 Sara Tirendi
Sara Tirendi Cinzia Domenicotti
Cinzia Domenicotti Anna Maria Bassi
Anna Maria Bassi Stefania Vernazza
Stefania Vernazza- 1Department of Experimental Medicine, University of Genoa, Genoa, Italy
- 2Inter-University Center for the Promotion of the 3Rs Principles in Teaching & Research (Centro 3R), Genoa, Italy
Glaucoma is the second leading cause of irreversible blindness worldwide. Although genetic background contributes differently to rare early-onset glaucoma (before age 40) or common adult-onset glaucoma, it is now considered an important factor in all major forms of the disease. Genetic and genomic studies, including GWAS, are contributing to identifying novel loci associated with glaucoma or to endophenotypes across ancestries to enrich the knowledge about glaucoma genetic susceptibility. Moreover, new high-throughput functional genomics contributes to defining the relevance of genetic results in the biological pathways and processes involved in glaucoma pathogenesis. Such studies are expected to advance significantly our understanding of glaucoma’s genetic basis and provide new druggable targets to treat glaucoma. This review gives an overview of the role of genetics in the pathogenesis or risk of glaucoma.
1 Introduction
Glaucoma is the second leading cause of blindness worldwide, and it is estimated that its incidence will increase in the next 20 years (1–3). The term glaucoma brings together a heterogeneous group of neurodegenerative diseases characterized by Retinal Ganglion Cell (RGC) degeneration as well as typical morphological changes in the Optic Nerve Head (ONH), i.e., the narrowing of the neuroretinal rim, the enlargement of the excavation, and the loss of prelaminar neural tissue (4, 5).
In general, all glaucoma forms are categorized into primary and secondary forms, according to their etiology. Primary glaucoma is defined as an isolated, idiopathic disease of the anterior chamber of the eye and the optic nerve whereas the secondary one is characteristic of specific syndromic frameworks (e.g., nail patella, exfoliation syndrome, aniridia), or associated with predisposing events, including systemic diseases (e.g., diabetes) and pharmacological treatment.
Primary glaucoma is further distinguished into three broad categories, based on the anatomy of the anterior chamber drainage angle, aqueous dynamics, and age at onset: (i) Primary Open Angle Glaucoma (POAG), (ii) Primary congenital glaucoma (PCG), and (iii) Primary Angle Closure Glaucoma (PACG) (3, 6).
Primary open angle glaucoma is responsible for most cases of adult-onset glaucoma (i.e., individuals aged over 40 years) in Africans, Latinos, Americans, and European populations (7, 8). However, POAG, to a lesser extent, may occur in young subjects ranging from 3 years old and 40 years of age, as in the case of Juvenile Open Angle Glaucoma (JOAG).
Primary congenital glaucoma is a rare form of glaucoma in infancy (before the age of 3 years) characterized by aqueous humor drainage obstruction due to an upstream non-development of neural crest-derived tissues, which results in developmental anomaly at the angle of the anterior chamber (9).
Finally, PACG is responsible for most cases of glaucoma-induced bilateral blindness in populations, such as Asians (10). In such glaucoma, the anatomic narrow-angle leads to trabecular meshwork (TM) obstruction and, consequently, to peripheral anterior synechiae, elevated intraocular pressure IOP, iris whirling or sectoral atrophy, and excessive pigment deposition on the trabecular surface (10–13).
Glaucoma can occur at all ages exhibiting a classic (Mendelian) inheritance pattern (i.e., onset before the age of 40) (14, 15) or a complex inheritance pattern [i.e., onset after the age of (16, 17)]. Therefore, genetic background is considered a precondition that can directly cause or contribute to glaucoma. This present review aims to summarize the main achievement in genetic studies of glaucoma, mainly focusing on glaucoma-causing genes.
2 Methods
The data in this present comprehensive review were collected using two different searches: PubMed1 and the online database: genome aggregation database,2 and the NHGRI-EBI Catalog of Human Genome-Wide Association Studies.3 The search of the references using PubMed identified a total of 157 hits with no time frame relative to keywords such as “Genetic of glaucoma” “POAG guidelines,” “JOAG management,” “MYOC mutations in glaucoma,” “CYP1B1 in glaucoma,” “PACG guidelines,” “clinical and genetic update of glaucoma,” “candidate genes involved in glaucoma.” Thus, the combined information obtained from articles relevant to the subject matter and the online database has represented the basis for writing the review.
3 Results
3.1 Primary open angle glaucoma
Primary open angle glaucoma is defined as a progressive chronic bilateral or asymmetric ocular disease (18). Clinical evidence in addition to visual field defects and the age of patients (i.e., > the 40s) includes structural abnormalities to the optic disk (OD) rim or parapapillary retinal nerve fiber layer (RNFL) (19). The severity of POAG damage can be distinguished as mild, moderate, and severe based on the presence or not of possible changes in the optic disk, RNFL damage, and degree of visual field abnormalities.
Although it is not an effective treatment for all patients, IOP management is the only clinically addressable risk factor to prevent or delay POAG progression (3). Primary open angle glaucoma guidelines (20) suggest the use of topical medications (e.g., Prostaglandin analogs, beta-blockers, alpha2 adrenergic agonists, parasympathomimetic, and topical and oral carbonic anhydrase inhibitors) as the first line of treatment to reach the target pressure. However, since treatment regimens require daily applications to control IOP, noncompliance, undesirable adverse effects, and cost limit their effectiveness (21, 22). Laser trabeculoplasty and glaucoma surgery (e.g., trabeculectomy, aqueous shunts, non-penetrating glaucoma surgery, micro-invasive glaucoma surgery, and so on…) are considered for further IOP reduction in eyes with inadequate initial responses (23).
Adult-onset POAG is a complex disease often caused by an intricate interplay at molecular levels between predisposing genes and evidence-based risk factors (e.g., ethnicity, gender, aging, IOP, central corneal thickness, migraine headache and peripheral vasospasm, myopia, systemic diseases, steroid responsiveness, positive history of glaucoma) (3, 24, 25). Primary open angle glaucoma may be inherited as a Mendelian trait, as in the case of causative mutations in genes such as the myocilin (MYOC), optineurin (OPTN), and TANK-binding kinase 1 (TBK1) (26, 27), where other genes or environmental factors play little role (See below).
Generally, POAG is unsuitable for linkage studies because they require many living POAG-affected relatives and this, for obvious reasons, is not always possible. Thus, highlighting an inheritance pattern for POAG represents a real challenge compared to the childhood-onset glaucomas (24, 28–31).
Genome-Wide Association Studies (GWAS) provide information about the common POAG genetic variants, i.e., Single Nucleotide Polymorphisms (SNPs), that are found among populations of diverse ancestry (Table 1). In this regard, recurring SNPs may constitute a quantifiable relationship between the genetic variability of POAG and its clinical manifestation (endophenotypes) (30, 41, 42). However, SNPs have limited predictive value in terms of risk assessment and prognosis of POAG due to its complex inheritance pattern (43).
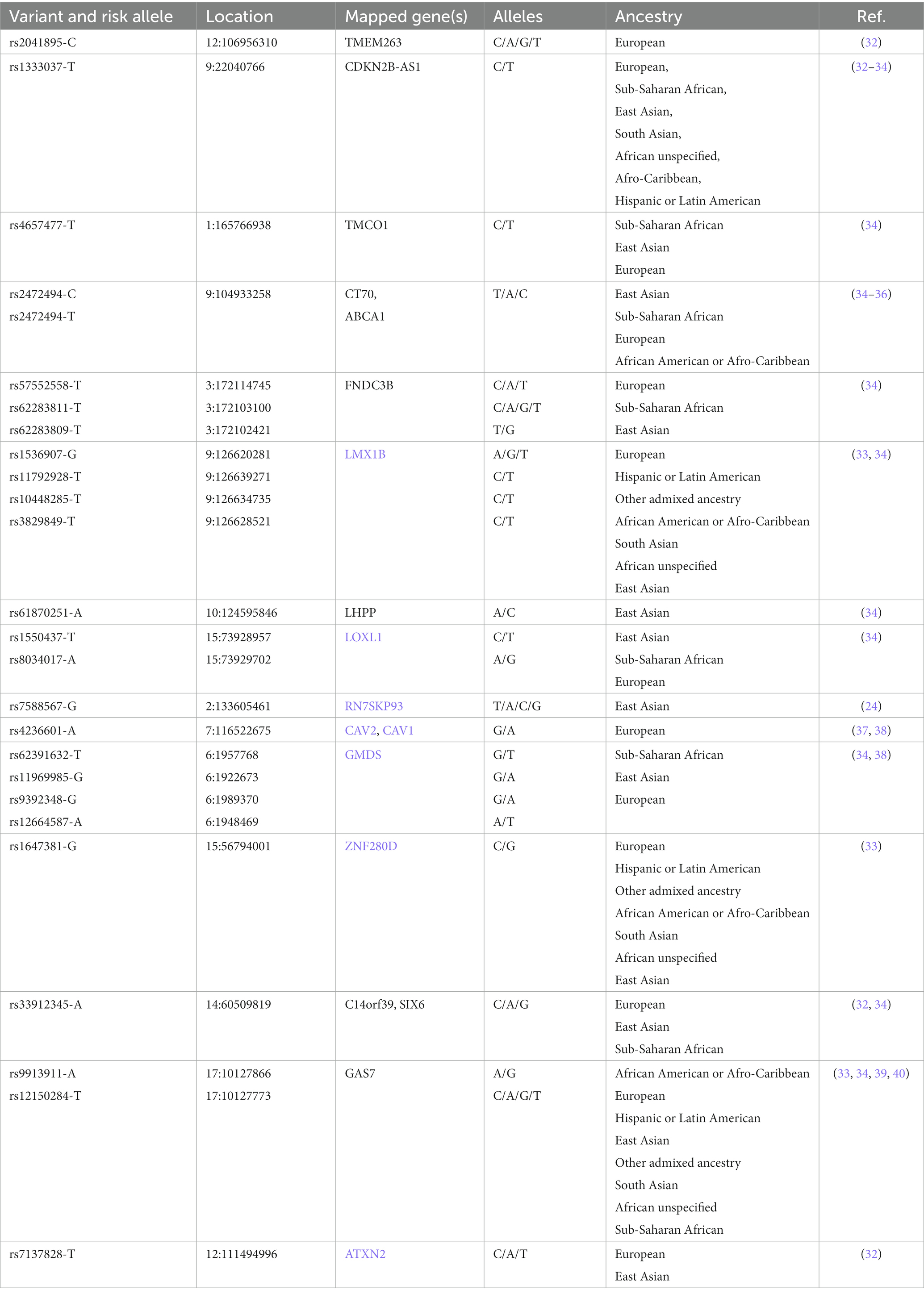
Table 1. List of some known SNPs identified in different glaucoma cohorts (source: GWAS Catalog).
3.1.1 Juvenile open angle glaucoma
Juvenile open angle glaucoma, as above mentioned, is an uncommon subset (0,38,100,000) of POAG that affects individuals during childhood or early adulthood.
Generally, JOAG is inherited in an autosomal dominant fashion in individuals with a strong family history of glaucoma. Nevertheless, it has been shown that in Iranian and Saudi Arabian populations, it occurs as an autosomal recessive or sporadic disease (17, 44–46). Therefore, genetically it is possible to identify different pathological mutations, including myocilin (MYOC), Cytochrome P450 subtype 1 Polypeptide 1 (CYP1B1), Optineurin (OPTN), WD Repeat domain 36 (WDR36), Neurotrophin 4 (NTF4), Latent Transforming growth factor-beta-binding Protein 2 (LTBP2), and Ankyrin repeat and SOCS-Box containing 10 (ASB10) (47–50). Recently, also the rare biallelic mutation CPAMD8 in both JOAG and PCG was identified as a unique form of autosomal recessive anterior segment dysgenesis with a variable range of clinical phenotypes (51).
In most cases, JOAG is characterized by severe IOP elevation (>40 mmHg) due to immaturity of the conventional outflow pathway, and thick corneas. However, the extent of clinical manifestations, including the IOP elevation, degree of goniodysgenesis, and the inheritance pattern, as well as the age at onset, are subject to individual variations. In this regard, four phenotype groups have been identified to classify affected individuals based on the age of onset, highest untreated IOP, gonioscopic findings, and iris features (52) (Table 2).
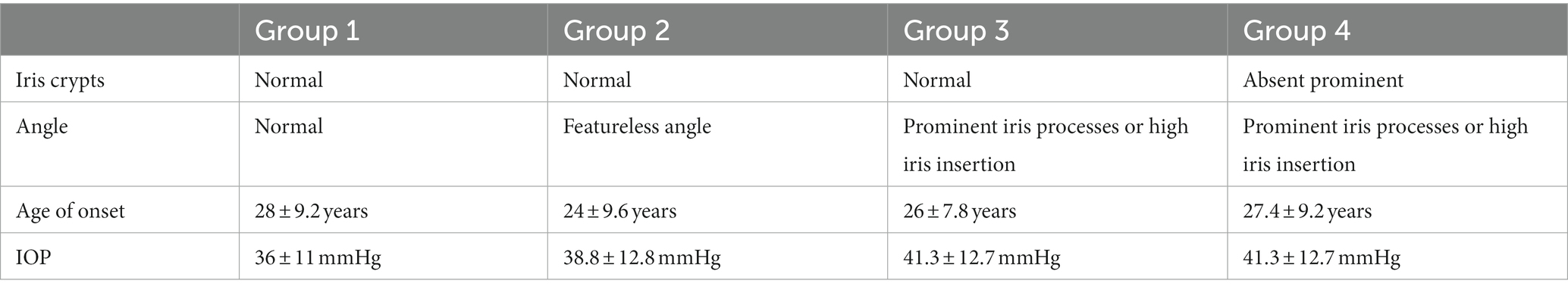
Table 2. Four phenotypic groups of JOAG.
Recent evidence also reported a JOAG subtype, i.e., Juvenile normal tension glaucoma (JNTG), in which glaucomatous damage occurs at normal-range IOP (≤ 21 mmHg) (53).
Juvenile open angle glaucoma differs from other early-onset glaucoma forms (e.g., congenital glaucoma) by the absence of buphthalmos, megalocornea, Haab’s striae, and ocular or other systemic developmental anomalies (17, 54).
Juvenile open angle glaucoma patients are often asymptomatic until an advanced stage, therefore, a prompt diagnosis is not always possible. When JOAG becomes symptomatic results in blurred vision, eye pain, decreased visual acuity, tearing, blinking, and glare (55). As known, an early JOAG diagnosis increases the chance of preserving vision and preventing permanent visual field loss. Surgery (i.e., trabeculectomy, external trabeculectomy, goniotomy, drainage implants, gonioscopy-assisted transluminal trabeculectomy, and so on…) is often required since the affected individuals are refractory to the standard primary drug approaches to manage the IOP (e.g., β blockers, topical carbonic anhydrase inhibitor, prostaglandin analogs, α-adrenergic agonists) (56, 57).
3.1.2 Myocilin
The MYOC gene, also known as Trabecular meshwork Inducible Glucocorticoid Response (TIGR), maps to the GLC1A (Glaucoma 1A) locus at chromosome 1q21– q31. It consists of three exons: the first encodes for amino-terminal region of the protein containing a peptide signal sequence and a leucine zipper-like motif (i.e., where the myocilin-myocilin interactions take place) with periodic arginine and leucine repeats arranged along an α-helix; the second encodes for the protein central region; the third encodes for the carboxyl-terminal half of myocilin, the olfactomedin (OLF)-domain, which includes the majority of the identified disease causing variants (58).
Myocilin encodes the myocilin glycoprotein expressed in several ocular tissues, such as the cornea, iris, ciliary body, retinal epithelium, and TM. Myocilin mutations can be responsible for the improper function of the eye tissue where the protein is commonly highly expressed, e.g., TM (59). Moreover, myocilin is found also in non-ocular tissues, such as the heart tissue and skeletal muscle but there is no evidence of its involvement in systemic disease (60, 61).
Myocilin in its wild-type (wt) state is a ubiquitous protein whose function in the eye has not been determined yet. One suggestion is that myocilin contributes to the maintenance of IOP homeostasis, although it has not been confirmed due to conflicting and contradictory data. While some studies report that myocilin up-regulation is responsible for IOP elevation (62, 63), other ones report that neither its overexpression nor its haploinsufficiency is the primary mechanism for glaucoma phenotype. (31, 64, 65). This latter evidence has challenged the pathological role of myocilin in promoting steroid-induced glaucoma (~40% of cases) after long-term glucocorticoid treatment (e.g., dexamethasone) (66). In this regard, previous studies on steroid-induced myocilin overexpression excluded its role in both ocular hypertension and endoplasmic reticulum (ER) stress (67, 68). However, a myocilin stress-related function in TM has been suggested because stressful stimuli, such as hydrogen peroxide and elevated IOP, increase its expression (69, 70).
The first evidence that showed a correlation between POAG and mutant myocilin date from 1997 (71). Linkage studies have shown that MYOC mutations, accounting for 8–34% of JOAG, 2–4% of adult-onset POAG cases, and in part for NTG, are mainly inherited in an autosomal dominant manner with incomplete penetrance (72). In this regard, disease severity varies between individuals due to a strong genotype–phenotype correlation (73). A meta-analysis of the penetrance of MYOC mutation revealed that except for a few variants (e.g., Pro370Leu, Thr377Arg, Asp380Ala, and Asn480Lys) having a stable penetrance, the penetrance of MYOC mutations can be influenced by factors such as aging, and ethnicity (74).
Under physiological conditions, wt myocilin after intracellular endoproteolytic processing, produces two fragments, i.e., the 35kD fragment containing the C-terminal OLF-like domain and the 20kD fragment containing the N-terminal leucine zipper-like domain (75). However, only the 35kD fragment is co-secreted with non-processed protein leading to assume that it may play a role in the regulation of the myocilin extracellular activity (76, 77). The best known-myocilin-induced disease phenotype is characterized by an intracellular abnormal buildup of myocilin in TM (26, 78, 79), resulting in a toxic gain-of-function disease mechanism. Indeed, most of the myocilin-associated glaucoma mutations are missense clustered in the OLF domain (Table 3). These mutations by inducing defects in myocilin stability, promote the mutant myocilin retention into the endoplasmic reticulum (ER) of TM cells with toxic consequences, such as ER stress, cytotoxicity, and TM apoptosis, which in turn affect the IOP control (61, 75, 91–93). However, also nonsense mutations are involved in about 5.7% of JOAG and adult-onset POAG cases. Notably, the nonsense mutation Gln368Stop is responsible for 40% of High Tension Glaucoma (HTG) (91, 94, 95).
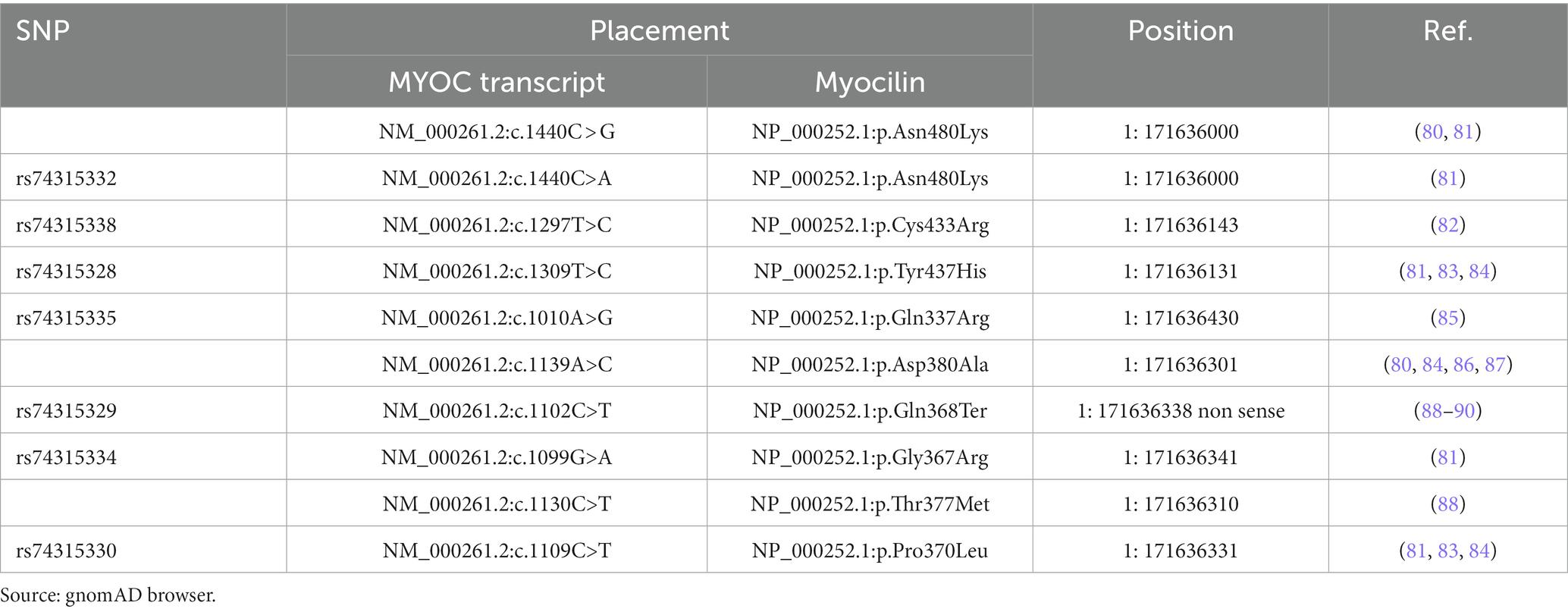
Table 3. List of some pathogenic/likely pathogenic glaucoma-associated myocilin missense mutations.
3.1.3 Familial normal tension glaucoma
Normal-tension glaucoma (NTG) is an OAG characterized by an IOP within a statistically normal range (≤21 mmHg). The NTG pattern of damage in the optic disk, retinal nerve fiber layer, and visual field significantly differs from that found in POAG, suggesting that the optic neuropathy is due to other mechanisms rather than elevating IOP (96). In this regard, a higher sensitivity to normal pressure, vascular dysregulation, an abnormally high translaminar pressure gradient, and a low cerebrospinal fluid pressure in the optic nerve sheath compartment, have been proposed to contribute to NTG pathogenesis (97).
Generally, NTG has a complex genetic basis, but the analyses of several pedigrees showed that about 2% of NTG forms (i.e., Familial NTG) are caused by a single-gene mutation. NTG genetic survey improved thanks to the GWAS since have identified several susceptible loci contributing to NTG on chromosomes 9p2, 2p21, 7q31, 1q24, and 6p21.1-p12.1 (37, 43, 96, 98).
Familial NTG refers to an early onset (at 20–35 years old) bilateral disease with variable severity (99). Optineurin, MYOC, and TBK1 are indicated as NTG-causing genes, according to the Mendelian autosomal dominant inheritance pattern.
3.1.4 Optineurin
The human OPTN gene, located on the short arm of chromosome 10 (10q13), encodes for optineurin, which is a multifunctional protein ubiquitously expressed in many tissues (e.g., the heart, brain, liver, skeletal muscle, kidney, pancreas, and the eye). In the eye, optineurin is expressed in the trabecular meshwork, non-pigmented ciliary epithelium, and remarkably in the retina (100). The well-characterized optineurin multiple cellular functions include selective autophagy (i.e., mitophagy, aggrephagy, xenophagy) through ubiquitin-signaling, NF-κB regulation, membrane trafficking, exocytosis, vesicle transport, transcriptional activation, and reorganizing of actin and microtubules. All these molecular functions arise from the interaction with a number of proteins, such as Rab8, huntingtin (Htt), transcription factor IIIA, myosin VI, and TANK binding protein 1 (TBK1) (101–103).
The optineurin-mediated autophagy-dysfunction by either overstimulation or by defects that block its function has attracted considerable attention due to its involvement in a variety of neurodegenerative diseases (glaucoma, amyotrophic lateral sclerosis, Parkinson’s disease, Huntington’s disease), inflammatory diseases (Crohn’s disease, acute liver failure, rheumatoid arthritis) cancer, and nephropathy (104). Notably, the autophagy-mediated RGC death in POAG, also known as autophagic cell death, seems to be promoted by an excess of catabolic activity induced by chronic IOP elevation (105) or alterations in OPTN and TBK1 (106). Under physiological conditions, OPTN to link structures targeted for elimination with proteins of an assembling autophagosome must be phosphorylated by TBK1. However, POAG-associated OPTN mutations may increase the autophagy activation by promoting OPTN phosphorylation via TBK1. Moreover, other OPTN functions may be altered by specific mutations (see below). On the other hand, the gain-of-function mutation and copy number variation (CNV) of the TBK1 gene have been proposed as possible Normal Tension Glaucoma (NTG, a POAG subtype) causing mechanism (107, 108).
Rezaie et al. (109) described four sequence alterations in OPTN involved in autosomal dominant adult-onset glaucoma cases, i.e., Glu50Lys, Arg545Gnl, 691_692insAG, and Met98Lys. Most of the affected families harboring these OPTN alterations had normal IOP, and only a small portion had high IOP. Among the OPTN polymorphisms, Glu50Lys, which is located within a putative bZIP motif, is the most recurrent likely disease-causing variant associated with hereditary NTG in Caucasian and Hispanic individuals. The Glu50Lys-induced functional alterations leading to RGC apoptosis are probably related to both autophagy and trafficking dysfunction and an increase in oxidative stress (110, 111).
Arg545Gnl may be considered a disease-causing variant only in Asian populations because this role was not shown in other studies (112). 691_692insAG variant was described as a dominant mutation responsible for familial POAG in Japan (109) and Eastern Europe (112). Met98Lys was initially proposed as an attributable risk factor for familial and sporadic POAG cases but due to conflicting results among diverse populations, it was not confirmed (113–115).
Interesting, semiquantitative, hierarchical evidence-based rules for locus interpretation (Sherloc), which are a refinement of the American College of Medical Genetics and Genomics–Association for Molecular Pathology (ACMG–AMP) guidelines, are providing an accurate clinical prediction of the functional consequences of OPTN mutations (116). The algorithms used to predict the effect of sequence change suggest that variants affecting protein sequence (e.g., missense mutation), its function (e.g., splicing mutation), or its abundance (e.g., frameshift or nonsense mutations) are presumed more likely to be deleterious (Please visit the National Library of Medicine for more information).4 Further studies on larger populations are required to confirm the incrimination of OPTN in POAG, to discriminate OPTN mutations causing POAG from those acting only as risk factors, and to ascertain the rate of loss-of-function OPTN variants in different populations.
3.2 PGC
Primary congenital glaucoma is considered a non-syndromic glaucoma form characterized by a childhood onset with variable incidence across countries and ethnic groups, but it is most widespread in countries where consanguineous marriages abound (e.g., Middle East) (117, 118).
This pediatric glaucoma occurs at birth or within the first 3 years of life, and the clinical outcomes vary depending on whether the neonatal/newborn-onset PGC (within a month after birth) or late-onset-PGC (after 2 years of age). Indeed, neonatal PGC has a poor visual prognosis compared to that diagnosis later, especially if accompanied by amblyopia (119, 120). Moreover, other elements strongly associated with poor long-term visual outcomes are unilateral disease, multiple surgeries, poor vision at diagnosis, and ocular co-morbidities.
The main mechanisms involved in PCG are related to trabecular dysgenesis due to mutations affecting normal TM development with or without iridodysgenesis and maldeveloped trabecular angle meshwork with or without dysgenesis of the Schlemm’s canal (121–123). Moreover, histopathological findings of the anterior chamber angle of PCG eyes reveal an immature anatomic relationship involving iris, TM, and ciliary body (119, 124, 125). Clinical features include high IOP, an increase in horizontal corneal diameter, the presence of corneal edema, Haab’s striae, the pupillary reflex, refraction, axial length, and keratometry (124).
High IOP and its fluctuations are managed through multiple or single surgical drainage procedures (e.g., goniotomy and trabeculotomy ab externo) aimed at targeting the abnormal trabecular angle. Topical hypotensive medications could be used post-op (126).
Genetically, PCG is considered heterogeneous. 10–40% of affected individuals show several pathogenetic variants of CYP1B1 or LTBP2 inherited in an autosomal recessive manner and with varying degrees of penetrance (127). Moreover, also autosomal dominant and pseudo-dominant inheritance patterns have been described as well (117, 128, 129).
Genetic linkage analysis has identified four chromosomal loci of recessively inherited PCG, namely GLC3A, GLC3B, GLC3C, and GLC3D. Among these, GLC3A on chromosome 2p22-p21, which harbors the CYP1B1 gene, is considered the most representative of the severe phenotypes (123) (Table 4).
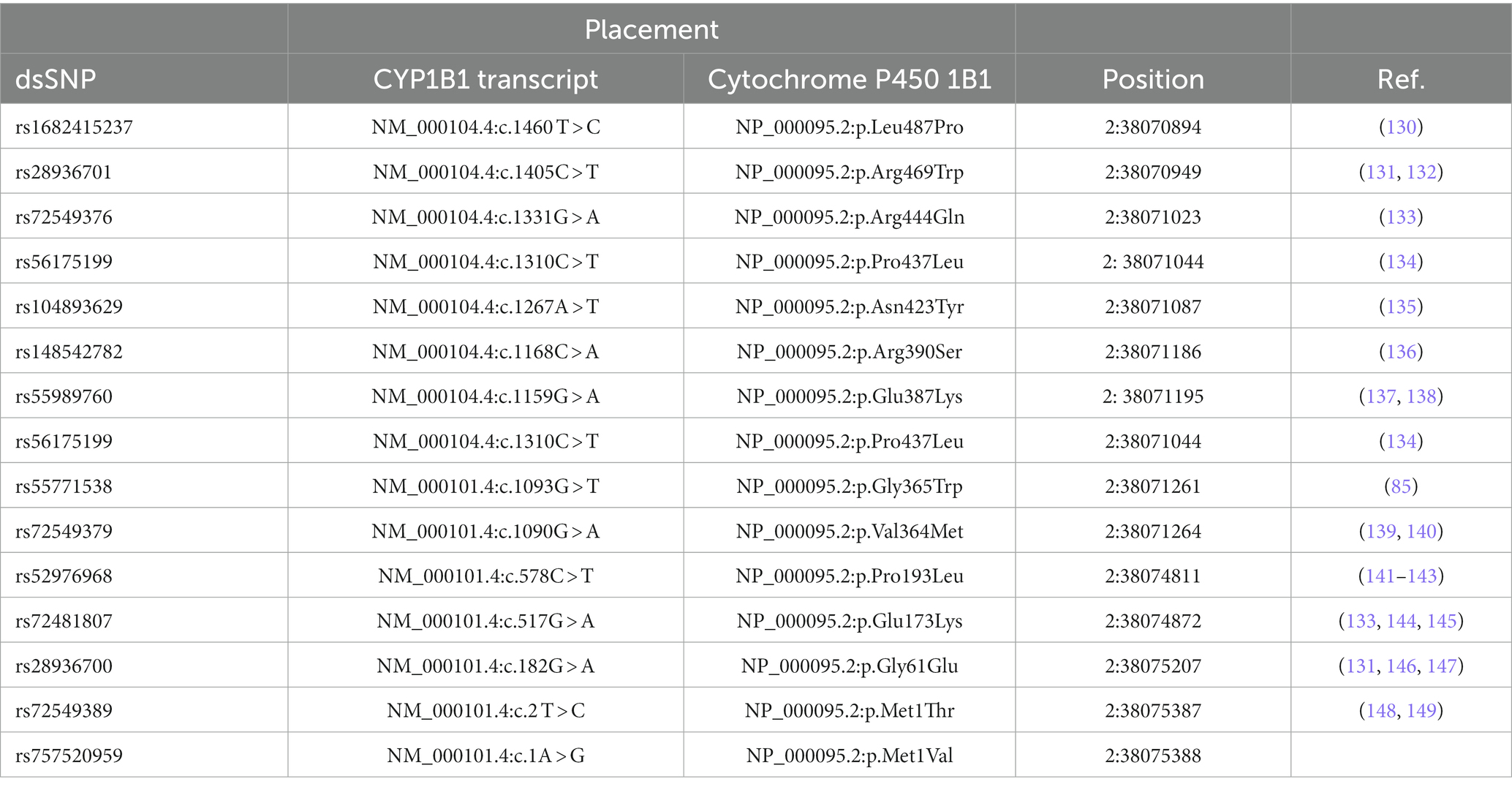
Table 4. List of some pathogenic/likely pathogenic glaucoma-associated CYP1B1 missense mutations (Source: gnomAD browser).
3.2.1 Cytochrome P450 subtype 1 polypeptide 1
Cytochrome P450 subtype 1 Polypeptide 1, belonging to the cytochrome P450 (CYP450) superfamily, is an enzyme mainly involved in oxidative metabolic activation (e.g., synthesis of retinoic acid) and detoxification of many compounds (e.g., several polycyclic aromatic hydrocarbons, endogenous steroids) (150).
Human Cyp1b1 consists of three exons and two introns, where the coding region is located within the second and the third exons. It is expressed constitutively in many parenchymal and stromal tissues, including the liver, brain, kidney, prostate, breast, cervix, uterus, ovary, lymph nodes, heart, placenta, lung, skeletal muscle, and eye (85, 151).
The role of CYP1B1 in PCG and JOAG is not clear, but it has been hypothesized that it plays a metabolic role in eye development and function, as well as cellular redox homeostasis maintenance (152–154). Therefore, the most accepted theory to explain the link between CYP1B1 mutation and PCG holds that mutated CYP1B1 is the leading cause of maladapted and dysfunctional TM due to defects in embryonic neural crest cell migration during development. This condition, in turn, leads to increased IOP, compression of the optic nerve, and blindness, if not treated surgically (155). Moreover, mutations in the Cyp1b1 gene, in addition to being strongly related to the pathophysiology of PCG, are associated with the most severe disease outcomes. Notably, a genotype–phenotype study in a large cohort of Moroccan PCG reports that both patients carrying CYP1B1 mutation and those with double CYP1B1 null alleles showed the most severe phenotype when compared with patients genetically negative (156).
Most of the CYP1B1 mutations responsible for a maldeveloped and dysfunctional TM are missense (Table 5), but how they affect disease phenotype and their frequency among different populations is still under investigation (152, 155). Recently, “in silico” methods (supported by databases, network analysis, and machine learning approaches) were used to identify possible high-risk deleterious variations by predicting the functional importance (neutral or deleterious) of the altered amino acid substitution on protein stability and the resulting phenotypic characteristics (e.g., disruption of the structural conformation), considering the allele frequency of variants (157). Based on such approaches, three novel missense mutations, i.e., Leu487Pro, Leu177Arg and Trp434Arg, and a transversion mutation Asp374Glu were identified in consanguineous Pakistani families as impacting the CYP1B1 protein stability, structure, and function (158). The results obtained from computational tools, functional assays, segregation analysis, genetic studies, and epidemiological data could potentially be helpful for the understanding and management of PCG.
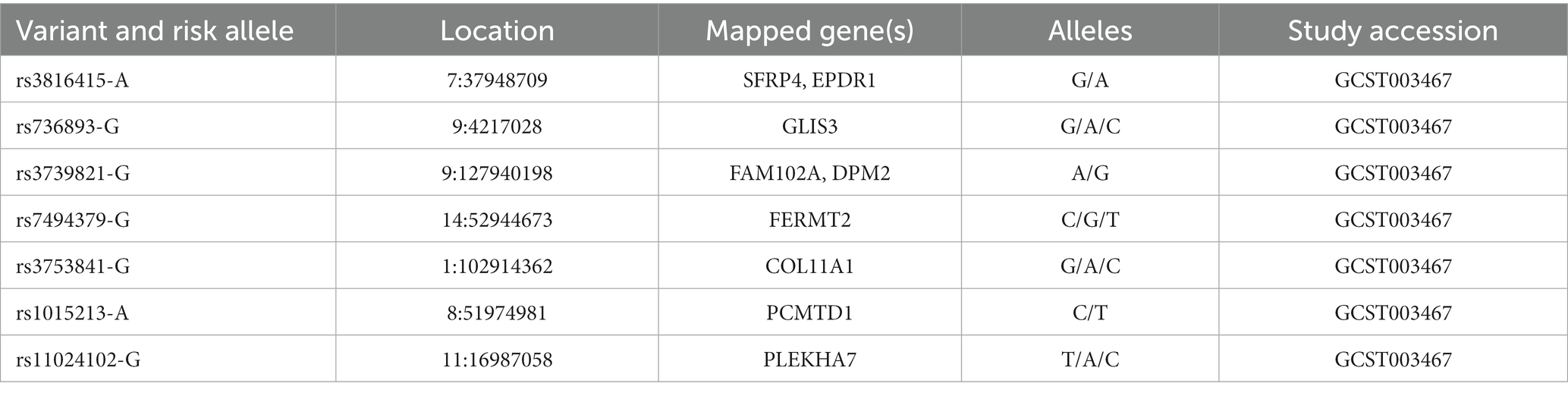
Table 5. SNPs of PACG susceptibility loci identified in different glaucoma cohorts (source: GWAS Catalog).
3.3 Primary angle closure glaucoma
Primary angle closure glaucoma (PACG) is characterized by at least 180° of iridotrabecular contact, resulting from mechanical obstruction of the TM in the anterior chamber angle and glaucomatous optic neuropathy due to elevated IOP. PACG pathogenesis can be explained as a pupillary block, which is the most common one, plateau iris or peripheral iris crowding, and multiple mechanism patterns, according to clinical phenotype, anatomic configurations, etiology and natural history (159).
From a clinical point of view, PACG is classified into acute, sub-acute, and chronic angle-closure glaucoma, based on the timing of onset, symptoms, and clinical findings. However, is not unusual to observe a combination of these clinical types in patients. Indeed, acute/subacute angle-closure glaucoma may progress to a chronic stage if it does not remit spontaneously or under medical intervention or vice-versa.
In acute glaucoma, the sudden elevation of IOP (usually more than 50 mmHg) leads to severe symptoms, including pressure-induced corneal edema (i.e., blurred vision and halos around lights), mid-dilated pupil, lens opacity (glaucomflecken), vascular congestion, headache, eye pain, and redness (160). The subacute presentation, which may occur before the acute attack or before peripheral anterior synechiae (in the case of chronic IOP elevation), can be asymptomatic or show mild prodromal symptoms, such as headache, blurred vision, and halos, coinciding with IOP spikes (161). Finally, in chronic angle-closure glaucoma, IOP is usually chronically raised and generally, no symptoms are experienced when vision loss occurs. The closure of the angle is due to peripheral anterior synechiae (more than 180 degrees), which occurs as a consequence of recurrent pupillary block or “creeping” angle closure (162, 163). Clinical goals of PACG management are reversing or preventing the angle-closure process, controlling IOP through topical ocular hypotensive agents, laser peripheral iridotomy or refractive lensectomy, and rescuing optic nerve from damage (164).
Diagnosis of PACG is associated with its natural course and is often delayed until an advanced phenotypic stage, i.e., when the patient has chronic visual loss or acute angle closure. Several risk factors, such as aging, female gender, Inuit, East Asian, and South Indians ethnicity, anatomical features (e.g., shallow anterior chamber depth, short axial length, thick and anteriorly positioned lens, plateau iris configuration, and hyperopia), family history (i.e., first-degree relatives), and environmental factors, are involved in promotion of the disease progression (165–167).
PACG is characterized by a complex inheritance, thus to increase knowledge of the common variant contribution, GWAS have genotyped eight SNPs of PACG susceptibility loci for the common adult-onset form of PACG within PLEKHA7 (pleckstrin homology domain containing A7), COL11A1 (collagen type XI alpha 1 chain), PCMTD1-ST18 [Protein-L-Isoaspartate (D-Aspartate) O-Methyltransferase Domain Containing] 1-ST18 C2H2C-type zinc finger transcription factor (PCMTD1-ST18), EPDR1 (ependymin related 1), CHAT (choline O-acetyltransferase), GLIS3 (GLIS family zinc finger 3), FERMT2 (fermitin family member 2), DPM2-FAM102A (dolichyl-phosphate mannosyltransferase subunit 2, regulatory and family sequence similarity 102 member A, respectively; Table 5) (14). However, these SNPs explain only 2% of the genetic variance in PACG. Thus, they could not be considered potential universal gene-dependent risk factors for PACG pathogenesis since the genotype distribution and allele frequency varies among populations of different ancestry. (168, 169).
4 Conclusion
Although impressive advancements in glaucoma study and treatment are progressing, it still represents the second leading cause of blindness worldwide. Only a small portion of rare early-onset glaucomas (before age 40) show Mendelian inheritance, while the common adult-onset forms are characterized by a complex inheritance.
GWAS studies aim to identify either genetic variants associated with disease or endophenotypes across ancestries and important functional insights underlying biological mechanisms of glaucoma pathogenesis. GWAS results are clinically important to identify subjects with an increased risk of the disease and they can be the starting point for studying the contribution of protein-changing, coding sequence genetic variants. Thus, next-generation sequencing technologies coupled with high throughput functional genomics can enrich our understanding of the disease pathophysiological mechanisms by identifying novel disease-associated or causative genetic variants for predicting glaucoma risk. However, although gene-based screening can be useful for genetic counseling and treatment and surveillance plans in families with a history of early-onset glaucoma, a limitation is a relatively low diagnostic yield after testing for the genes currently known (170). Moreover, gene-based screening for adult-onset glaucoma seems to be accurate when comprises a large number of risk alleles.
The study of the complex genetic background in glaucoma is in the making but in the future, the hope is that it will become a tool to assess personal risk for the disease and facilitate novel therapeutic strategies.
Author contributions
ST: Conceptualization, Writing – original draft, Writing – review & editing. CD: Supervision, Writing – review & editing. AB: Supervision, Writing – review & editing. SV: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
1. ^https://pubmed.ncbi.nlm.nih.gov/
2. ^https://gnomad.broadinstitute.org/
3. ^https://www.ebi.ac.uk/gwas/
4. ^https://www.ncbi.nlm.nih.gov/clinvar/?term=OPTN%5Bgene%5D&redir=gene
References
1. Tham, YC, Li, X, Wong, TY, Quigley, HA, Aung, T, and Cheng, CY. Global prevalence of Glaucoma and projections of Glaucoma burden through 2040. Ophthalmology. (2014) 121:2081–90. doi: 10.1016/j.ophtha.2014.05.013
2. Weinreb, RN, Aung, T, and Medeiros, FA. The pathophysiology and treatment of Glaucoma: a review. JAMA. (2014) 311:1901–11. doi: 10.1001/jama.2014.3192
3. Vernazza, S, Oddone, F, Tirendi, S, and Bassi, AM. Risk factors for retinal ganglion cell distress in Glaucoma and neuroprotective potential intervention. Int J Mol Sci. (2021) 22:7994. doi: 10.3390/ijms22157994
4. Gao, F, Wang, J, Chen, J, Wang, X, Chen, Y, and Sun, X. Etiologies and clinical characteristics of young patients with angle-closure glaucoma: a 15-year single-center retrospective study. Graefes Arch Clin Exp Ophthalmol. (2021) 259:2379–87. doi: 10.1007/s00417-021-05172-6
5. Sung, MS, Jin, HN, and Park, SW. Clinical features of advanced glaucoma with optic nerve head prelaminar schisis prelaminar schisis in advanced glaucoma. Am J Ophthalmol. (2021) 232:17–29. doi: 10.1016/j.ajo.2021.06.007
6. Elahi, E, Suri, F, and Yazdani, S. Glaucoma in Iran and contributions of studies in Iran to the understanding of the etiology of glaucoma. J Ophthalmic Vis Res. (2015) 10:68–76. doi: 10.4103/2008-322X.156120
7. Dastjerdi, S, Akgöz, B, and Civalek, Ö. On the shell model for human eye in Glaucoma disease. Int J Eng Sci. (2021) 158:103414. doi: 10.1016/j.ijengsci.2020.103414
8. Shin, HT, Yoon, BW, and Seo, JH. Analysis of risk allele frequencies of single nucleotide polymorphisms related to open-angle glaucoma in different ethnic groups. BMC Med Genet. (2021) 14:80. doi: 10.1186/s12920-021-00921-2
9. Sarfarazi, M, and Stoilov, I. Molecular genetics of primary congenital glaucoma. Eye. (2000) 14:422–8. doi: 10.1038/eye.2000.126
10. Varma, R, and Pan, Y. Natural history of glaucoma. Indian J Ophthalmol. (2011) 59:19–S23. doi: 10.4103/0301-4738.73682
11. Foster, PJ. The definition and classification of glaucoma in prevalence surveys. Br J Ophthalmol. (2002) 86:238–42. doi: 10.1136/bjo.86.2.238
12. Zhou, D, Mital, DP, Srinivasan, S, and Haque, S. Risk factors for open angle glaucoma â analyses using logistic regression. Int J Med Eng Inform. (2011) 3:203. doi: 10.1504/IJMEI.2011.042867
13. Dietze, J, Blair, K, and Havens, SJ. Glaucoma In:. StatPearls. Treasure Island, FL: StatPearls Publishing (2023)
14. Wiggs, JL, and Pasquale, LR. Genetics of glaucoma. Hum Mol Genet. (2017) 26:R21–7. doi: 10.1093/hmg/ddx184
15. Wiggs, JL. Genetic etiologies of Glaucoma. Arch Ophthalmol. (2007) 125:30. doi: 10.1001/archopht.125.1.30
16. Pagiatakis, C, Musolino, E, Gornati, R, Bernardini, G, and Papait, R. Epigenetics of aging and disease: a brief overview. Aging Clin Exp Res. (2021) 33:737–45. doi: 10.1007/s40520-019-01430-0
17. Turalba, AV, and Chen, TC. Clinical and genetic characteristics of primary juvenile-onset open-angle Glaucoma (JOAG). Semin Ophthalmol. (2008) 23:19–25. doi: 10.1080/08820530701745199
18. Jonas, JB, Budde, WM, and Panda-Jonas, S. Ophthalmoscopic evaluation of the optic nerve head. Surv Ophthalmol. (1999) 43:293–320. doi: 10.1016/S0039-6257(98)00049-6
19. Gedde, SJ, Vinod, K, Wright, MM, Muir, KW, Lind, JT, Chen, PP, et al. Primary open-angle Glaucoma preferred practice pattern®. Ophthalmology. (2021) 128:P71–P150. doi: 10.1016/j.ophtha.2020.10.022
20. Prum, BE, Rosenberg, LF, Gedde, SJ, Mansberger, SL, Stein, JD, Moroi, SE, et al. Primary open-angle Glaucoma preferred practice pattern® guidelines. Ophthalmology. (2016) 123:P41–P111. doi: 10.1016/j.ophtha.2015.10.053
21. Robin, A, and Covert, D. Does adjunctive Glaucoma therapy affect adherence to the initial primary therapy? Ophthalmology. (2005) 112:863–8. doi: 10.1016/j.ophtha.2004.12.026
22. Robin, AL, Novack, GD, Covert, DW, Crockett, RS, and Marcic, TS. Adherence in Glaucoma: objective measurements of once-daily and adjunctive medication use. Am J Ophthalmol. (2007) 144:533–540.e2. doi: 10.1016/j.ajo.2007.06.012
23. Wagner, IV, Stewart, MW, and Dorairaj, SK. Updates on the diagnosis and Management of Glaucoma. Mayo Clin Proc Innov Qual Outcomes. (2022) 6:618–35. doi: 10.1016/j.mayocpiqo.2022.09.007
24. Osman, W, Low, SK, Takahashi, A, Kubo, M, and Nakamura, Y. A genome-wide association study in the Japanese population confirms 9p21 and 14q23 as susceptibility loci for primary open angle glaucoma. Hum Mol Genet. (2012) 21:2836–42. doi: 10.1093/hmg/dds103
25. Welter, D, MacArthur, J, Morales, J, Burdett, T, Hall, P, Junkins, H, et al. The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. (2014) 42:D1001–6. doi: 10.1093/nar/gkt1229
26. Guevara-Fujita, ML, Perez-Grossmann, RA, Estrada-Cuzcano, A, Pawar, H, Vargas, E, Richards, JE, et al. Recurrent Myocilin Asn480Lys Glaucoma causative mutation arises De novo in a family of Andean descent. J Glaucoma. (2008) 17:67–72. doi: 10.1097/IJG.0b013e318133fc5b
27. Willoughby, CE, Chan, LLY, Herd, S, Billingsley, G, Noordeh, N, Levin, AV, et al. Defining the pathogenicity of Optineurin in juvenile open-angle Glaucoma. Investig Opthalmology Vis Sci. (2004) 45:3122–30. doi: 10.1167/iovs.04-0107
28. Trifan, OC, Traboulsi, EI, Stoilova, D, Alozie, I, Nguyen, R, Raja, S, et al. A third locus (GLC1D) for adult-onset primary open-angle glaucoma maps to the 8q23 region. Am J Ophthalmol. (1998) 126:17–28. doi: 10.1016/S0002-9394(98)00073-7
29. Liuska, PJ, Harju, M, Kivelä, TT, and Turunen, JA. Prevalence of MYOC risk variants for glaucoma in different populations. Acta Ophthalmol. (2021) 99:aos.14738. doi: 10.1111/aos.14738
30. Tran, JH, and Pasquale, LR. A comparison of genomic advances in exfoliation syndrome and primary open-angle Glaucoma. Curr Ophthalmol Rep. (2021) 9:96–106. doi: 10.1007/s40135-021-00270-4
31. Li, X, Xiao, X, Li, S, Sun, W, Wang, P, and Zhang, Q. Systemic genotype-phenotype analysis of MYOC variants based on exome sequencing and literature review. Asia-Pac J Ophthalmol. (2021) 10:173–82. doi: 10.1097/APO.0000000000000382
32. ANZRAG ConsortiumBailey, JNC, Loomis, SJ, Kang, JH, Allingham, RR, and Gharahkhani, P. Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat Genet. (2016) 48:189–94. doi: 10.1038/ng.3482
33. Choquet, H, Paylakhi, S, Kneeland, SC, Thai, KK, Hoffmann, TJ, Yin, J, et al. A multiethnic genome-wide association study of primary open-angle glaucoma identifies novel risk loci. Nat Commun. (2018) 9:2278. doi: 10.1038/s41467-018-04555-4
34. NEIGHBORHOOD consortium, ANZRAG consortium, Biobank Japan project, FinnGen study, UK Biobank Eye and Vision Consortium, GIGA study group. Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat Commun. (2021) 12:1–16. doi: 10.1038/s41467-020-20851-4
35. Shiga, Y, Akiyama, M, Nishiguchi, KM, Sato, K, Shimozawa, N, Takahashi, A, et al. Genome-wide association study identifies seven novel susceptibility loci for primary open-angle glaucoma. Hum Mol Genet. (2018) 27:1486–96. doi: 10.1093/hmg/ddy053
36. Huang, L, Chen, Y, Lin, Y, Tam, POS, Cheng, Y, Shi, Y, et al. Genome-wide analysis identified 17 new loci influencing intraocular pressure in Chinese population. Sci China Life Sci. (2019) 62:153–64. doi: 10.1007/s11427-018-9430-2
37. Thorleifsson, G, Walters, GB, Hewitt, AW, Masson, G, Helgason, A, DeWan, A, et al. Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma. Nat Genet. (2010) 42:906–9. doi: 10.1038/ng.661
38. Wellcome Trust Case Control Consortium 2NEIGHBORHOOD ConsortiumGharahkhani, P, Burdon, KP, Fogarty, R, Sharma, S, et al. Common variants near ABCA1, AFAP1 and GMDS confer risk of primary open-angle glaucoma. Nat Genet. (2014) 46:1120–5. doi: 10.1038/ng.3079,
39. NEIGHBORHOOD consortiumUK Biobank Eye and Vision ConsortiumCraig, JE, Han, X, Qassim, A, Hassall, M, et al. Multitrait analysis of glaucoma identifies new risk loci and enables polygenic prediction of disease susceptibility and progression. Nat Genet. (2020) 52:160–6. doi: 10.1038/s41588-019-0556-y
40. Zhou, W, Nielsen, JB, Fritsche, LG, Dey, R, Gabrielsen, ME, Wolford, BN, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. (2018) 50:1335–41. doi: 10.1038/s41588-018-0184-y
41. Yousefi, S, Gupta, K, Sun, J, Huang, X, Pasquale, L, Lu, L, et al. Novel genetic factors associated with primary open-angle Glaucoma identified using artificial intelligence. Invest Ophthalmol Vis Sci. (2021) 62:1491–1.
42. Petty, HR. Frontiers of complex disease mechanisms: membrane surface tension may link genotype to phenotype in Glaucoma. Front Cell Dev Biol. (2018) 6:32. doi: 10.3389/fcell.2018.00032
43. Burdon, KP, Macgregor, S, Hewitt, AW, Sharma, S, Chidlow, G, Mills, RA, et al. Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat Genet. (2011) 43:574–8. doi: 10.1038/ng.824
44. Bayat, B, Yazdani, S, Alavi, A, Chiani, M, Chitsazian, F, Tusi, BK, et al. Contributions of MYOC and CYP1B1 mutations to JOAG. Mol Vis. (2008) 14:508–17.
45. Souzeau, E, Burdon, KP, Ridge, B, Dubowsky, A, Ruddle, JB, and Craig, JE. A novel de novo Myocilin variant in a patient with sporadic juvenile open angle glaucoma. BMC Med Genet. (2016) 17:30. doi: 10.1186/s12881-016-0291-5
46. Kuchtey, J, Chowdhury, UR, Uptegraft, CC, Fautsch, MP, and Kuchtey, RW. A de novo MYOC mutation detected in juvenile open angle glaucoma associated with reduced myocilin protein in aqueous humor. Eur J Med Genet. (2013) 56:292–6. doi: 10.1016/j.ejmg.2013.03.002
47. Gupta, V, Somarajan, BI, Gupta, S, Chaurasia, AK, Kumar, S, Dutta, P, et al. The inheritance of juvenile onset primary open angle glaucoma: inheritance of JOAG. Clin Genet. (2017) 92:134–42. doi: 10.1111/cge.12906
48. Saeedi, O, Yousaf, S, Tsai, J, Palmer, K, Riazuddin, S, and Ahmed, Z. Delineation of novel compound heterozygous variants in LTBP2 associated with juvenile open angle Glaucoma. Gen. (2018) 9:527. doi: 10.3390/genes9110527
49. Somarajan, BI, Gupta, S, Mahalingam, K, Azmira, K, and Gupta, V. Digenic Inheritance in Juvenile Open-Angle Glaucoma. J Pediatr Genet. (2021) 12:150–4. doi: 10.1055/s-0040-1722213
50. Jafer Chardoub, AA, and Blair, K. StatPearls. Treasure Island (FL): StatPearls Publishing (2021).
51. Wiggs, JL. CPAMD8, a new gene for anterior segment dysgenesis and childhood Glaucoma. Ophthalmology. (2020) 127:767–8. doi: 10.1016/j.ophtha.2020.02.035
52. Birla, S, Gupta, D, Somarajan, BI, Gupta, S, Chaurasia, AK, Kishan, A, et al. Classifying juvenile onset primary open angle glaucoma using cluster analysis. Br J Ophthalmol. (2020) 104:827–35. doi: 10.1136/bjophthalmol-2019-314660
53. Selvan, H, Gupta, S, Wiggs, JL, and Gupta, V. Juvenile-onset open-angle glaucoma – a clinical and genetic update. Surv Ophthalmol. (2022) 67:1099–117. doi: 10.1016/j.survophthal.2021.09.001
54. Gupta, S, Singh, A, Mahalingam, K, Selvan, H, Gupta, P, Pandey, S, et al. Myopia and glaucoma progression among patients with juvenile onset open angle glaucoma: a retrospective follow up study. Ophthalmic Physiol Opt. (2021) 41:475–85. doi: 10.1111/opo.12805
55. Kwun, Y, Lee, EJ, Han, JC, and Kee, C. Clinical characteristics of juvenile-onset open angle Glaucoma. Korean J Ophthalmol. (2016) 30:127. doi: 10.3341/kjo.2016.30.2.127
56. Wang, Y, Wang, H, Han, Y, Shi, Y, Xin, C, Yin, P, et al. Outcomes of gonioscopy-assisted transluminal trabeculotomy in juvenile-onset primary open-angle glaucoma. Eye. (2021) 35:2848–54. doi: 10.1038/s41433-020-01320-0
57. Ishida, K, Mandal, A, and Netland, P. Glaucoma drainage implants in pediatric patients. Ophthalmol Clin North Am. (2005) 18:431–42. doi: 10.1016/j.ohc.2005.05.009
58. Hewitt, AW, Mackey, DA, and Craig, JE. Myocilin allele-specific glaucoma phenotype database. Hum Mutat. (2008) 29:207–11. doi: 10.1002/humu.20634
59. Sharma, R, and Grover, A. Myocilin-associated Glaucoma: A Historical Perspective and Recent Research Progress. Mol Vis. (2021) 14:480–493.
60. Zhang, Y, Li, S, Wen, X, Tong, H, Li, S, and Yan, Y. MYOC promotes the differentiation of C2C12 cells by regulation of the TGF-β signaling pathways via CAV1. Biology. (2021) 10:686. doi: 10.3390/biology10070686
61. Lieberman, RL. How does a protein’s structure spell the difference between health and disease? Our journey to understand glaucoma-associated myocilin. PLoS Biol. (2019) 17:e3000237. doi: 10.1371/journal.pbio.3000237
62. Fautsch, MP, Bahler, CK, Jewison, DJ, and Johnson, DH. Recombinant TIGR/MYOC increases outflow resistance in the human anterior segment. Invest Ophthalmol Vis Sci. (2000) 41:4163–8.
63. Fan, W, Li, W, Duan, C, Zhang, W, Guo, Y, and Chen, F. Characterization of a novel mutation in the MYOC gene in a Chinese family with primary open-angle glaucoma. Mol Med Rep. (2020) 22:3263–70. doi: 10.3892/mmr.2020.11441
64. Gould, DB, Miceli-Libby, L, Savinova, OV, Torrado, M, Tomarev, SI, Smith, RS, et al. Genetically increasing Myoc expression supports a necessary pathologic role of abnormal proteins in Glaucoma. Mol Cell Biol. (2004) 24:9019–25. doi: 10.1128/MCB.24.20.9019-9025.2004
65. Gould, DB, Reedy, M, Wilson, LA, Smith, RS, Johnson, RL, and John, SWM. Mutant Myocilin nonsecretion in vivo is not sufficient to cause Glaucoma. Mol Cell Biol. (2006) 26:8427–36. doi: 10.1128/MCB.01127-06
66. Fingert, JH, Stone, EM, Sheffield, VC, and Alward, WLM. Myocilin Glaucoma. Surv Ophthalmol. (2002) 47:547–61. doi: 10.1016/S0039-6257(02)00353-3
67. Patel, GC, Phan, TN, Maddineni, P, Kasetti, RB, Millar, JC, Clark, AF, et al. Dexamethasone-induced ocular hypertension in mice. Am J Pathol. (2017) 187:713–23. doi: 10.1016/j.ajpath.2016.12.003
68. Kasetti, RB, Maddineni, P, Millar, JC, Clark, AF, and Zode, GS. Increased synthesis and deposition of extracellular matrix proteins leads to endoplasmic reticulum stress in the trabecular meshwork. Sci Rep. (2017) 7:14951. doi: 10.1038/s41598-017-14938-0
69. Polansky, JR, Fauss, DJ, Chen, P, Chen, H, Lütjen-Drecoll, E, Johnson, D, et al. Cellular pharmacology and molecular biology of the trabecular meshwork inducible glucocorticoid response gene product. Ophthalmologica. (1997) 211:126–39. doi: 10.1159/000310780
70. Borrás, T, Rowlette, LLS, Tamm, ER, Gottanka, J, and Epstein, DL. Effects of elevated intraocular pressure on outflow facility and TIGR/MYOC expression in perfused human anterior segments. Invest Ophthalmol Vis Sci. (2002) 43:33–40.
71. Souzeau, E, Burdon, KP, Dubowsky, A, Grist, S, Usher, B, Fitzgerald, JT, et al. Higher prevalence of Myocilin mutations in advanced Glaucoma in comparison with less advanced disease in an Australasian disease registry. Ophthalmology. (2013) 120:1135–43. doi: 10.1016/j.ophtha.2012.11.029
72. Yaqoob Shahani, M, Memon, S, Ahmed Sheikh, S, Bano, U, Shaikh, P, Gul Memon, S, et al. Genetic heterogeneity of primary open-angle glaucoma in Pakistan. Saudi J Biol Sci. (2023) 30:103488. doi: 10.1016/j.sjbs.2022.103488
73. Craig, JE, Baird, PN, Healey, DL, McNaught, AI, McCartney, PJ, Rait, JL, et al. Evidence for genetic heterogeneity within eight glaucoma families, with the GLC1A Gln368STOP mutation being an important phenotypic modifier11None of the authors has a financial interest relating to this article. Ophthalmology. (2001) 108:1607–20. doi: 10.1016/S0161-6420(01)00654-6
74. Xie, JJ, Zhang, GW, Cui, HY, Li, N, Liu, XX, Hao, MY, et al. Penetrance of MYOC gene mutation in primary open-angle glaucoma: a systematic review and meta-analysis. Ophthalmic Genet. (2022) 43:240–7. doi: 10.1080/13816810.2021.2021427
75. Aroca-Aguilar, JD, Sánchez-Sánchez, F, Ghosh, S, Coca-Prados, M, and Escribano, J. Myocilin mutations causing Glaucoma inhibit the intracellular Endoproteolytic cleavage of Myocilin between amino acids Arg226 and Ile227. J Biol Chem. (2005) 280:21043–51. doi: 10.1074/jbc.M501340200
76. Zhou, A, Webb, G, Zhu, X, and Steiner, DF. Proteolytic processing in the secretory pathway. J Biol Chem. (1999) 274:20745–8. doi: 10.1074/jbc.274.30.20745
77. Thomas, G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. (2002) 3:753–66. doi: 10.1038/nrm934
78. Vincent, AL, Billingsley, G, Buys, Y, Levin, AV, Priston, M, Trope, G, et al. Digenic inheritance of early-onset Glaucoma: CYP1B1, a potential modifier gene. Am J Hum Genet. (2002) 70:448–60. doi: 10.1086/338709
79. Alward, WLM, Fingert, JH, Coote, MA, Johnson, AT, Lerner, SF, Junqua, D, et al. Clinical features associated with mutations in the chromosome 1 open-angle Glaucoma gene (GLC1A). N Engl J Med. (1998) 338:1022–7. doi: 10.1056/NEJM199804093381503
80. Gobeil, S, Letartre, L, and Raymond, V. Functional analysis of the glaucoma-causing TIGR/myocilin protein: integrity of amino-terminal coiled-coil regions and olfactomedin homology domain is essential for extracellular adhesion and secretion. Exp Eye Res. (2006) 82:1017–29. doi: 10.1016/j.exer.2005.11.002
81. Nakahara, E, and Hulleman, JD. A simple secretion assay for assessing new and existing Myocilin variants. Curr Eye Res. (2022) 47:918–22. doi: 10.1080/02713683.2022.2047205
82. Vasconcellos, JPC. Novel mutation in the MYOC gene in primary open angle glaucoma patients. J Med Genet. (2000) 37:301–3. doi: 10.1136/jmg.37.4.301
83. Jia, LY, Gong, B, Pang, CP, Huang, Y, Lam, DSC, Wang, N, et al. Correction of the disease phenotype of Myocilin-causing Glaucoma by a natural Osmolyte. Investig Opthalmology Vis Sci. (2009) 50:3743. doi: 10.1167/iovs.08-3151
84. Liu, Y, and Vollrath, D. Reversal of mutant myocilin non-secretion and cell killing: implications for glaucoma. Hum Mol Genet. (2004) 13:1193–204. doi: 10.1093/hmg/ddh128
85. Stoilov, I, Akarsu, AN, Alozie, I, Child, A, Barsoum-Homsy, M, Turacli, ME, et al. Sequence analysis and homology modeling suggest that primary congenital Glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved Core structures of cytochrome P4501B1. Am J Hum Genet. (1998) 62:573–84. doi: 10.1086/301764
86. Stothert, AR, Fontaine, SN, Sabbagh, JJ, and Dickey, CA. Targeting the ER-autophagy system in the trabecular meshwork to treat glaucoma. Exp Eye Res. (2016) 144:38–45. doi: 10.1016/j.exer.2015.08.017
87. Vollrath, D, and Liu, Y. Temperature sensitive secretion of mutant myocilins. Exp Eye Res. (2006) 82:1030–6. doi: 10.1016/j.exer.2005.10.007
88. Shimizu, S, Lichter, PR, Johnson, AT, Zhou, Z, Higashi, M, Gottfredsdottir, M, et al. Age-dependent prevalence of mutations at the GLC1A locus in primary open-angle glaucoma. Am J Ophthalmol. (2000) 130:165–77. doi: 10.1016/S0002-9394(00)00536-5
89. Angius, A. Myocilin Gln368stop mutation and advanced age as risk factors for late-onset primary open-angle Glaucoma. Arch Ophthalmol. (2000) 118:674–9. doi: 10.1001/archopht.118.5.674
90. Cheng, JW, Cheng, SW, Ma, XY, Cai, JP, Li, Y, Lu, GC, et al. Myocilin polymorphisms and primary open-angle Glaucoma: a systematic review and Meta-analysis. PloS One. (2012) 7:e46632. doi: 10.1371/journal.pone.0046632
91. Nag, A, Lu, H, Arno, M, Iglesias, AI, Bonnemaijer, P, Broer, L, et al. Evaluation of the Myocilin mutation Gln368Stop demonstrates reduced penetrance for Glaucoma in European populations. Ophthalmology. (2017) 124:547–53. doi: 10.1016/j.ophtha.2016.11.018
92. Burns, JN, Turnage, KC, Walker, CA, and Lieberman, RL. The stability of Myocilin Olfactomedin domain variants provides new insight into Glaucoma as a protein Misfolding disorder. Biochemistry. (2011) 50:5824–33. doi: 10.1021/bi200231x
93. Wang, H, Li, M, Zhang, Z, Xue, H, Chen, X, and Ji, Y. Physiological function of myocilin and its role in the pathogenesis of glaucoma in the trabecular meshwork (review). Int J Mol Med. (2018) 43:671–81. doi: 10.3892/ijmm.2018.3992
94. Fingert, JH. Penetrance of Myocilin mutations—who gets Glaucoma? JAMA Ophthalmol. (2019) 137:35–7. doi: 10.1001/jamaophthalmol.2018.4470
95. Liu, Y, and Allingham, RR. Major review: molecular genetics of primary open-angle glaucoma. Exp Eye Res. (2017) 160:62–84. doi: 10.1016/j.exer.2017.05.002
96. Takamoto, M, Kaburaki, T, Mabuchi, A, Araie, M, Amano, S, Aihara, M, et al. Common variants on chromosome 9p21 are associated with Normal tension Glaucoma. PloS One. (2012) 7:e40107. doi: 10.1371/journal.pone.0040107
97. Killer, H, and Pircher, A. Normal tension glaucoma: review of current understanding and mechanisms of the pathogenesis. Eye. (2018) 32:924–30. doi: 10.1038/s41433-018-0042-2
98. The Writing Committee fot the Normal Tension Glaucoma Genetic Study Group of Japan Glaucoma Society. Genome-wide association study of Normal tension Glaucoma: common variants in SRBD1 and ELOVL5 contribute to disease susceptibility. Ophthalmology. (2010) 117:1331–1338.e5. doi: 10.1016/j.ophtha.2009.12.001
99. Fox, AR, and Fingert, JH. Familial normal tension glaucoma genetics. Prog Retin Eye Res. (2023) 96:101191. doi: 10.1016/j.preteyeres.2023.101191
100. Rezaie, T, and Sarfarazi, M. Molecular cloning, genomic structure, and protein characterization of mouse optineurin☆. Genomics. (2005) 85:131–8. doi: 10.1016/j.ygeno.2004.10.011
101. Qiu, Y, Wang, J, Li, H, Yang, B, Wang, J, He, Q, et al. Emerging views of OPTN (optineurin) function in the autophagic process associated with disease. Autophagy. (2022) 18:73–85. doi: 10.1080/15548627.2021.1908722
102. Bansal, M, Swarup, G, and Balasubramanian, D. Functional analysis of optineurin and some of its disease-associated mutants: functional analysis of Optineurin and its disease-associated mutants. IUBMB Life. (2015) 67:120–8. doi: 10.1002/iub.1355
103. Minegishi, Y, Nakayama, M, Iejima, D, Kawase, K, and Iwata, T. Significance of optineurin mutations in glaucoma and other diseases. Prog Retin Eye Res. (2016) 55:149–81. doi: 10.1016/j.preteyeres.2016.08.002
104. Sirohi, K, and Swarup, G. Defects in autophagy caused by glaucoma-associated mutations in optineurin. Exp Eye Res. (2016) 144:54–63. doi: 10.1016/j.exer.2015.08.020
105. Park, HYL, Kim, JH, and Park, CK. Activation of autophagy induces retinal ganglion cell death in a chronic hypertensive glaucoma model. Cell Death Dis. (2012) 3:e290–11. doi: 10.1038/cddis.2012.26
106. Scheetz, TE, Roos, BR, Solivan-Timpe, F, Miller, K, DeLuca, AP, Stone, EM, et al. SQSTM1 mutations and Glaucoma. PloS One. (2016) 11:e0156001. doi: 10.1371/journal.pone.0156001
107. Wei, X, Cho, K, Thee, EF, Jager, MJ, and Chen, DF. Neuroinflammation and microglia in glaucoma: time for a paradigm shift. J Neurosci Res. (2019) 97:70–6. doi: 10.1002/jnr.24256
108. Ahmad, L, Zhang, SY, Casanova, JL, and Sancho-Shimizu, V. Human TBK1: a gatekeeper of Neuroinflammation. Trends Mol Med. (2016) 22:511–27. doi: 10.1016/j.molmed.2016.04.006
109. Rezaie, T, Child, A, Hitchings, R, Brice, G, Miller, L, Coca-Prados, M, et al. Adult-onset primary open-angle Glaucoma caused by mutations in Optineurin. Science. (2002) 295:1077–9. doi: 10.1126/science.1066901
110. Minegishi, Y, Iejima, D, Kobayashi, H, Chi, ZL, Kawase, K, Yamamoto, T, et al. Enhanced optineurin E50K–TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum Mol Genet. (2013) 22:3559–67. doi: 10.1093/hmg/ddt210
111. De Marco, N, Buono, M, Troise, F, and Diez-Roux, G. Optineurin increases cell survival and Translocates to the nucleus in a Rab8-dependent manner upon an apoptotic stimulus. J Biol Chem. (2006) 281:16147–56. doi: 10.1074/jbc.M601467200
112. Ayala-Lugo, RM, Pawar, H, Reed, DM, Lichter, PR, Moroi, SE, Page, M, et al. Variation in optineurin (OPTN) allele frequencies between and within populations. Mol Vis. (2007) 13:151–63.
113. Cheng, JW, Li, P, and Wei, RL. Meta-analysis of association between optineurin gene and primary open-angle glaucoma. Med Sci Monit. (2010) 16:CR369–77.
114. He, JN, Lu, S, Chen, LJ, Tam, POS, Zhang, BN, Leung, CKS, et al. Coding region mutation screening in Optineurin in Chinese Normal-tension Glaucoma patients. Dis Markers. (2019) 2019:1–5. doi: 10.1155/2019/5820537
115. Sears, NC, Boese, EA, Miller, MA, and Fingert, JH. Mendelian genes in primary open angle glaucoma. Exp Eye Res. (2019) 186:107702. doi: 10.1016/j.exer.2019.107702
116. Nykamp, K, Anderson, M, Powers, M, Garcia, J, Herrera, B, Ho, YY, et al. Sherloc: a comprehensive refinement of the ACMG–AMP variant classification criteria. Genet Med. (2017) 19:1105–17. doi: 10.1038/gim.2017.37
117. Abu-Amero, KK, Osman, EA, Mousa, A, Wheeler, J, Whigham, B, Allingham, RR, et al. Screening of CYP1B1 and LTBP2 genes in Saudi families with primary congenital glaucoma: genotype-phenotype correlation. Mol Vis. (2011) 17:2911–9.
118. Elder, MJ. Congenital glaucoma in the West Bank and Gaza strip. Br J Ophthalmol. (1993) 77:413–6. doi: 10.1136/bjo.77.7.413
119. Kipp, MA. Childhood glaucoma. Pediatr Clin North Am. (2003) 50:89–104. doi: 10.1016/S0031-3955(02)00110-4
120. Lee, HJ, Kim, YK, Jeoung, JW, Park, KH, and Kim, SJ. Visual outcomes and associated factors of primary congenital glaucoma in children. Graefes Arch Clin Exp Ophthalmol. (2021) 259:3445–51. doi: 10.1007/s00417-021-05232-x
121. Lewis, CJ, Hedberg-Buenz, A, DeLuca, AP, Stone, EM, Alward, WLM, and Fingert, JH. Primary congenital and developmental glaucomas. Hum Mol Genet. (2017) 26:R28–36. doi: 10.1093/hmg/ddx205
122. Sarfarazi, M, Akarsu, NA, Hossain, A, Turacli, EM, Aktan, GS, Barsoum-Homsy, M, et al. Assignment of a locus (GLC3A) for primary congenital Glaucoma (Buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics. (1995) 30:171–7. doi: 10.1006/geno.1995.9888
123. Akarsu, A. A second locus (GLC3B) for primary congenital glaucoma (Buphthalmos) maps to the 1p36 region. Hum Mol Genet. (1996) 5:1199–203. doi: 10.1093/hmg/5.8.1199
124. Yu Chan, JY, Choy, BN, Ng, AL, and Shum, JW. Review on the Management of Primary Congenital Glaucoma. J Curr Glaucoma Pract. (2015) 9:92–9. doi: 10.5005/jp-journals-10008-1192
125. Hoskins, HD, Shaffer, RN, and Hetherington, J. Anatomical classification of the developmental Glaucomas. Arch Ophthalmol. (1984) 102:1331–6. doi: 10.1001/archopht.1984.01040031081030
126. Mocan, MC, Mehta, AA, and Aref, AA. Update in genetics and surgical Management of Primary Congenital Glaucoma. Turk J Ophthalmol. (2019) 49:347–55. doi: 10.4274/tjo.galenos.2019.28828
127. Genčík, A, Genčíkova, A, and Ferák, V. Population genetical aspects of primary congenital glaucoma. I. Incidence, prevalence, gene frequency, and age of onset. Hum Genet. (1982) 61:193–7. doi: 10.1007/BF00296440
128. Leysen, L, Cassiman, C, Vermeer, S, Casteels, I, and Balikova, I. Genetics in primary congenital glaucoma: implications in disease management and counseling. Eur J Med Genet. (2022) 65:104378. doi: 10.1016/j.ejmg.2021.104378
129. Chakrabarti, S, Rao, K, and Nagireddy, S. Complex genetic mechanisms in glaucoma: an overview. Indian J Ophthalmol. (2011) 59:31–S42. doi: 10.4103/0301-4738.73685
130. Lang, E, Koller, S, Bähr, L, Töteberg-Harms, M, Atac, D, Roulez, F, et al. Exome sequencing in a Swiss childhood Glaucoma cohort reveals CYP1B1 and FOXC1 variants as Most frequent causes. Transl Vis Sci Technol. (2020) 9:47. doi: 10.1167/tvst.9.7.47
131. Bejjani, BA. Multiple CYP1B1 mutations and incomplete penetrance in an inbred population segregating primary congenital glaucoma suggest frequent de novo events and a dominant modifier locus. Hum Mol Genet. (2000) 9:367–74. doi: 10.1093/hmg/9.3.367
132. Bejjani, BA, Lewis, RA, Tomey, KF, Anderson, KL, Dueker, DK, Jabak, M, et al. Mutations in CYP1B1, the gene for cytochrome P4501B1, are the predominant cause of primary congenital Glaucoma in Saudi Arabia. Am J Hum Genet. (1998) 62:325–33. doi: 10.1086/301725
133. Banerjee, A, Chakraborty, S, Chakraborty, A, Chakrabarti, S, and Ray, K. Functional and structural analyses of CYP1B1 variants linked to congenital and adult-onset Glaucoma to investigate the molecular basis of these diseases. PloS One. (2016) 11:e0156252. doi: 10.1371/journal.pone.0156252
134. Chakrabarti, S, Ghanekar, Y, Kaur, K, Kaur, I, Mandal, AK, Rao, KN, et al. A polymorphism in the CYP1B1 promoter is functionally associated with primary congenital glaucoma. Hum Mol Genet. (2010) 19:4083–90. doi: 10.1093/hmg/ddq309
135. Melki, R. CYP1B1 mutations in French patients with early-onset primary open-angle glaucoma. J Med Genet. (2004) 41:647–51. doi: 10.1136/jmg.2004.020024
136. Panicker, SG, Mandal, AK, Reddy, ABM, Gothwal, VK, and Hasnain, SE. Correlations of genotype with phenotype in Indian patients with primary congenital Glaucoma. Investig Opthalmology Vis Sci. (2004) 45:1149–56. doi: 10.1167/iovs.03-0404
137. Plásilová, M, Stoilov, I, Sarfarazi, M, Kádasi, L, Feráková, E, and Ferák, V. Identification of a single ancestral CYP1B1 mutation in Slovak gypsies (Roms) affected with primary congenital glaucoma. J Med Genet. (1999) 36:290–4. doi: 10.1136/jmg.36.4.290
138. Lim, SH, Tran-Viet, KN, Yanovitch, TL, Freedman, SF, Klemm, T, Call, W, et al. CYP1B1, MYOC, and LTBP2 mutations in primary congenital glaucoma patients in the United States. Am J Ophthalmol. (2013) 155:508–517.e5. doi: 10.1016/j.ajo.2012.09.012
139. Chen, Y. CYP1B1 and MYOC mutations in 116 Chinese patients with primary congenital Glaucoma. Arch Ophthalmol. (2008) 126:1443. doi: 10.1001/archopht.126.10.1443
140. Waryah, YM, Iqbal, M, Sheikh, SA, Baig, MA, Narsani, AK, Atif, M, et al. Two novel variants in CYP1B1 gene: a major contributor of autosomal recessive primary congenital glaucoma with allelic heterogeneity in Pakistani patients. Int J Ophthalmol. (2019) 12:8–15. doi: 10.18240/ijo.2019.01.02
141. Chakrabarti, S, Kaur, K, Kaur, I, Mandal, AK, Parikh, RS, Thomas, R, et al. Globally, CYP1B1 mutations in primary congenital Glaucoma are strongly structured by geographic and haplotype backgrounds. Investig Opthalmology Vis Sci. (2006) 47:43. doi: 10.1167/iovs.05-0912
142. Kumar, A, Basavaraj, MG, Gupta, SK, Qamar, I, Ali, AM, Bajaj, V, et al. Role of CYP1B1, MYOC, OPTN, and OPTC genes in adult-onset primary open-angle glaucoma: predominance of CYP1B1 mutations in Indian patients. Mol Vis. (2007) 13:667–76.
143. Panicker, SG, Reddy, ABM, Mandal, AK, Ahmed, N, Nagarajaram, HA, Hasnain, SE, et al. Identification of novel mutations causing familial primary congenital glaucoma in Indian pedigrees. Invest Ophthalmol Vis Sci. (2002) 43:1358–66.
144. Chen, X, Chen, Y, Wang, L, Jiang, D, Wang, W, Xia, M, et al. CYP1B1 genotype influences the phenotype in primary congenital glaucoma and surgical treatment. Br J Ophthalmol. (2014) 98:246–51. doi: 10.1136/bjophthalmol-2013-303821
145. Chen, L, Huang, L, Zeng, A, and He, J. CYP1B1 gene mutations with incomplete penetrance in a Chinese pedigree with primary congenital glaucoma: a case report and review of literatures. Int J Clin Exp Med. (2015) 8:14538–41.
146. Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
147. Belmouden, A, Melki, R, Hamdani, M, Zaghloul, K, Amraoui, A, Nadifi, S, et al. A novel frameshift founder mutation in the cytochrome P450 1B1 (CYP1B1) gene is associated with primary congenital glaucoma in Morocco. Clin Genet. (2002) 62:334–9. doi: 10.1034/j.1399-0004.2002.620415.x
148. Al-Haddad, C, Abdulaal, M, Badra, R, Barikian, A, Noureddine, B, and Farra, C. Genotype/phenotype correlation in primary congenital Glaucoma patients in the Lebanese population: a pilot study. Ophthalmic Genet. (2014) 37:1–6. doi: 10.3109/13816810.2014.924015
149. Wang, LL, Li, Y, and Zhou, SF. A bioinformatics approach for the phenotype prediction of nonsynonymous single nucleotide polymorphisms in human cytochromes P450. Drug Metab Dispos. (2009) 37:977–91. doi: 10.1124/dmd.108.026047
150. Chakrabarti, S, Kaur, K, and Mandal, A. Primary congenital glaucoma and the involvement of CYP1B1. Middle East Afr J Ophthalmol. (2011) 18:7–16. doi: 10.4103/0974-9233.75878
151. Muskhelishvili, L, Thompson, PA, Kusewitt, DF, Wang, C, and Kadlubar, FF. In situ hybridization and Immunohistochemical analysis of cytochrome P450 1B1 expression in human Normal tissues. J Histochem Cytochem. (2001) 49:229–36. doi: 10.1177/002215540104900210
152. Sheibani, N, Zhao, Y, and Sorenson, C. Cytochrome P450 1B1 and primary congenital glaucoma. J Ophthalmic Vis Res. (2015) 10:60–7. doi: 10.4103/2008-322X.156116
153. Falero-Perez, J, Larsen, MC, Teixeira, LBC, Zhang, HF, Lindner, V, Sorenson, CM, et al. Targeted deletion of Cyp1b1 in pericytes results in attenuation of retinal neovascularization and trabecular meshwork dysgenesis. Trends Dev Biol. (2019) 12:1–12.
154. Haddad, A, Ait Boujmia, OK, El Maaloum, L, and Dehbi, H. Analysis of CYP1B1 gene mutations in primary congenital glaucoma patients. Eur J Ophthalmol. (2021) 31:2796–807. doi: 10.1177/11206721211016308
155. Shah, M, Bouhenni, R, and Benmerzouga, I. Geographical variability in CYP1B1 mutations in primary congenital Glaucoma. J Clin Med. (2022) 11:2048. doi: 10.3390/jcm11072048
156. Berraho, A, Serrou, A, Fritez, N, El Annas, A, Bencherifa, F, Gaboun, F, et al. Genotype-phenotype correlation in Moroccan patients with primary congenital glaucoma. J Glaucoma. (2015) 24:297–305. doi: 10.1097/IJG.0b013e31829f99b7
157. Shahid, M, Azfaralariff, A, Tufail, M, Hussain Khan, N, Abdulkareem Najm, A, Firasat, S, et al. Screening of high-risk deleterious missense variations in the CYP1B1 gene implicated in the pathogenesis of primary congenital glaucoma: a comprehensive in silico approach. PeerJ. (2022) 10:e14132. doi: 10.7717/peerj.14132
158. Firasat, S, Kaul, H, Ashfaq, UA, and Idrees, S. In silico analysis of five missense mutations in CYP1B1 gene in Pakistani families affected with primary congenital glaucoma. Int Ophthalmol. (2018) 38:807–14. doi: 10.1007/s10792-017-0508-4
159. Salmon, JF. Predisposing factors for chronic angle-closure glaucoma. Prog Retin Eye Res. (1999) 18:121–32. doi: 10.1016/S1350-9462(98)00007-X
160. Chan, PP, Pang, JC, and Tham, CC. Acute primary angle closure–treatment strategies, evidences and economical considerations. Eye. (2019) 33:110–9. doi: 10.1038/s41433-018-0278-x
161. Sihota, R. An Indian perspective on primary angle closure and glaucoma. Indian J Ophthalmol. (2011) 59:76–S81. doi: 10.4103/0301-4738.73687
162. Foulds, WS, and Phillips, CI. Some observations on chronic closed-angle GLAUCOMA. Br J Ophthalmol. (1957) 41:208–13. doi: 10.1136/bjo.41.4.208
163. Lowe, RF. Aetiology of the anatomical basis for primary angle-closure glaucoma. Biometrical comparisons between normal eyes and eyes with primary angle-closure glaucoma. Br J Ophthalmol. (1970) 54:161–9. doi: 10.1136/bjo.54.3.161
164. American Academy of ophthalmology. (2020). Primary Angle-Closure Disease PPP 2020. Available from: https://www.aao.org/education/preferred-practice-pattern/primary-angle-closure-disease-ppp
165. Amerasinghe, N, and Aung, T. Angle-closure: risk factors, diagnosis and treatment In:. Progress in brain research. Netherlands: Elsevier (2008). 31–45.
166. Thangavelu, L, Che Mat Nor, SM, Abd Aziz, D, Sulong, S, Tin, A, and Ahmad Tajudin, LS. Genetic markers PLEKHA7, ABCC5, and KALRN are not associated with the progression of primary angle closure Glaucoma (PACG) in Malays. Cureus. (2021). Available from: https://www.cureus.com/articles/73959-genetic-markers-plekha7-abcc5-and-kalrn-are-not-associated-with-the-progression-of-primary-angle-closure-glaucoma-pacg-in-malays
167. Shi, H, Chen, Y, Lu, H, Zhu, R, Zhang, J, He, M, et al. In-depth analysis of eight susceptibility loci of primary angle closure glaucoma in Han Chinese. Exp Eye Res. (2021) 202:108350. doi: 10.1016/j.exer.2020.108350
168. Aswa, M, Helmy, H, Noweir, S, Ismail, S, Taha, A, and Atef, A. Impact of rs11024102 PLEKHA7, rs3753841 COL11A1 single nucleotide polymorphisms, and serum levels of oxidative stress markers on the risk of primary angle-closure glaucoma in Egyptians. J Genet Eng Biotechnol. (2022) 20:126. doi: 10.1186/s43141-022-00400-w
169. Shuai, P, Yu, M, Li, X, Zhou, Y, Liu, X, Liu, Y, et al. Genetic associations in PLEKHA 7 and COL 11 a 1 with primary angle closure glaucoma: a meta-analysis: Meta-analysis of association in glaucoma. Clin Exp Ophthalmol. (2015) 43:523–30. doi: 10.1111/ceo.12516
Keywords: genetics, Glaucoma, GWAS, MYOC, OPTN, CYP1B1
Citation: Tirendi S, Domenicotti C, Bassi AM and Vernazza S (2023) Genetics and Glaucoma: the state of the art. Front. Med. 10:1289952. doi: 10.3389/fmed.2023.1289952
Edited by:
Gemma Caterina Maria Rossi, San Matteo Hospital Foundation (IRCCS), ItalyReviewed by:
Francesca Lazzara, University of Catania, ItalyRatnakar Tripathi, University of Missouri, United States
Copyright © 2023 Tirendi, Domenicotti, Bassi and Vernazza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefania Vernazza, c3RlZmFuaWEudmVybmF6emFAdW5pZ2UuaXQ=