 Andrew A. Umansky
Andrew A. Umansky Louis Charles Fortier
Louis Charles Fortier- Department of Microbiology and Infectious Diseases, Faculty of Medicine and Health Sciences, Université de Sherbrooke, Sherbrooke, QC, Canada
With the antibiotic crisis and the rise in antimicrobial resistance worldwide, new therapeutic alternatives are urgently needed. Phage therapy represents one of the most promising alternatives but for some pathogens, such as Clostridioides difficile, important challenges are being faced. The perspective of phage therapy to treat C. difficile infections is complicated by the fact that no strictly lytic phages have been identified so far, and current temperate phages generally have a narrow host range. C. difficile also harbors multiple antiphage mechanisms, and the bacterial genome is often a host of one or multiple prophages that can interfere with lytic phage infection. Nevertheless, due to recent advances in phage host receptor recognition and improvements in genetic tools to manipulate phage genomes, it is now conceivable to genetically engineer C. difficile phages to make them suitable for phage therapy. Other phage-based alternatives such as phage endolysins and phage tail-like bacteriocins (avidocins) are also being investigated but these approaches also have their own limitations and challenges. Last but not least, C. difficile produces spores that are resistant to phage attacks and all current antibiotics, and this complicates therapeutic interventions. This mini-review gives a brief historical overview of phage work that has been carried out in C. difficile, presents recent advances in the field, and addresses the most important challenges that are being faced, with potential solutions.
Introduction
Clostridioides difficile is one of the top priority pathogens according to the US CDC (1). This Gram-positive, strictly anaerobic spore-forming bacillus is the main cause of antibiotic-associated diarrhea. In the early 2000s, major outbreaks occurred in North America and Europe (2). One group of strains, designated as ribotype 027 (R027), has been associated with increased disease severity, poorer clinical outcome, and more frequent relapses (3–5). Current treatments rely on antibiotics, which further disrupt the protective gut microbiota. Spores are resistant to all antibiotics, and once antibiotherapy is stopped, residual spores within the gut or spores ingested from contaminated environments can germinate due to the permissive microbiota. Consequently, many patients experience one or more relapses leading to recurrent C. difficile infections (rCDI) (6). The best intervention to treat rCDI is the fecal microbiota transplant (FMT) that swiftly restores the gut microbiota diversity, which is associated with colonization resistance (7). However, this approach presents several limitations including the risk for potential transfer of unwanted microbes and the lack of knowledge on the long-term impact of FMT on health (8). Therefore, other therapeutic strategies are urgently needed. Phage therapy is the administration of bacteriophages (or phages) that specifically kill target bacteria and is a promising alternative or complement to antibiotherapy in the fight against multidrug-resistant pathogens (9–12). The main advantage of therapeutic phages is their great specificity toward target bacteria, thus sparing other beneficial bacteria. A targeted approach like phage therapy could be a very powerful solution in the case of rCDI. Over the last two decades, the potential of phages or phage derivatives to treat or prevent CDI has been explored. This mini-review summarizes the current advances in phage-based approaches to fight against C. difficile. The most urgent challenges that must be addressed and mitigating strategies are also discussed.
Whole phage treatment
Several phages infecting C. difficile have been isolated, but only 33 of them have been sequenced and characterized more deeply. It is important to stress that all C. difficile phages described to date have a temperate lifestyle, i.e., these phages lead to lysogeny (13, 14). Upon incorporation of a prophage, a bacterial host becomes automatically resistant to further lytic reinfection by the same or a related phage. It is, therefore, generally discouraged to use temperate phages for therapy, although genetic engineering has the potential to transform temperate phages into an important source of therapeutic agents (15, 16). Moreover, phage resistance due to lysogeny can be mitigated or even eliminated by the temperate phage antibiotic synergy (tPAS) phenomenon, which consists of combining temperate phages with sub-inhibitory concentrations of antibiotics to activate the SOS response and prevent phage entry into the lysogenic cycle (17). tPAS has not been tested in C. difficile, but the efficacy of several unmodified temperate phages was assessed under different laboratory settings and in preclinical animal models. Table 1 summarizes these studies and those in which phage-derived antimicrobials were tested.
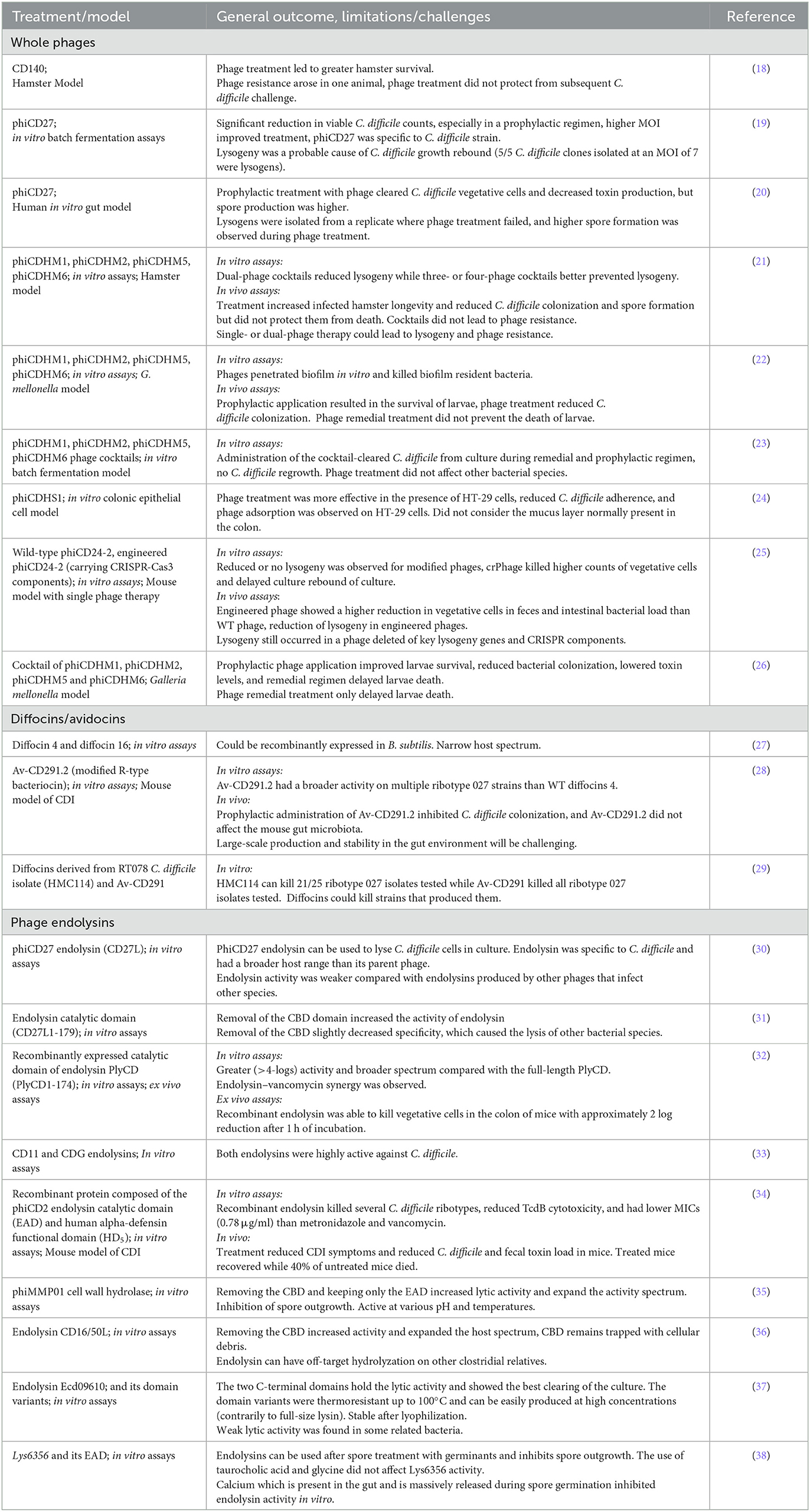
Table 1. Relevant studies investigating the use of phage-based therapeutics against C. difficile.
Phage cocktails are more efficient than single phage treatments
The lytic potential of different C. difficile phages has been assessed in several in vitro assays. The main findings are that phages kill vegetative cells efficiently, reducing bacterial counts by several logs (19, 21, 23, 25). However, one common observation with single phage treatments has been culture rebound due to lysogeny. This was well described with phage phiCD27 used in batch fermentations and in vitro gut models (19, 20). A well-known method to limit the rise of phage resistance is to use phage cocktails (9). The administration of single or multiple phage combinations was compared in vitro (21, 23). Cocktails comprising 3–4 different phages better prevented the culture regrowth compared with cocktails comprising a single phage. Interestingly, some cocktails were shown to prevent C. difficile biofilm formation (22). Biofilms are complex ecosystems generally comprising multiple bacterial species embedded into a matrix composed of extracellular polymeric substances (EPS), such as polysaccharides, DNA, amyloids, lipids, and proteins (39). Some phages possess depolymerase activity at the tip of their tail that can degrade EPS and biofilms (40). Although C. difficile phages with depolymerase activity have never been described, some phages were shown to penetrate and destabilize already-formed biofilms. However, complete eradiction of C. difficile from an already established biofilm was not observed (22). It is worth mentioning that the results varied depending on the targeted C. difficile strain, suggesting that optimization of the cocktails would be necessary on a strain-specific basis. The behavior of phages in the presence of human colonic cells in culture was also investigated, and a higher lytic activity of phage phiCDHS1 was observed in the presence of cultured HT-29 cells. This was explained by the high-phage adsorption to the cell line on which C. difficile also adheres, promoting phage–bacteria interactions (24).
Temperate phages are generally unable to completely cure CDI in animal models
The hamster model of CDI has been used to assess the efficacy of temperate phages. The first study was reported by Ramesh et al. (18). The authors found that simultaneous administration of ϕCD140 along with C. difficile spores greatly increased the survival of infected hamsters, as opposed to untreated animals that died within 72 h (18). While this first study was globally successful, one of the treated hamsters died due to the rise of phage resistance, which had been attributed to phage receptor mutation or lysogeny. In vivo lysogeny was later confirmed in hamsters during treatment with phage phiCD119. The proportion of lysogenic C. difficile clones was shown to increase over time, and by day 4 of the experiment, only lysogens could be isolated (41). These results revealed the incapacity of single-temperate phages to treat CDI in hamsters. To circumvent the problem of lysogeny, two- to four-phage cocktails were tested (22). While the luminal bacterial loads were reduced by at least 4 logs, the four-phage cocktail, administered every 8 h, prolonged hamster survival by 3 days compared with untreated animals. Ultimately, all animals died of CDI. Due to hamsters being highly sensitive to C. difficile and its toxins, the relevance of this model to the human condition has been questioned (42). Therefore, alternative CDI models, such as the wax moth larvae Galleria mellonella, have been recently developed. An optimized four-phage cocktail was tested against infection with a ribotype 014/020 C. difficile strain in G. mellonella larvae. Prophylactic single-dose cocktail administration (106 PFU) prior to bacterial inoculation with 105 CFU of vegetative cells led to complete protection and survival of all insects 60 h post-infection, though ~2-log bacterial counts were still detected at the end of the experiment. When phages were administered simultaneously with bacteria, survival dropped to 72%, whereas treatment with phages 2 h post-infection led to 30% survival after 60 h, and all larvae died at the end of the experiment. Multiple phage doses as well as vancomycin prophylaxis before infection and prior to phage treatment improved the outcome. These results show that the timing of phage inoculation is crucial for the efficacy of phage therapy, and that prophylactic regimens are more effective than remedial regimens, as observed in vitro (22, 26). The G. mellonella model is easier to manipulate than hamsters or mice and can be useful to test different hypotheses. Whether the observations made with this model are readily transferable to a more complex ecosystem like the mammalian gut remains to be demonstrated.
Genetically engineered phages
The first case of a genetically engineered C. difficile phage involved the deletion of the genes coding for the cI repressor and the integrase in phage phiCD24-2 (43), therefore creating the first strictly lytic C. difficile phage (25). The authors also produced a crPhage carrying a mini-CRISPR array targeting the toxin locus, as well as a phage combining both modifications. The crPhages had significantly increased killing power and showed reduced lysogeny while the strictly lytic phages did not lead to lysogeny in vitro. The strictly lytic phage was not better than the wild-type phage at reducing bacterial counts in vitro, contrary to the in vivo condition where it performed better. Unexpectedly, regrowth of C. difficile was observed in the mouse gut with the recombinant phages. Clones recovered from fecal samples were lysogens of the modified phages. This unexpected result could not be clearly explained, but it was hypothesized that endogenous prophages could potentially have complemented the lost functions in the recombinant phages (25).
Phage tail-like particles are potent antimicrobials
Phage tail-like particles are high molecular weight bacteriocins produced by several bacteria as a mechanism to compete with closely related species. Several strains of C. difficile produce these particles, which have been called diffocins (29, 44–47). Diffocins are genetically and structurally related to contractile phage tails but without a capsid. Hence, they do not carry genetic material and cannot replicate. Once adsorbed to a susceptible host via a specific receptor, contraction of the tail sheath leads to perforation of the cell membrane by the inner tail tube, causing leakage of the cell content and death (Figure 1) (48). Heterologous expression of functional recombinant diffocins has been successful in Bacillus subtilis (27). However, akin to their phage homologs, the host range of diffocins is generally narrow. To circumvent this, a genetically engineered diffocin was created by replacing its receptor-binding protein (RBP) with another from phi027, a prophage conserved in the genome of the R20291 and other R027 epidemic strains (28). This hybrid diffocin, called Avidocin-CD291 (or Av-CD291), was redirected toward strain R20291 and was able to kill all ribotype 027 strains tested in vitro, in addition to one or more isolates of ribotype 001, 015, and 087 strains (28). A turning point was reached when diffocins were shown to recognize their bacterial host via binding to the surface layer protein A (SlpA) (49). The authors demonstrated that diffocins' RBPs specifically bind to certain SlpA isoforms (or SLCTs, for surface layer cassette types). Interestingly, they also showed the interchangeability of the diffocin RBPs, and genetically engineered avidocins could be redirected toward different C. difficile strains based on their SLCT status (49). Most importantly, the efficacy of Av-CD291 was tested in a mouse model of C. difficile spore transmission that mimics the natural human-to-human transmission. Prophylactic administration of purified Av-CD291 in the drinking water completely prevented the colonization of mice, and no C. difficile could be detected in fecal samples, as opposed to the placebo group (28). There was no significant change in the composition of the microbiota, suggesting that avidocins are very specific to C. difficile and do not disturb the microbiota (28, 29). This study also revealed that Av-CD291 administration in mice did not disturb colonization resistance to C. difficile or vancomycin-resistant Enterococcus faecium (28).
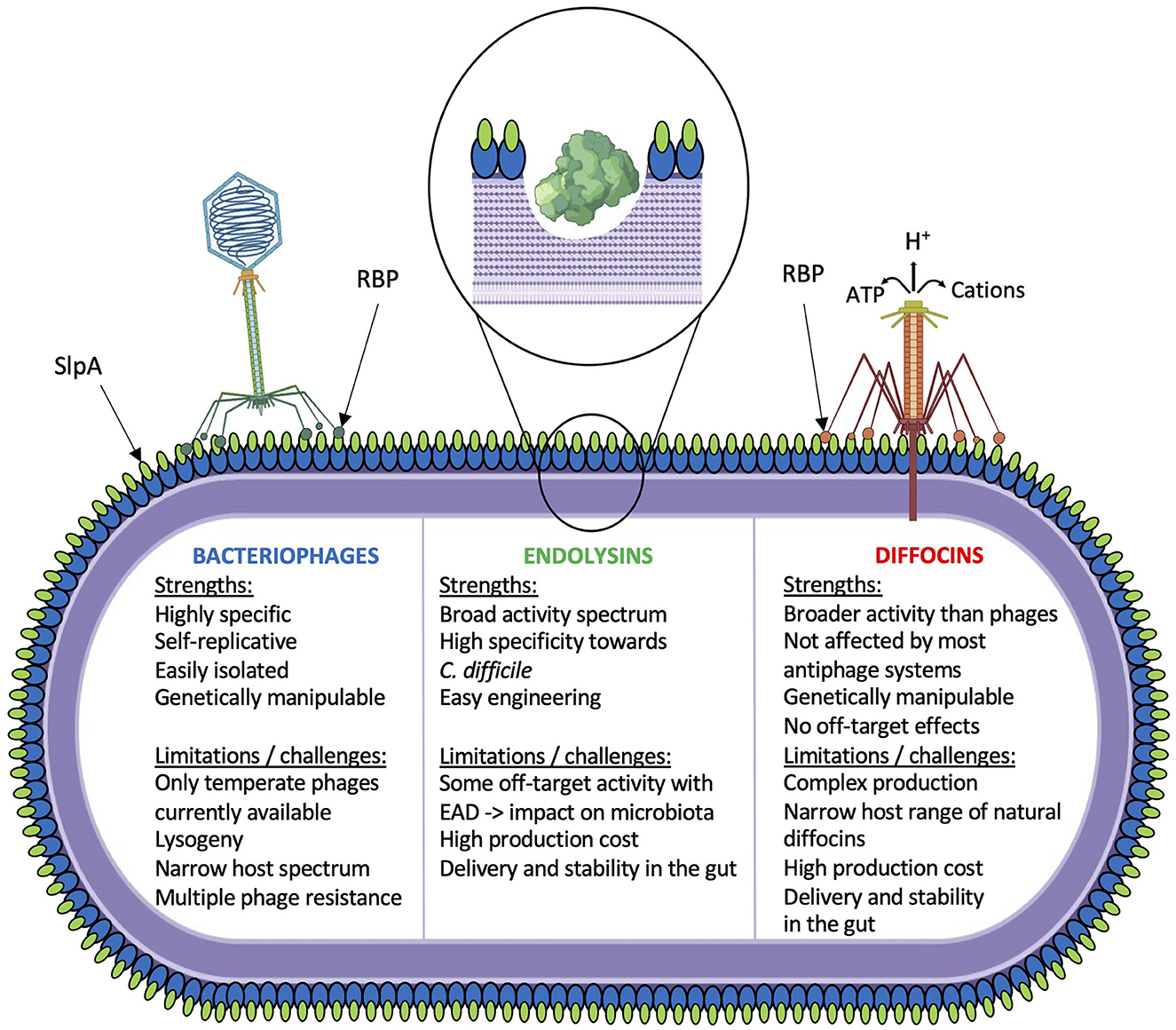
Figure 1. Schematic overview of current phage-based investigations. Strengths and limitations/challenges for each strategy are indicated.
Cell wall-degrading enzymes as antimicrobials
Phage endolysins are also promising alternatives to whole phages (Figure 1). Tailed phages must break up the cell wall to escape their host at the end of the lytic cycle. The canonical holin–endolysin pathway involves a small protein, the holin, that accumulates into the cytoplasmic membrane until a programmed time of the lytic cycle at which point it forms pores into the cell membrane. This allows the endolysin to escape the cytoplasm and reach the peptidoglycan layer that it hydrolyzes from within until lysis (50). Gram-positive phage endolysins are generally composed of an enzymatically active domain (EAD) and a cell wall-binding domain (CBD). Most C. difficile phage endolysins are N-acetylmuramoyl-L-alanine amidases (35, 51). When purified endolysin is added extracellularly to a bacterial suspension in vitro, rapid lysis occurs in a matter of 10–20 min. A few C. difficile phage endolysins have been cloned and characterized (Table 1), CD27L being the first to be described (30). Of note, one common observation that has been reported with all C. difficile phage endolysins is that the catalytic domain of the enzyme alone (EAD) is sufficient for full activity. In fact, the removal of the CBD was reported to increase the lytic activity of C. difficile (30–32, 36, 38). It was also shown that the CBD from the CD16/50L endolysin was responsible for trapping the endolysin within cellular debris after lysis. It was hypothesized that the CBD domain prevents the endolysin from being released freely into the environment upon cell lysis, therefore preserving uninfected bacteria that can serve as hosts for subsequent phage infection (36). Full-length endolysins were shown to be very specific toward C. difficile, as little or no activity was noted on other commensal bacteria, including related Clostridia. However, most truncated endolysins comprising only the EAD displayed a slightly broader host range, killing all C. difficile isolates tested, in addition to a few other species, in particular Clostridium sordellii, Clostridium bifermentans, Bacillus subtilis, and Listeria monocytogenes (30–32, 36, 38).
An interesting feature of gram-positive endolysins is their modular architecture that allows interchanging EAD and CBD. The LHD is a lysin–human defensin fusion protein that results from the fusion of the CBD from phage phiC2 endolysin and the functional domain from the human α-defensin 5 (HD5) (34). LHD was very active on several C. difficile strains of different ribotypes, and the minimum inhibitory concentration (0.78 μg/ml) was >4 times lower than that of metronidazole and vancomycin. Interestingly, LHD inhibited the glycosylation activity and toxicity of TcdB, as shown with HD5 (52). Furthermore, treatment of mice infected with the R20291 epidemic strain twice a day for 7 consecutive days with LHD rescued all treated mice from death, as opposed to 60% survival for control mice. Toxin levels and the number of spores were also reduced in the treated group. Interestingly, pre-treatment of bacteria with vancomycin increased the lytic activity of the endolysin PlyCD1 − 174, suggesting a synergistic effect as described with phages (53). In addition, PlyCD1 − 174 endolysin was shown to be active ex vivo in a complex mouse fecal environment (32).
Discussion
Aside from the common challenges that phage therapy faces in general, such as safety, efficacy, the lack of data from clinical trials, resistance, regulatory hurdles, and patentability, several limitations are particularly relevant to CDI. The most urgent problems to address are as follows: (i) the lack of strictly lytic phages, (ii) the narrow host range of current phages, and (iii) the problem of phage resistance. Alternatives to whole phages also have their limitations, such as (vi) the difficulty to produce avidocins or endolysins on a large scale, (vii) the stability of avidocins and endolysins in the intestinal environment, and (viii) the off-target activity of genetically engineered endolysins. However, there are potential solutions to these limitations.
The lack of strictly lytic phages is the most critical problem with C. difficile phages, and further screening of environmental samples is not the solution. The most reasonable strategy is the genetic engineering of temperate phages. Such phages have been successfully created and tested in other phage–host models (54, 55), and a human patient infected by Mycobacterium abscessus has been treated with one of them (16). The first report of a genetically engineered C. difficile phage has proven the feasibility of this approach, although further research is required to better characterize the behavior of genetically engineered phages in vivo. It will be particularly important to investigate how therapeutic phages interact with the highly prevalent and diverse endogenous prophages in C. difficile genomes (56).
Another important limitation of current C. difficile phages is their narrow host range, implying that multiple phages will need to be combined into cocktails to cover the most clinically relevant strains of C. difficile (21). Due to recent advances in our understanding of host recognition by C. difficile phages, the surface layer protein A (SlpA) seems to be a general receptor used by many phages and diffocins (28, 49, 57–59). It is, therefore, reasonable to foresee the selection of phages based on their RBP to target C. difficile strains expressing specific SlpA isoforms. Although RBP can be identified in phage genomes using bioinformatics tools, it will be important to determine if additional phage proteins participate in host recognition or if other receptors exist, as suggested in one study (58).
Resistance is always a critical concern when undertaking phage therapy (12, 60). Several mechanisms of phage resistance exist (61), and an important one is the mutation of the phage receptor. Work on diffocins led to the isolation of two spontaneous C. difficile mutants that had an SNP causing severe truncation of the SlpA protein, leading to full resistance to diffocins (49). If strictly lytic phages are developed for CDI treatment, phage resistance through mutation of SlpA might occur as well. However, in vitro and in vivo data showed that loss of slpA comes with a huge fitness cost. Indeed, slpA mutants produce less toxins, sporulate less, are more sensitive to antimicrobial peptides, and are avirulent (49, 62). Therefore, phage resistance through receptor mutation would be compensated by loss of virulence, facilitating patient recovery.
Additional factors that can affect the success of a phage infection will also need to be considered. For instance, active antiphage mechanisms have been described in C. difficile, including restriction-modification systems (63) and a type I CRISPR-Cas system (64). A superinfection exclusion mechanism mediated by the phase-variable cell wall protein CwpV has also been described, although its impact on phage resistance in vivo needs to be investigated (65). The recent identification of functional anti-CRISPR systems in several C. difficile phage genomes (66) suggests that CRISPR-mediated interference could potentially be short-circuited by selecting anti-CRISPR-containing phages or by incorporating anti-CRISPR genes into genetically modified phages. Importantly, incorporation of CRISPR cassettes into cargo phage genomes to target the host cell [e.g., toxin or other virulence genes (25)] will require considering the fact that some naturally occurring lysogens might express anti-CRISPR systems and therefore negatively interfere with the engineered phages.
Avidocins are very appealing alternatives to whole phages, but their large-scale production will be challenging because they are a complex assemblage of multiple components. On the other hand, endolysins are much simpler to produce and have a broader host range, which is a clear advantage. However, off-target killing might be a problem, as some of the other clostridial species that can be lysed by certain recombinant endolysins are beneficial species, such as C. scindens which has been shown to protect against CDI through primary bile acid conversion (67).
Sporulation of C. difficile is also one important hurdle in the treatment of CDI. Spores are naturally produced in the gut during infection and become resistant to most antimicrobials. Hence, combination therapy that includes spore germinants could promote germination, therefore ensuring maximum killing of the infected strain. This strategy has, however, been shown to interfere with endolysin activity, as the massive release of calcium during germination is shown to inhibit the activity of LysCD6356, at least in vitro (38). The importance of this observation to the in vivo condition requires further investigation. Nevertheless, even if residual spores remain after phage-based treatment, the risk of relapse should be lower than with conventional antibiotics because the gut microbiota will be spared in the process.
In conclusion, we are still a few steps from a viable phage-based product to fight CDI, but recent progress in our understanding of phage–host interactions and the development of more efficient molecular tools to genetically engineer phages will certainly lead to exciting advances in the next few years.
Author contributions
AU: Conceptualization, Writing—original draft, Writing—review and editing. L-CF: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing—original draft, Writing—review and editing.
Funding
This work was funded by an NSERC discovery grant (RGPIN RGPIN-2020-05776).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. CDC. COVID-19: U.S. Impact on Antimicrobial Resistance, Special Report 2022. Atlanta, GA: U.S. Department of Health and Human Services, CDC (2022). Available online at: https://www.cdc.gov/drugresistance/covid19.html
2. He M, Miyajima F, Roberts P, Ellison L, Pickard DJ, Martin MJ, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet. (2013) 45:109–13. doi: 10.1038/ng.2478
3. Chakra CNA, Pépin J, Sirard S, Valiquette L. Risk factors for recurrence, complications and mortality in Clostridium difficile infection: a systematic review. PLoS One. (2014) 9:e98400. doi: 10.1371/journal.pone.0098400
4. Almutairi MS, Gonzales-Luna AJ, Alnezary FS, Fallatah SB, Alam MJ, Begum K, et al. Comparative clinical outcomes evaluation of hospitalized patients infected with Clostridioides difficile ribotype 106 vs. other toxigenic strains. Anaerobe. (2021) 72:102440. doi: 10.1016/j.anaerobe.2021.102440
5. Ofori E, Ramai D, Dhawan M, Mustafa F, Gasperino J, Reddy M. Community-acquired Clostridium difficile: epidemiology, ribotype, risk factors, hospital and intensive care unit outcomes, and current and emerging therapies. J Hosp Infect. (2018) 99:436–42. doi: 10.1016/j.jhin.2018.01.015
6. Eyre DW, Walker AS, Wyllie D, Dingle KE, Griffiths D, Finney J, et al. Predictors of first recurrence of Clostridium difficile infection: implications for initial management. Clin Infect Dis. (2012) 55:S77–87. doi: 10.1093/cid/cis356
7. Yadegar A, Pakpoor S, Ibrahim FF, Nabavi-Rad A, Cook L, Walter J, et al. Beneficial effects of fecal microbiota transplantation in recurrent Clostridioides difficile infection. Cell Host Microbe. (2023) 31:695–711. doi: 10.1016/j.chom.2023.03.019
8. DeFilipp Z, Bloom PP, Soto MT, Mansour MK, Sater MRA, Huntley MH, et al. Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med. (2019) 381:2043–50. doi: 10.1056/NEJMoa1910437
9. Hatfull GF, Dedrick RM, Schooley RT. Phage therapy for antibiotic-resistant bacterial infections. Annu Rev Med. (2021) 73:1–15. doi: 10.1146/annurev-med-080219-122208
10. Moelling K, Broecker F, Willy C. A wake-up call: we need phage therapy now. Viruses. (2018) 10:688. doi: 10.3390/v10120688
11. Jones JD, Trippett C, Suleman M, Clokie MRJ, Clark JR. The future of clinical phage therapy in the United Kingdom. Viruses. (2023) 15:721. doi: 10.3390/v15030721
12. Schmidt C. Phage therapy's latest makeover. Nat Biotechnol. (2019) 37:581–6. doi: 10.1038/s41587-019-0133-z
13. Heuler J, Fortier LC, Sun X. Clostridioides difficile phage biology and application. Fems Microbiol Rev. (2021) 45:fuab012. doi: 10.1093/femsre/fuab012
14. Nale JY, Thanki AM, Rashid SJ, Shan J, Vinner GK, Dowah ASA, et al. Diversity, dynamics and therapeutic application of Clostridioides difficile bacteriophages. Viruses. (2022) 14:2772. doi: 10.3390/v14122772
15. Monteiro R, Pires DP, Costa AR, Azeredo J. Phage therapy: going temperate? Trends Microbiol. (2018) 27:368–78. doi: 10.1016/j.tim.2018.10.008
16. Dedrick RM, Guerrero-Bustamante CA, Garlena RA, Russell DA, Ford K, Harris K, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. (2019) 25:730–3. doi: 10.1038/s41591-019-0437-z
17. Al-Anany AM, Fatima R, Hynes AP. Temperate phage-antibiotic synergy eradicates bacteria through depletion of lysogens. Cell Rep. (2021) 35:109172. doi: 10.1016/j.celrep.2021.109172
18. Ramesh V, Fralick JA, Rolfe RD. Prevention of Clostridium difficile-induced ileocecitis with Bacteriophage. Anaerobe. (1999) 5:69–78. doi: 10.1006/anae.1999.0192
19. Meader E, Mayer MJ, Gasson MJ, Steverding D, Carding SR, Narbad A. Bacteriophage treatment significantly reduces viable Clostridium difficile and prevents toxin production in an in vitro model system. Anaerobe. (2010) 16:549–54. doi: 10.1016/j.anaerobe.2010.08.006
20. Meader E, Mayer MJ, Steverding D, Carding SR, Narbad A. Evaluation of bacteriophage therapy to control Clostridium difficile and toxin production in an in vitro human colon model system. Anaerobe. (2013) 22:25–30. doi: 10.1016/j.anaerobe.2013.05.001
21. Nale JY, Spencer J, Hargreaves KR, Buckley AM, Trzepiński P, Douce GR, et al. Bacteriophage combinations significantly reduce Clostridium difficile growth in vitro and proliferation in vivo. Antimicrob Agents Ch. (2016) 60:968–81. doi: 10.1128/AAC.01774-15
22. Nale JY, Chutia M, Carr P, Hickenbotham PT, Clokie MRJ. ‘Get in early'; biofilm and wax moth (Galleria mellonella) models reveal new insights into the therapeutic potential of Clostridium difficile bacteriophages. Front Microbiol. (2016) 7:1383. doi: 10.3389/fmicb.2016.01383
23. Nale JY, Redgwell TA, Millard A, Clokie MRJ. Efficacy of an optimised bacteriophage cocktail to clear Clostridium difficile in a batch fermentation model. Antibiotics. (2018) 7:13. doi: 10.3390/antibiotics7010013
24. Shan J, Ramachandran A, Thanki AM, Vukusic FBI, Barylski J, Clokie MRJ. Bacteriophages are more virulent to bacteria with human cells than they are in bacterial culture; insights from HT-29 cells. Sci Rep-UK. (2018) 8:5091. doi: 10.1038/s41598-018-23418-y
25. Selle K, Fletcher JR, Tuson H, Schmitt DS, McMillan L, Vridhambal GS, et al. In vivo targeting of Clostridioides difficile using phage-delivered CRISPR-Cas3 antimicrobials. MBio. (2020) 11:e00019–20. doi: 10.1128/mBio.00019-20
26. Nale JY, Chutia M, Cheng JKJ, Clokie MRJ. Refining the Galleria mellonella model by using stress marker genes to assess Clostridioides difficile infection and recuperation during phage therapy. Microorg. (2020) 8:1306. doi: 10.3390/microorganisms8091306
27. Gebhart D, Williams SR, Bishop-Lilly KA, Govoni GR, Willner KM, Butani A, et al. Novel high-molecular-weight, R-type bacteriocins of Clostridium difficile. J Bacteriol. (2012) 194:6240–7. doi: 10.1128/JB.01272-12
28. Gebhart D, Lok S, Clare S, Tomas M, Stares M, Scholl D, et al. A modified R-type bacteriocin specifically targeting Clostridium difficile prevents colonization of mice without affecting gut microbiota diversity. MBio. (2015) 6:e02368–14. doi: 10.1128/mBio.02368-14
29. Sangster W, Hegarty JP, Stewart DB. Phage tail-like particles kill Clostridium difficile and represent an alternative to conventional antibiotics. Surgery. (2015) 157:96–103. doi: 10.1016/j.surg.2014.06.015
30. Mayer MJ, Narbad A, Gasson MJ. Molecular characterization of a Clostridium difficile bacteriophage and its cloned biologically active endolysin. J Bacteriol. (2008) 190:6734–40. doi: 10.1128/JB.00686-08
31. Mayer MJ, Garefalaki V, Spoerl R, Narbad A, Meijers R. Structure-based modification of a Clostridium difficile-targeting endolysin affects activity and host range. J Bacteriol. (2011) 193:5477–86. doi: 10.1128/JB.00439-11
32. Wang Q, Euler CW, Delaune A, Fischetti VA. Using a novel lysin to help control Clostridium difficile infections. Antimicrob Agents Chemother. (2015) 59:7447–57. doi: 10.1128/AAC.01357-15
33. Mehta KK, Paskaleva EE, Wu X, Grover N, Mundra RV, Chen K, et al. Newly identified bacteriolytic enzymes that target a wide range of clinical isolates of Clostridium difficile. Biotechnol Bioeng. (2016) 113:2568–76. doi: 10.1002/bit.26029
34. Peng Z, Wang S, Gide M, Zhu D, Patabendige HMLW Li C, et al. A novel bacteriophage lysin-human defensin fusion protein is effective in treatment of Clostridioides difficile infection in mice. Front Microbiol. (2019) 9:3234. doi: 10.3389/fmicb.2018.03234
35. Mondal SI, Akter A, Draper LA, Ross RP, Hill C. Characterization of an endolysin targeting Clostridioides difficile that affects spore outgrowth. Int J Mol Sci. (2021) 22:5690. doi: 10.3390/ijms22115690
36. Phothichaisri W, Chankhamhaengdecha S, Janvilisri T, Nuadthaisong J, Phetruen T, Fagan RP, et al. Potential role of the host-derived cell-wall binding domain of endolysin CD16/50L as a molecular anchor in preservation of uninfected Clostridioides difficile for new rounds of phage infection. Microbiol Spectr. (2022) 10:e0236121. doi: 10.1128/spectrum.02361-21
37. Sekiya H, Yamaji H, Yoshida A, Matsunami R, Kamitori S, Tamai E. Biochemical characterizations of the putative endolysin Ecd09610 catalytic domain from Clostridioides difficile. Antibiotics. (2022) 11:1131. doi: 10.3390/antibiotics11081131
38. Alyahya K, Baillie L. Assessing the feasibility of employing a combination of a bacteriophage-derived endolysin and spore germinants to treat relapsing Clostridioides difficile infection. Microorganisms. (2023) 11:1651. doi: 10.3390/microorganisms11071651
39. Flemming HC, Hullebusch ED, van Neu TR, Nielsen PH, Seviour T, Stoodley P, et al. The biofilm matrix: multitasking in a shared space. Nat Rev Microbiol. (2023) 21:70–86. doi: 10.1038/s41579-022-00791-0
40. Knecht LE, Veljkovic M, Fieseler L. Diversity and function of phage encoded depolymerases. Front Microbiol. (2020) 10:2949. doi: 10.3389/fmicb.2019.02949
41. Revathi G, Fralick JA, Rolfe RD. In vivo lysogenization of a Clostridium difficile bacteriophage ϕCD119. Anaerobe. (2011) 17:125–9. doi: 10.1016/j.anaerobe.2011.05.012
42. Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, et al. A mouse model of Clostridium difficile-associated disease. Gastroenterology. (2008) 135:1984–92. doi: 10.1053/j.gastro.2008.09.002
43. Sekulovic O, Garneau JR, Néron A, Fortier LC. Characterization of temperate phages infecting Clostridium difficile isolates of human and animal origins. Appl Environ Microb. (2014) 80:2555–63. doi: 10.1128/AEM.00237-14
44. Nagy E, Földes J. Electron microscopic investigation of lysogeny of Clostridium difficile strains isolated from antibiotic-associated diarrhea cases and from healthy carriers. APMIS. (1991) 99:321–6. doi: 10.1111/j.1699-0463.1991.tb05156.x
45. Fortier LC, Moineau S. Morphological and genetic diversity of temperate phages in Clostridium difficile. Appl Environ Microb. (2007) 73:7358–66. doi: 10.1128/AEM.00582-07
46. Hargreaves KR, Colvin HV, Patel KV, Clokie JJP, Clokie MRJ. Genetically diverse Clostridium difficile strains harboring abundant prophages in an estuarine environment. Appl Environ Microbiol. (2013) 79:6236–43. doi: 10.1128/AEM.01849-13
47. Nale JY, Shan J, Hickenbotham PT, Fawley WN, Wilcox MH, Clokie MRJ. Diverse temperate bacteriophage carriage in Clostridium difficile 027 strains. PLoS ONE. (2012) 7:e37263. doi: 10.1371/journal.pone.0037263
48. Patz S, Becker Y, Richert-Pöggeler KR, Berger B, Ruppel S, Huson DH, et al. Phage tail-like particles are versatile bacterial nanomachines—a mini-review. J Adv Res. (2019) 19:75–84. doi: 10.1016/j.jare.2019.04.003
49. Kirk JA, Gebhart D, Buckley AM, Lok S, Scholl D, Douce GR, et al. New class of precision antimicrobials redefines role of Clostridium difficile S-layer in virulence and viability. Sci Transl Med. (2017) 9:eaah6813. doi: 10.1126/scitranslmed.aah6813
50. Cahill J, Young R. Phage lysis: multiple genes for multiple barriers. Adv virus Res. (2018) 103:33–70. doi: 10.1016/bs.aivir.2018.09.003
51. Mondal SI, Draper LA, Ross RP, Hill C. Bacteriophage endolysins as a potential weapon to combat Clostridioides difficile infection. Gut Microbes. (2020) 12:1813533. doi: 10.1080/19490976.2020.1813533
52. Giesemann T, Guttenberg G, Aktories K. Human α-defensins inhibit Clostridium difficile toxin B. Gastroenterology. (2008) 134:2049–58. doi: 10.1053/j.gastro.2008.03.008
53. Nieuwenhuyse BV, Linden DV, der Chatzis O, Lood C, Wagemans J, Lavigne R, et al. Bacteriophage-antibiotic combination therapy against extensively drug-resistant Pseudomonas aeruginosa infection to allow liver transplantation in a toddler. Nat Commun. (2022) 13:5725. doi: 10.1038/s41467-022-33294-w
54. Park JY, Moon BY, Park JW, Thornton JA, Park YH, Seo KS. Genetic engineering of a temperate phage-based delivery system for CRISPR/Cas9 antimicrobials against Staphylococcus aureus. Sci Rep. (2017) 7:44929. doi: 10.1038/srep44929
55. Lynch KH, Seed KD, Stothard P, Dennis JJ. Inactivation of Burkholderia cepacia complex phage KS9 gp41 identifies the phage repressor and generates lytic virions. J Virol. (2010) 84:1276–88. doi: 10.1128/JVI.01843-09
56. Labrie SJ, Moineau S. Abortive infection mechanisms and prophage sequences significantly influence the genetic makeup of emerging lytic lactococcal phages. J Bacteriol. (2006) 189:1482–7. doi: 10.1128/JB.01111-06
57. Royer ALM, Umansky AA, Allen MM, Garneau JR, Ospina-Bedoya M, Kirk JA, et al. Clostridioides difficile S-layer protein A (SlpA) serves as a general phage receptor. Microbiol Spectr. (2023) 11:e03894–22. doi: 10.1128/spectrum.03894-22
58. Phetruen T, Chanarat S, Janvilisri T, Phanchana M, Charoensutthivarakul S, Phothichaisri W, et al. Receptor binding protein of prophage reversibly recognizes the low-molecular weight subunit of the surface-layer protein SlpA in Clostridioides difficile. Front Microbiol. (2022) 13:998215. doi: 10.3389/fmicb.2022.998215
59. Whittle MJ, Bilverstone TW, Esveld RJ, van Lücke AC, Lister MM, Kuehne SA, et al. A novel bacteriophage with broad host range against Clostridioides difficile ribotype 078 supports SlpA as the likely phage receptor. Microbiol Spectr. (2022) 10:e02295–21. doi: 10.1128/spectrum.02295-21
60. Nick JA, Dedrick RM, Gray AL, Vladar EK, Smith BE, Freeman KG, et al. Host and pathogen response to bacteriophage engineered against Mycobacterium abscessus lung infection. Cell. (2022) 185:1860–74.e12. doi: 10.1016/j.cell.2022.04.024
61. Rostøl JT, Marraffini L. (Ph)ighting phages: how bacteria resist their parasites. Cell Host Microbe. (2019) 25:184–94. doi: 10.1016/j.chom.2019.01.009
62. Ormsby MJ, Vaz F, Kirk JA, Barwinska-Sendra A, Hallam JC, Lanzoni-Mangutchi P, et al. An intact S-layer is advantageous to Clostridioides difficile within the host. PLoS Pathog. (2023) 19:e1011015. doi: 10.1371/journal.ppat.1011015
63. Purdy D, O'Keeffe TAT, Elmore M, Herbert M, McLeod A, Bokori-Brown M, et al. Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol Microbiol. (2002) 46:439–52. doi: 10.1046/j.1365-2958.2002.03134.x
64. Boudry P, Semenova E, Monot M, Datsenko KA, Lopatina A, Sekulovic O, et al. Function of the CRISPR-Cas system of the human pathogen Clostridium difficile. MBio. (2015) 6:e01112–15. doi: 10.1128/mBio.01112-15
65. Sekulovic O, Bedoya MO, Fivian-Hughes AS, Fairweather NF, Fortier L. The Clostridium difficile cell wall protein CwpV confers phase-variable phage resistance. Mol Microbiol. (2015) 98:329–42. doi: 10.1111/mmi.13121
66. Muzyukina P, Shkaruta A, Guzman NM, Andreani J, Borges AL, Bondy-Denomy J, et al. Identification of an Anti-CRISPR protein that inhibits the CRISPR-Cas Type I-B system in clostridioides difficile. BioRxiv [Preprint]. (2023). doi: 10.1101/2023.05.22.541795
Keywords: Clostridioides (Clostridium) difficile infection, bacteriophage (phage), phage therapy, endolysin, diffocin, avidocin, phage engineering
Citation: Umansky AA and Fortier LC (2023) The long and sinuous road to phage-based therapy of Clostridioides difficile infections. Front. Med. 10:1259427. doi: 10.3389/fmed.2023.1259427
Received: 15 July 2023; Accepted: 03 August 2023;
Published: 23 August 2023.
Edited by:
Mark Willcox, University of New South Wales, AustraliaReviewed by:
Prasanth Manohar, Texas A&M University, United StatesCopyright © 2023 Umansky and Fortier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Louis Charles Fortier, TG91aXMtQ2hhcmxlcy5Gb3J0aWVyQFVTaGVyYnJvb2tlLmNh