
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 10 October 2023
Sec. Pulmonary Medicine
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1275684
This article is part of the Research TopicPulmonary Hypertension in the Modern Era: Science and Clinical Practice, Volume IIIView all 5 articles
 Abdullah A. Alqarni1,2*
Abdullah A. Alqarni1,2* Abdulelah M. Aldhahir3
Abdulelah M. Aldhahir3 Sara A. Alghamdi4
Sara A. Alghamdi4 Jaber S. Alqahtani5
Jaber S. Alqahtani5 Rayan A. Siraj6Hassan Alwafi7Abdulkareem A. AlGarni8,9
Rayan A. Siraj6Hassan Alwafi7Abdulkareem A. AlGarni8,9 Mansour S. Majrshi10,11
Mansour S. Majrshi10,11 Saad M. Alshehri12Linhua Pang13
Saad M. Alshehri12Linhua Pang13Pulmonary hypertension (PH) due to chronic obstructive pulmonary disease (COPD) is classified as Group 3 PH, with no current proven targeted therapies. Studies suggest that cigarette smoke, the most risk factor for COPD can cause vascular remodelling and eventually PH as a result of dysfunction and proliferation of pulmonary artery smooth muscle cells (PASMCs) and pulmonary artery endothelial cells (PAECs). In addition, hypoxia is a known driver of pulmonary vascular remodelling in COPD, and it is also thought that the presence of hypoxia in patients with COPD may further exaggerate cigarette smoke-induced vascular remodelling; however, the underlying cause is not fully understood. Three main pathways (prostanoids, nitric oxide and endothelin) are currently used as a therapeutic target for the treatment of patients with different groups of PH. However, drugs targeting these three pathways are not approved for patients with COPD-associated PH due to lack of evidence. Thus, this review aims to shed light on the role of impaired prostanoids, nitric oxide and endothelin pathways in cigarette smoke- and hypoxia-induced pulmonary vascular remodelling and also discusses the potential of using these pathways as therapeutic target for patients with PH secondary to COPD.
Pulmonary hypertension (PH) is a clinical condition defined based on haemodynamic instabilities presented in an increased vascular resistance within pulmonary arteries and is mainly characterised by a mean pulmonary artery pressure (mPAP) that is greater than 20 mmHg and a pulmonary vascular resistance (PVR) greater than 2 Wood Units, as per the new updated ESC/ERS guidelines (1). PH could occur in association with other diseases or isolated for idiopathic purposes, based on which PH is classified (2, 3). This classification divides PH into 5 main groups.
PH due to lung diseases, such as chronic obstructive pulmonary disease (COPD) or interstitial lung disease, is classified as group 3 PH (4). Studies on group 3 PH are not very extensive which makes it an area suitable for further exploration (5–8). COPD is one of the lung diseases that commonly develop into PH (9). Additionally, COPD is a leading cause of morbidity and mortality worldwide and a major cause of disability-adjusted life-years lost worldwide (10, 11). The American Thoracic Society defines COPD as a preventable and treatable disease state characterised by airflow limitation that is not fully reversible, which is progressive and associated with a chronic inflammation response of the lungs to noxious particles or gases (10). COPD is mainly caused by cigarette smoking, but it could also be caused by exposure to biomass fuel combustion, autoimmunity, or chronic infection (10, 12, 13).
Previous studies have reported that the prevalence of PH in patients with COPD varies between 20.5 and 90.8% (14–22). PH is found in advanced COPD, and it is a serious complication involving haemodynamic instabilities leading to frequent exacerbation episodes and decrease in survival rate (5, 9, 23). There is no complete understanding of the pathogenesis of PH in COPD patients; however, studies have shown that PH is characterised in COPD patients by hypertrophy of the pulmonary arterial walls and loss of capillary beds (24). Medium to small pulmonary vasculatures have not been vastly studied in regards of their remodelling. Nevertheless, it is known that such remodelling is linked to maladaptation of endothelial cells to chronic cigarette smoking with increased release of vasoconstrictors and reduced releases of vasodilators and it occurs in all affected pulmonary vessels with varying severities regardless of their size (25).
Hypoxia and inflammation are also drivers of vasculature remodelling and development of PH (26). Due to the damage in alveolar walls caused by alveolar hypoxia, vasoconstriction of the surrounding arteries occurs to maintain ventilation and perfusion balance (8). Consequently, developing PH. However, PH can still occur in COPD patients without hypoxemia (6). Alongside the main symptoms COPD patients experience, which include cough, dyspnea, shortness of breath, and sputum production, the development of PH will cause some haemodynamic symptoms as well (23). Moreover, PH in COPD is known to increase mPAP up to 35 mmHg and higher in some severe cases. Consequently, increasing morbidity and mortality and significantly reducing the quality of life (27).
Together with the treatments of COPD and pulmonary rehabilitation, different COPD-associated PH therapies have been experimented with including supplemental oxygen, calcium channel blockers, statins, and type 1 PH targeted therapies. To date, there has not been any type of therapy to be recognised as the therapy of choice for COPD-associated PH patients. However, some therapies have only presented hemodynamic improvement with inconsistent functional or clinical effects (28). Clinicians have adopted medications approved for group 1 PH, including endothelin receptor antagonist (ERA) and phosphodiesterase-5 inhibitors (PDE5i), nitric oxide, and prostaglandin I2 (PGI2) analogue, for COPD-associated PH (8, 29). Randomised clinical trials have been done to evaluate the efficacy of group 1 PH therapy on COPD-associated PH patients, most of which were done on PH cases of mild to moderate severity. However, results have not been effective enough as minimal to no effect was seen on exercise capacities as well as limited efficacy on improving the quality of life in such patients (27, 28). Furthermore, hemodynamic status and survival have been found to improve in moderate to severe cases in studies with mixed populations including multiple pulmonary diseases (27, 28). Such results of group 1 PH medications in COPD-associated PH cases are insufficient and conflicting making further exploration of this topic an absolute necessity (6, 27, 28).
The hallmark of all forms of PH is thought to be pulmonary artery vasoconstriction and remodelling of the pulmonary vessel wall, including dysfunction and proliferation of pulmonary artery cells. Although the pathophysiological mechanisms of COPD in PH are not fully understood, there has been a re-emerging interest in studying the leading causes of PH in COPD with the focus on trying to find the best therapeutic targets for such cases. It is suggested that PH complicating COPD occurs as a consequence of the combined effects of inflammation, cigarette smoke (CS), and hypoxia, which could then lead to elevated PVR and eventually contribute to PH in COPD patients (30). Given that the role of inflammation in inducing pulmonary vascular remodelling has been described elsewhere (31, 32), this review mainly focuses on the roles of hypoxia and CS in the process of pulmonary vascular remodelling in COPD.
Pulmonary artery vasoconstriction induced by alveolar hypoxia is a reflex contraction of pulmonary artery smooth muscle cells (PASMCs). This hypoxic pulmonary vasoconstriction is a vasoprotective response to hypoxia to maintain ventilation-perfusion balance, as blood flow in the pulmonary arteries of COPD patients is shifted from hypoxic alveoli toward normoxic alveoli to help minimise ventilation-perfusion mismatch. As the disease progresses, persistent hypoxia (resulting from inadequate delivery of oxygen to the tissues) is thought to lead to elevated PVR and PH in COPD (33).
Over the past two decades, substantially more information has become available and the traditional view that suggests the elevated PVR in COPD can only be a result of hypoxic pulmonary vasoconstriction has been challenged (34, 35). Recent evidence shows that endothelial dysfunction (driven by CS, inflammation, and chronic hypoxia in COPD) also plays a key role in the cause of pulmonary vasoconstriction and pulmonary vascular remodelling in COPD patients (35–38). This suggests that CS can be the primary driver of pulmonary vascular remodelling in COPD (39), although hypoxia is definitely known to induce pulmonary vascular remodelling. Today, the exact mechanistic basis of CS and hypoxia, either individually or in combination, on the induction of pulmonary artery dysfunction and remodelling in COPD is still unclear.
Pulmonary vascular remodelling refers to the key structural changes in the vascular wall that eventually contribute to elevated PAP by increasing PVR in all forms of PH, including COPD-associated PH (40, 41). Pulmonary vascular abnormalities have been attributed to the proliferation of endothelial, smooth muscle, and fibroblast cells, which are cellular components of the three layers of pulmonary vascular wall: intima, media, and adventitia, respectively (42).
The intima is composed predominantly of endothelial cells, and plays an important role in regulating the vascular tone and controlling cell growth. Thickness of the intima layer has been associated with endothelial dysfunction in the pulmonary arteries of COPD patients, which may initiate the process of vascular remodelling (43). In addition, medial wall thickening, caused by proliferative smooth muscle cells, has been observed in patients with COPD (44). More importantly, histochemical and immunohistochemical studies performed in the vasculature of patients with COPD have shown increased number of smooth muscle cells (but not fibroblasts) in the enlarged intima as a result of staining the intimal layer by specific smooth muscle cell and fibroblast markers (31, 41, 43). The proliferation of smooth muscle cells observed in the thickened intima of patients with COPD occurred in an inward direction, thus reducing the vascular lumen (31). This suggests that PASMCs play an important role, in addition to PAECs, in the development of COPD-associated PH.
Although the key signalling pathways involved in the remodelling process are unknown, the pathophysiological changes reported in the innermost two layers of pulmonary artery in COPD, the media and intima, are believed to occur as a result of PASMC proliferation due to PAEC dysfunction (41, 43). Therefore, PASMCs and PAECs are considered the two key cell types that play a major role in the pathophysiology of COPD-associated PH.
PAECs are located in the intimal layer of pulmonary arteries that line the vascular lumen and play a key role in controlling cell growth and regulating vascular tone. Apoptosis-resistant and hyper-proliferative PAECs have been reported in patients with group 1 PH (45). It has also been shown that vascular endothelial growth factor (VEGF) receptor inhibitor in combination with chronic hypoxia can cause vascular endothelial cells apoptosis. This initial apoptosis is followed by proliferative vascular endothelial cells in rat models of PH (46), suggesting that the emergence of apoptosis-resistant proliferating vascular endothelial cells may play an important role in vascular remodelling and the development of PH. This is also supported by a more recent study showing that the initial apoptosis, induced by VEGF receptor inhibitor, is followed by increased proliferation of apoptosis-resistant human pulmonary microvascular endothelial cells (47). The accumulative evidence suggests that proliferative PAECs play a role in the pulmonary vascular remodelling in group 1 PH. However, whether PAEC proliferation plays a role in the development of COPD-associated vascular remodelling and PH is largely unknown.
It has been reported that pulmonary vascular remodelling is observed in pulmonary vessels of COPD patients with hypoxemia (48). It has also been suggested that the degree of PH in COPD is directly related to the severity of hypoxemia, and the alteration of pulmonary vessels in COPD at a late stage of the disease is thought to result from chronic hypoxia (44), indicating that hypoxia is an important contributor to vascular remodelling in COPD. This is supported by human and animal cellular experimental studies that demonstrate induction of PASMC proliferation in response to hypoxia. For example, hypoxia has been shown to induce proliferation of human PASMCs (48–50) and rat PASMCs (51). Similarly, experimental studies have also shown that hypoxia can induce proliferation of human PAECs (52, 53). However, there are conflicting data with regards to the direct effect of hypoxia on the proliferation of PAECs in human and animal cellular experimental models. It has been shown that hypoxia alone does not stimulate proliferation of PAECs in human cellular experimental models of hypoxia-induced PH, as well as in hypoxic models of mice and rats (49). These rather contradictory observations may be due to methodological factors such as differences in exposure time and oxygen concentrations. Although the most recent data suggest that hypoxia is a direct stimulus of PASMC and PAEC proliferation, the underlying cause that leads to hypoxia-induced proliferation of PASMCs and PAECs is unclear.
CS is considered the most common etiological factor in developing COPD (54). Although its role in COPD-associated PH is not well characterised, the traditional view that PH in COPD is secondary to chronic hypoxia and emphysema is challenged by clinical and experimental evidence (55). Apparent vascular remodelling has been demonstrated in “healthy” smokers and patients with mild-to-moderate COPD, despite the fact that PH is usually diagnosed in patients with advanced COPD (56). Furthermore, thickness of vascular layers has been observed in patients with COPD and smokers with no sign of airway obstruction (41, 57, 58), suggesting CS plays an important role in vascular remodelling in COPD. These findings are supported by experimental studies showing that CS extract directly stimulates proliferation of human PASMCs (59, 60) as well as PAECs (60), which ultimately contributes to pulmonary vascular remodelling. Interestingly, more prominent remodelling in the pulmonary arteries is present in animals exposed to CS and hypoxia, in comparison to either CS or hypoxia alone (61). These observations support the idea that CS can initiate vascular remodelling, which may be further amplified by chronic hypoxia in advanced COPD. However, the possible contribution of PASMC and PAEC dysfunction in this process remain to be explored.
Pulmonary vascular remodelling, as a consequence of PASMC, PAEC, and fibroblasts hypertrophy and proliferation, is an important pathological feature of PH. Current data show that there is significant contribution of PASMC, PAEC, and fibroblasts in the vascular remodelling leading to type 1 PH. However, there has not been enough data showing the contribution of fibroblasts in the vascular remodelling developing COPD-PH (62, 63). Although there is still no specific marker for pulmonary vascular remodelling in all forms of PH, targeting mediators of vascular dysfunction has been shown to be effective in reversing vascular remodelling in group 1 PH. Early evidence suggests that an imbalance of reduced anti-proliferative mediators as opposed to increased proliferative mediators may be critical for the aberrant PASMC proliferation caused by CS and chronic hypoxia in COPD-associated PH (64). However, the extent of the imbalance, the role of the imbalanced individual mediator in vascular remodelling, the contribution of PAECs (particularly PASMCs) in this process, and the effect of CS and hypoxia on the imbalance are still unclear.
Nitric oxide, a potent pulmonary vasodilator, anti-proliferative mediator, and endothelial-1 synthesis inhibitor (65), is generated by nitric oxide synthase (NOS) in vasculature. NOS is known to have three different isoforms named according to their roles: neuronal nitric oxide synthase (nNOS or NOS1), inducible (iNOS) or NOS2, and endothelial NOS (eNOS) or NOS3 (66). nNOS is constitutively expressed in neuronal cells and skeletal muscle, while iNOS is induced at sites of inflammation in response to inflammatory mediators (e.g., TNF-α and IL-1β) in many cell types (67). eNOS is constitutively expressed in vascular endothelial cells and is considered the main source of nitric oxide production in the pulmonary circulation system (68). After being produced by eNOS, nitric oxide is known to activate soluble guanylyl cyclase (sGC), the primary receptor for nitric oxide, resulting in the formation of the second messenger cyclic guanosine monophosphate (cGMP), which causes vasorelaxation and inhibits PASMC proliferation by decreasing the intracellular calcium concentration (56).
Deficiency of eNOS expression has been reported in the pulmonary arteries of smokers (69), in the lung tissues of patients with group 1 PH and non-COPD related PH (70), as well as in lung homogenates of guinea pigs exposed to CS (61). Furthermore, levels of nitric oxide in the plasma of guinea pigs exposed to CS (61) and in CS extract-treated human PAECs (71) are decreased. In addition, hypoxia, a pathological stimulus leading to vascular remodelling in COPD (42), has been shown to reduce eNOS expression and nitric oxide production in the lungs of piglets exposed to chronic hypoxia (72), as well as in human saphenous vein endothelial cells (73). Although there is limited information in the literature concerning the effect of hypoxia and CS on eNOS expression and nitric oxide production in human vascular cells, studies so far suggest an association of reduced nitric oxide bioavailability with PH development and a role for reduced eNOS expression in CS- and hypoxia-induced vascular remodelling. However, the underlying cause and role of CS-induced effects and the synergies between CS and hypoxia in vascular remodelling in COPD remain unclear.
Addressing the nitric oxide bioavailability dysfunction through stimulating sGC together with the inhibition of the cGMP-degrading enzyme phosphosiestrase type 5 (PDE5) can consequently aid in restoring nitric oxide-mediated protective effects in vascular cells which has been widely studied in group 1 PH and has shown anti-proliferative and vasodilatory effects in the pulmonary arteries of patients with group 1 PH (74–76). Recent preclinical evidence showed that treatment with sGC stimulator riociguat for 3 months (following 8 months of CS exposure) can reduce pulmonary vascular remodelling and mean vessel wall thickness, and fully reverse PH in a mouse model of CS-induced PH (77). Similarly, treatment with riociguat has been reported to decrease PVR in retrospective analysis of seven patients with COPD-associated PH (77). These preliminary data, together with the potential effectiveness of inhaled pulmonary vasodilators led to the currently ongoing clinical trial conducted to assess the effect of inhaled sGC stimulator on exercise capacity in COPD-associated PH (78).
In addition to the beneficial effect of sGC stimulator, the use of the PDE5 inhibitor sildenafil was demonstrated to prevent CS-induced PH in a guinea-pig model by reducing PAP (79). Sildenafil has also been shown to return PAP to normal levels, to attenuate vascular remodelling, and prevent hypoxia-induced PH in a rat model (80). Although the long-term effect of sildenafil on pulmonary haemodynamics in patients with COPD is unknown, treatment with sildenafil significantly increases plasma cGMP levels, reduces PAP increase in healthy volunteers breathing 11% O2, and attenuates pulmonary vascular remodelling in mice exposed to hypoxia (81). In addition, sildenafil has been reported to produce significant vasodilation in pulmonary circulation and improve pulmonary haemodynamics by reducing PAP and PVR in patients with COPD-associated PH (82). However, no improvement in exercise capacity has been observed with the use of sildenafil in COPD-related PH (83–85). The apparent absence of a beneficial effect on exercise capacity could be because PDE5 inhibitors possibly worsen gas exchange (84) as consequence of the inhibition of pulmonary hypoxic vasoconstriction (81). Due to the absence of large randomised trials and lack of sufficient evidence, these vasodilator drugs are currently not approved for the treatment of PH in COPD (1).
To minimise the risk associated with the use of systemic vasodilators (e.g., sildenafil) in those with group 3 PH, it has been suggested that inhaled therapies are preferred over oral therapies for this particular group of patients as these drugs can selectively improve perfusion where ventilation is best. Inhaled treprostinil is the first approved therapy for patients with group 3 PH (only due to interstitial lung disease) as it improves exercise capacity (86). Our recent systematic review demonstrated that inhaled therapies targeting prostacyclin pathway can reduce pulmonary vascular resistance without worsening pulmonary gas exchange (87). Although the use inhaled nitric oxide is not currently approved for adults with all forma of PH, limited studies assessed the short-term efficacy of using inhaled nitric oxide in patents with PH due to COPD and demonstrated potential benefits in improving hemodynamic parameters in COPD (88, 89). However, it should be noted that these studies are limited by very small sample size. The findings of the currently ongoing study assessing inhaled sGC stimulator use in PH due to COPD may also provide further evidence on the efficacy and safety of inhaled therapies targeting nitric oxide pathway (78). Given the small number of studies available that have assessed the role of eNOS expression and the release of nitric oxide in the development of COPD-associated PH, the potential usefulness of targeting the nitric oxide pathway, particularly inhaled therapies in the treatment of PH associated with COPD needs further attention. Furthermore, an enhanced understanding of the pathway may lead to a targeted therapy worthy of investigation for the treatment of PH in COPD.
Arachidonic acid (AA) and its metabolite pathway play a key role in the homeostasis of vascular smooth muscle and endothelial cells. Abnormalities in this pathway have been shown to be associated with vascular remodelling and PH (90, 91). Vasoactive prostanoids, including prostaglandins (PGs) and thromboxane (TX) are the major metabolites of AA and are formed by COX or prostaglandin G/H synthase. As shown in Figure 1, the synthesis of PGs and TX involves the hydrolysis of cellular phospholipids via the action of phospholipase A2 (PLA2) enzyme to produce free AA. AA is converted to unstable prostaglandin H2 (PGH2) by COX activity, and then to main prostanoids PGI2, TXA2, PGD2, PGE2 and PGF2 via their respective synthases PGIS, thromboxane A synthase (TXAS), prostaglandin D synthase (PGDS), prostaglandin E2 synthases (PGESs) and prostaglandin F synthase (PGFS) (92, 93). Currently, there are nine known prostanoid receptors expressed in different cell types, namely IP for PGI2, TP for TXA2, DP1-2 for PGD2, EP1-4 for PGE2, and FP for PGF2. Activation of these receptors leads to a wide variety of biological effects, particularly vasodilation or vasoconstriction in vascular cells. IP, EP2/4 and DP1-2 receptors are considered relaxant receptors, while the activation of contractile receptors (TP, EP1/3, and FP) is known to induce vasoconstriction (94).
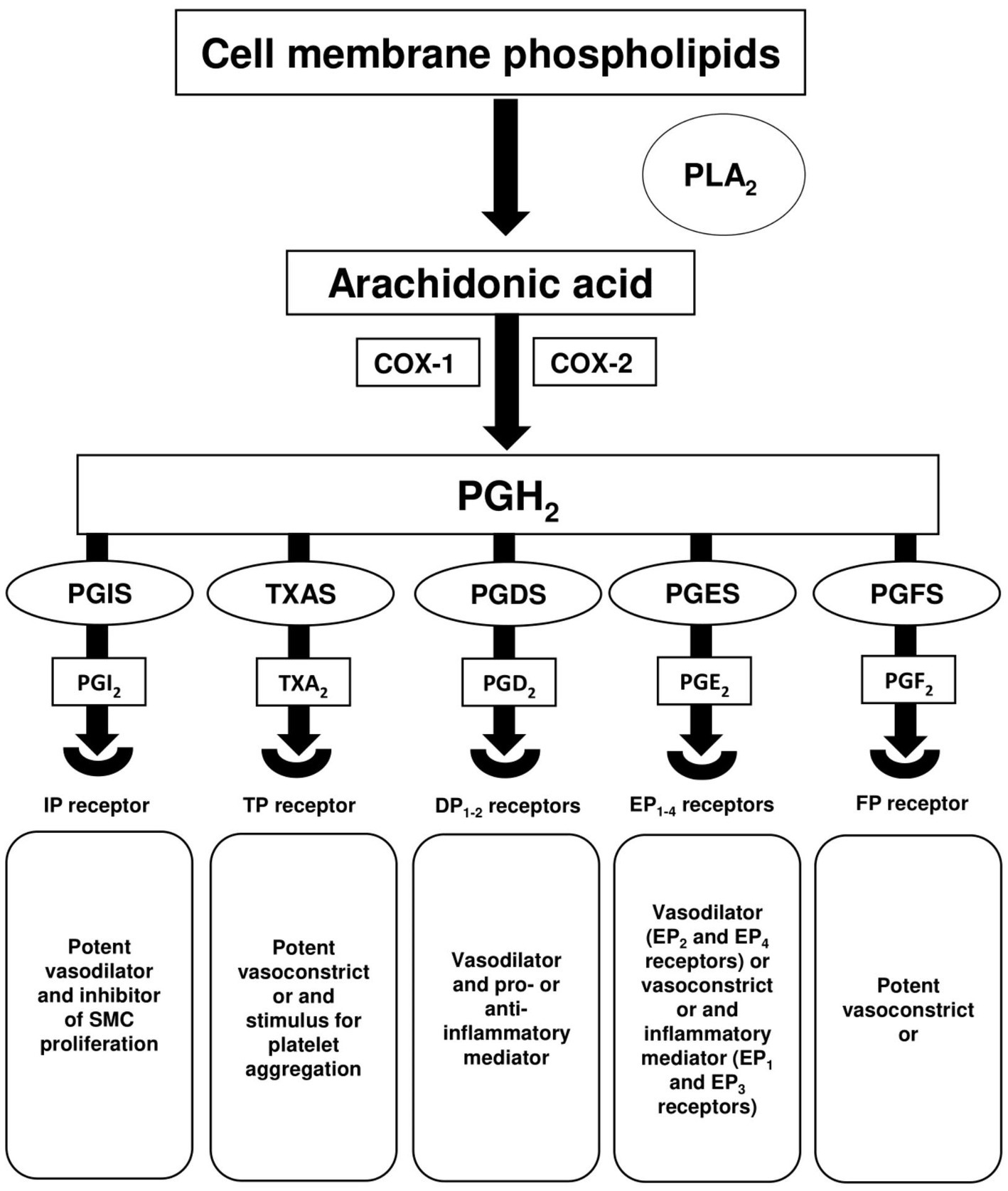
Figure 1. Arachidonic acid metabolism. Hydrolysis of cell membrane phospholipids via phospholipase A2 (PLA2) releases arachidonic acid (AA), which is then converted by cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) to the intermediate prostaglandin H2 (PGH2). PGH2 is converted to multiple prostanoids; prostacyclin (PGI2), thromboxane A2 (TXA2), prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), and prostaglandin F2 (PGF2) via their respective synthases; prostaglandin I synthase (PGIS), thromboxane A synthase (TXAS), prostaglandin D synthase (PGDS), prostaglandin E2 synthases (PGESs), and prostaglandin F synthase (PGFS).
There are no reports to date showing the involvement of PGF2 and PGD2 in pulmonary vascular remodelling in all forms of PH. Conversely, PGI2, TXA2, and PGE2 play an important role in maintaining balance in cardiovascular homeostasis (95). Although PGI2/TXA2 imbalance (96) and PGE2 deficiency (97) have been implicated in the development of group 1 PH, studies to date do not provide clear evidence about the role of these vasoactive prostanoids (PGI2, TXA2, and PGE2) in the development of PH in COPD.
There are two isoforms of cyclooxygenase (COX): COX-1 and COX-2. COX-1 is expressed constitutively in many cells and serves as a housekeeping gene, while COX-2 is a highly inducible gene, and its expression is increased in response to inflammation (98–101). In addition to the role of COX-2 induction in both inflammation and airway remodelling in COPD (102), it has long been suggested that COX-2 upregulation is involved in pulmonary vascular remodelling (90). Recently, increased COX-2 protein expression has been reported in both vascular endothelial cells in lung tissues of patients with COPD and CS extract-treated human umbilical vein endothelial cells (103). CS extract treatment has also been shown to induce mRNA expression of COX-2 in human pulmonary microvascular endothelial cells (104). Importantly, the COX-2 expression upregulation following CS extract stimulation in human umbilical vein endothelial cells and in patients with COPD is associated with an increased cell apoptosis rate (103). In addition, our recent findings demonstrated that CS extract induced COX-2 in both human PASMCs and PAECs and that COX-2 inhibitor can reduce CS-induced PASMC and PAEC proliferation (60). These findings suggest a role of COX-2 induction in CS-induced pulmonary vascular remodelling in PH.
Besides the effect of CS on COX-2 expression, hypoxia (a key stimulus for vascular remodelling in PH) has also been reported to induce COX-2 expression in human PASMCs (93), in rat lungs (90), PASMCs (93), and in human umbilical vein endothelial cells (105, 106). Although the use of selective COX-2 inhibitor has been shown to increase endothelin-1 release in human PASMCs (107), it has been demonstrated that selective inhibition of COX-2 induction can prevent hypoxic pulmonary vasoconstriction (108), the development of PH in a rat model (109) and chronic hypoxia-induced PH in new-born pigs (110). It has also been reported that the use of selective COX-2 inhibitor can completely block hypoxia-induced proliferation of human umbilical vein endothelial cells (105). These findings suggest the anti-proliferative effect of COX-2 inhibitor in umbilical vein endothelial cells is likely via the inhibition of prostanoid production. Nevertheless, more studies are needed to determine whether the increase in the expression of COX-2 and the production of its downstream vasoactive prostanoid mediators and cell proliferation by hypoxia observed in human umbilical vein endothelial cells exist in human PAECs, the major regulators of vascular function in PH. In addition, COX-2 induction in response to hypoxia in experimental human and animal cellular studies may represent an important mechanism by which aortic smooth muscle cells can increase their capacity for prostanoid production (111).
Collectively, the induction of COX-2 in CS-and hypoxia-treated vascular cells (as well as in the lungs of COPD patients and smokers) suggests a potentially critical role of its downstream vasoactive prostanoid products in vascular remodelling in COPD-associated PH. It is therefore necessary to understand the effect of CS and hypoxia, either individually or in combination, on the expression of COX-2 and its downstream vasoactive prostanoid synthases, and on the release of vasoactive prostanoids in human PASMCs and PAECs, as well as the subsequent impact on cell proliferation and apoptosis.
PGE2 is a known inflammatory mediator (112) and plays a role in regulating blood pressure in pulmonary circulation (113). PGE2 is mainly formed via the metabolism of AA by COX-1 and COX-2. The free AA is converted into PGE2 via COX enzymes and terminal PGESs. Three different types of PGES may be involved in regulating PGE2 production: microsomal prostaglandin E synthase-1 (mPGES-1), microsomal prostaglandin E synthase-2 (mPGES-2), and cytosolic PGE2 synthase (cPGES). mPGES-2 and cPGES are constitutively expressed in several tissues and are known to have a housekeeping role (114), whereas mPGES-1 is expressed at relatively low basal levels and acts as the terminal enzyme downstream of COX enzymes in producing PGE2 from PGH2 (115). Amongst the three different types of PGES, studies have shown that mPGES-1 as a key regulator for induced PGE2 under inflammatory condition (116–119). Although it is believed that mPGES-1 may have inflammatory effects (120) and deletion of mPGES-1 in the vasculature can be a potential novel target for development of anti-inflammatory drugs (112), the role of mPGES-1 in vascular remodelling in smokers and all forms of PH (including COPD-associated PH) is largely unknown.
PGE2 acts on four specific G-protein-coupled receptors in various cell types: EP1, EP2, EP3, and EP4. PGE2 production in the vascular cells may lead to either vasodilation or vasoconstriction, depending on its binding receptors (115). It is suggested that the stimulation of EP2 and EP4 receptors could lead to vasodilation by increasing intracellular cyclic adenosine monophosphate (cAMP) concentrations. Vasoconstriction is mediated via the activation of EP1 and EP3 receptors by increasing intracellular calcium and decreasing intracellular cAMP concentrations, respectively (97). Recently, it has been reported that circulating levels of PGE2 are reduced in a chronic hypoxia rat model of PH and in patients with group 1 PH (97). Convincingly, the use of highly selective EP2 receptor agonist butaprost has been shown to cause a significant reduction in proliferation of human PASMCs derived from patients with group 1 PH (121). These observations suggest that PGE2 can induce a vasodilatory/anti-proliferative effect via EP2 in human PASMCs; however, the potential role of PGE2 in vascular remodelling in COPD-associated PH has not been studied.
We have previously found that CS extract reduced PGE2 levels in human PASMCs extract as a result of the downregulated mPGES-1 expression (60). This suggests that CS-induced a decrease in the levels of PGE2 may contribute to PASMC proliferation, leading to pulmonary vascular remodelling in COPD-associated PH. Hypoxia has also been reported to induce PGE2 in human PASMCs (93) as well as in human umbilical vein endothelial cells (105). Importantly, exogenous PGE2 and hypoxia can stimulate proliferation of human umbilical vein endothelial cells, and this cell proliferation can be inhibited by the combined EP1/2 receptor antagonist AH6809 (105). These observations suggest that PGE2 may modulate vascular remodelling via EP1/2 receptors. However, whether hypoxia affects PGE2 production in human PAECs, and whether PGE2 can play the same role in hypoxia-induced proliferation of PAECs are both unknown.
Taken together, the role of the mPGES-1/PGE2 pathway in vascular abnormalities in PH remains controversial, due to the capability of PGE2 and its receptors to either induce or suppress vasodilatory/anti-proliferative and vasoconstrictive/proliferative effects. To date, the effect of hypoxia on mPGES-1 expression and PGE2 production in human PASMCs and PAECs, and the potential contribution of PGE2 to CS- and hypoxia-induced vascular remodelling have not been determined.
PGI2, also called prostacyclin, a potent vasodilator and an inhibitor of PASMC proliferation, was first reported in 1976 by Needleman and Vane (122). PGI2 is a downstream product from the sequential enzymatic actions of COX and PGIS and is considered the predominant prostanoid in vasculature. Within the lungs, PGI2 is primarily produced in vascular smooth muscle and endothelial cells (123, 124) and is quickly transformed by non-enzymatic procedures to a latent hydrolysis item, 6-keto-PGF1α (125). PGI2 acts on the IP receptor to induce vasodilatory and anti-proliferative effects as a result of increased intracellular cAMP concentrations.
In addition to the importance of PGIS/PGI2 in group 1 PH, reduced PGIS/PGI2 has also been reported in the lungs of smokers with COPD, as well as in CS extract-treated human umbilical vein endothelial cells (126). The study also showed that umbilical vein endothelial cell stimulation with CS extract can induce apoptosis, whilst the use of the PGI2 analogue beraprost sodium prevents CS extract-induced cell apoptosis (126). The findings of the same group also demonstrated decreased PGI2 production and induced apoptosis in the lungs of CS extract-treated emphysematous rats (127), suggesting that the deficiency of PGI2 in CS extract-treated umbilical vein endothelial cells may contribute to cell apoptosis and vascular remodelling. This view is supported by another research group, who showed a downregulation of PGIS mRNA and protein expression and a reduction of PGI2 production in lung tissue extracts from patients with emphysema (104). Consistently, we showed that CS extract reduced PGIS mRNA and protein expression and PGI2 production in human PASMCs and more importantly, PGI2 analogue inhibited CS extract-induced PASMC and PAEC proliferation (60). In primary human pulmonary microvascular endothelial cells, CS extract treatment has also been shown to reduce the mRNA expression of PGIS (104). These observations suggest PGI2 may play a crucial role in CS-induced pulmonary vascular cell dysfunction and remodelling in COPD.
Hypoxia has been demonstrated to induce protective effects by further inducing the constitutively expressed PGIS in human aortic smooth muscle cells and human umbilical vein endothelial cells (111), as well as increasing PGI2 production in human aortic smooth muscle cells (111) and in human PASMCs (128). Although hypoxia can also increase the mRNA expression of PGIS and the stable metabolite of PGI2 in mice chronically exposed to hypoxia (111), increased PGIS expression and PGI2 production following the use of PGIS gene transfer has been shown to attenuate medial thickening of small pulmonary arteries and improve hypoxic PH in a mouse model (129). Consistently, PGIS overexpression by gene transfer has been reported to provide protection against the development of hypoxic PH, and to help prevent pulmonary vascular remodelling in a transgenic mouse model (130). In addition, it has been demonstrated that PGI2 analogue beraprost sodium can inhibit hypoxia-induced human PASMC proliferation (131). The ability of PASMCs to produce more PGI2 following hypoxia exposure, and the role of PGI2 analogue in preventing proliferation of PASMCs induced by hypoxia, provide early evidence of the protective effect of PGI2 in hypoxia-induced vascular remodelling. However, hypoxia effect on PGIS expression and PGI2 production in human PAECs is unknown. In addition, the contribution of hypoxia effect on PGIS/PGI2 to the functions of both human PASMCs and PAECs remains to be investigated.
The deficiency of endogenous prostacyclin represents the rationale for targeting the prostacyclin pathway for the treatment of patients with group 1 PH (91). It is suggested that exogenous and endogenous PGI2 can induce the relaxation of vascular smooth muscle and inhibit platelet activation (132). Currently, PGI2 analogues and PGI2 receptor agonists are used as part of the clinical management for treatment of patients with group 1 PH and have been shown to improve exercise capacity, symptoms, and haemodynamics, although the mortality rate associated with group 1 PH has not been significantly reduced with long-term use (133–138). The significant improvements in signs and symptoms of patients with group 1 PH after administration of PGI2 analogues and PGI2 receptor agonists are likely due to the inhibition of smooth muscle cell proliferation and vasodilating effects on pulmonary arteries (132). However, the efficacy of such treatment in patients with PH in COPD is still unknown due to lack of studies. Recently, the PERFECT trial was conducted to evaluate the effect of inhaled prostacyclin on exercise capacity in patients with PH due to COPD (139). Unfortunately, the PERFECT trial was terminated by the data safety monitoring committee following a routine safety and efficacy analysis (139). Given that the findings of the PERFECT trial have not yet been published, more studies are needed to assess the role of PGI2 in COPD-associated PH and the efficacy of PGI2 analogues and PGI2 receptor agonists use to treat patients with PH due to COPD.
TXA2, named after its role in thrombosis, is a potent vasoconstrictor, platelet aggregator, and proliferative mediator with opposing effects to the vasoprotective PGI2 in regulating vascular tone (140, 141). Together with PGI2, TXA2 plays an important role in maintaining homeostatic balance in pulmonary circulation. TXA2 is one of the downstream products from COX-2 activity and its production is mediated by the enzyme TXAS. TXA2 is unstable and rapidly converted to the inactive metabolite, thromboxane B2 (TXB2) (142). After being produced by TXAS, TXA2 binds to the TP receptor, stimulates the activation of platelet aggregation, and causes pulmonary vasoconstriction as a consequence of intracellular calcium concentration increase (143, 144). Traditionally, platelets were considered the only cellular source of TXA2. It is now known that TXA2 is produced in a variety of cells including vascular smooth muscle and endothelial cells.
We have previously demonstrated that TXA2 is increased in human PASMCs isolated from smokers with COPD compared with TXA2 levels in healthy PASMCs (145). More importantly, the balance between vasoconstrictive TXA2 and vasoprotective PGI2 is found to be important in the homeostasis of vascular function. For example, it has been reported that the balance between these two vasoactive mediators in patients with group 1 PH is shifted away from PGI2 toward TXA2 (96, 146–148). Although the role Like CS, hypoxia of TXA2 in COPD-associated PH is unknown, previous studies suggest CS is a major contributing factor to TXA2 increase in COPD. Elevated levels of TXA2 have been reported in the urine of smokers and ex-smokers COPD patients, compared with healthy subjects (149). In addition, it has been demonstrated that TXA2 is increased in both bronchoalveolar lavage fluid and the lung tissue of rat models exposed to CS (150). Consistent with this animal study, CS extract has been shown to induce TXA2 levels in human PAECs (60, 71) as well as PASMCs (60), suggesting that TXA2 is a key feature of CS-induced pulmonary artery cell dysfunction.
Like CS, hypoxia can induce pulmonary vasoconstriction and vascular remodelling. However, the involvement of TXA2 in this process is not understood. It has been reported that TXA2 levels are increased in arterial and venous plasma of hypoxic rats (151). Increased TXA2 contributes to pulmonary artery constriction in piglet models of hypoxia-induced PH. Furthermore, the use of a COX-2 inhibitor diminishes the production of downstream COX-2-dependent constrictors, TXA2, without adversely affecting other prostanoid production, including PGI2 release (110). Concordant with this, using selective COX-2 inhibitors and selective TP receptor antagonists reduces hyperresponsiveness of pulmonary arteries from mice exposed to chronic hypoxia, through blocking TXA2-TP receptor signalling (152). In addition, the use of TXAS inhibitor has been shown to blunt the development of hypoxia-induced PH in a neonatal piglet model (153). Although these in vivo data suggest that the induction of TXA2 production by hypoxia is of critical importance in PH, there are no cellular studies on the effect of hypoxia on TXAS expression and TXA2 production in human PAECs and PASMCs. In addition, whether TXAS expression and TXA2 production effect plays a role in hypoxia-induced vascular remodelling is largely unknown.
As increased TXA2 has been implicated in the pathogenesis of group 1 PH (96), blocking the enhanced TXA2 effect could restore PGI2/TXA2 balance and lead to a therapeutic approach for the treatment of patients with group 1 PH (154, 155). The use of PGI2 analogues to compensate for the loss of PGI2 production in patients with group 1 PH has been clinically approved and has demonstrated significant improvements in the symptoms of group 1 PH (156). Although early evidence from preclinical animal studies suggests that the inhibition of TXA2 by synthase inhibition or receptor antagonism can be effective for hypoxic PH (152, 153), TXAS inhibitor and TXA2 receptor antagonist are not clinically approved for the treatment of patients with group 1 PH and other forms of PH, due to lack of any evidence for their efficacy.
Although targeting TXA2-TP receptor signalling is not currently used as a therapeutic target for patients with all forms of PH (due to lack of evidence), the inhibition of COX-2-derived TXA2 production and blocking TXA2 effects using TXAS inhibitors or TP receptor antagonists, have been shown to prevent PH in animal model studies (110, 151–153). Our previous novel findings suggest that blocking increased TXA2 effects by the use of TXA2 receptor antagonist (daltroban) can exert anti-proliferative effects (60). Furthermore, we showed that the addition of PGI2 analogue (beraprost sodium) and the inhibition of CS extract- and hypoxia-induced COX-2-derived TXA2 production by COX-2 inhibitor (celecoxib) can restore the balance of prostanoids and help reduce pulmonary vascular remodelling in COPD-associated PH via the inhibition of PASMC and PAEC proliferation. To the best of our knowledge, no clinical trial has been conducted to evaluate the effect of drugs targeting TXA2-TP receptor signalling. Thus, a well powered multicentre, randomised, double-blind, placebo-controlled crossover trial to assess the effect of drugs targeting TXA2 pathway on clinical outcomes in patients with PH due to COPD is needed.
Endothelin is known as a potent vasoconstrictor and proliferative mediator and was first identified by Yanagisawa and colleagues in 1988 (157). The initial endothelin gene product, prepro-endothelin is cleaved by endopeptidase to pro-endothelin or big endothelin. The big endothelin is subsequently converted into endothelin isoforms via a specific enzyme, called an endothelin-converting enzyme. There are three different isoforms of endothelin: endothelin-1, endothelin-2, and endothelin-3. Endothelin-1 is a 21-amino acid peptide and is found mainly in the cardiovascular system (158). Endothelin-2 differs from endothelin-1 by only two amino acids, shows similar endothelin pharmacology to endothelin-1, and is found primarily in the myocardium, kidney, and placental tissues (159). Endothelin-3 is found mainly in the nervous system, differs by six amino acids from endothelin-1, and is considered a weaker vasoconstrictor when compared with endothelin-1 (159, 160).
Endothelin, latterly named endothelin-1, is the most studied peptide of the endothelin family. Although the primary source of endothelin-1 is considered to be endothelial cells (157), numerous cell types (e.g., PASMCs) can release this peptide in vitro upon stimulation with TGF-β1 and pro-inflammatory mediators (161, 162). Once produced, endothelin-1 then binds to either an endothelin-A (ET-A) or endothelin-B (ET-B) receptors (156, 163). ET-B is most often found in vascular endothelial cells, while vascular smooth muscle cells can express both ET-A and ET-B receptors (159). Activation of the ET-B receptor in pulmonary vascular endothelial cells can promote vasodilation by enhancing the release of nitric oxide (146, 164). However, the activation of ET-A and ET-B receptors in vascular smooth muscle cells can lead to vasoconstriction (165, 166).
Elevated endothelin-1 levels in plasma and lung tissues have been shown to be associated with the pathogenesis of groups 1 and 2 PH (167, 168). However, previous studies investigating the role of endothelin-1 on smokers and patients with COPD have been inconsistent and contradictory. It has recently been reported that ET-1 is related to the pathological process of onest as well as development of PH due to COPD (169). Although it has been reported that the levels of endothelin-1 are unchanged in the lung tissue samples of smokers compared with those of non-smokers (69), increased ET-A and ET-B receptors expression has been observed in pulmonary arteries from smokers and COPD patients (170). In addition, an in vitro study has shown that CS extract induces endothelin-1 mRNA expression and endothelin-1 secretion in both bovine and human PAECs (171). It also been reported that CS extract induces endothelin release and ET-B receptor protein and mRNA expression in human PAECs (71). The same group has also shown that the use of bosentan (an ET-A and ET-B receptors antagonist) can inhibit CS extract-induced endothelin receptors expression and CS extract-induced proliferation of human PASMCs (170).
In addition to CS, hypoxia plays a key role in the development of PH in COPD (44). However, the contribution of endothelin-1 in this process remains unclear, as results from studies in cultured cells are conflicting. For example, hypoxia induces endothelin-1 mRNA expression in human PAECs (172) and in human pulmonary microvascular endothelial cells (173), whereas hypoxia reduces endothelin-1 production in cultured rat lung endothelial cells (174). Interestingly, CS has been shown to reduce plasma levels of endothelin-1 under hypoxic conditions in guinea pigs, although CS and hypoxia individually have been shown to induce plasma levels of endothelin-1 (61). Thus, there is a need for further in vitro investigations to improve our understanding of the effect of CS and hypoxia, either individually or in combination, on the endothelin pathway and the impact of the possible effect in pulmonary vascular remodelling in COPD.
Clinically, the use of bosentan has been shown to foster significant improvement in exercise capacity and haemodynamics in patients with group 1 PH (175–178). This is supported by in vitro studies showing that the stimulation of ET-A and ET-B receptors by endothelin-1 treatment in human PASMCs can promote the proliferation of the cells, and may eventually contribute to vascular remodelling and PH (179). Bosentan has also been shown to inhibit the proliferation of human PASMCs from group 1 PH patients (180). These observations suggest that targeting ET-A and ET-B receptors may be a promising therapeutic target for the treatment of group 1 PH. Although the use of bosentan has failed clinically to improve exercise capacity, and hypoxaemia has become progressively worse in COPD without severe PH (181), it has been suggested that the use of bosentan to treat patients with severe or very severe COPD-associated PH can be beneficial (182).
Taken together, evidence from in vitro studies showing the inhibitory effect of ET-A and ET-B receptors antagonist on CS extract -induced proliferation of human PASMCs suggests that there may be an important role for the endothelin pathway in CS-induced vascular remodelling in COPD-associated PH. However, the effect of hypoxia with or without CS extract on endothelin release in human PASMCs and PAECs has not been explored. In addition, the contribution of this possible effect to the function of PASMCs and PAECs is yet to be identified.
To date, there is currently no treatment approved for patients with COPD-associated PH due to lack of evidence and no proven benefits. While the available evidence suggests that CS and hypoxia, known stimuli of vascular remodelling in COPD, can cause imbalanced vasoactive gene expression and mediator release, the association between pulmonary vascular remodelling and dysregulated prostanoids, nitric oxide and endothelin in PH due to COPD is still not well understood. Given that drugs targeting these three pathways are not currently used for COPD-associated PH and inhaled therapies are preferred over oral therapies to minimise the risk associated with the use of systemic vasodilators in this particular group of patients, more research is urgently needed to assess the safety and efficacy of drugs particularly targeting prostanoids and nitric oxide through inhalation route in patients with COPD-associated PH.
AAlq: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing. AAld: Conceptualization, Writing – original draft, Writing – review & editing. SAlg: Writing – original draft, Writing – review & editing. JA: Conceptualization, Software, Writing – original draft, Writing – review & editing. RS: Conceptualization, Software, Writing – original draft, Writing – review & editing. HA: Investigation, Software, Visualization, Writing – review & editing. AAlG: Investigation, Software, Visualization, Writing – review & editing. MM: Funding acquisition, Investigation, Software, Visualization, Writing – review & editing. SAls: Investigation, Writing – original draft, Writing – review & editing. LP: Conceptualization, Resources, Supervision, Validation, Writing – original draft, Writing –review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Humbert, M, Kovacs, G, Hoeper, MM, Badagliacca, R, Berger, RMF, Brida, M, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. (2022):3618–731. doi: 10.1183/13993003.00455-2022
2. Walther, CP, Nambi, V, Hanania, NA, and Navaneethan, SD. Diagnosis and management of pulmonary hypertension in patients with CKD. Am J Kidney Dis. (2020) 75:935–45. doi: 10.1053/j.ajkd.2019.12.005
3. Mandras, SA, Mehta, HS, and Vaidya, A. Pulmonary hypertension: a brief guide for clinicians. Mayo Clin Proc. (2020) 95:1978–88. doi: 10.1016/j.mayocp.2020.04.039
4. Piccari, L, Aguilar-Colindres, R, and Rodríguez-Chiaradía, DA. Pulmonary hypertension in interstitial lung disease and in chronic obstructive pulmonary disease: different entities? Curr Opin Pulm Med. (2023) 10:370–9. doi: 10.1097/MCP.0000000000000984
5. Humbert, M, Kovacs, G, Hoeper, MM, Badagliacca, R, Berger, RM, Brida, M, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: developed by the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European reference network on rare respiratory diseases (ERN-LUNG). Eur Heart J. (2022) 43:3618–731. doi: 10.1093/eurheartj/ehac237
6. Blanco, I, Tura-Ceide, O, Peinado, VI, and Barberà, JA. Updated perspectives on pulmonary hypertension in COPD. Int J Chron Obstruct Pulmon Dis. (2020) 15:1315–24. doi: 10.2147/COPD.S211841
7. Kurakula, K, Smolders, VF, Tura-Ceide, O, Jukema, JW, Quax, PH, and Goumans, M-J. Endothelial dysfunction in pulmonary hypertension: cause or consequence? Biomedicine. (2021) 9:57. doi: 10.3390/biomedicines9010057
8. Sakao, S. Chronic obstructive pulmonary disease and the early stage of cor pulmonale: a perspective in treatment with pulmonary arterial hypertension-approved drugs. Respir Investig. (2019) 57:325–9. doi: 10.1016/j.resinv.2019.03.013
9. Gredic, M, Blanco, I, Kovacs, G, Helyes, Z, Ferdinandy, P, Olschewski, H, et al. Pulmonary hypertension in chronic obstructive pulmonary disease. Br J Pharmacol. (2021) 178:132–51. doi: 10.1111/bph.14979
10. Celli, BR, Decramer, M, Wedzicha, JA, Wilson, KC, Agustí, A, Criner, GJ, et al. An official American Thoracic Society/European Respiratory Society statement: research questions in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2015) 191:e4–e27. doi: 10.1164/rccm.201501-0044ST
11. Li, X, Cao, X, Guo, M, Xie, M, and Liu, X. Trends and risk factors of mortality and disability adjusted life years for chronic respiratory diseases from 1990 to 2017: systematic analysis for the global burden of disease study 2017. BMJ. (2020) 368:m234. doi: 10.1136/bmj.m234
12. Jindal, S, and Jindal, A. COPD in biomass exposed nonsmokers: a different phenotype. Expert Rev Respir Med. (2021) 15:51–8. doi: 10.1080/17476348.2021.1835476
13. Saeed, ZH, El Hakim, MAA, and Mohamed, NR. Chronic obstructive pulmonary disease in non-smokers: role of oxidative stress. Egypt J Bronchol. (2021) 15:1–6. doi: 10.1186/s43168-021-00088-5
14. Burrows, B, Kettel, LJ, Niden, AH, Rabinowitz, M, and Diener, CF. Patterns of cardiovascular dysfunction in chronic obstructive lung disease. N Engl J Med. (1972) 286:912–8. doi: 10.1056/NEJM197204272861703
15. Weitzenblum, E, Hirth, C, Ducolone, A, Mirhom, R, Rasaholinjanahary, J, and Ehrhart, M. Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease. Thorax. (1981) 36:752–8. doi: 10.1136/thx.36.10.752
16. Weitzenblum, E, Sautegeau, A, Ehrhart, M, Mammosser, M, Hirth, C, and Roegel, E. Long-term course of pulmonary arterial pressure in chronic obstructive pulmonary disease. Am Rev Respir Dis. (1984) 130:993–8. doi: 10.1164/arrd.1984.130.6.993
17. Oswald-Mammosser, M, Apprill, M, Bachez, P, Ehrhart, M, and Weitzenblum, E. Pulmonary hemodynamics in chronic obstructive pulmonary disease of the emphysematous type. Respiration. (1991) 58:304–10.
18. Scharf, SM, Iqbal, M, Keller, C, Criner, G, Lee, S, Fessler, HE, et al. Hemodynamic characterization of patients with severe emphysema. Am J Respir Crit Care Med. (2002) 166:314–22. doi: 10.1164/rccm.2107027
19. Arcasoy, SM, Christie, JD, Ferrari, VA, Sutton, MS, Zisman, DA, Blumenthal, NP, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. (2003) 167:735–40. doi: 10.1164/rccm.200210-1130OC
20. Chaouat, A, Bugnet, AS, Kadaoui, N, Schott, R, Enache, I, Ducolone, A, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2005) 172:189–94. doi: 10.1164/rccm.200401-006OC
21. Thabut, G, Dauriat, G, Stern, JB, Logeart, D, Levy, A, Marrash-Chahla, R, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. (2005) 127:1531–6. doi: 10.1378/chest.127.5.1531
22. Katiyar, V, and Khare, R. Prevalence of pulmonary hypertension in COPD. Int J Adv Med. (2018) 5:356–60. doi: 10.18203/2349-3933.ijam20181061
23. Kalkan, F, Ucar, EY, Kalkan, K, and Araz, O. Comparison of functional capacity and symptoms of COPD patients with and without pulmonary hypertension. Eurasian J Med. (2020) 52:166. doi: 10.5152/eurasianjmed.2020.19391
24. Goel, K, Egersdorf, N, Gill, A, Cao, D, Collum, SD, Jyothula, SS, et al. Characterization of pulmonary vascular remodeling and MicroRNA-126-targets in COPD-pulmonary hypertension. Respir Res. (2022) 23:349. doi: 10.1186/s12931-022-02267-4
25. Zhang, Y, and Xu, C-B. The roles of endothelin and its receptors in cigarette smoke-associated pulmonary hypertension with chronic lung disease. Pathol Res Pract. (2020) 216:153083. doi: 10.1016/j.prp.2020.153083
26. Pullamsetti, SS, Mamazhakypov, A, Weissmann, N, Seeger, W, and Savai, R. Hypoxia-inducible factor signaling in pulmonary hypertension. J Clin Invest. (2020) 130:5638–51. doi: 10.1172/JCI137558
27. Dauriat, G, Reynaud-Gaubert, M, Cottin, V, Lamia, B, Montani, D, Canuet, M, et al. Severe pulmonary hypertension associated with chronic obstructive pulmonary disease: a prospective French multicenter cohort. J Heart Lung Transplant. (2021) 40:1009–18. doi: 10.1016/j.healun.2021.04.021
28. Arif, R, Pandey, A, Zhao, Y, Arsenault-Mehta, K, Khoujah, D, and Mehta, S. Treatment of pulmonary hypertension associated with COPD: a systematic review. ERJ Open Res. (2022) 8:00348–2021. doi: 10.1183/23120541.00348-2021
29. Mandras, S, Kovacs, G, Olschewski, H, Broderick, M, Nelsen, A, Shen, E, et al. Combination therapy in pulmonary arterial hypertension—targeting the nitric oxide and prostacyclin pathways. J Cardiovasc Pharmacol Ther. (2021) 26:453–62. doi: 10.1177/10742484211006531
30. Chaouat, A, Naeije, R, and Weitzenblum, E. Pulmonary hypertension in COPD. Eur Respir J. (2008) 32:1371–85. doi: 10.1183/09031936.00015608
31. Barbera, JA, Peinado, VI, and Santos, S. Pulmonary hypertension in chronic obstructive pulmonary disease. Eur Respir J. (2003) 21:892–905. doi: 10.1183/09031936.03.00115402
32. Rudyk, O, and Aaronson, PI. Redox regulation, oxidative stress, and inflammation in group 3 pulmonary hypertension. Adv Exp Med Biol. (2021) I:209–41. doi: 10.1007/978-3-030-63046-1_13
33. Shujaat, A, Bajwa, AA, and Cury, JD. Pulmonary hypertension secondary to COPD. Pulm Med. (2012) 2012:203952. doi: 10.1155/2012/203952
34. Piccari, L, Blanco, I, Torralba, Y, Arismendi, E, Gistau, C, Ramirez, A, et al. Mechanisms of hypoxaemia in severe pulmonary hypertension associated with COPD. Eur Respir J. (2023) 62:2300463. doi: 10.1183/13993003.00463-2023
35. Kovacs, G, Agusti, A, Barbera, JA, Celli, B, Criner, G, Humbert, M, et al. Pulmonary vascular involvement in chronic obstructive pulmonary disease. Is there a pulmonary vascular phenotype? Am J Respir Crit Care Med. (2018) 198:1000–11. doi: 10.1164/rccm.201801-0095PP
36. Bogaard, HJ. Hypoxic pulmonary vasoconstriction in COPD-associated pulmonary hypertension: been there, done that? Eur Respir J. (2017) 50:1701191. doi: 10.1183/13993003.01191-2017
37. Peinado, VI, Santos, S, Ramirez, J, Roca, J, Rodriguez-Roisin, R, and Barbera, JA. Response to hypoxia of pulmonary arteries in chronic obstructive pulmonary disease: an in vitro study. Eur Respir J. (2002) 20:332–8. doi: 10.1183/09031936.02.00282002
38. Klings, ES, and Lee, JS. Pulmonary hypertension due to lung disease and/or hypoxemia (group 3 pulmonary hypertension): epidemiology, pathogenesis, and diagnostic evaluation in adults. UpToDate. (2022):4.
39. Wright, JL, Levy, RD, and Churg, A. Pulmonary hypertension in chronic obstructive pulmonary disease: current theories of pathogenesis and their implications for treatment. Thorax. (2005) 60:605–9. doi: 10.1136/thx.2005.042994
40. Wright, JL, Petty, T, and Thurlbeck, WM. Analysis of the structure of the muscular pulmonary arteries in patients with pulmonary hypertension and COPD: National Institutes of Health nocturnal oxygen therapy trial. Lung. (1992) 170:109–24. doi: 10.1007/BF00175982
41. Santos, S, Peinado, VI, Ramirez, J, Melgosa, T, Roca, J, Rodriguez-Roisin, R, et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J. (2002) 19:632–8. doi: 10.1183/09031936.02.00245902
42. Pak, O, Aldashev, A, Welsh, D, and Peacock, A. The effects of hypoxia on the cells of the pulmonary vasculature. Eur Respir J. (2007) 30:364–72. doi: 10.1183/09031936.00128706
43. Peinado, VI, Barbera, JA, Ramirez, J, Gomez, FP, Roca, J, Jover, L, et al. Endothelial dysfunction in pulmonary arteries of patients with mild COPD. Am J Phys. (1998) 274:L908–13. doi: 10.1152/ajplung.1998.274.6.L908
44. Sakao, S, Voelkel, NF, and Tatsumi, K. The vascular bed in COPD: pulmonary hypertension and pulmonary vascular alterations. Eur Respir Rev. (2014) 23:350–5. doi: 10.1183/09059180.00007913
45. Masri, FA, Xu, W, Comhair, SA, Asosingh, K, Koo, M, Vasanji, A, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L548–54. doi: 10.1152/ajplung.00428.2006
46. Taraseviciene-Stewart, L, Kasahara, Y, Alger, L, Hirth, P, Mc Mahon, G, Waltenberger, J, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. (2001) 15:427–38. doi: 10.1096/fj.00-0343com
47. Sakao, S, Taraseviciene-Stewart, L, Lee, JD, Wood, K, Cool, CD, and Voelkel, NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J. (2005) 19:1178–80. doi: 10.1096/fj.04-3261fje
48. Wang, CG, Li, C, Lei, W, Jiang, JH, Huang, JA, and Zeng, DX. The association of neuron-derived orphan receptor 1 with pulmonary vascular remodeling in COPD patients. Int J Chron Obstruct Pulmon Dis. (2018) 13:1177–86. doi: 10.2147/COPD.S151820
49. Yu, L, and Hales, CA. Hypoxia does neither stimulate pulmonary artery endothelial cell proliferation in mice and rats with pulmonary hypertension and vascular remodeling nor in human pulmonary artery endothelial cells. J Vasc Res. (2011) 48:465–75. doi: 10.1159/000327005
50. Raghavan, A, Zhou, G, Zhou, Q, Ibe, JC, Ramchandran, R, Yang, Q, et al. Hypoxia-induced pulmonary arterial smooth muscle cell proliferation is controlled by forkhead box M1. Am J Respir Cell Mol Biol. (2012) 46:431–6. doi: 10.1165/rcmb.2011-0128OC
51. Li, X, Peng, K, Zhou, Y, Deng, F, and Ma, J. Inhibitory effect of bailing capsule on hypoxia-induced proliferation of rat pulmonary arterial smooth muscle cells. Saudi Med J. (2016) 37:498–505. doi: 10.15537/smj.2016.5.14953
52. Porter, KM, Kang, BY, Adesina, SE, Murphy, TC, Hart, CM, and Sutliff, RL. Chronic hypoxia promotes pulmonary artery endothelial cell proliferation through H2O2-induced 5-lipoxygenase. PLoS One. (2014) 9:e98532. doi: 10.1371/journal.pone.0098532
53. Dabral, S, Tian, X, Kojonazarov, B, Savai, R, Ghofrani, HA, Weissmann, N, et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur Respir J. (2016) 48:1137–49. doi: 10.1183/13993003.00773-2015
54. MacNee, W. Pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc. (2005) 2:258–66. doi: 10.1513/pats.200504-045SR
55. Wright, JL, Tai, H, and Churg, A. Vasoactive mediators and pulmonary hypertension after cigarette smoke exposure in the guinea pig. J Appl Physiol. (2006) 100:672–8. doi: 10.1152/japplphysiol.00274.2005
56. Barbera, JA, and Blanco, I. Pulmonary hypertension in patients with chronic obstructive pulmonary disease: advances in pathophysiology and management. Drugs. (2009) 69:1153–71. doi: 10.2165/00003495-200969090-00002
57. Wright, JL, Lawson, L, Pare, PD, Hooper, RO, Peretz, DI, Nelems, JM, et al. The structure and function of the pulmonary vasculature in mild chronic obstructive pulmonary disease. The effect of oxygen and exercise. Am Rev Respir Dis. (1983) 128:702–7. doi: 10.1164/arrd.1983.128.4.702
58. Naeije, R, and Barbera, JA. Pulmonary hypertension associated with COPD. Crit Care. (2001) 5:286–9. doi: 10.1186/cc1049
59. Xiang, M, Xu, YJ, Liu, XS, and Zeng, DX. Cigarette smoke extract promotes human pulmonary artery smooth muscle cells proliferation through protein kinase C alpha-dependent induction of cyclin D1. Chin Med J. (2010) 123:3663–70.
60. Alqarni, AA, Brand, OJ, Pasini, A, Alahmari, M, Alghamdi, A, and Pang, L. Imbalanced prostanoid release mediates cigarette smoke-induced human pulmonary artery cell proliferation. Respir Res. (2022) 23:136. doi: 10.1186/s12931-022-02056-z
61. Ferrer, E, Peinado, VI, Castaneda, J, Prieto-Lloret, J, Olea, E, Gonzalez-Martin, MC, et al. Effects of cigarette smoke and hypoxia on pulmonary circulation in the guinea pig. Eur Respir J. (2011) 38:617–27. doi: 10.1183/09031936.00105110
62. Sakao, S, Tatsumi, K, and Voelkel, NF. Reversible or irreversible remodeling in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. (2010) 43:629–34. doi: 10.1165/rcmb.2009-0389TR
63. Tuder, RM. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. (2017) 367:643–9. doi: 10.1007/s00441-016-2539-y
64. Zakynthinos, E, Daniil, Z, Papanikolaou, J, and Makris, D. Pulmonary hypertension in COPD: pathophysiology and therapeutic targets. Curr Drug Targets. (2011) 12:501–13. doi: 10.2174/138945011794751483
65. Labonte, J, Brochu, I, Simard, E, and D'Orleans-Juste, P. Distinct modulation of the endothelin-1 pathway in iNOS−/− and eNOS−/− mice. Can J Physiol Pharmacol. (2008) 86:516–25. doi: 10.1139/Y08-042
66. Umar, S, and van der Laarse, A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol Cell Biochem. (2010) 333:191–201. doi: 10.1007/s11010-009-0219-x
67. Newby, AC, Southgate, KM, and Assender, JW. Inhibition of vascular smooth muscle cell proliferation by endothelium-dependent vasodilators. Herz. (1992) 17:291–9.
68. Coggins, MP, and Bloch, KD. Nitric oxide in the pulmonary vasculature. Arterioscler Thromb Vasc Biol. (2007) 27:1877–85. doi: 10.1161/ATVBAHA.107.142943
69. Barbera, JA, Peinado, VI, Santos, S, Ramirez, J, Roca, J, and Rodriguez-Roisin, R. Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. Am J Respir Crit Care Med. (2001) 164:709–13. doi: 10.1164/ajrccm.164.4.2101023
70. Giaid, A, and Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. (1995) 333:214–21. doi: 10.1056/NEJM199507273330403
71. Milara, J, Ortiz, JL, Juan, G, Guijarro, R, Almudever, P, Martorell, M, et al. Cigarette smoke exposure up-regulates endothelin receptor B in human pulmonary artery endothelial cells: molecular and functional consequences. Br J Pharmacol. (2010) 161:1599–615. doi: 10.1111/j.1476-5381.2010.00979.x
72. Fike, CD, Kaplowitz, MR, Thomas, CJ, and Nelin, LD. Chronic hypoxia decreases nitric oxide production and endothelial nitric oxide synthase in newborn pig lungs. Am J Phys. (1998) 274:L517–26. doi: 10.1152/ajplung.1998.274.4.L517
73. Takemoto, M, Sun, J, Hiroki, J, Shimokawa, H, and Liao, JK. Rho-kinase mediates hypoxia-induced downregulation of endothelial nitric oxide synthase. Circulation. (2002) 106:57–62. doi: 10.1161/01.CIR.0000020682.73694.AB
74. Tonelli, AR, Haserodt, S, Aytekin, M, and Dweik, RA. Nitric oxide deficiency in pulmonary hypertension: pathobiology and implications for therapy. Pulm Circ. (2013) 3:20–30. doi: 10.4103/2045-8932.109911
75. Humbert, M, Lau, EM, Montani, D, Jais, X, Sitbon, O, and Simonneau, G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. (2014) 130:2189–208. doi: 10.1161/CIRCULATIONAHA.114.006974
76. Jeremy, JY, Rowe, D, Emsley, AM, and Newby, AC. Nitric oxide and the proliferation of vascular smooth muscle cells. Cardiovasc Res. (1999) 43:580–94. doi: 10.1016/S0008-6363(99)00171-6
77. Pichl, A, Sommer, N, Bednorz, M, Seimetz, M, Hadzic, S, Kuhnert, S, et al. Riociguat for treatment of pulmonary hypertension in COPD – a translational study. Eur Respir J. (2019) 53:1802445. doi: 10.1183/13993003.02445-2018
78. Sharp, Merck, and LLC, Dohme, MK-5475-013 INSIGNIA-PH-COPD: a study of the efficacy and safety of MK-5475 (an inhaled sGC stimulator) in adults with PH-COPD. (2023). Available at: https://classic.clinicaltrials.gov/show/NCT05612035
79. Dominguez-Fandos, D, Valdes, C, Ferrer, E, Puig-Pey, R, Blanco, I, Tura-Ceide, O, et al. Sildenafil in a cigarette smoke-induced model of COPD in the guinea-pig. Eur Respir J. (2015) 46:346–54. doi: 10.1183/09031936.00139914
80. Sebkhi, A, Strange, JW, Phillips, SC, Wharton, J, and Wilkins, MR. Phosphodiesterase type 5 as a target for the treatment of hypoxia-induced pulmonary hypertension. Circulation. (2003) 107:3230–5. doi: 10.1161/01.CIR.0000074226.20466.B1
81. Zhao, L, Mason, NA, Morrell, NW, Kojonazarov, B, Sadykov, A, Maripov, A, et al. Sildenafil inhibits hypoxia-induced pulmonary hypertension. Circulation. (2001) 104:424–8. doi: 10.1161/hc2901.093117
82. Blanco, I, Gimeno, E, Munoz, PA, Pizarro, S, Gistau, C, Rodriguez-Roisin, R, et al. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am J Respir Crit Care Med. (2010) 181:270–8. doi: 10.1164/rccm.200907-0988OC
83. Rietema, H, Holverda, S, Bogaard, HJ, Marcus, JT, Smit, HJ, Westerhof, N, et al. Sildenafil treatment in COPD does not affect stroke volume or exercise capacity. Eur Respir J. (2008) 31:759–64. doi: 10.1183/09031936.00114207
84. Lederer, DJ, Bartels, MN, Schluger, NW, Brogan, F, Jellen, P, Thomashow, BM, et al. Sildenafil for chronic obstructive pulmonary disease: a randomized crossover trial. COPD. (2012) 9:268–75. doi: 10.3109/15412555.2011.651180
85. Blanco, I, Santos, S, Gea, J, Guell, R, Torres, F, Gimeno-Santos, E, et al. Sildenafil to improve respiratory rehabilitation outcomes in COPD: a controlled trial. Eur Respir J. (2013) 42:982–92. doi: 10.1183/09031936.00176312
86. Waxman, A, Restrepo-Jaramillo, R, Thenappan, T, Ravichandran, A, Engel, P, Bajwa, A, et al. Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med. (2021) 384:325–34. doi: 10.1056/NEJMoa2008470
87. Alqarni, AA, Aldhahir, AM, Bintalib, HM, Alqahtani, JS, Siraj, RA, Majrshi, M, et al. Inhaled therapies targeting prostacyclin pathway in pulmonary hypertension due to COPD: systematic review. Front Med. (2023) 10:1217156. doi: 10.3389/fmed.2023.1217156
88. Adnot, S, Kouyoumdjian, C, Defouilloy, C, Andrivet, P, Sediame, S, Herigault, R, et al. Hemodynamic and gas exchange responses to infusion of acetylcholine and inhalation of nitric oxide in patients with chronic obstructive lung disease and pulmonary hypertension. Am Rev Respir Dis. (1993) 148:310–6. doi: 10.1164/ajrccm/148.2.310
89. Hajian, B, De Backer, J, Vos, W, Van Holsbeke, C, Ferreira, F, Quinn, DA, et al. Pulmonary vascular effects of pulsed inhaled nitric oxide in COPD patients with pulmonary hypertension. Int J Chron Obstruct Pulmon Dis. (2016) 11:1533–41. doi: 10.2147/COPD.S106480
90. Chida, M, and Voelkel, NF. Effects of acute and chronic hypoxia on rat lung cyclooxygenase. Am J Phys. (1996) 270:L872–8. doi: 10.1152/ajplung.1996.270.5.L872
91. Gomberg-Maitland, M, and Olschewski, H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur Respir J. (2008) 31:891–901. doi: 10.1183/09031936.00097107
92. Profit, L, and Chrisp, P. Lumiracoxib: the evidence of its clinical impact on the treatment of osteoarthritis. Core Evid. (2007) 2:131–50. doi: 10.2147/CE.S7425
93. Bradbury, DA, Newton, R, Zhu, YM, Stocks, J, Corbett, L, Holland, ED, et al. Effect of bradykinin, TGF-beta1, IL-1beta, and hypoxia on COX-2 expression in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. (2002) 283:L717–25. doi: 10.1152/ajplung.00070.2002
94. Lang, IM, and Gaine, SP. Recent advances in targeting the prostacyclin pathway in pulmonary arterial hypertension. Eur Respir Rev. (2015) 24:630–41. doi: 10.1183/16000617.0067-2015
95. Majed, BH, and Khalil, RA. Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol Rev. (2012) 64:540–82. doi: 10.1124/pr.111.004770
96. Christman, BW, McPherson, CD, Newman, JH, King, GA, Bernard, GR, Groves, BM, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. (1992) 327:70–5. doi: 10.1056/NEJM199207093270202
97. Fan, F, Tian, H, Geng, J, Deng, J, Liu, Y, Chen, C, et al. Mechanism of Beraprost effects on pulmonary hypertension: contribution of cross-binding to PGE2 receptor 4 and modulation of O2 sensitive voltage-gated K(+) channels. Front Pharmacol. (2018) 9:1518. doi: 10.3389/fphar.2018.01518
98. Smith, WL, DeWitt, DL, and Garavito, RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. (2000) 69:145–82. doi: 10.1146/annurev.biochem.69.1.145
99. DuBois, RN, Awad, J, Morrow, J, Roberts, LJ, and Bishop, PR. Regulation of eicosanoid production and mitogenesis in rat intestinal epithelial cells by transforming growth factor-alpha and phorbol ester. J Clin Investig. (1994) 93:493–8. doi: 10.1172/JCI116998
100. Subbaramaiah, K, Telang, N, Ramonetti, JT, Araki, R, DeVito, B, Weksler, BB, et al. Transcription of cyclooxygenase-2 is enhanced in transformed mammary epithelial cells. Cancer Res. (1996) 56:4424–9.
101. Sheng, H, Williams, CS, Shao, J, Liang, P, DuBois, RN, and Beauchamp, RD. Induction of cyclooxygenase-2 by activated ha-ras oncogene in Rat-1 fibroblasts and the role of mitogen-activated protein kinase pathway. J Biol Chem. (1998) 273:22120–7. doi: 10.1074/jbc.273.34.22120
102. Chen, Y, Chen, P, Hanaoka, M, Droma, Y, and Kubo, K. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology. (2008) 13:1014–21. doi: 10.1111/j.1440-1843.2008.01365.x
103. Shi, Z, Chen, Y, Pei, Y, Long, Y, Liu, C, Cao, J, et al. The role of cyclooxygenase-2 in the protection against apoptosis in vascular endothelial cells induced by cigarette smoking. J Thorac Dis. (2017) 9:30–41. doi: 10.21037/jtd.2017.01.23
104. Nana-Sinkam, SP, Lee, JD, Sotto-Santiago, S, Stearman, RS, Keith, RL, Choudhury, Q, et al. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med. (2007) 175:676–85. doi: 10.1164/rccm.200605-724OC
105. Zhao, L, Wu, Y, Xu, Z, Wang, H, Zhao, Z, Li, Y, et al. Involvement of COX-2/PGE2 signalling in hypoxia-induced angiogenic response in endothelial cells. J Cell Mol Med. (2012) 16:1840–55. doi: 10.1111/j.1582-4934.2011.01479.x
106. Schmedtje, JF Jr, Ji, YS, Liu, WL, DuBois, RN, and Runge, MS. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J Biol Chem. (1997) 272:601–8. doi: 10.1074/jbc.272.1.601
107. Wort, SJ, Woods, M, Warner, TD, Evans, TW, and Mitchell, JA. Cyclooxygenase-2 acts as an endogenous brake on endothelin-1 release by human pulmonary artery smooth muscle cells: implications for pulmonary hypertension. Mol Pharmacol. (2002) 62:1147–53. doi: 10.1124/mol.62.5.1147
108. Kylhammar, D, and Radegran, G. Cyclooxygenase-2 inhibition and thromboxane a(2) receptor antagonism attenuate hypoxic pulmonary vasoconstriction in a porcine model. Acta Physiol (Oxf). (2012) 205:507–19. doi: 10.1111/j.1748-1716.2012.02437.x
109. Rakotoniaina, Z, Guerard, P, Lirussi, F, Rochette, L, Dumas, M, Goirand, F, et al. Celecoxib but not the combination of celecoxib+atorvastatin prevents the development of monocrotaline-induced pulmonary hypertension in the rat. Naunyn Schmiedeberg's Arch Pharmacol. (2008) 378:241–51. doi: 10.1007/s00210-008-0298-3
110. Fike, CD, Kaplowitz, MR, Zhang, Y, and Pfister, SL. Cyclooxygenase-2 and an early stage of chronic hypoxia-induced pulmonary hypertension in newborn pigs. J Appl Physiol. (2005) 98:1111–8. doi: 10.1152/japplphysiol.00810.2004
111. Camacho, M, Rodriguez, C, Guadall, A, Alcolea, S, Orriols, M, Escudero, JR, et al. Hypoxia upregulates PGI-synthase and increases PGI(2) release in human vascular cells exposed to inflammatory stimuli. J Lipid Res. (2011) 52:720–31. doi: 10.1194/jlr.M011007
112. Yin, J, Xia, W, Zhang, Y, Ding, G, Chen, L, Yang, G, et al. Role of dihydroartemisinin in regulating prostaglandin E2 synthesis cascade and inflammation in endothelial cells. Heart Vessel. (2018) 33:1411–22. doi: 10.1007/s00380-018-1190-9
113. Swan, CE, and Breyer, RM. Prostaglandin E2 modulation of blood pressure homeostasis: studies in rodent models. Prostaglandins Other Lipid Mediat. (2011) 96:10–3. doi: 10.1016/j.prostaglandins.2011.07.001
114. Murakami, M, Nakashima, K, Kamei, D, Masuda, S, Ishikawa, Y, Ishii, T, et al. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem. (2003) 278:37937–47. doi: 10.1074/jbc.M305108200
115. Yang, G, and Chen, L. An update of microsomal prostaglandin E Synthase-1 and PGE2 receptors in cardiovascular health and diseases. Oxidative Med Cell Longev. (2016) 2016:5249086. doi: 10.1155/2016/5249086
116. Sasaki, Y, Kamiyama, S, Kamiyama, A, Matsumoto, K, Akatsu, M, Nakatani, Y, et al. Genetic-deletion of cyclooxygenase-2 downstream prostacyclin synthase suppresses inflammatory reactions but facilitates carcinogenesis, unlike deletion of microsomal prostaglandin E synthase-1. Sci Rep. (2015) 5:17376. doi: 10.1038/srep17376
117. Chen, L, Yang, G, Xu, X, Grant, G, Lawson, JA, Bohlooly, YM, et al. Cell selective cardiovascular biology of microsomal prostaglandin E synthase-1. Circulation. (2013) 127:233–43. doi: 10.1161/CIRCULATIONAHA.112.119479
118. Martin, EM, and Jones, SL. Inhibition of microsomal prostaglandin E-synthase-1 (mPGES-1) selectively suppresses PGE2 in an in vitro equine inflammation model. Vet Immunol Immunopathol. (2017) 192:33–40. doi: 10.1016/j.vetimm.2017.09.008
119. Lundequist, A, Nallamshetty, SN, Xing, W, Feng, C, Laidlaw, TM, Uematsu, S, et al. Prostaglandin E(2) exerts homeostatic regulation of pulmonary vascular remodeling in allergic airway inflammation. J Immunol. (2010) 184:433–41. doi: 10.4049/jimmunol.0902835
120. Yin, J, Xia, W, Li, Y, Guo, C, Zhang, Y, Huang, S, et al. COX-2 mediates PM2.5-induced apoptosis and inflammation in vascular endothelial cells. Am J Transl Res. (2017) 9:3967–76.
121. Patel, JA, Shen, L, Hall, SM, Benyahia, C, Norel, X, McAnulty, RJ, et al. Prostanoid EP(2) receptors are up-regulated in human pulmonary arterial hypertension: a key anti-proliferative target for Treprostinil in smooth muscle cells. Int J Mol Sci. (2018) 19:2372. doi: 10.3390/ijms19082372
122. Needleman, P, Moncada, S, Bunting, S, Vane, JR, Hamberg, M, and Samuelsson, B. Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature. (1976) 261:558–60. doi: 10.1038/261558a0
123. Kawabe, J, Ushikubi, F, and Hasebe, N. Prostacyclin in vascular diseases. - recent insights and future perspectives. Circ J. (2010) 74:836–43. doi: 10.1253/circj.CJ-10-0195
124. Liou, JY, Shyue, SK, Tsai, MJ, Chung, CL, Chu, KY, and Wu, KK. Colocalization of prostacyclin synthase with prostaglandin H synthase-1 (PGHS-1) but not phorbol ester-induced PGHS-2 in cultured endothelial cells. J Biol Chem. (2000) 275:15314–20. doi: 10.1074/jbc.275.20.15314
125. Wu, KK, and Liou, JY. Cellular and molecular biology of prostacyclin synthase. Biochem Biophys Res Commun. (2005) 338:45–52. doi: 10.1016/j.bbrc.2005.08.021
126. Chen, Y, Luo, H, Kang, N, Guan, C, Long, Y, Cao, J, et al. Beraprost sodium attenuates cigarette smoke extract-induced apoptosis in vascular endothelial cells. Mol Biol Rep. (2012) 39:10447–57. doi: 10.1007/s11033-012-1924-1
127. Chen, Y, Hanaoka, M, Chen, P, Droma, Y, Voelkel, NF, and Kubo, K. Protective effect of beraprost sodium, a stable prostacyclin analog, in the development of cigarette smoke extract-induced emphysema. Am J Physiol Lung Cell Mol Physiol. (2009) 296:L648–56. doi: 10.1152/ajplung.90270.2008
128. Yang, X, Sheares, KK, Davie, N, Upton, PD, Taylor, GW, Horsley, J, et al. Hypoxic induction of cox-2 regulates proliferation of human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. (2002) 27:688–96. doi: 10.1165/rcmb.2002-0067OC
129. Kawakami, T, Kanazawa, H, Satoh, T, Ieda, M, Ieda, Y, Kimura, K, et al. AAV-PGIS gene transfer improves hypoxia-induced pulmonary hypertension in mice. Biochem Biophys Res Commun. (2007) 363:656–61. doi: 10.1016/j.bbrc.2007.09.039
130. Geraci, MW, Gao, B, Shepherd, DC, Moore, MD, Westcott, JY, Fagan, KA, et al. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J Clin Investig. (1999) 103:1509–15. doi: 10.1172/JCI5911
131. Kadowaki, M, Mizuno, S, Demura, Y, Ameshima, S, Miyamori, I, and Ishizaki, T. Effect of hypoxia and Beraprost sodium on human pulmonary arterial smooth muscle cell proliferation: the role of p27kip1. Respir Res. (2007) 8:77. doi: 10.1186/1465-9921-8-77
132. Barst, RJ. A review of pulmonary arterial hypertension: role of ambrisentan. Vasc Health Risk Manag. (2007) 3:11–22. doi: 10.1007/978-1-84628-653-7
133. Sitbon, O, Delcroix, M, Bergot, E, Boonstra, AB, Granton, J, Langleben, D, et al. EPITOME-2: an open-label study assessing the transition to a new formulation of intravenous epoprostenol in patients with pulmonary arterial hypertension. Am Heart J. (2014) 167:210–7. doi: 10.1016/j.ahj.2013.08.007
134. Simonneau, G, Barst, RJ, Galie, N, Naeije, R, Rich, S, Bourge, RC, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. (2002) 165:800–4. doi: 10.1164/ajrccm.165.6.2106079
135. Simonneau, G, Torbicki, A, Hoeper, MM, Delcroix, M, Karlocai, K, Galie, N, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. (2012) 40:874–80. doi: 10.1183/09031936.00137511
136. Olschewski, H, Simonneau, G, Galie, N, Higenbottam, T, Naeije, R, Rubin, LJ, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. (2002) 347:322–9. doi: 10.1056/NEJMoa020204
137. Galie, N, Humbert, M, Vachiery, JL, Vizza, CD, Kneussl, M, Manes, A, et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. (2002) 39:1496–502. doi: 10.1016/s0735-1097(02)01786-2
138. Hiremath, J, Thanikachalam, S, Parikh, K, Shanmugasundaram, S, Bangera, S, Shapiro, L, et al. Exercise improvement and plasma biomarker changes with intravenous treprostinil therapy for pulmonary arterial hypertension: a placebo-controlled trial. J Heart Lung Transplant. (2010) 29:137–49. doi: 10.1016/j.healun.2009.09.005
139. Therapeutics, U, and PBC, LB. Inhaled treprostinil in participants with pulmonary hypertension due to chronic obstructive pulmonary disease (PH-COPD) Available at: https://classic.clinicaltrials.gov/show/NCT03794583 (2018).
140. Wilson, SJ, Roche, AM, Kostetskaia, E, and Smyth, EM. Dimerization of the human receptors for prostacyclin and thromboxane facilitates thromboxane receptor-mediated cAMP generation. J Biol Chem. (2004) 279:53036–47. doi: 10.1074/jbc.M405002200
141. Ruan, C-H, Dixon, RAF, Willerson, JT, and Ruan, K-H. Prostacyclin therapy for pulmonary arterial hypertension. Tex Heart Inst J. (2010) 37:391–9.
142. Ellis, EF, Oelz, O, Roberts, LJ 2nd, Payne, NA, Sweetman, BJ, Nies, AS, et al. Coronary arterial smooth muscle contraction by a substance released from platelets: evidence that it is thromboxane A2. Science. (1976) 193:1135–7. doi: 10.1126/science.959827
143. Chen, H. Role of thromboxane A2 signaling in endothelium-dependent contractions of arteries. Prostaglandins Other Lipid Mediat. (2018) 134:32–7. doi: 10.1016/j.prostaglandins.2017.11.004
144. Touyz, RM, Alves-Lopes, R, Rios, FJ, Camargo, LL, Anagnostopoulou, A, Arner, A, et al. Vascular smooth muscle contraction in hypertension. Cardiovasc Res. (2018) 114:529–39. doi: 10.1093/cvr/cvy023
145. Alqarni, AA. Increased thromboxane A(2) levels in pulmonary artery smooth muscle cells isolated from patients with chronic obstructive pulmonary disease. Medicina (Kaunas). (2023) 59:165. doi: 10.3390/medicina59010165
146. Rubin, LJ, and Badesch, DB. Evaluation and management of the patient with pulmonary arterial hypertension. Ann Intern Med. (2005) 143:282–92. doi: 10.7326/0003-4819-143-4-200508160-00009
147. Badesch, DB, McLaughlin, VV, Delcroix, M, Vizza, CD, Olschewski, H, Sitbon, O, et al. Prostanoid therapy for pulmonary arterial hypertension. J Am Coll Cardiol. (2004) 43:56S–61S. doi: 10.1016/j.jacc.2004.02.036
148. Jones, DA, Benjamin, CW, and Linseman, DA. Activation of thromboxane and prostacyclin receptors elicits opposing effects on vascular smooth muscle cell growth and mitogen-activated protein kinase signaling cascades. Mol Pharmacol. (1995) 48:890–6.
149. Davi, G, Basili, S, Vieri, M, Cipollone, F, Santarone, S, Alessandri, C, et al. Enhanced thromboxane biosynthesis in patients with chronic obstructive pulmonary disease. The Chronic Obstructive Bronchitis and Haemostasis Study Group. Am J Respir Crit Care Med. (1997) 156:1794–9. doi: 10.1164/ajrccm.156.6.9706026
150. An, J, Li, JQ, Wang, T, Li, XO, Guo, LL, Wan, C, et al. Blocking of thromboxane A(2) receptor attenuates airway mucus hyperproduction induced by cigarette smoke. Eur J Pharmacol. (2013) 703:11–7. doi: 10.1016/j.ejphar.2013.01.042
151. Su, YC, and Jin, XR. Relation of TXA2 and PGI2 to the difference in hypoxic pulmonary vasoconstriction between different strains of rats. J Tongji Med Univ. (1989) 9:148–52. doi: 10.1007/BF02908965
152. Delannoy, E, Courtois, A, Freund-Michel, V, Leblais, V, Marthan, R, and Muller, B. Hypoxia-induced hyperreactivity of pulmonary arteries: role of cyclooxygenase-2, isoprostanes, and thromboxane receptors. Cardiovasc Res. (2010) 85:582–92. doi: 10.1093/cvr/cvp292
153. Hirenallur, SD, Detweiler, ND, Haworth, ST, Leming, JT, Gordon, JB, and Rusch, NJ. Furegrelate, a thromboxane synthase inhibitor, blunts the development of pulmonary arterial hypertension in neonatal piglets. Pulm Circ. (2012) 2:193–200. doi: 10.4103/2045-8932.97605
154. Kawut, SM, Bagiella, E, Shimbo, D, Lederer, DJ, Al-Naamani, N, Roberts, KE, et al. Rationale and design of a phase II clinical trial of aspirin and simvastatin for the treatment of pulmonary arterial hypertension: ASA-STAT. Contemp Clin Trials. (2011) 32:280–7. doi: 10.1016/j.cct.2010.12.005
155. Robbins, IM, Kawut, SM, Yung, D, Reilly, MP, Lloyd, W, Cunningham, G, et al. A study of aspirin and clopidogrel in idiopathic pulmonary arterial hypertension. Eur Respir J. (2006) 27:578–84. doi: 10.1183/09031936.06.00095705
156. Galie, N, Humbert, M, Vachiery, JL, Gibbs, S, Lang, I, Torbicki, A, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. (2016) 37:67–119. doi: 10.1093/eurheartj/ehv317
157. Yanagisawa, M, Kurihara, H, Kimura, S, Tomobe, Y, Kobayashi, M, Mitsui, Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. (1988) 332:411–5. doi: 10.1038/332411a0
158. Compeer, MG, Suylen, DPL, Hackeng, TM, and De Mey, JGR. Endothelin-1 and -2: two amino acids matter. Life Sci. (2012) 91:607–12. doi: 10.1016/j.lfs.2012.02.011
159. Fagan, KA, McMurtry, IF, and Rodman, DM. Role of endothelin-1 in lung disease. Respir Res. (2001) 2:90–101. doi: 10.1186/rr44
160. Bondurand, N, Dufour, S, and Pingault, V. News from the endothelin-3/EDNRB signaling pathway: role during enteric nervous system development and involvement in neural crest-associated disorders. Dev Biol. (2018) 444:S156–69. doi: 10.1016/j.ydbio.2018.08.014
161. Kowalczyk, A, Kleniewska, P, Kolodziejczyk, M, Skibska, B, and Goraca, A. The role of endothelin-1 and endothelin receptor antagonists in inflammatory response and sepsis. Arch Immunol Ther Exp. (2015) 63:41–52. doi: 10.1007/s00005-014-0310-1
162. Markewitz, BA, Farrukh, IS, Chen, Y, Li, Y, and Michael, JR. Regulation of endothelin-1 synthesis in human pulmonary arterial smooth muscle cells. Effects of transforming growth factor-beta and hypoxia. Cardiovasc Res. (2001) 49:200–6. doi: 10.1016/S0008-6363(00)00221-2
163. Galie, N, Manes, A, and Branzi, A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res. (2004) 61:227–37. doi: 10.1016/j.cardiores.2003.11.026
164. Hirata, Y, Emori, T, Eguchi, S, Kanno, K, Imai, T, Ohta, K, et al. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J Clin Invest. (1993) 91:1367–73. doi: 10.1172/JCI116338
165. Xiong, PY, Potus, F, Chan, W, and Archer, SL. Models and molecular mechanisms of World Health Organization Group 2 to 4 pulmonary hypertension. Hypertension. (2018) 71:34–55. doi: 10.1161/HYPERTENSIONAHA.117.08824
166. Fukuroda, T, Kobayashi, M, Ozaki, S, Yano, M, Miyauchi, T, Onizuka, M, et al. Endothelin receptor subtypes in human versus rabbit pulmonary arteries J Appl Physiol (1994) 76:1976–82. doi: 10.1152/jappl.1994.76.5.1976,
167. Giaid, A, Yanagisawa, M, Langleben, D, Michel, RP, Levy, R, Shennib, H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. (1993) 328:1732–9. doi: 10.1056/NEJM199306173282402
168. Stewart, DJ, Levy, RD, Cernacek, P, and Langleben, D. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med. (1991) 114:464–9. doi: 10.7326/0003-4819-114-6-464
169. Yang, D, Wang, L, Jiang, P, Kang, R, and Xie, Y. Correlation between hs-CRP, IL-6, IL-10, ET-1, and chronic obstructive pulmonary disease combined with pulmonary hypertension. J Healthc Eng. (2022) 2022:3247807. doi: 10.1155/2022/3247807
170. Milara, J, Gabarda, E, Juan, G, Ortiz, JL, Guijarro, R, Martorell, M, et al. Bosentan inhibits cigarette smoke-induced endothelin receptor expression in pulmonary arteries. Eur Respir J. (2012) 39:927–38. doi: 10.1183/09031936.00021411
171. Lee, SD, Lee, DS, Chun, YG, Shim, TS, Lim, CM, Koh, Y, et al. Cigarette smoke extract induces endothelin-1 via protein kinase C in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. (2001) 281:L403–11. doi: 10.1152/ajplung.2001.281.2.L403
172. Zhang, J, Narayan, VM, Juedes, N, and Patel, JM. Hypoxic upregulation of preproendothelin-1 gene expression is associated with protein tyrosine kinase-PI3K signaling in cultured lung vascular endothelial cells. Int J Clin Exp Med. (2009) 2:87–94.
173. Minchenko, A, and Caro, J. Regulation of endothelin-1 gene expression in human microvascular endothelial cells by hypoxia and cobalt: role of hypoxia responsive element. Mol Cell Biochem. (2000) 208:53–62. doi: 10.1023/a:1007042729486
174. Markewitz, BA, Kohan, DE, and Michael, JR. Hypoxia decreases endothelin-1 synthesis by rat lung endothelial cells. Am J Phys. (1995) 269:L215–20. doi: 10.1152/ajplung.1995.269.2.L215
175. Galie, N, Hinderliter, AL, Torbicki, A, Fourme, T, Simonneau, G, Pulido, T, et al. Effects of the oral endothelin-receptor antagonist bosentan on echocardiographic and doppler measures in patients with pulmonary arterial hypertension. J Am Coll Cardiol. (2003) 41:1380–6. doi: 10.1016/s0735-1097(03)00121-9
176. Galie, N, Rubin, L, Hoeper, M, Jansa, P, Al-Hiti, H, Meyer, G, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. (2008) 371:2093–100. doi: 10.1016/S0140-6736(08)60919-8
177. Channick, RN, Simonneau, G, Sitbon, O, Robbins, IM, Frost, A, Tapson, VF, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. (2001) 358:1119–23. doi: 10.1016/S0140-6736(01)06250-X
178. Rubin, LJ, Badesch, DB, Barst, RJ, Galie, N, Black, CM, Keogh, A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. (2002) 346:896–903. doi: 10.1056/NEJMoa012212
179. Davie, N, Haleen, SJ, Upton, PD, Polak, JM, Yacoub, MH, Morrell, NW, et al. ET(a) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am J Respir Crit Care Med. (2002) 165:398–405. doi: 10.1164/ajrccm.165.3.2104059
180. Kunichika, N, Landsberg, JW, Yu, Y, Kunichika, H, Thistlethwaite, PA, Rubin, LJ, et al. Bosentan inhibits transient receptor potential channel expression in pulmonary vascular myocytes. Am J Respir Crit Care Med. (2004) 170:1101–7. doi: 10.1164/rccm.200312-1668OC
181. Stolz, D, Rasch, H, Linka, A, Di Valentino, M, Meyer, A, Brutsche, M, et al. A randomised, controlled trial of bosentan in severe COPD. Eur Respir J. (2008) 32:619–28. doi: 10.1183/09031936.00011308
Keywords: nitric oxide, prostanoid, pulmonary hypertension, COPD, endothelin, type 3 pulmonary hypertension, COPD-associated pulmonary hypertension, pulmonary hypertension in COPD
Citation: Alqarni AA, Aldhahir AM, Alghamdi SA, Alqahtani JS, Siraj RA, Alwafi H, AlGarni AA, Majrshi MS, Alshehri SM and Pang L (2023) Role of prostanoids, nitric oxide and endothelin pathways in pulmonary hypertension due to COPD. Front. Med. 10:1275684. doi: 10.3389/fmed.2023.1275684
Edited by:
Elena Goncharova, University of California, Davis, United StatesReviewed by:
Imad Al Ghouleh, University of Pittsburgh, United StatesCopyright © 2023 Alqarni, Aldhahir, Alghamdi, Alqahtani, Siraj, Alwafi, AlGarni, Majrshi, Alshehri and Pang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdullah A. Alqarni, YWFhbHFhcm5pMUBrYXUuZWR1LnNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.