 Xidan Zou1†
Xidan Zou1† Liping Ren2†
Liping Ren2† Peiling Cai3
Peiling Cai3 Yang Zhang4
Yang Zhang4 Hui Ding1
Hui Ding1 Kejun Deng1
Kejun Deng1 Xiaolong Yu5*
Xiaolong Yu5* Hao Lin1*
Hao Lin1* Chengbing Huang6*
Chengbing Huang6*- 1School of Life Science and Technology, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu, China
- 2School of Healthcare Technology, Chengdu Neusoft University, Chengdu, China
- 3School of Basic Medical Sciences, Chengdu University, Chengdu, China
- 4Innovative Institute of Chinese Medicine and Pharmacy, Academy for Interdiscipline, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 5School of Materials Science and Engineering, Hainan University, Haikou, China
- 6School of Computer Science and Technology, Aba Teachers University, Aba, China
Introduction: Hemagglutinin (HA) is responsible for facilitating viral entry and infection by promoting the fusion between the host membrane and the virus. Given its significance in the process of influenza virus infestation, HA has garnered attention as a target for influenza drug and vaccine development. Thus, accurately identifying HA is crucial for the development of targeted vaccine drugs. However, the identification of HA using in-silico methods is still lacking. This study aims to design a computational model to identify HA.
Methods: In this study, a benchmark dataset comprising 106 HA and 106 non-HA sequences were obtained from UniProt. Various sequence-based features were used to formulate samples. By perform feature optimization and inputting them four kinds of machine learning methods, we constructed an integrated classifier model using the stacking algorithm.
Results and discussion: The model achieved an accuracy of 95.85% and with an area under the receiver operating characteristic (ROC) curve of 0.9863 in the 5-fold cross-validation. In the independent test, the model exhibited an accuracy of 93.18% and with an area under the ROC curve of 0.9793. The code can be found from https://github.com/Zouxidan/HA_predict.git. The proposed model has excellent prediction performance. The model will provide convenience for biochemical scholars for the study of HA.
1. Introduction
Influenza is a contagious respiratory disease, posing a significant threat to human health and causing varying degrees of disease burden globally (1, 2). Hemagglutinin (HA), a glycoprotein on the surface of influenza viruses, mediates viral entry and infection by binding to host sialic acid receptors (3). The highly conserved stem or stalk region of HA has been identified as a promising target for the development of a universal influenza vaccine (4). Accurate identification of HA is crucial for targeted vaccine and drug development.
With the increasing maturity of protein sequence coding methods and machine learning algorithms, sequence-based protein recognition has been an effective approach for rapid identification of protein. It achieves classification and identification of specific proteins using protein sequence coding methods and machine learning algorithms, which has been widely used in the prediction studies of cell-penetrating peptides (5), hemolytic peptide (6), anti-cancer peptides (7), hormone proteins (8), autophagy proteins (9), and Anti-CRISPR proteins (10), etc., because of its high recognition accuracy in the protein identification study.
Despite the pivotal role of HA in influenza virus infection, existing machine learning-based research on HA has primarily focused on influenza virus subtype classification (11, 12), influenza virus host prediction (13), influenza virus mutation and evolution prediction (14), HA structure–function analysis (15), and influenza virus pathogenicity and prevalence prediction (16). However, there are currently no approaches for HA identification based on HA sequence information and machine learning techniques.
In this study, we proposed a machine learning-based prediction model for HA to achieve effective identification. Firstly, we constructed a benchmark dataset based on existing protein databases. Next, we employed feature extraction methods to encode the protein sequences. Subsequently, we fused all the extracted features and utilized the analysis of variance (ANOVA) combined with incremental feature selection (IFS) strategies to obtain the most informative feature subset. Finally, the HA prediction model was developed based on this optimal feature subset. The workflow is shown in Figure 1.
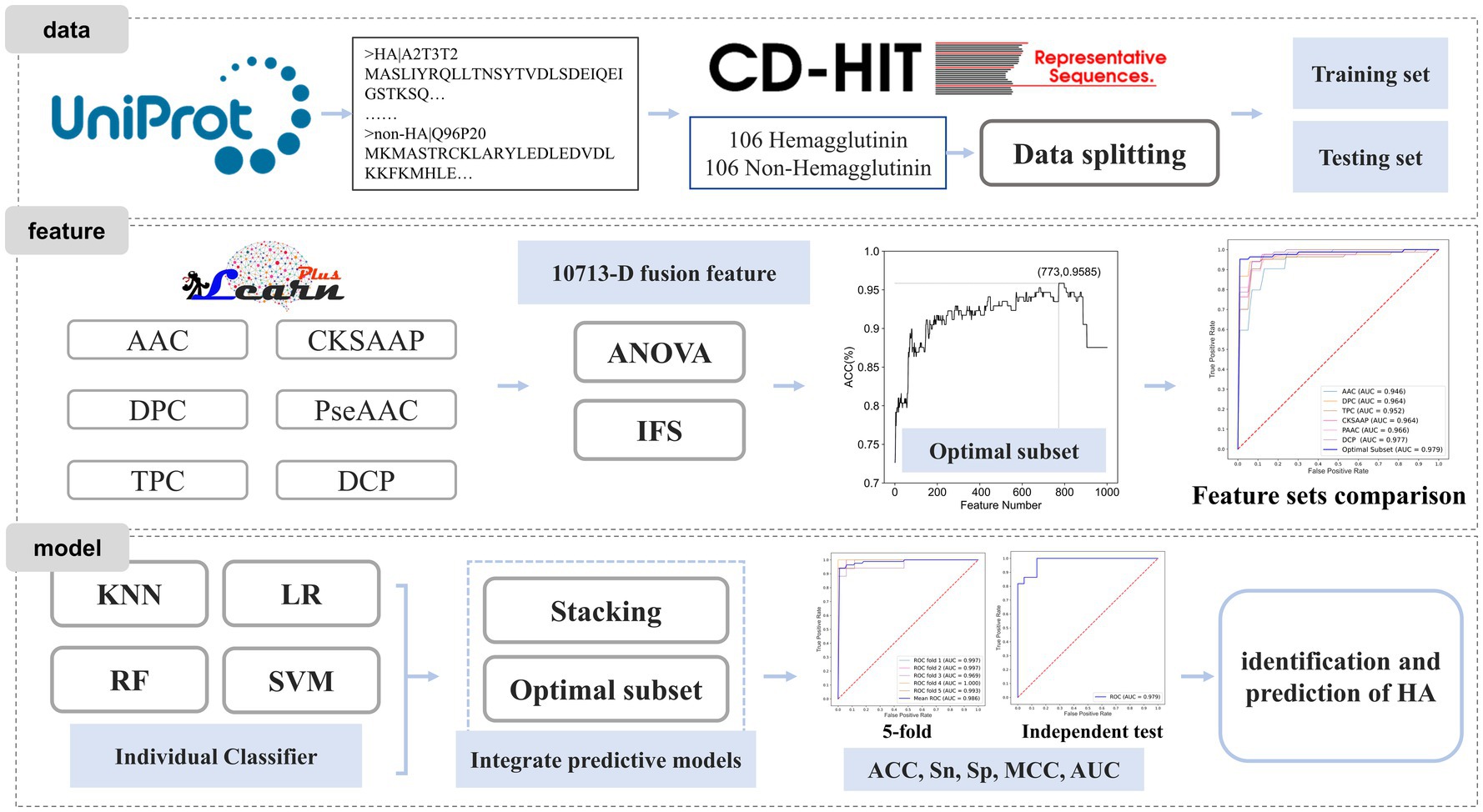
Figure 1. Workflow diagram for constructing the HA prediction model.
2. Method and materials
2.1. Benchmark dataset
A benchmark dataset is essential for bioinformatics analysis (17, 18). The dataset used in this study was collected from the Universal Protein Resource (UniProt) (19). To ensure the quality of the dataset, several pre-processing steps were performed. Protein sequences containing nonstandard letters (e.g., ‘B’, ‘U’, ‘X’, ‘Z’) were eliminated. Redundancy removal was done using CD-HIT (20) to remove sequences with high similarity. The cutoff value was set to 80%, and sequences with a similarity higher than 80% were removed. The non-HA dataset was down-sampled to ensure a balanced dataset with equal positive and negative samples. The final benchmark dataset consisted of 212 protein sequences, including 106 HA and 106 non-HA samples. The dataset was randomly split into a training dataset and a test dataset in a 4:1 ratio. The above-mentioned model training set data and test set data are included in https://github.com/Zouxidan/HA_predict.git. At the same time, a dataset named ‘predict_data.txt’ for testing is also included.
2.2. Feature extraction
Feature extraction plays a crucial role in protein identification and prediction (10, 21–25). However, machine learning algorithms cannot directly process protein sequence information for computation and model construction. Therefore, it is necessary to convert protein sequence information into numerical data that can be understood and utilized by machine learning algorithms (26–29). Here, we employed various methods for feature extraction of protein sequences, including Amino Acid Composition (AAC), Dipeptide Composition (DPC), Tripeptide Composition (TPC), Composition of k-spaced Amino Acid Pairs (CKSAAP), Pseudo-Amino Acid Composition (PseAAC), PseAAC of Distance-Pairs and Reduced Alphabet (DCP). These sequence feature extraction approaches have been widely adopted in the field of bioinformatics (30–32). The implementation of these feature extraction methods was based on iLearnPlus (33).
A protein sequence P of length L can be represented as:
where R1 denotes the first amino acid of the sequence, R2 denotes the second amino acid, and so on.
2.2.1. AAC
AAC is a commonly used method for protein sequence feature extraction, which involves 20 feature vectors. AAC was defined as:
where ai denotes the i-th natural amino acid and N(ai) denotes the frequency of amino acid ai in the protein sequence.
2.2.2. DPC
Similar to AAC, DPC counts the frequency of amino acids, but it focuses on the frequency of two adjacent amino acids in a protein sequence. DPC was defined as:
where (ai, aj) denotes two adjacent amino acids and N(ai, aj) denotes the frequency of the amino acid pair (ai, aj) in the protein sequence.
2.2.3. TPC
TPC is another feature extraction method that considers the relationship among three adjacent amino acids, providing more protein sequence information compared to AAC and DPC. TPC was defined as:
where (ai, aj, az) denotes the combination of three adjacent amino acids, and N(ai, aj, az) denotes the frequency of the tripeptide combination (ai, aj, az) in the protein sequence.
2.2.4. CKSAAP
To obtain further sequence information, Chen et al. proposed CKSAAP (34) which was defined as:
where k denotes the number of amino acids spaced between two amino acids, xk denotes k arbitrary amino acids, (ai, xk, aj) denotes the spaced amino acid pair, and N(ai, xk, aj) denotes the frequency of the spaced amino acid pair (ai, xk, aj) in the protein sequence.
2.2.5. PseAAC
To incorporate protein sequence ordinal information and improve prediction quality, a powerful feature, called PseAAC, was proposed, which incorporated the physicochemical characteristics of amino acids. PseAAC was defined as:
where xi denotes the normalized amino acid frequency, ω denotes the weight factor for short-range and long-range, and θj denotes the j-th sequence correlation factor.
θj was calculated as:
Θ(Ri + Ri + j) was defined as:
where H1(Ri), H2(Ri), and M(Ri) denote the standardized hydrophobicity, standardized hydrophilicity, and standardized side chain mass of the amino acid Ri, respectively.
The hydrophobicity, hydrophilicity, and side chain mass of amino acids were standardized using the following equations:
where H1(Ri), H2(Ri), and M(Ri) denote the standardized hydrophobicity, standardized hydrophilicity, and standardized side chain mass of amino acids, respectively, and , , and denote the corresponding raw physicochemical properties of amino acids.
2.2.6. DCP
To incorporate more protein sequence order information and reduce the impact of high-dimensional features, Liu et al. proposed DCP (35). Based on a validated amino acid simplification alphabet scheme (36), three simplified amino acid alphabets were defined as:
For any simplified amino acid alphabet, DCP was defined as:
where z denotes the number of amino acid clusters in the simplified alphabet, and denotes the frequency of any two amino acid clusters with distance d in the protein sequence.
In this study, the following parameters were used for protein sequence feature extraction: k = 1 for CKSAAP (amino acid spacing value), λ = 10 for PseAAC (number of amino acid theoretical properties), and ω = 0.7 for the weight factor for short-range and long-range. Consequently, we extracted features that include 20-dimensional AAC, 400-dimensional DPC, 8000-dimensional TPC, 800-dimensional CKSAAP, 30-dimensional PseAAC, and 1,463-dimensional DCP.
2.3. Feature fusion and selection
Different feature extraction methods offer diverse interpretations and representations of protein sequences. Relying solely on a single feature extraction method may limit the information provided by a single feature. To obtain a more comprehensive and reliable interpretation of protein sequences, we fused all features to create a fused feature set, resulting in a 10,713-dimensional feature set (20 + 400 + 8,000 + 800 + 30 + 1,463). We then selected the optimal feature subset using ANOVA and IFS.
ANOVA, a widely used feature selection tool, tests the difference in means between groups to determine whether the independent variable influences the dependent variable. Its high accuracy has made it an effective choice for feature selection (8). For a feature f, its F-value was calculated based on the principle of ANOVA as follows:
where F(f) represents the F-value of feature f, SSA represents the sum of squares between groups, SSE represents the sum of squares within groups, K-1 and N-K denote the degrees of freedom between and within groups, respectively. N is the total number of samples, and K is the number of groups.
SSA and SSE were calculated as follows:
where denotes the j-th feature of the i-th group, K represents the number of groups, and ki represents the total number of samples in the i-th group.
A larger F-value indicates a stronger influence of the feature on data classification, thereby contributing more to the data classification results. In the feature set, the large amount of data, redundant data and noise will not only result in higher computational costs, but also cause the phenomenon of overfitting or reduced accuracy of the prediction model. The above fusion feature set contains 10,713 features, which is a large number of features. For saving computational time and reducing computational cost, we firstly use ANOVA to initially filter to obtain the 1,000 features which have the greatest influence on the classification results.
Next, the optimal subset of features was determined by searching the top 1,000 features ranked by F-value using IFS. IFS is a frequently employed feature selection method in the field of bioinformatics (37, 38). The specific process of IFS is as follows. Firstly, all features were sorted in descending order according to their F-values obtained from ANOVA. Then, each feature was sequentially added to the feature set, and a model was constructed using support vector machine (SVM) for each newly formed feature subset. Grid search was utilized to obtain optimal models, and their performance was evaluated using 5-fold cross-validation. The optimal feature subset was defined as the set of features that maximized the model’s accuracy.
2.4. Machine learning methodology and modeling
The advancement of machine learning has provided an effective approach to solving biological problems (39–42). Utilizing machine learning techniques to identify proteins based on sequence features has proven to be a rapid and widely applied method in various studies (43–45).
Constructing appropriate models is crucial for achieving accurate and robust predictions. In this study, we selected four commonly used machine learning algorithms, namely K-nearest neighbor (KNN) (46), logistic regression (LR) (47), random forest (RF) (48), and SVM (49), to build the fundamental classifier model for the HA dataset. The optimal parameters for each algorithm were obtained using grid search. To further enhance the model’s accuracy and generalization ability, we developed an integrated classifier model by combining the four basic classifier models. The Stacking algorithm was employed, with logistic regression serving as the second-layer classifier. All the machine learning models utilized in this study were implemented using scikit-learn (50).
KNN is a simple yet effective machine learning algorithm based on the implementation of the distance between data and data. LR is a binary classification algorithm based on the sigmoid function, which classifies samples by their corresponding output values. In RF, the result of prediction is determined by the vote or average of decision trees. The basic principle of SVM is to separate two classes of training data by defining a hyperplane and maximizing the distance between the two classes.
The Stacking algorithm is one of the widely used integrated learning methods, which obtains predictive models with higher accuracy and better generalization ability by combining basic classifier models. The Stacking algorithm was initially proposed by Wolpert (51). Its basic idea is to obtain an optimal integrated classifier model by training and combining multiple basic classifier models. In the Stacking algorithm, machine learning algorithms with strong learning and fitting capabilities are frequently used to construct basic classifier models for adequate learning and interpretation of training data. To reduce the degree of overfitting, simple algorithms with strong interpretations are commonly used to construct integrated classifier models.
2.5. Performance evaluation
To assess the effectiveness of the constructed models, we employed 5-fold cross-validation and independent testing. The performance of the proposed model was evaluated using several metrics, including accuracy (ACC), sensitivity (Sn), specificity (Sp), Matthew’s correlation coefficient (MCC), and the area under the receiver operating characteristic curve (AUC) (27, 52–56). ACC, Sn, Sp, and MCC were expressed as:
where TP, TN, FP, and FN represent the following respectively: correctly identified positive samples, correctly identified negative samples, incorrectly identified negative samples, and incorrectly identified positive samples.
Additionally, we utilized the receiver operating characteristic (ROC) curve to evaluate model performance. A higher AUC value indicates better model performance, as it reflects the proximity to 1 according to the underlying principle.
3. Results and discussion
3.1. Optimal feature subset
We constructed optimal feature subsets using ANOVA and IFS and evaluated the models for each subset using the ACC. Figure 2A shows the IFS curve for the fusion feature set. When the feature set contained 773 features, the prediction model achieved a maximum ACC value of 0.9585.
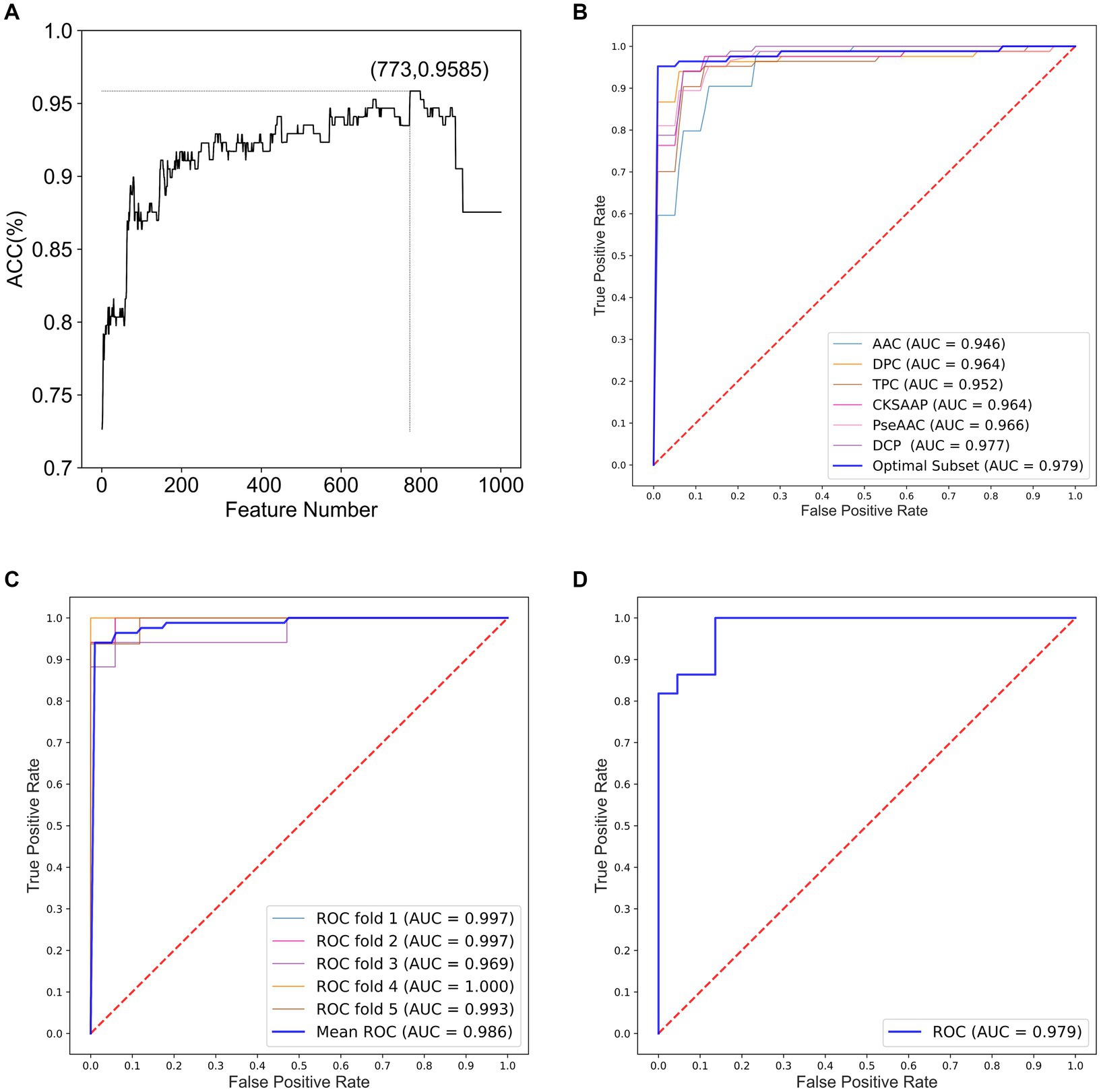
Figure 2. Performance analysis for optimal feature subsets and HA prediction models. (A) IFS curve for fusion features. (B) ROC curves of models constructed based on optimal feature subsets and six single feature sets. (C) ROC curves of the integrated classifier model with 5-fold cross-validation. (D) ROC curves of the integrated classifier model with independent testing.
In optimal feature subset, 13-dimensional AAC, 73-dimensional DPC, 629-dimensional TPC, 29-dimensional CKSAAP, and 29-dimensional DCP features are included. Notably, PseAAC is not included in this subset, suggesting that it is less effective in classifying HA compared to the other features. Furthermore, TPC has the highest proportion in optimal feature subset, indicating that TPC provides the best identification and differentiation ability among the six methods for feature extraction.
To demonstrate the impact of optimal feature subsets on model performance, we compared the performance of SVM prediction models constructed with optimal feature subsets to those constructed with six single feature sets. Each model was optimized using grid search within the same parameter range, and all models were evaluated using 5-fold cross-validation. Table 1 presents the results of the comparison, and Figure 2B shows the ROC curves for the 5-fold cross-validation of these models. The model constructed with the optimal feature subset achieved an ACC of 94.06% and an AUC of 0.970, outperforming the models constructed with other single feature sets. These results indicate that the optimal feature subset significantly improved the model’s prediction performance.
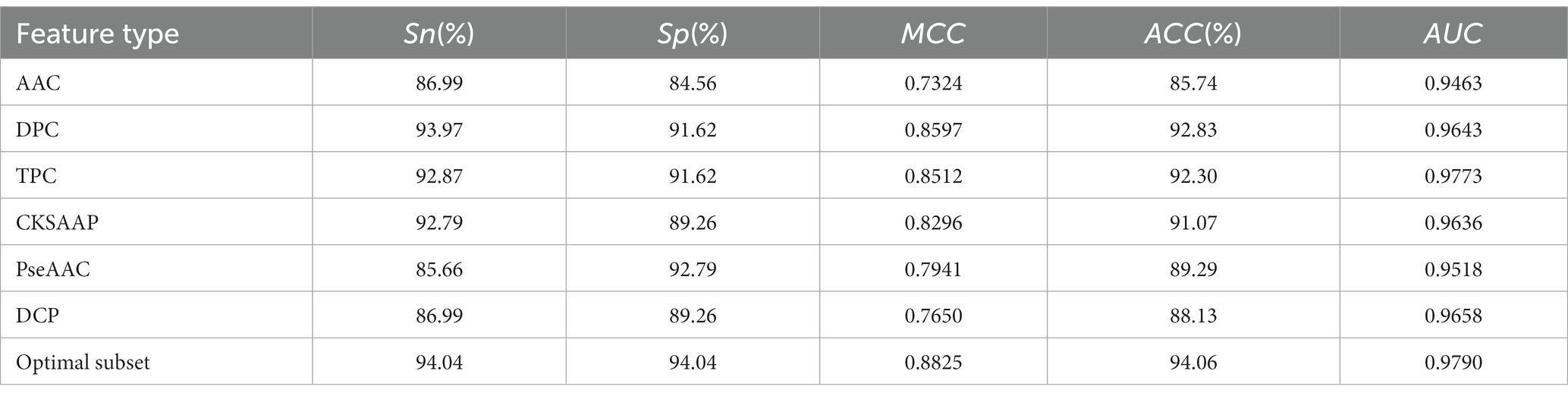
Table 1. Performance of models constructed based on optimal feature subsets and six single feature sets.
3.2. Model construction and evaluation
We constructed four basic models and an integrated model based on the optimal feature subsets. The optimal parameters for each algorithm were as follows: K = 52 for KNN, n = 62 for RF, f = 6 for the number of features considered during best-split search, ξ = 4 for the SVM kernel parameter, and C = 32 for the regularization parameter.
Table 2 presents the performance comparison of different classifier models using two testing methods. Figures 2C,D show the ROC curve of the constructed integration model using these wo testing methods. With 5-fold cross-validation, the proposed integrated model achieved an ACC of 95.85% and an AUC of 0.9863. On the independent test set, the integrated model achieved an ACC of 93.18% and an AUC of 0.9793. These results demonstrate that the proposed integrated model exhibited better HA prediction capability, improved model performance, and enhanced generalization ability compared to a single model.
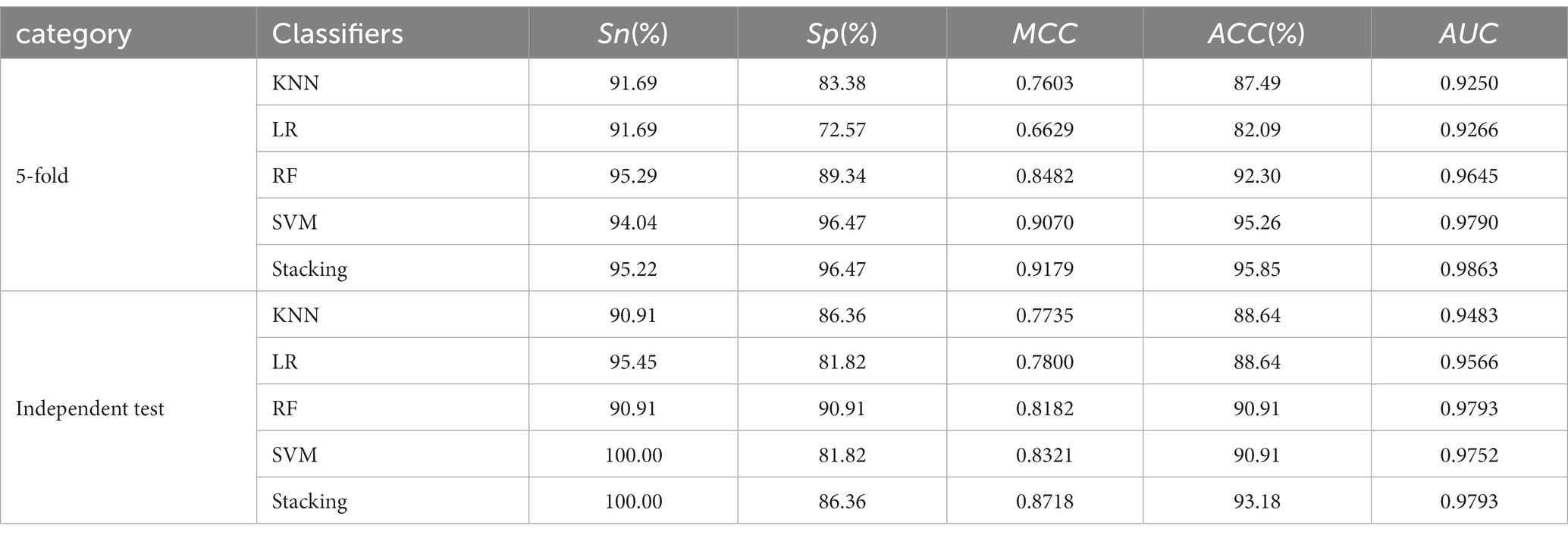
Table 2. Performance of the integrated classifier model and the four basic classifier models.
3.3. Comparison of other machine learning algorithms
We have created two models based on optimal feature subsets and compared their performance to demonstrate the superiority of our proposed model. The comparison results are presented in Table 3, where we compared the model constructed with the XGboost algorithm with our proposed model. The main parameters of the model constructed based on the XGboost algorithm are as follows: max_depth = 3, learning_rate = 0.16, colsample_bytree = 0.85, subsample = 0.75. The results in Table 3 show that our model has good classification performance.
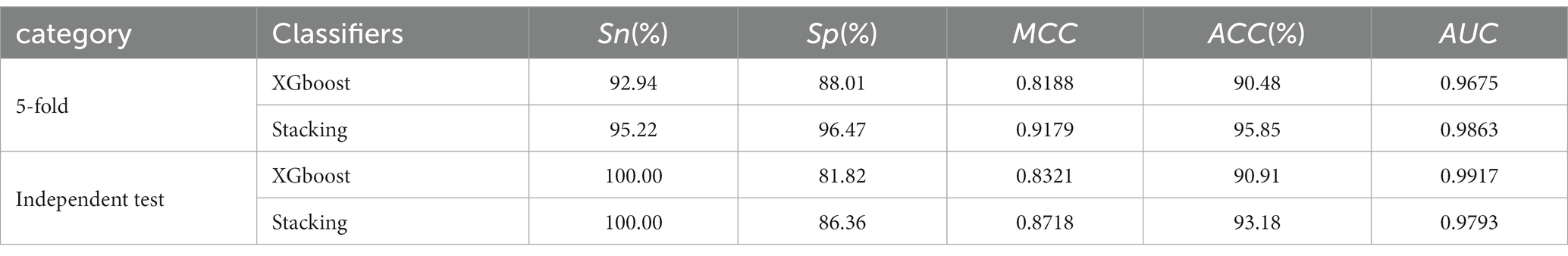
Table 3. Performance of the stacking classifier model and the XGboost classifier models.
3.4. Leave-one-out validation of the model
Due to the small sample data size, model robustness may be questioned. To ensure credible results, we use the leave-one-out method to re-validate model performance. The results of the model performance evaluation based on the leave-one-out method are shown in Table 4. In the performance evaluation of the model using the leave-one-out method, the model achieves an ACC of 93.45% and an AUC of 0.9846. The model shows good performance on both cross-validation methods, signifying its stability and the reliability of its classification outcomes.

Table 4. Performance evaluation based on the leave-one-out method.
4. Conclusion
Hemagglutinin (HA) is a vital glycoprotein found on the surface of influenza viruses, and accurately identifying HA is crucial for the development of targeted vaccine drugs. In this study, we proposed a prediction model based on HA protein sequence features. The model was constructed using the Stacking algorithm, incorporating an optimal subset of features and a basic classifier model. Our results demonstrated that the constructed model exhibits excellent predictive capacity and generalization ability.
We anticipate that the model will prove valuable in the effective identification and prediction of HA. Moving forward, we plan to explore additional feature extraction methods and optimize our prediction model to further enhance its performance. Additionally, we are committed to developing an accessible web server to facilitate the identification and prediction of HA.
In summary, our research provides a promising approach to accurately identifying HA and lays the foundation for the development of targeted vaccine drugs. We believe that our findings contribute to the advancement of influenza research and offer valuable insights for future studies in this field.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Author contributions
XZ: Data curation, Formal analysis, Methodology, Visualization, Writing – original draft. LR: Formal analysis, Investigation, Validation, Writing – review & editing. PC: Investigation, Validation, Writing – review & editing. YZ: Investigation, Software, Validation, Writing – review & editing. HD: Supervision, Validation, Writing – review & editing. KD: Data curation, Investigation, Supervision, Writing – review & editing. XY: Conceptualization, Writing – review & editing. HL: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing. CH: Conceptualization, Methodology, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (62250028 and 62202069), the Natural Science Foundation of Sichuan Province (2022NSFSC1610 and 2023NSFSC0678), and the China Postdoctoral Science Foundation (2023M730507).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer QT declared a shared affiliation with the author YZ to the handling editor at the time of review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Krammer, F, Smith, GJD, Fouchier, RAM, Peiris, M, Kedzierska, K, Doherty, PC, et al. Influenza. Nat Rev Dis Primers. (2018) 4:21. doi: 10.1038/s41572-018-0002-y
2. Uyeki, TM, Hui, DS, Zambon, M, Wentworth, DE, and Monto, AS. Influenza. Lancet. (2022) 400:693–706. doi: 10.1016/S0140-6736(22)00982-5
3. Skehel, JJ, and Wiley, DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. (2000) 69:531–69. doi: 10.1146/annurev.biochem.69.1.531
4. Nuwarda, RF, Alharbi, AA, and Kayser, V. An overview of influenza viruses and vaccines. Vaccine. (2021) 9:27. doi: 10.3390/vaccines9091032
5. Qiang, X, Zhou, C, Ye, X, Du, PF, Su, R, and Wei, L. CPPred-FL: a sequence-based predictor for large-scale identification of cell-penetrating peptides by feature representation learning. Brief Bioinform. (2020) 21:11–23. doi: 10.1093/bib/bby091
6. Hasan, MM, Schaduangrat, N, Basith, S, Lee, G, Shoombuatong, W, and Manavalan, B. HLPpred-fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics. (2020) 36:3350–6. doi: 10.1093/bioinformatics/btaa160
7. Wei, L, Zhou, C, Chen, H, Song, J, and Su, R. ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics. (2018) 34:4007–16. doi: 10.1093/bioinformatics/bty451
8. Tang, H, Zhao, YW, Zou, P, Zhang, CM, Chen, R, Huang, P, et al. HBPred: a tool to identify growth hormone-binding proteins. Int J Biol Sci. (2018) 14:957–64. doi: 10.7150/ijbs.24174
9. Jiao, S, Chen, Z, Zhang, L, Zhou, X, and Shi, L. ATGPred-FL: sequence-based prediction of autophagy proteins with feature representation learning. Amino Acids. (2022) 54:799–809. doi: 10.1007/s00726-022-03145-5
10. Dao, FY, Liu, ML, Su, W, Lv, H, Zhang, ZY, Lin, H, et al. AcrPred: A hybrid optimization with enumerated machine learning algorithm to predict anti-CRISPR proteins. Int J Biol Macromol. (2023) 228:706–14. doi: 10.1016/j.ijbiomac.2022.12.250
11. Cacciabue, M, and Marcone, DN. INFINITy: A fast machine learning-based application for human influenza A and B virus subtyping. Influenza Other Respir Viruses. (2023) 17:e13096. doi: 10.1111/irv.13096
12. Ao, C, Jiao, S, Wang, Y, Yu, L, and Zou, Q. Biological sequence classification: A review on data and general methods. Research. (2022) 2022:0011. doi: 10.34133/research.0011
13. Xu, Y, and Wojtczak, D. Dive into machine learning algorithms for influenza virus host prediction with hemagglutinin sequences. Biosystems. (2022) 220:104740. doi: 10.1016/j.biosystems.2022.104740
14. Yin, R, Thwin, NN, Zhuang, P, Lin, Z, and Kwoh, CK. IAV-CNN: A 2D convolutional neural network model to predict antigenic variants of influenza a virus. IEEE/ACM Trans Comput Biol Bioinform. (2022) 19:3497–506. doi: 10.1109/tcbb.2021.3108971
15. Wang, H, Zang, Y, Zhao, Y, Hao, D, Kang, Y, Zhang, J, et al. Sequence matching between hemagglutinin and neuraminidase through sequence analysis using machine learning. Viruses. (2022) 14:469. doi: 10.3390/v14030469
16. Kargarfard, F, Sami, A, Hemmatzadeh, F, and Ebrahimie, E. Identifying mutation positions in all segments of influenza genome enables better differentiation between pandemic and seasonal strains. Gene. (2019) 697:78–85. doi: 10.1016/j.gene.2019.01.014
17. Su, W, Liu, ML, Yang, YH, Wang, JS, Li, SH, Lv, H, et al. PPD: A manually curated database for experimentally verified prokaryotic promoters. J Mol Biol. (2021) 433:166860. doi: 10.1016/j.jmb.2021.166860
18. Wei, Y, Zou, Q, Tang, F, and Yu, L. WMSA: a novel method for multiple sequence alignment of DNA sequences. Bioinformatics. (2022) 38:5019–25. doi: 10.1093/bioinformatics/btac658
19. Bateman, A, Martin, MJ, Orchard, S, Magrane, M, Ahmad, S, Alpi, E, et al. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. (2022) 51:D523–31. doi: 10.1093/nar/gkac1052
20. Fu, LM, Niu, BF, Zhu, ZW, Wu, ST, and Li, WZ. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. (2012) 28:3150–2. doi: 10.1093/bioinformatics/bts565
21. Manavalan, B, and Patra, MC. MLCPP 2.0: an updated cell-penetrating peptides and their uptake efficiency predictor. J Mol Biol. (2022) 434:167604. doi: 10.1016/j.jmb.2022.167604
22. Shoombuatong, W, Basith, S, Pitti, T, Lee, G, and Manavalan, B. THRONE: A new approach for accurate prediction of human RNA N7-Methylguanosine sites. J Mol Biol. (2022) 434:167549. doi: 10.1016/j.jmb.2022.167549
23. Thi Phan, L, Woo Park, H, Pitti, T, Madhavan, T, Jeon, YJ, and Manavalan, B. MLACP 2.0: an updated machine learning tool for anticancer peptide prediction. Comput Struct Biotechnol J. (2022) 20:4473–80. doi: 10.1016/j.csbj.2022.07.043
24. Bupi, N, Sangaraju, VK, Phan, LT, Lal, A, Vo, TTB, Ho, PT, et al. An effective integrated machine learning framework for identifying severity of tomato yellow leaf curl virus and their experimental validation. Research. (2023) 6:0016. doi: 10.34133/research.0016
25. Ao, C, Ye, X, Sakurai, T, Zou, Q, and Yu, L. m5U-SVM: identification of RNA 5-methyluridine modification sites based on multi-view features of physicochemical features and distributed representation. BMC Biol. (2023) 21:93. doi: 10.1186/s12915-023-01596-0
26. Wang, YH, Zhang, YF, Zhang, Y, Gu, ZF, Zhang, ZY, Lin, H, et al. Identification of adaptor proteins using the ANOVA feature selection technique. Methods. (2022) 208:42–7. doi: 10.1016/j.ymeth.2022.10.008
27. Lv, H, Dao, F-Y, and Lin, H. DeepKla: an attention mechanism-based deep neural network for protein lysine lactylation site prediction. iMeta. (2022) 1:e11. doi: 10.1002/imt2.11
28. Yang, K, Li, M, Yu, L, and He, X. Repositioning linifanib as a potent anti-necroptosis agent for sepsis. bioRxiv. (2023) 9:57. doi: 10.1101/2022.03.24.485557
29. Wang, Y, Zhai, Y, Ding, Y, and Zou, Q. SBSM-pro: support bio-sequence machine for proteins. arXiv Preprint. (2023). doi: 10.48550/arXiv.2308.10275
30. Zhang, D, Xu, ZC, Su, W, Yang, YH, Lv, H, Yang, H, et al. iCarPS: a computational tool for identifying protein carbonylation sites by novel encoded features. Bioinformatics. (2020) 37:171–7. doi: 10.1093/bioinformatics/btaa702
31. Manavalan, B, Basith, S, Shin, TH, Wei, L, and Lee, G. Meta-4mCpred: A sequence-based Meta-predictor for accurate DNA 4mC site prediction using effective feature representation. Mol Ther Nucleic Acids. (2019) 16:733–44. doi: 10.1016/j.omtn.2019.04.019
32. Hasan, MM, Manavalan, B, Khatun, MS, and Kurata, H. i4mC-ROSE, a bioinformatics tool for the identification of DNA N4-methylcytosine sites in the Rosaceae genome. Int J Biol Macromol. (2020) 157:752–8. doi: 10.1016/j.ijbiomac.2019.12.009
33. Chen, Z, Zhao, P, Li, C, Li, FY, Xiang, DX, Chen, YZ, et al. iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. (2021) 49:e60. doi: 10.1093/nar/gkab122
34. Chen, K, Kurgan, L, and Rahbari, M. Prediction of protein crystallization using collocation of amino acid pairs. Biochem Biophys Res Commun. (2007) 355:764–9. doi: 10.1016/j.bbrc.2007.02.040
35. Liu, B, Xu, JH, Lan, X, Xu, RF, Zhou, JY, Wang, XL, et al. iDNA-Prot vertical bar dis: identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general Pseudo amino acid composition. PLoS One. (2014) 9:12. doi: 10.1371/journal.pone.0106691
36. Peterson, EL, Kondev, J, Theriot, JA, and Phillips, R. Reduced amino acid alphabets exhibit an improved sensitivity and selectivity in fold assignment. Bioinformatics. (2009) 25:1356–62. doi: 10.1093/bioinformatics/btp164
37. Dao, FY, Lv, H, Zhang, ZY, and Lin, H. BDselect: A package for k-mer selection based on the binomial distribution. Curr Bioinforma. (2022) 17:238–44. doi: 10.2174/1574893616666211007102747
38. Yang, H, Luo, Y, Ren, X, Wu, M, He, X, Peng, B, et al. Risk prediction of diabetes: big data mining with fusion of multifarious physical examination indicators. Inf. Fusion. (2021) 75:140–9. doi: 10.1016/j.inffus.2021.02.015
39. Charoenkwan, P, Chiangjong, W, Nantasenamat, C, Hasan, MM, Manavalan, B, and Shoombuatong, W. StackIL6: a stacking ensemble model for improving the prediction of IL-6 inducing peptides. Brief Bioinform. (2021) 22:bbab172. doi: 10.1093/bib/bbab172
40. Basith, S, Lee, G, and Manavalan, B. STALLION: a stacking-based ensemble learning framework for prokaryotic lysine acetylation site prediction. Brief Bioinform. (2022) 23:bbab376. doi: 10.1093/bib/bbab376
41. Hasan, MM, Tsukiyama, S, Cho, JY, Kurata, H, Alam, MA, Liu, X, et al. Deepm5C: A deep-learning-based hybrid framework for identifying human RNA N5-methylcytosine sites using a stacking strategy. Mol Ther. (2022) 30:2856–67. doi: 10.1016/j.ymthe.2022.05.001
42. Jeon, YJ, Hasan, MM, Park, HW, Lee, KW, and Manavalan, B. TACOS: a novel approach for accurate prediction of cell-specific long noncoding RNAs subcellular localization. Brief Bioinform. (2022) 23:bbac243. doi: 10.1093/bib/bbac243
43. Yuan, SS, Gao, D, Xie, XQ, Ma, CY, Su, W, Zhang, ZY, et al. IBPred: A sequence-based predictor for identifying ion binding protein in phage. Comput Struct Biotechnol J. (2022) 20:4942–51. doi: 10.1016/j.csbj.2022.08.053
44. Zhang, ZY, Ning, L, Ye, X, Yang, YH, Futamura, Y, Sakurai, T, et al. iLoc-miRNA: extracellular/intracellular miRNA prediction using deep BiLSTM with attention mechanism. Brief Bioinform. (2022) 23:bbac395. doi: 10.1093/bib/bbac395
45. Yang, Y, Gao, D, Xie, X, Qin, J, Li, J, Lin, H, et al. DeepIDC: A prediction framework of injectable drug combination based on heterogeneous information and deep learning. Clin Pharmacokinet. (2022) 61:1749–59. doi: 10.1007/s40262-022-01180-9
46. Cover, T, and Hart, P. Nearest neighbor pattern classification. IEEE Trans Inf Theory. (1967) 13:21–7. doi: 10.1109/TIT.1967.1053964
47. Freedman, DA. Statistical models: theory and practice. New York: Cambridge University Press (2005).
49. Cortes, C, and Vapnik, V. Support-Vector Networks. Mach Learn. (1995) 20:273–97. doi: 10.1007/bf00994018
50. Pedregosa, F, Varoquaux, G, Gramfort, A, Michel, V, Thirion, B, Grisel, O, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. (2011) 12:2825–30.
52. Sun, Z, Huang, Q, Yang, Y, Li, S, Lv, H, Zhang, Y, et al. PSnoD: identifying potential snoRNA-disease associations based on bounded nuclear norm regularization. Brief Bioinform. (2022) 23:bbac240. doi: 10.1093/bib/bbac240
53. Basith, S, Hasan, MM, Lee, G, Wei, L, and Manavalan, B. Integrative machine learning framework for the identification of cell-specific enhancers from the human genome. Brief Bioinform. (2021) 22:bbab252. doi: 10.1093/bib/bbab252
54. Manavalan, B, Basith, S, Shin, TH, and Lee, G. Computational prediction of species-specific yeast DNA replication origin via iterative feature representation. Brief Bioinform. (2021) 22:bbaa304. doi: 10.1093/bib/bbaa304
55. Wei, L, He, W, Malik, A, Su, R, Cui, L, and Manavalan, B. Computational prediction and interpretation of cell-specific replication origin sites from multiple eukaryotes by exploiting stacking framework. Brief Bioinform. (2021) 22:bbaa275. doi: 10.1093/bib/bbaa275
Keywords: hemagglutinin, machine learning, sequence features, feature extraction, stacking
Citation: Zou X, Ren L, Cai P, Zhang Y, Ding H, Deng K, Yu X, Lin H and Huang C (2023) Accurately identifying hemagglutinin using sequence information and machine learning methods. Front. Med. 10:1281880. doi: 10.3389/fmed.2023.1281880
Edited by:
J. Francis Borgio, Imam Abdulrahman Bin Faisal University, Saudi ArabiaReviewed by:
Qiang Tang, Chengdu University of Traditional Chinese Medicine, ChinaJiesi Luo, Southwest Medical University, China
Ke Han, Harbin University of Commerce, China
Zhijun Liao, Fujian Medical University, China
Copyright © 2023 Zou, Ren, Cai, Zhang, Ding, Deng, Yu, Lin and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaolong Yu, eXV4aWFvbG9uZ0BoYWluYW51LmVkdS5jbg==; Hao Lin, aGxpbkB1ZXN0Yy5lZHUuY24=; Chengbing Huang, MjAwNDk2MDdAYWJ0dS5lZHUuY24=
†These authors have contributed equally to this work