in benchmark dataset, these computational tools cannot correctly recognize snake toxin proteins. Therefore, it is essential to establish a computational tool to recognize snake toxin proteins.
To fill the gap, we proposed the first predictor named Deep-STP based on deep learning to recognize snake toxin proteins. The graphical illustration of the entire study was shown in Figure 2. First, the snake toxin protein sequences were encoded by three different kinds of descriptors, namely, word to vector (10), g-gap and natural vector (11). Subsequently, the feature set was optimized by combining ANOVA (11) and GBDT (12) with IFS procedure. By inputting the optimal feature into deep learning, the snake toxin proteins can be recognized. The performance of the anticipated model was evaluated by 10-fold CV and independent data.
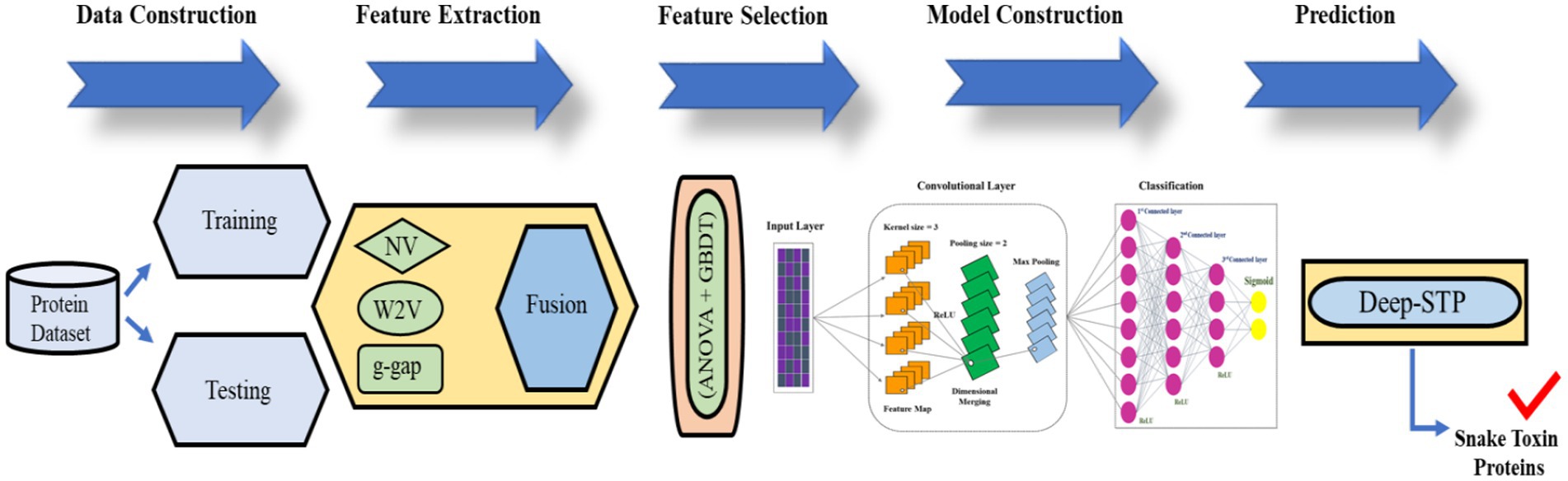
Figure 2. The graphical illustration of the entire study.
2 Materials and methods
A real and reliable data is crucial for the establishment of prediction model. In this work, positive and negative samples were collected from open-source database UniProt (13) and RefSeq (14). We have excluded the similar sequences using 80% as cutoff of sequence identity (15). After the elimination process, we finally obtained the dataset of 270 positive and 339 negative sequences of the prominent protein families of snake toxin. Subsequently, the data were separated into 80% training data and 20% independent data to objectively estimate the efficiencies and performances of the models, as shown in Supplementary Table S1.
2.1 Feature descriptors
It is an important step for protein function prediction to express the sequence information with effective mathematical descriptors (16). Here, three kinds of feature descriptors were used to encode the snake toxin protein sequences.
2.1.1 g-gap dipeptide composition
The relationship between the two end-to-end 2-D amino acid residues can be expressed using this feature encoding approach. Consequently, important links between two residues are found using g-gap dipeptide composition. Thus, a protein ‘F’ can be described as
where ‘t’ is the transposition vector and is the i-th occurrence of g-gap dipeptide which is define as
where ‘p’ is the number of amino acid residues, is the i-th value number of g-gap and ‘L’ is the length of ‘F’ protein.
2.1.2 Natural vector
As a starting point for phylogenetic and evolutionary study, the natural vector scheme (NV) was created by Deng et al. (17). Here, we have also used NV to formulate the samples. A 60-dimensional vector can be created using this approach to plot biological sequences. The NV scheme has a significant ability to classify proteins because it has no parameters (18).
Let us say a protein ‘P’ with a length of ‘L’ residues can be expressed as.
where Qi (i = (1, 2, … L)) indicates the i-th amino acid of protein ‘P’. The NV is expressed as.
wk (.): (A, C, D, E…W, Y) → (0,1).where wk (Qi) = 1, if Qi = k. otherwise, wk (Qi) = 0.
In protein ‘P’, mk is the number of k-th amino acid which can be computed as
Let T(k)(i) is the gap between the first and i-th amino acid, ηk is the mean of the amino acids k and Sk is the overall distance which is shown in equation (5).
Let ‘ ’ is the 2nd order regularized moment, which is computed as
Thus, ‘P’ can be termed as
where ‘T’ is the vector transposition.
2.1.3 Word2Vector
The ‘word2vector’ (W2V) is a NLP (Natural language processing) technique which has the ability to utilize neural networks to produce illustrations of the distribution of words (19, 20). In this method, word embeddings are utilized to illustrate of words. Indeed, the vectors which have the ability to encode the words closer in the vector space are supposed to be an identical meaning. The ‘word2vector’ consists of two different kinds of models, namely, continuous bag of words (21) and the other one is continuous skip gram (22). The main idea of the continuous skip gram is to utilize the words to predict its adjoining words (23). The quantified intelligence of continuous bag of words uses context words from a nearby booth to predict words. The continuous bag of words model structure logically implies the advantage of consistently condensing the scattered information in the data. Thus, in this work, we employed the continuous bag of words to train the appropriate resemblance of protein sequences. The dimension of the word2vector embedding is 200.
2.2 Feature selection
The redundancy in the feature vectors can produce unsatisfactory performance (24). Therefore, selecting the ideal features is a significant step to eliminate the irrelevant features and enhance the efficiency of the model (25). There are many feature selection and ranking methods to optimize the features, such as ANOVA (26, 27), F-score (28), mRMR (29), GBDT and LGBM (12). ANOVA is a reputable choice to overcome these complications, because it takes short time and yield effective outcomes. The merging of top-performing features does not guarantee that the best outcomes can be achieved. These features are conceivably to have a higher level of redundancy, which leads to another unnecessary knowledge in the feature. Hence, GBDT is an ideal choice to conquer these hitches. In this work, ANOVA and GBDT with IFS were employed to achieve the best feature subset which could produce the maximum accuracy. The whole procedure for feature selection has been already elucidated in our previous study (12). The prediction accuracy of models constructed with different numbers of features and contribution of feature descriptors have been shown in Figures 3A,B.
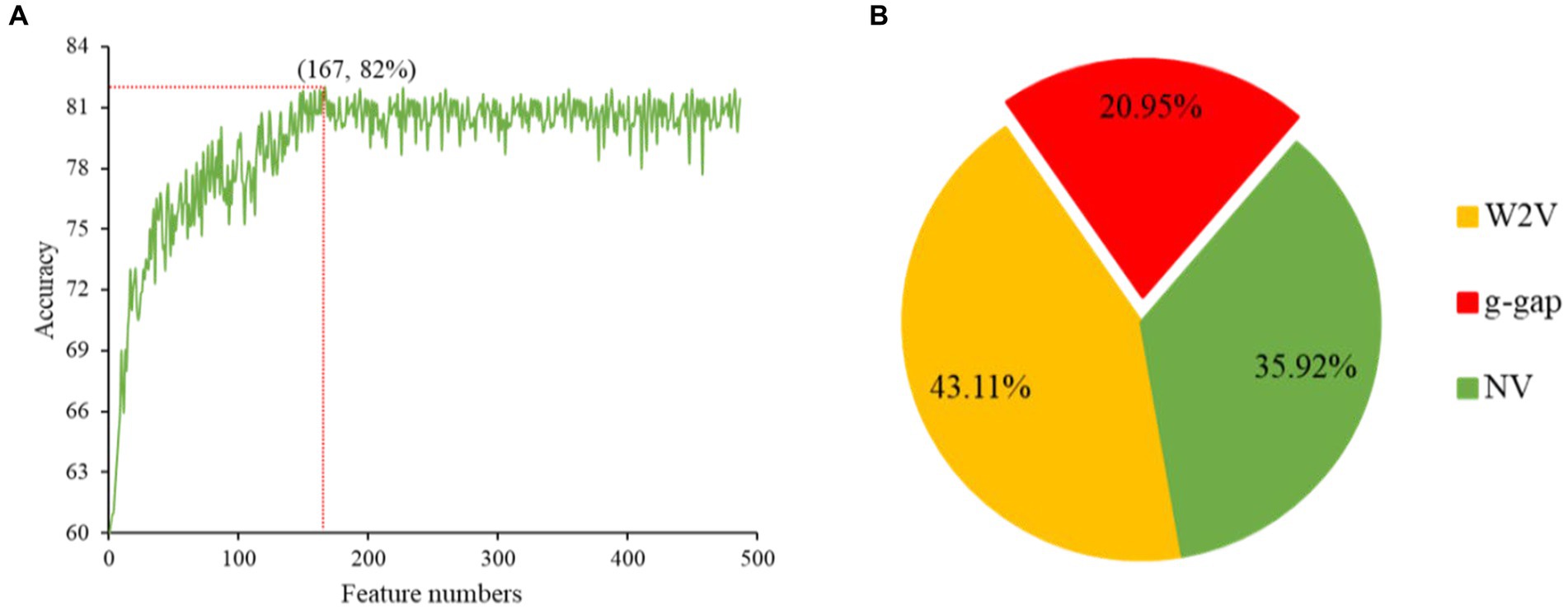
Figure 3. The prediction accuracy of models constructed with different numbers of features (A). Contribution of descriptors in CNN-based fusion model to classify snake toxin proteins (B).
2.3 Convolutional neural network
Convolutional neural networks (CNN) was first developed by LeCun et al. (30) and are now largely used in the developments of biology and bioinformatics (31). The core idea behind CNN is to use layer-wise convolutions and pooling techniques to build a large number of filters that can extract hidden topological properties from input. The performance of CNN on 2-D image and matrix data has been excellent (32). Moreover, 1-D CNN has been utilized to overcome the natural language processing and biomedical sequence data recognition problems (33). In this work, we executed 1-D CNN to recognize snake toxin proteins. We utilized Keras 2.3.1 (34), Python 3.5.4 and Tensor Flow 2.1.0 to execute this experimentation.
2.4 Metrics evaluation
Accuracy, precision, recall and F1-score (35) were used to assess the efficiency of the projected model and can be expressed as
where ‘TP’ represents the truly predicted snake toxin protein sequences and ‘FP’ indicates the non-snake toxin protein sequences predicted as snake toxin protein sequence. ‘TN’ symbolizes the truly predicted non-snake toxin protein sequences and ‘FN’ demonstrate the snake toxin protein sequences which were predicted as non-snake toxin protein sequence.
3 Results and discussion
3.1 Performance evaluation
Initially, we converted the sequence data into feature vectors by using three types of feature encoding schemes. Then, each feature vector was assessed by CNN-based classifier by employing a 10-fold CV. Subsequently, ANOVA and GBDT were implemented to select the optimal feature. Figure 3A displays the prediction accuracy of models constructed with different numbers of features. The maximum accuracy of 82.00% was achieved on 167 optimal features. Figure 3B shows the contribution of feature descriptors in CNN-based fusion model. The optimal model was trained on the data with 167 features derived from three kinds of descriptors. In final optimized-fusion model, NV, W2V and g-gap dipeptide descriptors account for 35.92, 43.11, and 20.95%, respectively. We have also visualized the feature fusions by using t-SNE (t-distributed stochastic neighbor embedding) technique. The t-SNE visualization of feature fusion before and after the feature selection are shown in Figures 4A,B. Figure 4C shows the single-encoding performance on different machine learning-based (ML-based) classifiers before the selection of features (36) and Figure 4D shows the performance of single-encoding after feature selections on different ML-based classifiers. Table 1 also shows the performance of feature fusion models before and after the feature selection on different ML-based classifiers by utilizing 10-fold CV.
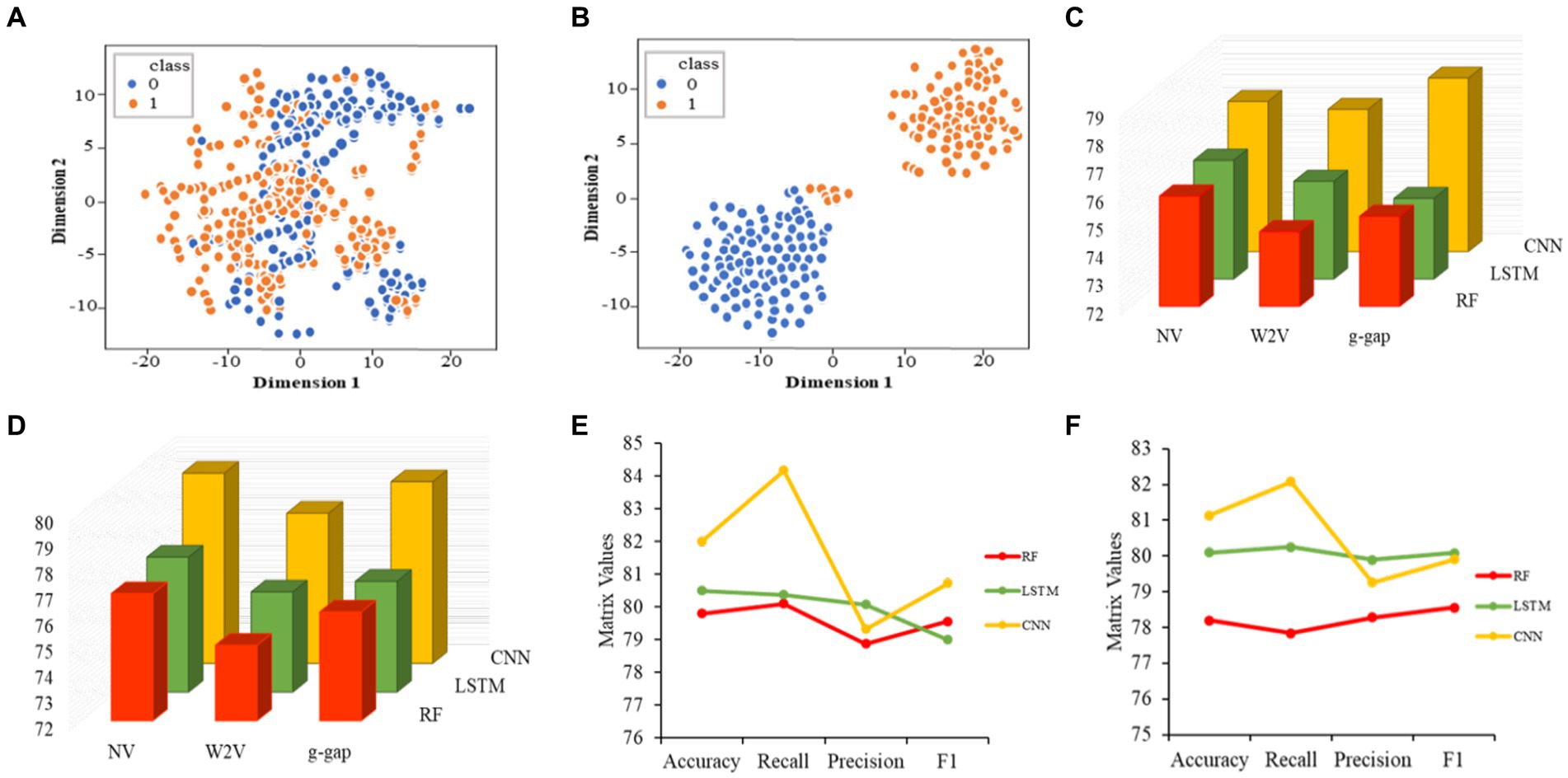
Figure 4. Visualization of feature fusion before the feature selection (A). Visualization of feature fusion after the feature selection (B). Performance of single-encoded features on different classifiers before the feature selection (C). Performance of single-encoded features on different classifiers after the feature selection (D). Comparison of proposed CNN-based fusion model with different machine learning-based fusion models on the basis of 10-fold CV (E). Comparison of proposed CNN-based fusion model with different machine learning-based fusion models on independent data (F).
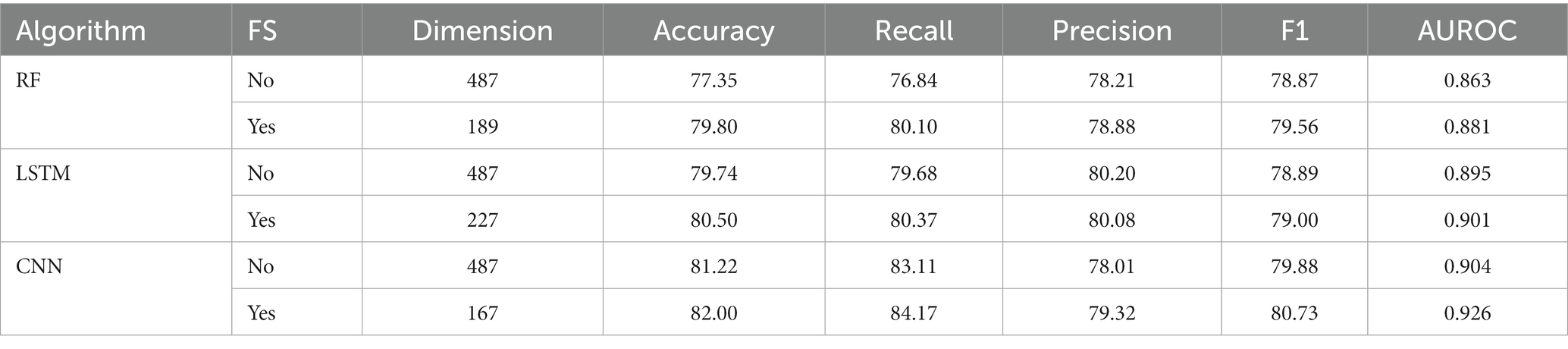
Table 1. Performance of fusion models by using different algorithms.
The comparisons of proposed CNN-based fusion model with different machine learning-based fusion models on 10-fold CV as well as on independent dataset are shown in Figures 4E,F. From these comparisons, we may conclude that the best model is based on the CNN with 167 optimal features. The model could produce the AUROC of 0.926 and 0.917 on training and independent dataset.
3.2 Performance evaluation of different ML algorithms
Various single feature and their fusion were inputted into other ML-based classifiers, such as long short-term memory (LSTM) and random forest (RF), for determining which machine learning method is the best for snake toxin prediction. The 10-fold CV and independent dataset test were employed to estimate the efficiency of these models. The comparison outcomes have been shown in Tables 1, 2. We noticed that the AUROC of CNN-based prediction model was 2.5–4.5% higher than that of other classifiers on 10-fold CV and 1.7–4.1% higher than that of other classifiers on independent test. Figures 5A–D displayed that the CNN-based prediction model is best among all classifiers.
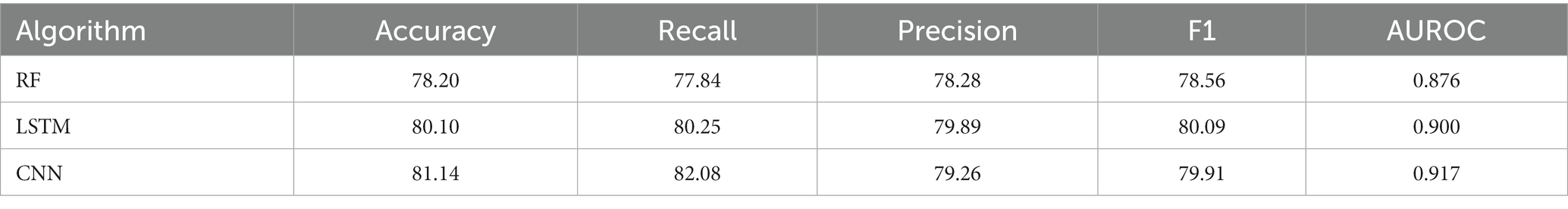
Table 2. Performance of fusion models on independent data.
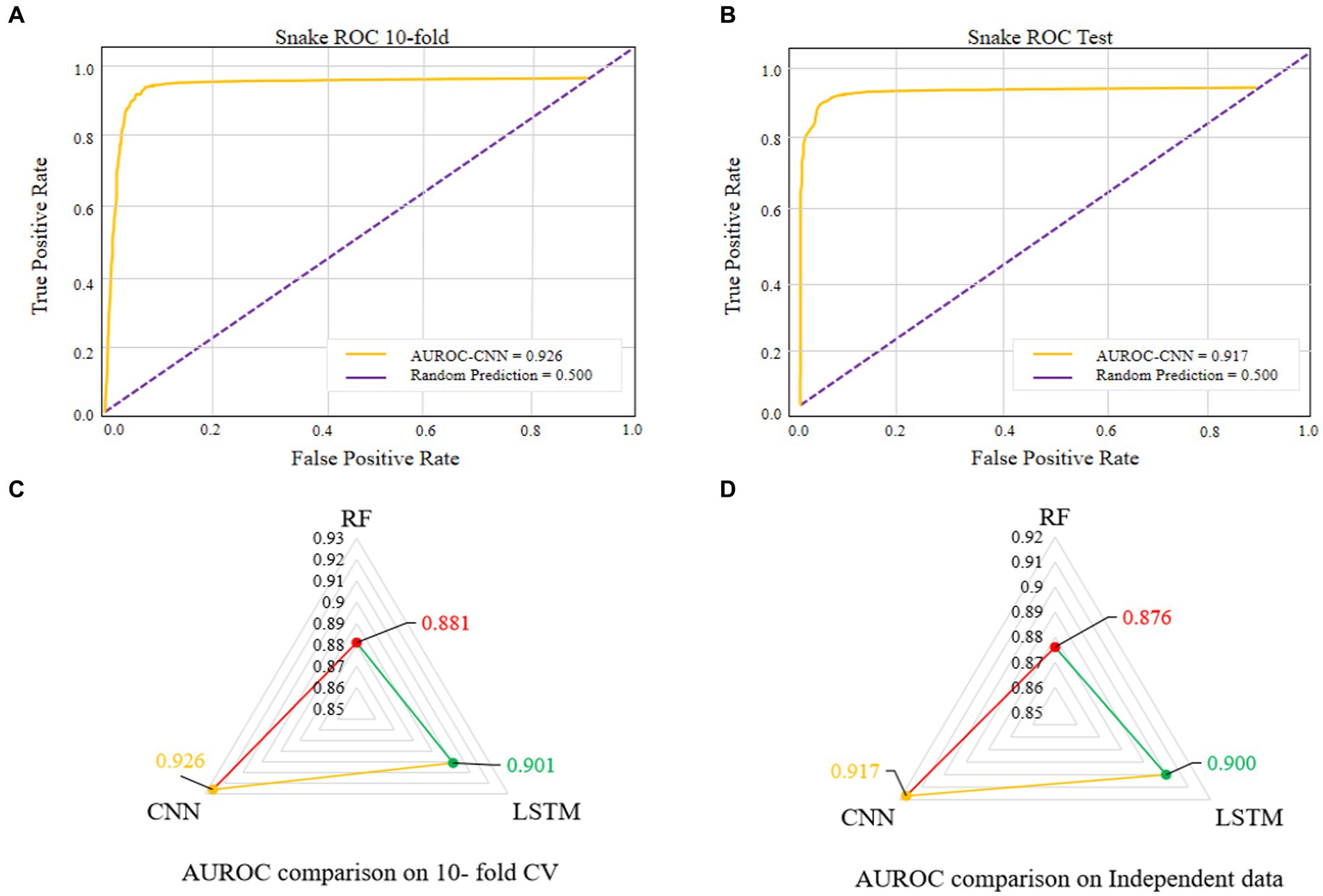
Figure 5. AUROC of the best performing model on 10-fold CV (A). AUROC of the best performing model on independent data (B). Comparison of different machine learning-based models on 10-fold CV (C). Comparison of different machine learning-based models on independent data (D).
4 Conclusion
Snake venom is a mixture of deadly proteins that can anesthetize and kill prey. Scientists have found a variety of proteins with potential pharmacological uses from snake venom. Further research on snake venom protein will contribute to drug development. In this work, an innovative computational model was constructed to classify snake toxin proteins. NV, W2V, and g-gap were utilized to encode the protein sequences. Subsequently, optimal feature subset was obtained by ANOVA and GBDT with IFS. By comparing different machine learning-based models, the best model was attained by the CNN-based classifier. Furthermore, the results showed that the proposed model could provide spectacular generalization ability. The dataset and codes are available at https://github.com/linDing-groups/Deep-STP. Further studies will focus on constructing a web application for the anticipated model. Moreover, other advance feature selection techniques and algorithms will be employed to further increase the efficiency of classification.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
HZ: Conceptualization, Experimentation, Methodology, Visualization, Writing—original draft preparation. ZG: Data curation, Methodology, Experimentation. RMA: Data curation, Experimentation, Methodology, Visualization. ZA: Data curation, Methodology, Visualization. PC: Data curation, Visualization. XC: Methodology. YZ: Methodology, Writing – review & editing. HL: Conceptualization, Supervision, Writing – review & editing. ZS: Conceptualization, Writing – review & editing. All authors have read and agreed to the published version of the manuscript.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work has been supported by the National Nature Scientific Foundation of China (62302079), the Natural Science Foundation of Sichuan Province (2022NSFSC1610) and funding of Cells and Regenerative Medicine Innovation Team (CDFYCX202208).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2023.1291352/full#supplementary-material
References
1. Osipov, AV, and Utkin, YN. Snake venom toxins targeted at the nervous system. Snake Venoms Toxinol. (2017):189–214. doi: 10.1007/978-94-007-6410-1_23
2. Yamazaki, Y, and Morita, T. Structure and function of snake venom cysteine-rich secretory proteins. Toxicon. (2004) 44:227–31. doi: 10.1016/j.toxicon.2004.05.023
3. Nirthanan, S. Snake three-finger α-neurotoxins and nicotinic acetylcholine receptors: molecules, mechanisms and medicine. Biochem Pharmacol. (2020) 181:114168. doi: 10.1016/j.bcp.2020.114168
4. Okuda, J, and Kiyokawa, R. Snake as a symbol in medicine and pharmacy-a historical study. Yakushigaku Zasshi. (2000) 35:25–40.
5. Bordon, KDCF, Cologna, CT, Fornari-Baldo, EC, Pinheiro-Junior, EL, Cerni, FA, Amorim, FG, et al. From animal poisons and venoms to medicines: achievements, challenges and perspectives in drug discovery. Front Pharmacol. (2020) 11:1132. doi: 10.3389/fphar.2020.01132
6. Pearson, WR. Finding protein and nucleotide similarities with FASTA. Curr Protoc Bioinformatics. (2016) 53:3.9.1–3.9.25. doi: 10.1002/0471250953.bi0309s53
7. Zou, Q, Hu, Q, Guo, M, and Wang, G. HAlign: fast multiple similar DNA/RNA sequence alignment based on the Centre star strategy. Bioinformatics. (2015) 31:2475–81. doi: 10.1093/bioinformatics/btv177
8. Wan, S, and Zou, Q. HAlign-II: efficient ultra-large multiple sequence alignment and phylogenetic tree reconstruction with distributed and parallel computing. Algorithms Mol Biol. (2017) 12:25. doi: 10.1186/s13015-017-0116-x
9. Madden, T. The BLAST sequence analysis tool, the NCBI handbook. 2nd ed. Bethesda, MD: National Center for Biotechnology Information (US) (2013).
10. Zulfiqar, H, Sun, Z-J, Huang, Q-L, Yuan, S-S, Lv, H, Dao, F-Y, et al. Deep-4mCW2V: a sequence-based predictor to identify N4-methylcytosine sites in Escherichia coli. Methods. (2021) 203:558–63. doi: 10.1016/j.ymeth.2021.07.011
11. Tang, H, Zhao, YW, Zou, P, Zhang, CM, Chen, R, Huang, P, et al. HBPred: a tool to identify growth hormone-binding proteins. Int J Biol Sci. (2018) 14:957–64. doi: 10.7150/ijbs.24174
12. Zulfiqar, H, Yuan, S-S, Huang, Q-L, Sun, Z-J, Dao, F-Y, Yu, X-L, et al. Identification of cyclin protein using gradient boost decision tree algorithm. Comput Struct Biotechnol J. (2021) 19:4123–31. doi: 10.1016/j.csbj.2021.07.013
13. UniProt Consortium. Uni Prot: a worldwide hub of protein knowledge. Nucleic Acids Res. (2019) 47:D506–15. doi: 10.1093/nar/gky1049
14. O'Leary, NA, Wright, MW, Brister, JR, Ciufo, S, Haddad, D, McVeigh, R, et al. Reference sequence (ref Seq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. (2016) 44:D733–45. doi: 10.1093/nar/gkv1189
15. Fu, L, Niu, B, Zhu, Z, Wu, S, and Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. (2012) 28:3150–2. doi: 10.1093/bioinformatics/bts565
16. Lv, H, Dao, F-Y, Zulfiqar, H, and Lin, H. Deep IPs: comprehensive assessment and computational identification of phosphorylation sites of SARS-CoV-2 infection using a deep learning-based approach. Brief Bioinform. (2021) 22:244. doi: 10.1093/bib/bbab244
17. Deng, M, Yu, C, Liang, Q, He, RL, and Yau, SS-T. A novel method of characterizing genetic sequences: genome space with biological distance and applications. PloS One. (2011) 6:e17293. doi: 10.1371/journal.pone.0017293
18. Zhang, D, Chen, H-D, Zulfiqar, H, Yuan, S-S, Huang, Q-L, Zhang, Z-Y, et al. iBLP: an XGBoost-based predictor for identifying bioluminescent proteins. Comput Math Methods Med. (2021) 2021:1–15. doi: 10.1155/2021/6664362
19. Zou, Q, Xing, P, Wei, L, and Liu, B. Gene 2vec: gene subsequence embedding for prediction of mammalian N6-Methyladenosine sites from mRNA. RNA. (2019) 25:205–18. doi: 10.1261/rna.069112.118
20. Charoenkwan, P, Nantasenamat, C, Hasan, MM, Manavalan, B, and Shoombuatong, W. BERT4Bitter: a bidirectional encoder representations from transformers (BERT)-based model for improving the prediction of bitter peptides. Bioinformatics. (2021) 37:2556–62. doi: 10.1093/bioinformatics/btab133
21. Deho, B.O., Agangiba, A.W., Aryeh, L.F., and Ansah, A.J., Sentiment analysis with word embedding, 2018 IEEE 7th international conference on Adaptive Science & Technology (ICAST), Accra, Ghana. (2018), pp. 1–4.
22. McCormick, C. (2016). Word 2vec tutorial-the skip-gram model. Available at: http://mccormickml.com/2016/04/19/word2vec-tutorial-the-skip-gram-model
24. Zulfiqar, H, Dao, F-Y, Lv, H, Yang, H, Zhou, P, Chen, W, et al. Identification of potential inhibitors against SARS-Cov-2 using computational drug repurposing study. Curr Bioinforma. (2021) 16:1320–7. doi: 10.2174/1574893616666210726155903
25. Dao, F-Y, Lv, H, Zulfiqar, H, Yang, H, Su, W, Gao, H, et al. A computational platform to identify origins of replication sites in eukaryotes. Brief Bioinform. (2021) 22:1940–50. doi: 10.1093/bib/bbaa017
26. Zou, X, Ren, L, Cai, P, Zhang, Y, Ding, H, Deng, K, et al. Accurately identifying hemagglutinin using sequence information and machine learning methods. Front Med. (2023) 10:1281880. doi: 10.3389/fmed.2023.1281880
27. Zhu, W, Yuan, SS, Li, J, Huang, CB, Lin, H, and Liao, B. A first computational frame for recognizing heparin-binding protein. Diagnostics. (2023) 13:2465. doi: 10.3390/diagnostics13142465
28. Dao, FY, Lv, H, Wang, F, Feng, CQ, Ding, H, Chen, W, et al. Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics. (2019) 35:2075–83. doi: 10.1093/bioinformatics/bty943
29. De Jay, N, Papillon-Cavanagh, S, Olsen, C, El-Hachem, N, Bontempi, G, and Haibe-Kains, B. mRMRe: an R package for parallelized mRMR ensemble feature selection. Bioinformatics. (2013) 29:2365–8. doi: 10.1093/bioinformatics/btt383
30. LeCun, Y, Bottou, L, Bengio, Y, and Haffner, P. Gradient-based learning applied to document recognition. Proc IEEE. (1998) 86:2278–324. doi: 10.1109/5.726791
31. Niu, M, Lin, Y, and Zou, Q. sgRNACNN: identifying sgRNA on-target activity in four crops using ensembles of convolutional neural networks. Plant Mol Biol. (2021) 105:483–95. doi: 10.1007/s11103-020-01102-y
32. Kwon, Y-H, Shin, S-B, and Kim, S-D. Electroencephalography based fusion two-dimensional (2D)-convolution neural networks (CNN) model for emotion recognition system. Sensors. (2018) 18:1383. doi: 10.3390/s18051383
33. Lv, H, Dao, F-Y, Zulfiqar, H, Su, W, Ding, H, Liu, L, et al. A sequence-based deep learning approach to predict CTCF-mediated chromatin loop. Brief Bioinform. (2021) 22:bbab031. doi: 10.1093/bib/bbab031
34. Chollet, F.. (2015). Keras: Deep learning library for theano and tensorflow. Available at: https://keras.io/k
35. Cao, R, Freitas, C, Chan, L, Sun, M, Jiang, H, and Chen, Z. ProLanGO: protein function prediction using neural machine translation based on a recurrent neural network. Molecules. (2017) 22:1732. doi: 10.3390/molecules22101732
Keywords: snake toxin, deep learning, feature vectors, word embedding, feature selection, ANOVA
Citation: Zulfiqar H, Guo Z, Ahmad RM, Ahmed Z, Cai P, Chen X, Zhang Y, Lin H and Shi Z (2024) Deep-STP: a deep learning-based approach to predict snake toxin proteins by using word embeddings. Front. Med. 10:1291352. doi: 10.3389/fmed.2023.1291352
Edited by:
Waqas Nazeer, University of Education, Winneba, GhanaReviewed by:
Santosh Panjikar, Australian Synchrotron, AustraliaWang-Ren Qiu, Jingdezhen Ceramic Institute, China
Copyright © 2024 Zulfiqar, Guo, Ahmad, Ahmed, Cai, Chen, Zhang, Lin and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Zhang, eWFuZ3poYW5nQGNkdXRjbS5lZHUuY24=; Hao Lin, aGxpbkB1ZXN0Yy5lZHUuY24=; Zheng Shi, ZHJzaGl6MTAwMkBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work