 Ahmed Ahmed Touni1,2*
Ahmed Ahmed Touni1,2* Rohan S. Shivde2
Rohan S. Shivde2 Harika Echuri3Rasha T. A. Abdel-Aziz1
Harika Echuri3Rasha T. A. Abdel-Aziz1 Hossam Abdel-Wahab1
Hossam Abdel-Wahab1 Roopal V. Kundu1
Roopal V. Kundu1 I. Caroline Le Poole2*
I. Caroline Le Poole2*- 1Department of Dermatology, Faculty of Medicine, Minia University, Minia, Egypt
- 2Department of Dermatology, Feinberg School of Medicine, Northwestern University, Chicago, IL, United States
- 3Department of Dermatology, Emory University, Atlanta, GA, United States
Vitiligo is a common acquired pigmentary disorder that presents as progressive loss of melanocytes from the skin. Epidermal melanocytes and keratinocytes are in close proximity to each other, forming a functional and structural unit where keratinocytes play a pivotal role in supporting melanocyte homeostasis and melanogenesis. This intimate relationship suggests that keratinocytes might contribute to ongoing melanocyte loss and subsequent depigmentation. In fact, keratinocyte dysfunction is a documented phenomenon in vitiligo. Keratinocyte apoptosis can deprive melanocytes from growth factors including stem cell factor (SCF) and other melanogenic stimulating factors which are essential for melanocyte function. Additionally, keratinocytes control the mobility/stability phases of melanocytes via matrix metalloproteinases and basement membrane remodeling. Hence keratinocyte dysfunction may be implicated in detachment of melanocytes from the basement membrane and subsequent loss from the epidermis, also potentially interfering with repigmentation in patients with stable disease. Furthermore, keratinocytes contribute to the autoimmune insult in vitiligo. Keratinocytes express MHC II in perilesional skin and may present melanosomal antigens in the context of MHC class II after the pigmented organelles have been transferred from melanocytes. Moreover, keratinocytes secrete cytokines and chemokines including CXCL-9, CXCL-10, and IL-15 that amplify the inflammatory circuit within vitiligo skin and recruit melanocyte-specific, skin-resident memory T cells. In summary, keratinocytes can influence vitiligo development by a combination of failing to produce survival factors, limiting melanocyte adhesion in lesional skin, presenting melanocyte antigens and enhancing the recruitment of pathogenic T cells.
1. Melanocytes are not the only affected cells in vitiligo: keratinocyte apoptosis
1.1. Evidence of keratinocyte apoptosis
Vitiligo is a pigmentary disease characterized by white patches of skin expanding in size over time. The pathological hallmark of vitiligo is absence of melanocytes from lesional skin, which can be highlighted with special stains labelling melanocytes (1). Although keratinocytes appear to be normal as shown by routine hematoxylin and eosin staining, the basal and parabasal keratinocytes show features of apoptosis by electron microscopic examination. Apoptotic features are found not only in depigmented but also in normally pigmented skin (2, 3). Those keratinocytes demonstrate swelling of membrane-bound organelles; intracellular edema, and formation of vacuoles with clearing of the cytoplasmic matrix, and swollen mitochondria with disruption of cristae (4).
1.2. Mechanism of action
It is unclear how keratinocyte apoptosis occurs in patients with vitiligo (5). One proposed mechanism involves increased levels of lesional Tumor Necrosis Factor- α (TNF-α) (4). Indeed TNF-α is increased within vitiligo lesions when compared to normally pigmented skin (6). TNF-α is at the center of the extrinsic pathway of apoptosis, and increased TNF-α levels could lead to a reduced activation of NF-κB via impaired PI3K/AKT activation, possibly contributing to keratinocyte apoptosis (7). Moreover, in addition to melanocyte-reactive antibodies, anti-keratinocyte antibodies have been detected in the sera of patients. It remains to be seen whether these antibodies are a cause or consequence of keratinocyte damage (8). Taken together, keratinocyte apoptosis can indirectly influence melanocyte viability and support depigmentation. This prompts the question how keratinocytes might influence melanocyte viability in the skin.
2. Keratinocyte apoptosis negatively impacts melanocyte functions
2.1. Keratinocytes affect melanocyte viability
Keratinocyte apoptosis in vitiligo negatively impacts melanogenesis and melanocyte homeostasis (9). Keratinocyte integrity is essential for melanocyte function within the epidermal melanin unit (10). The main function of melanocytes is to synthesize melanin through oxidation of tyrosine, supported by enzymes uniquely found in melanocytic cells (8). Resulting melanin is stored in melanosomes, specialized lysosome-related organelles that move from the nucleus towards melanocyte dendrites upon melanin deposition (9, 10). Pigmented melanosomes are then transferred to adjacent keratinocytes, providing a supranuclear cap-like shield to protect the cell against UV radiation (11–13). Melanin synthesis and melanosome distribution within the epidermis determine skin pigmentation (14, 15). Pigment synthesis is influenced by genetics, UV exposure, hormones, and chemical mediators (11, 12). Keratinocytes are one of the main sources of soluble mediators that control melanogenesis (Figure 1). Ultraviolet light activates a p53-dependent pathway resulting in the release of paracrine factors from keratinocytes (13, 14). These factors act on neighboring melanocytes by receptor interaction to induce melanin synthesis.
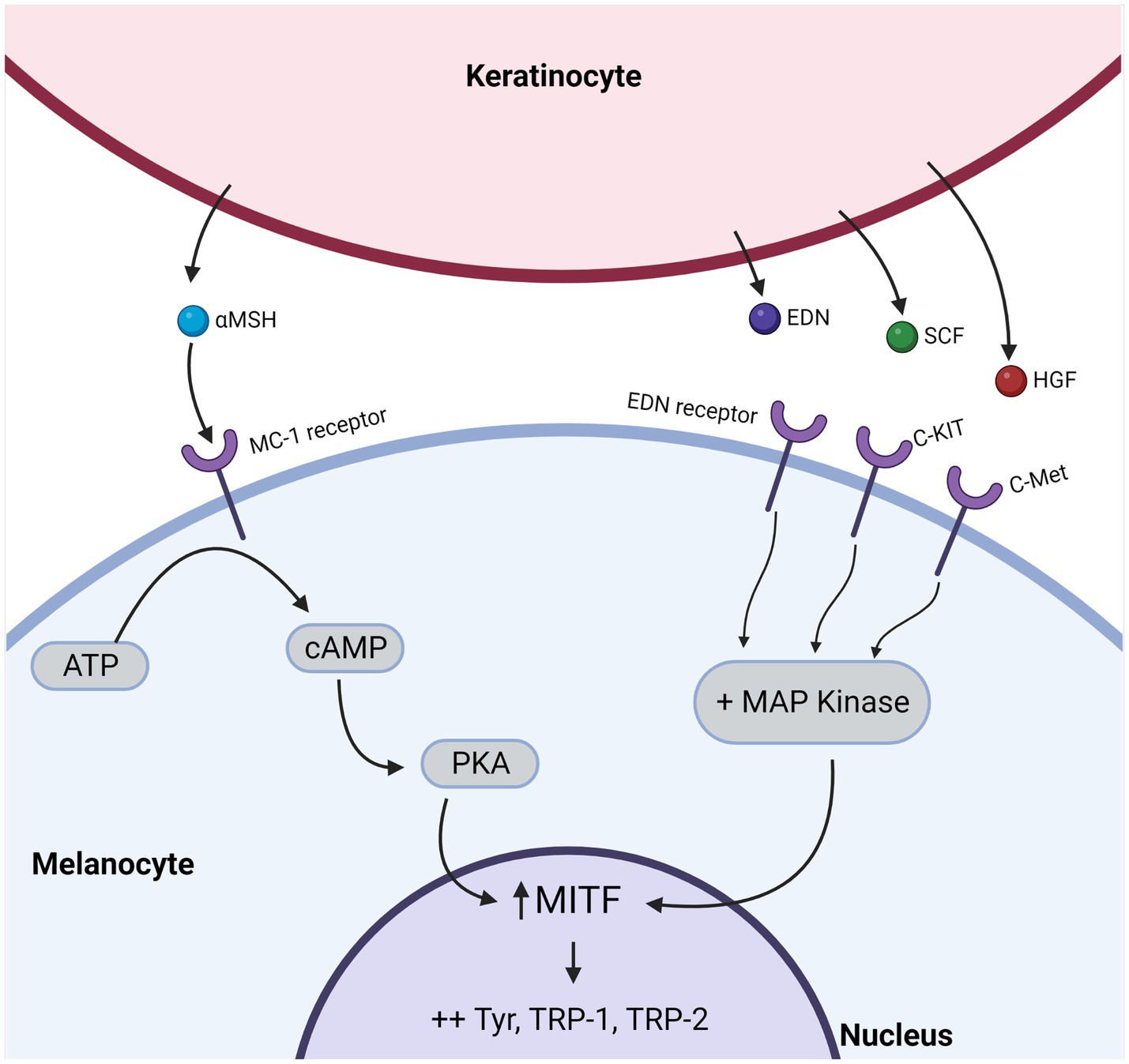
Figure 1. Keratinocytes secrete growth and melanogenic factors that influence signal transduction pathways within melanocytes. Keratinocyte-derived, melanocyte-stimulating factors include alpha-melanocyte stimulating hormone (α-MSH), Endothelin-1 (EDN-1), Stem Cell Factor (SCF), and Hepatocyte Growth Factor (HGF) that bind to their corresponding receptors on the surface of melanocytes. Among these receptors, c-kit, c-Met, and EDN receptors activate Mitogen-Activated Protein (MAP) kinases, while the melanocortin-1 (MC-1) receptor activates protein kinase A (PKA). In turn, MAP kinase or PKA will activate microphthalmia-associated transcription factor (MITF), which regulates the expression of multiple pigmentation-related genes.
2.2. Keratinocyte Activation of MITF
Stimulation of melanogenesis occurs through activation of Microphthalmia-Associated Transcription Factor (MITF) and its downstream targets, including the melanogenic enzymes Tyrosinase (TYR), Tyrosinase-Related Protein 1 (TYRP1), and Dopachrome tautomerase (DCT) with subsequent proliferation, differentiation of melanocytes and increased melanin production (15, 16). Stem Cell Factor (SCF) also promotes melanocyte proliferation (17) while treating melanocytes with anti-SCF leads to a dose–dependent decline in melanocyte numbers (17).
2.3. Influence of cell-to-cell contact
Keratinocytes control melanocyte proliferation and differentiation not only by soluble mediators but also by direct cell-to-cell contact (18). The direct contact between melanocytes and basal undifferentiated keratinocytes influences melanocyte proliferation capacity along with the adhesion molecules pattern (19). Therefore, keratinocyte apoptosis deprives melanocytes of growth factors, melanogenic stimuli, and direct cell contact, resulting in reduced melanocyte health and increased sensitivity to apoptotic signals (17). By responding to UV-induced changes, MITF is activated and supports melanin formation, resulting in transfer of melanin-containing organelles to neighboring keratinocytes. Successful UV-induced pigmentation requires a coordinated effort between melanocytes and keratinocytes in the skin. This prompted further questions into the melanosome transfer process.
3. Keratinocyte protease-activated receptor-2 is dysregulated in vitiligo
3.1. Impact of PAR-2 on the epidermal melanin unit
PAR-2 is thought to be involved in development of vitiligo through a reduced melanosome uptake by keratinocytes (20). The distribution of melanin within the epidermal melanin unit is regulated in part by expression of the keratinocyte receptor PAR-2, which is markedly reduced in the depigmented skin of vitiligo patients when compared with the skin of healthy subjects (21). PAR-2 is involved in melanosome phagocytosis and consequently, modulation of PAR-2 activation affects melanosome transfer, contributing to the regulation of skin pigmentation (20).
3.2. PAR-2 activity in vivo
In vivo, PAR-2 activation results in increased melanin deposition and enhanced keratinocyte cap formation, while its inhibition results in aberrant melanosome packaging, abnormal melanosome dynamics, and atypical melanosome distribution, leading to depigmentation (20, 22). This PAR-2 reduction seems to be a particular feature of vitiligo because a similar reduction was not observed in non-vitiligo leukoderma (21). With PAR-2 as a master regulator of melanosome transfer, we may however be overlooking additional molecules mediating cell to cell contact that also influence the normal pigmentation process.
4. Adhesion molecules connecting keratinocytes and melanocytes
4.1. The epidermal melanin unit defines the interaction between keratinocytes and melanocytes
The epidermis is made up of a mesh of keratinocytes interconnected with melanocytes and scattered Langerhans cells. The epidermis and dermis are separated and held together by the basement membrane. Each melanocyte within the epidermis is in contact with 30–40 neighboring keratinocytes through long dendritic extensions that form adhesive structures (23). Melanocytes are securely attached to the basement membrane, and their adhesion, and migration are under control of neighboring keratinocytes (18).
4.2. Intercellular contact molecules mediate melanocyte-keratinocyte adhesion
Unlike keratinocyte–keratinocyte adhesion, melanocyte–keratinocyte interaction does not involve specific adhesive structures such as desmosomes but is instead mediated by simple adhesion molecules such as integrins and cadherins (24). Integrins are also crucial for melanocyte adhesion to the basement membrane as they attach to vitronectin, fibronectin, and type I and IV collagen in the basement membrane zone (25, 26).
4.3. Keratinocytes regulate integrin expression
Keratinocytes can modulate integrin expression through releasing SCF to facilitate a more motile status of melanocytes as needed in the repigmentation process (27). Moreover, the pro-inflammatory cytokine IL-1β, which is released from UV-exposed keratinocytes, acts directly on melanocytes. In response, melanocytes release Cellular Communication Network Factor 3 (CCN3) to up-regulate Discoidin Domain Receptor (DDR) tyrosine kinase activity within melanocytes (28, 29). This increases adherence to the basement membrane through contact with collagen I. Several studies showed that genetic variants of the DDR1 gene are associated with vitiligo, and the expression of DDR1 is decreased in vitiligo lesions (30–32).
4.4. Tenascin is upregulated in vitiligo lesional skin
Vitiligo skin displays elevated tenascin expression in the basal membrane and papillary dermis (33). Tenascin has anti-adhesive properties, as it decreases the ability of melanocytes to bind to the basement membrane (34, 35).
4.5. Interaction with inflammatory cells is affected by keratinocytes
Harnessing the role played by keratinocytes in controlling the homeostasis of melanocytes is an interesting therapeutic strategy for vitiligo and attempts have been developed for that purpose. For example, afamelanotide is a potent synthetic linear analog of alpha-Melanocyte Stimulating Hormone (α-MSH) that can stimulate melanogenesis and facilitate the transfer of melanosomes (36). It may also restore balance of the cytokine environment by acting on immune cells expressing melanocortin-1 receptor (MC1R) (including macrophages and T cells) to mediate anti-inflammatory effects (37). In an initial randomized controlled trail, combination therapy in the form of Afamelanotide implants plus narrow-band ultraviolet B (NB-UVB) demonstrated a statistically superior rate of repigmentation over NB-UVB treatment alone (38). A plethora of cell membrane molecules can thus influence pigmentation and potentially contribute to loss of pigmentation in vitiligo. Yet another category of a intercellular communication molecules can influence melanocyte physiology: introducing the extracellular matrix.
5. Extracellular matrix deposits impacting melanocyte release
5.1. MMP expression is induced in response to UV
Matrix metalloproteinases (MMPs) are a family of zinc-dependent proteases that have an ability to cleave components of the extracellular matrix, preparing an optimal setting for cell migration (39, 40). Cell migration is essential in inflammatory reactions, remodeling, and healing. Keratinocytes express MMP-9 and MMP-2 in response to ultraviolet light or proinflammatory cytokines such as IFN-γ, TNF-α, IL-1β, and IL-6 (41, 42).
5.2. Melanocytes are lost by sloughing off
MMPs induce the decoupling of melanocytes from their neighboring keratinocytes and from the basement membrane through E-cadherin (43). Lateral melanocyte migration involves a process of detachment and re-attachment to neighboring keratinocytes (44). If the re-attachment process fails, melanocytes would be released and slough off alongside keratinocytes (45). This is highlighted in vitiligo since MMPs have a role in melanocyte death or migration in active or stable disease, respectively (45).
5.3. MMPs affect repigmentation
In active disease the levels of MMPs, especially MMP-9, are elevated in the lesional skin of vitiligo patients under the influence of IFN-γ and TNF-α. MMP-9 induces a decoupling of melanocytes from the basement membrane via disruption of E-cadherin with subsequent death of melanocytes (45). On the other hand, significantly lower expression of metalloproteinases has been observed in perilesional skin of patients with stable disease (39, 42). This deficiency could interfere with the ability of melanocytes to migrate and to repigment vitiligo skin (42). Pro-MMP activation occurs at the cell surface, thus proteolysis is greatest in the immediate pericellular environment, where it can influence cell–cell and cell–extracellular matrix (ECM) interactions (40, 46). It is therefore reasonable to assume that migration of melanocytes (or their precursor melanoblasts) from the outer root sheath of hair follicles or hair bulge into clinically depigmented epidermis would require their release from those adhesion sites and subsequent penetration of existing basement membrane barriers in vivo (47). These processes are directed, at least in part, by limited proteolysis of the ECM by MMPs (47). This aligns with the impaired expression of MMP9 in perilesional skin of patients, correlating with a poor response to UVB-based phototherapy.
5.4. Beta-catenin is involved in vitiligo
The E-cadherin cytoplasmic domain in keratinocytes forms a complex with β-catenin (48). Loss of E-cadherin leads to release of β-catenin which can induce MMP-1, MMP-2, and MMP-9 to stimulate melanocyte migration (49–52). Melanocyte migration is also stimulated by α-MSH-MC1R and stem cell factor (SCF)/ signaling pathways, endothelin-1 (ET-1), basic fibroblast growth factor (bFGF) and hepatocyte growth factor (HGF) (53–55).
5.5. Stress-jnduced Koebnerization
Melanocyte dendrites may facilitate attachment of melanocytes to the basal layer of the epidermis and are considered a major component of the melanocyte adhesion system independent of the structural junctions (56). Gauthier et al. (57) reported detachment and trans-epidermal elimination of melanocytes following minor mechanical trauma in vitiligo patients. Stress-induced detachment may then help explain the Koebner phenomenon observed in patients with vitiligo (58). Also, in vitiliginous skin, the expression of cadherins is decreased while tenascin, an extracellular matrix molecule that inhibits adhesion of melanocytes to fibronectin, is increased (33). These changes can be explained by increased production of metalloproteinase-9 (MMP-9) by keratinocytes under the influence of IFN-γ and TNF-α, characteristic vitiligo-associated cytokines (45). By mediating altered adhesion of melanocytes in lesional skin, the role of keratinocytes can be significant in vitiligo. The specific physiology of keratinocytes in vitiligo that would influence vitiligo development is however better understood when accounting for vitiligo-associated mutations in the pigmentary disorder.
6. Disease-associated mutations suggesting a role for keratinocytes in vitiligo
6.1. Vitiligo-associated gene mutations have been identified
Vitiligo is a complex genetic disease as supported by the familial clustering of vitiligo cases in 6–8% of first-degree relatives. However, the concordance of vitiligo in monozygotic twins is only ~23%, suggesting that environmental factors also contribute to disease development (59). Multiple vitiligo susceptibility genes have been identified in genome-wide association studies, showing that besides immunomodulatory genes, some melanocyte specific genes (60–62) are associated with progressive depigmentation. Interestingly, keratinocyte-related genes have been linked to vitiligo pathogenesis as well, which could point to a functional link between both cell types. Examples are found in the endothelin-1 (EDN1), cyclooxygenase (COX2), NLR family pyrin domain containing 1 (NALP1), and Liver X receptor alpha (LXR-α) genes.
6.2. A role for EDN1
The EDN1 gene is expressed by epidermal keratinocytes (63) and encodes a potent vasoconstrictor peptide that acts on neighboring melanocytes to modulate their function and survival (64, 65). However, EDN1 polymorphisms were only identified as a risk factor for the development of segmental vitiligo (66, 67).
6.3. COX2 involvement as a ultraviolet light-induced gene
The COX2 gene encodes a key enzyme in the production of prostaglandin E2 (PGE2). Its production by epidermal keratinocytes is induced by ultraviolet radiation (68). PGE2 supports melanocyte proliferation and melanogenesis (69). A functional polymorphism of the COX2 gene is associated with an increased risk of vitiligo development (70, 71). This polymorphism reduces the mRNA levels encoding COX2 and affects the subsequent production of PGE2, which further impairs melanocyte survival and melanogenesis.
6.4. NALP1 is associated with vitiligo
Jin et al. (72) identified NALP1 as a vitiligo-associated gene. This group found an association between the expression of specific vitiligo-associated NALP1 variants and an extended autoimmune and autoinflammatory disease phenotype. NALP1 is a key regulator of the innate skin immune system, and a principal inflammasome sensor in human keratinocytes (73). Ultraviolet radiation and cellular stress can induce NALP1 activation. Subsequent caspase-1–dependent processing of pro-interleukin-1β (IL-1β) leads to release of IL-1β and downstream inflammatory responses that recruit T cells to the skin (74, 75). Single nucleotide polymorphisms of NALP1 are associated with generalized vitiligo (76, 77). Pharmacological targeting of NALP1 activation in epidermal keratinocytes may thus represent a promising strategy for the treatment of inflammatory autoimmune skin diseases such as vitiligo.
6.5. LXR-α polymorphisms are potentially involved in disease
The LXR-α gene contributes to melanocyte proliferation and differentiation (78). A polymorphism of the LXR-α gene is linked to the development of vitiligo in some populations (79, 80). LXR-α upregulation is associated with keratinocyte damage in vitiliginous skin. Such damage leads to decreased keratinocyte-derived mediators and growth factors that otherwise support the growth and/or melanization of surrounding melanocytes, leaving them more prone to apoptosis (81, 82). Interestingly, LXR-α expression decreases or inhibits the expression MMPs, and this decrease in MMPs in turn inhibit the migration or replacement of melanocytes from hair outer root sheath melanoblasts in perilesional vitiligo skin (41, 83). Several genes have been postulated as major players in vitiligo development. As vitiligo is first and foremost a T cell mediated autoimmune disorder, the specific alterations we encounter in active disease should impact the autoimmune response. Below are some findings that can provide insight in the process.
7. Keratinocytes generate chaperokines and cytokines involved in vitiligo
7.1. Oxidative stress leads to cytokine and chaperokine expression
Oxidative stress has been observed in both melanocytes and keratinocytes (84). Oxidative stress can trigger the release of inducible heat shock protein 70 (HSP70i) from dying cells and from melanocytes under stress (85). Additionally, this heat shock protein can associate with melanosomes, suggesting that HSP70i can chaperone melanosomal proteins (85). Once released, HSP70i can potentiate maturation and activation of plasmacytoid dendritic cells (pDCs) as well natural killer (NK) cells (86).
7.2. Keratinocytes are involved in T cell recruitment
Enhanced type 1 polarizing cytokines released from pDCs can potentiate expression of CXCL9 and CXCL10 by keratinocytes (86). These chemokines are primarily responsible for recruiting cytotoxic T cells to the skin of vitiligo patients (87). Modified HSP70 (HSP70iQ435A) was proposed as a treatment for vitiligo; the modified HSP70i binds human DCs and reduces their activation. In vivo the modified HSP70i induced a shift from inflammatory to tolerogenic DCs in mice (88). Also, ex vivo treatment of human skin averted the disease-related shift from quiescent to effector T cell phenotype (88), while in vivo application of HSP70iQ435A caused repigmentation of vitiligo lesions in a swine model of the disease (89).
7.3. Proposing a role for antigen transfer from melanocytes
MHC Class II expression is not normally found on healthy tissue cells, however, it is expressed in vitiligo skin, and it includes both melanocytes and surrounding keratinocytes in the perilesional area. This expression occurs in response to IL-22 or IFN-γ. As immunogenic melanosomal antigens are transferred with pigmented melanosomes and melanosomes can form a source of antigens to be presented in the context of Major Histocompatibility Complex (MHC) II (90), keratinocytes in vitiligo skin can activate a CD4+ T-helper response by de novo presentation of melanosomal antigens to them, together with MHC II + melanocytes. In turn, these CD4+ T cells recruit MHC class I-restricted cytotoxic T cells to the skin which deliver a lethal melanocyte-specific attack, sparing keratinocytes. The resulting focal immune infiltrate is a hallmark of vitiligo pathogenesis (Figure 2).
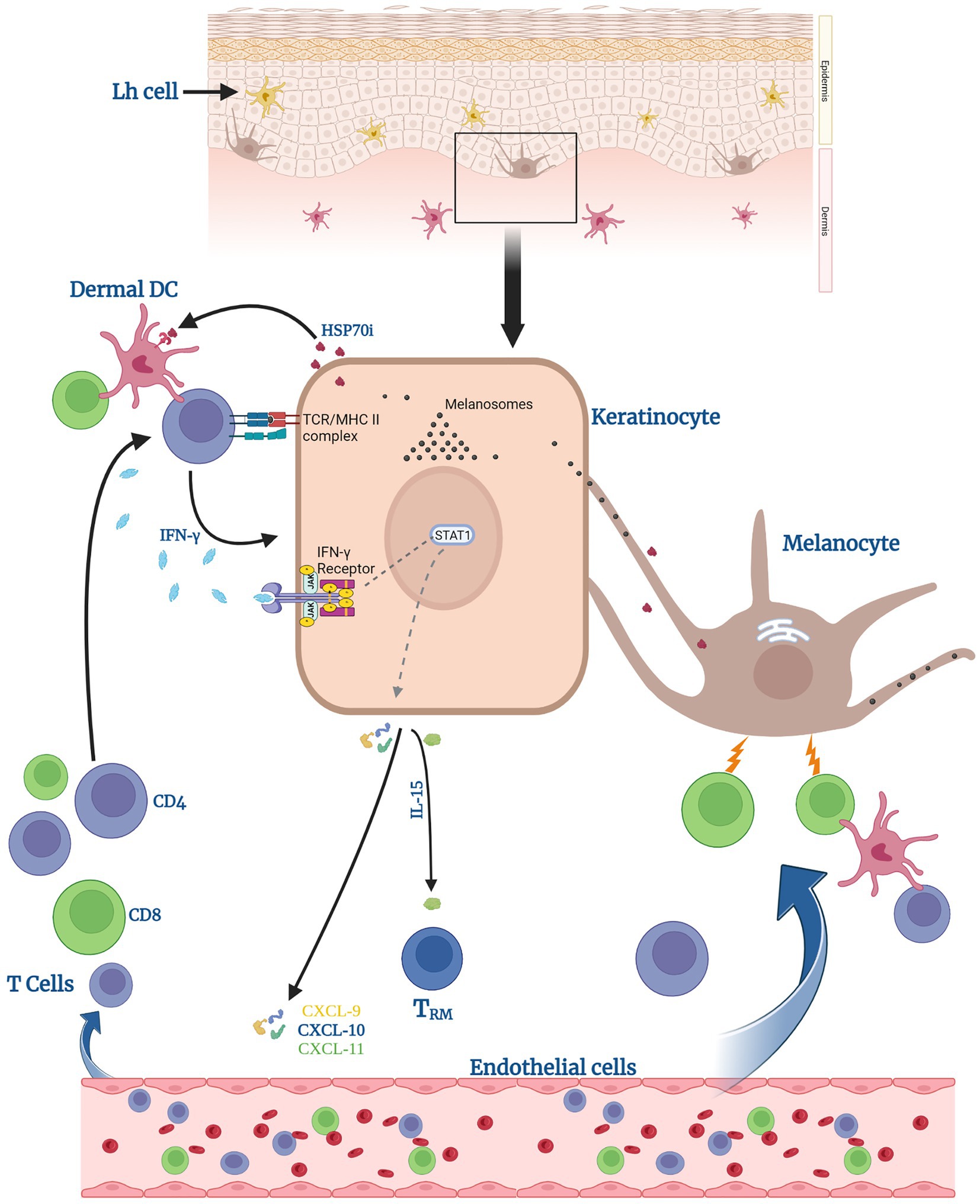
Figure 2. Keratinocytes can help induce anti-melanocyte immune responses. Keratinocytes can activate dermal dendritic cells (DCs) and CD4 T cells by presenting processed melanosomal antigens in the context of MHC II, supported by surrounding HSP70. Activated CD4 T cells secrete IFN-γ. Upon receptor binding, this leads to signal transducer and activator of transcription (STAT1) phosphorylation by Janus Kinases (JAKs) and release of CXCL chemokines 9–11 and of IL-15. Said chemokines help recruit T cells to eradicate melanocytes. IL-15 specifically supports the generation of tissue-resident memory T cells (TRM) within the lesional skin.
7.4. IL-22 can mediate inflammasome activation
Patients with vitiligo exhibit greater serum levels of IL-22 when compared to control individuals without vitiligo (91). IL-22 can also trigger the release IL-1β via the involvement of the NLRP3 inflammasome in keratinocytes (92).
7.5. Keratinocytes activate IFNγ inducible genes
Vitiligo patients carry increased numbers of circulating, melanocyte-reactive cytotoxic T cells that partially migrate to the skin. T cells can be seen infiltrating a very restricted area surrounding the lesional skin (5). These T cells are held responsible for the expansion of vitiligo and mediate progressive melanocyte destruction. IFN-γ inducible genes compose the dominant cytokine pathway expressed in lesional skin. IFN-γ mediates the recruitment of melanocyte-specific, autoreactive CD8 T cells to the skin (93) by stimulating keratinocyte-derived CXCL10 and Chemokine C-X-C Motif Ligand (CXCL)9 to recruit Chemokine C-X-C Motif Receptor (CXCR)3- expressing autoreactive CD8+ T cells to the skin (94, 95) and creating an environment that delays repigmentation (96). This is in part due to a shift in energy utilization by keratinocytes toward oxidative phosphorylation (96, 97).
7.6. Alarmin involvement blocks growth factor release
IL-33 is an alarmin which is produced by keratinocytes under the influence of TNFα and IFN-γ. IL-33 inhibits melanocyte growth in vitiligo, blocking growth factors and increasing the release of pro-inflammatory cytokines IL-6 and TNFα (98, 99).
7.7. IL-15 supports resident-memory T cell differentiation
IL-15 is an important cytokine in vitiligo pathogenesis due to its ability to generate and maintain signals of tissue-resident memory T cells (TRM) (100). Keratinocytes are a source of IL-15, which promotes differentiation of self-reactive T cells into TRM cells (101). The latter T cells contribute to disease maintenance and the recurrence of disease in the same site (101).
7.8. A potential role can be assigned to CXCL16
Stressed keratinocytes release CXCL16 upon activation of the unfolded protein response (102). CXCL16 acts on CXCR6 expressed on CD8+ T cells. CXCL16 mediates trafficking of these CD8+ T cells to the skin, furthering melanocyte destruction (102).
7.9. Inhibiting the IFN-γ axis
Keratinocytes are considered a key source of cytokines and chemokines involved in vitiligo (103). Hence, interfering with the activity of these cytokines has emerged as a promising target for vitiligo treatment. For example, targeted therapeutics are now available to interfere with the interferon (IFN)-γ-CXCL10 axis. For example, Janus kinase (JAK) 1/3 inhibitors, and JAK 1/2 inhibitors directly inhibit IFN-γ signaling, and both revealed favorable outcomes in clinical trials (104, 105). Additionally, in vitiligo mouse models, an anti-CD122 antibody that targets IL-15 signaling was reported to effectively reverse depigmentation (100). Anti-CD122 therapy, either systemically or locally, decreased TRM-induced IFN-γ production and resulted in long-term repigmentation (100). These findings raised enthusiasm for CD122-targeted drugs for vitiligo and other tissue-specific autoimmune disorders. Taken together, genetic alterations that occur in vitiligo development are expected to influence, at least in part, the autoimmune process that follows. The true culprit may vary among patients, yet there is ample reason to assign a role for keratinocytes in vitiligo disease development.
8. Conclusion
Recently, there have been tremendous advances in understanding the interactions between keratinocytes and melanocytes in the pathogenesis of vitiligo, to the degree that vitiligo should be not only be considered a disease of melanocytes alone. Keratinocytes release growth- and melanogenic factors essential for melanocyte survival and function. Hence, disrupting the synthesis of these growth factors from keratinocytes can jeopardize melanocyte viability and function. Additionally, keratinocytes have the machinery to process and present melanosomal antigens from transferred melanosomes to T cells within the epidermis, thus initiating the attack directed against melanocytes. Finally, keratinocytes secrete cytokines that activate and recruit various immune cells into the skin to deliver a lethal hit to melanocytes. Recognizing this type of keratinocyte-melanocyte cross-talk helps to design new treatment strategies that can be implemented in vitiligo.
Author contributions
AT and HE performed the literature review and drafted the manuscript. ICLP initiated and guided the manuscript preparation. RS, ICLP, and AT prepared the figures. All authors contributed to the article and approved the submitted version.
Acknowledgments
AT has a Joint-supervision scholarship from the Ministry of Higher Education and Scientific Research, Arab Republic of Egypt. ICLP is supported in part by NIH P30 AR075049 to Amy S. Paller, PI, that is devoted to studying keratinocytes and their microenvironment.
Conflict of interest
ICLP is a CSO for Temprian Therapeutics, seeking a clinical application for modified HSP70i to treat vitiligo.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kim, YC, Kim, YJ, Kang, HY, Sohn, S, and Lee, ES. Histopathologic features in vitiligo. Am J Dermatopathol. (2008) 30:112–6. doi: 10.1097/DAD.0b013e3181651511
2. Moellmann, G, Klein-Angerer, S, Scollay, DA, Nordlund, JJ, and Lerner, AB. Extracellular granular material and degeneration of keratinocytes in the normally pigmented epidermis of patients with vitiligo. J Invest Dermatol. (1982) 79:321–30. doi: 10.1111/1523-1747.ep12500086
3. Bhawan, J, and Bhutani, LK. Keratinocyte damage in vitiligo. J Cutan Pathol. (1983) 10:207–12. doi: 10.1111/j.1600-0560.1983.tb00328.x
4. Moretti, S, Fabbri, P, Baroni, G, Berti, S, Bani, D, Berti, E, et al. Keratinocyte dysfunction in vitiligo epidermis: cytokine microenvironment and correlation to keratinocyte apoptosis. Histol Histopathol. (2009) 24:849–57. doi: 10.14670/HH-24.849
5. van den Boorn, JG, Konijnenberg, D, Dellemijn, TA, van der Veen, JP, Bos, JD, Melief, CJ, et al. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol. (2009) 129:2220–32. doi: 10.1038/jid.2009.32
6. Birol, A, Kisa, U, Kurtipek, GS, Kara, F, Kocak, M, Erkek, E, et al. Increased tumor necrosis factor alpha (TNF-alpha) and interleukin 1 alpha (IL1-alpha) levels in the lesional skin of patients with nonsegmental vitiligo. Int J Dermatol. (2006) 45:992–3. doi: 10.1111/j.1365-4632.2006.02744.x
7. Kim, NH, Jeon, S, Lee, HJ, and Lee, AY. Impaired PI3K/Akt activation-mediated NF-kappaB inactivation under elevated TNF-alpha is more vulnerable to apoptosis in vitiliginous keratinocytes. J Invest Dermatol. (2007) 127:2612–7. doi: 10.1038/sj.jid.5700900
8. Yu, HS, Kao, CH, and Yu, CL. Coexistence and relationship of antikeratinocyte and antimelanocyte antibodies in patients with non-segmental-type vitiligo. J Invest Dermatol. (1993) 100:823–8. doi: 10.1111/1523-1747.ep12476728
9. Lee, AY. Role of keratinocytes in the development of vitiligo. Ann Dermatol. (2012) 24:115–25. doi: 10.5021/ad.2012.24.2.115
10. Wu, X, Yang, Y, Xiang, L, and Zhang, C. The fate of melanocyte: mechanisms of cell death in vitiligo. Pigment Cell Melanoma Res. (2021) 34:256–67. doi: 10.1111/pcmr.12955
11. Takeuchi, S, Fukumoto, T, Nishigori, C, Declercq, L, Yarosh, DB, Mammone, T, et al. Dynamic visualization of melanosome endo/phagocytosis during melanin transfer using melanosomes pre-stained with carbocyanine dyes. J Dermatol Sci. (2022) 105:65–7. doi: 10.1016/j.jdermsci.2021.12.004
12. Videira, IF, Moura, DF, and Magina, S. Mechanisms regulating melanogenesis. An Bras Dermatol. (2013) 88:76–83. doi: 10.1590/S0365-05962013000100009
13. Abdel-Malek, ZA, Kadekaro, AL, and Swope, VB. Stepping up melanocytes to the challenge of UV exposure. Pigment Cell Melanoma Res. (2010) 23:171–86. doi: 10.1111/j.1755-148X.2010.00679.x
14. Kim, M, Shibata, T, Kwon, S, Park, TJ, and Kang, HY. Ultraviolet-irradiated endothelial cells secrete stem cell factor and induce epidermal pigmentation. Sci Rep. (2018) 8:4235. doi: 10.1038/s41598-018-22608-y
15. Busca, R, and Ballotti, R. Cyclic AMP a key messenger in the regulation of skin pigmentation. Pigment Cell Res. (2000) 13:60–9. doi: 10.1034/j.1600-0749.2000.130203.x
16. Park, HY, Kosmadaki, M, Yaar, M, and Gilchrest, BA. Cellular mechanisms regulating human melanogenesis. Cell Mol Life Sci. (2009) 66:1493–506. doi: 10.1007/s00018-009-8703-8
17. Lee, AY, Kim, NH, Choi, WI, and Youm, YH. Less keratinocyte-derived factors related to more keratinocyte apoptosis in depigmented than normally pigmented suction-blistered epidermis may cause passive melanocyte death in vitiligo. J Invest Dermatol. (2005) 124:976–83. doi: 10.1111/j.0022-202X.2005.23667.x
18. Wang, JX, Fukunaga-Kalabis, M, and Herlyn, M. Crosstalk in skin: melanocytes, keratinocytes, stem cells, and melanoma. J Cell Commun Signal. (2016) 10:191–6. doi: 10.1007/s12079-016-0349-3
19. Shih, IM, Elder, DE, Hsu, MY, and Herlyn, M. Regulation of Mel-CAM/MUC18 expression on melanocytes of different stages of tumor progression by normal keratinocytes. Am J Pathol. (1994) 145:837–45.
20. Seiberg, M, Paine, C, Sharlow, E, Andrade-Gordon, P, Costanzo, M, Eisinger, M, et al. Inhibition of melanosome transfer results in skin lightening. J Invest Dermatol. (2000) 115:162–7. doi: 10.1046/j.1523-1747.2000.00035.x
21. Moretti, S, Nassini, R, Prignano, F, Pacini, A, Materazzi, S, Naldini, A, et al. Protease-activated receptor-2 downregulation is associated to vitiligo lesions. Pigment Cell Melanoma Res. (2009) 22:335–8. doi: 10.1111/j.1755-148X.2009.00562.x
22. Paine, C, Sharlow, E, Liebel, F, Eisinger, M, Shapiro, S, and Seiberg, M. An alternative approach to depigmentation by soybean extracts via inhibition of the PAR-2 pathway. J Invest Dermatol. (2001) 116:587–95. doi: 10.1046/j.1523-1747.2001.01291.x
23. Jimbow, K. Current update and trends in melanin pigmentation and melanin biology. Keio J Med. (1995) 44:9–18. doi: 10.2302/kjm.44.9
24. Haass, NK, Smalley, KS, Li, L, and Herlyn, M. Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res. (2005) 18:150–9. doi: 10.1111/j.1600-0749.2005.00235.x
25. Hara, M, Yaar, M, Tang, A, Eller, MS, Reenstra, W, and Gilchrest, BA. Role of integrins in melanocyte attachment and dendricity. J Cell Sci. (1994) 107:2739–48. doi: 10.1242/jcs.107.10.2739
26. Kim, SJ, Kim, MS, Park, HJ, Lee, H, Yun, JI, Lim, HW, et al. Screening of integrins localized on the surface of human epidermal melanocytes. In Vitro Cell Dev Biol Anim. (2020) 56:435–43. doi: 10.1007/s11626-020-00471-4
27. Scott, G, Ewing, J, Ryan, D, and Abboud, C. Stem cell factor regulates human melanocyte-matrix interactions. Pigment Cell Res. (1994) 7:44–51. doi: 10.1111/j.1600-0749.1994.tb00017.x
28. Fukunaga-Kalabis, M, Martinez, G, Liu, ZJ, Kalabis, J, Mrass, P, Weninger, W, et al. CCN3 controls 3D spatial localization of melanocytes in the human skin through DDR1. J Cell Biol. (2006) 175:563–9. doi: 10.1083/jcb.200602132
29. Ricard, AS, Pain, C, Daubos, A, Ezzedine, K, Lamrissi-Garcia, I, Bibeyran, A, et al. Study of CCN3 (NOV) and DDR1 in normal melanocytes and vitiligo skin. Exp Dermatol. (2012) 21:411–6. doi: 10.1111/j.1600-0625.2012.01473.x
30. Almasi-Nasrabadi, M, Amoli, MM, Robati, RM, Rajabi, F, Ghalamkarpour, F, and Gauthier, Y. CDH1 and DDR1 common variants confer risk to vitiligo and autoimmune comorbidities. Gene. (2019) 700:17–22. doi: 10.1016/j.gene.2019.03.026
31. Silva de Castro, CC, Do Nascimento, LM, Walker, G, Werneck, RI, Nogoceke, E, and Mira, MT. Genetic variants of the DDR1 gene are associated with vitiligo in two independent Brazilian population samples. J Invest Dermatol. (2010) 130:1813–8. doi: 10.1038/jid.2010.34
32. Kim, HJ, Uhm, YK, Yun, JY, Im, SH, Yim, SV, Chung, JH, et al. Association between polymorphisms of discoidin domain receptor tyrosine kinase 1 (DDR1) and non-segmental vitiligo in the Korean population. Eur J Dermatol. (2010) 20:231–2. doi: 10.1684/ejd.2010.0853
33. Le Poole, IC, van den Wijngaard, RM, Westerhof, W, and Das, PK. Tenascin is overexpressed in vitiligo lesional skin and inhibits melanocyte adhesion. Br J Dermatol. (1997) 137:171–8. doi: 10.1046/j.1365-2133.1997.18011894.x
34. Esmat, SM, Hadidi, HHE, Hegazy, RA, Gawdat, HI, Tawdy, AM, Fawzy, MM, et al. Increased tenascin C and DKK1 in vitiligo: possible role of fibroblasts in acral and non-acral disease. Arch Dermatol Res. (2018) 310:425–30. doi: 10.1007/s00403-018-1830-z
35. Abdou, AG, Maraee, AH, Shoeib, MA, and Elbana, R. Immunolocalization of tenascin-C in vitiligo. Appl Immunohistochem Mol Morphol. (2012) 20:501–11. doi: 10.1097/PAI.0b013e318246c793
36. McNeil, MM, Nahhas, AF, Braunberger, TL, and Hamzavi, IH. Afamelanotide in the treatment of dermatologic disease. Skin Therapy Lett. (2018) 23:6–10.
37. Minder, EI, Barman-Aksoezen, J, and Schneider-Yin, X. Pharmacokinetics and pharmacodynamics of Afamelanotide and its clinical use in treating dermatologic disorders. Clin Pharmacokinet. (2017) 56:815–23. doi: 10.1007/s40262-016-0501-5
38. Lim, HW, Grimes, PE, Agbai, O, Hamzavi, I, Henderson, M, Haddican, M, et al. Afamelanotide and narrowband UV-B phototherapy for the treatment of vitiligo: a randomized multicenter trial. JAMA Dermatol. (2015) 151:42–50. doi: 10.1001/jamadermatol.2014.1875
39. Esmat, S, Abdel Halim, DM, Hegazy, RA, Sayed, S, and Saleh, MA. Matrix metalloproteinase in acral and non-acral vitiligo. Photodermatol Photoimmunol Photomed. (2018) 34:211–3. doi: 10.1111/phpp.12352
40. Sternlicht, MD, and Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. (2001) 17:463–516. doi: 10.1146/annurev.cellbio.17.1.463
41. Lei, TC, Vieira, WD, and Hearing, VJ. In vitro migration of melanoblasts requires matrix metalloproteinase-2: implications to vitiligo therapy by photochemotherapy. Pigment Cell Res. (2002) 15:426–32. doi: 10.1034/j.1600-0749.2002.02044.x
42. Kumar, R, Parsad, D, Kanwar, AJ, and Kaul, D. Altered levels of Ets-1 transcription factor and matrix metalloproteinases in melanocytes from patients with vitiligo. Br J Dermatol. (2011) 165:285–91. doi: 10.1111/j.1365-2133.2011.10324.x
43. Wagner, RY, Luciani, F, Cario-Andre, M, Rubod, A, Petit, V, Benzekri, L, et al. Altered E-cadherin levels and distribution in melanocytes precede clinical manifestations of vitiligo. J Invest Dermatol. (2015) 135:1810–9. doi: 10.1038/jid.2015.25
44. Bishnoi, A, and Parsad, D. Repigmentation patterns in vitiligo: where do we stand? Br J Dermatol. (2016) 175:460–1. doi: 10.1111/bjd.14890
45. Boukhedouni, N, Martins, C, Darrigade, AS, Drullion, C, Rambert, J, Barrault, C, et al. Type-1 cytokines regulate MMP-9 production and E-cadherin disruption to promote melanocyte loss in vitiligo. JCI Insight. (2020) 5:e133772. doi: 10.1172/jci.insight.133772
46. Yamamoto, K, Murphy, G, and Troeberg, L. Extracellular regulation of metalloproteinases. Matrix Biol. (2015) 44-46:255–63. doi: 10.1016/j.matbio.2015.02.007
47. Su, M, Miao, F, Jiang, S, Shi, Y, Luo, L, He, X, et al. Role of the p53TRPM1/miR211MMP9 axis in UVBinduced human melanocyte migration and its potential in repigmentation. Int J Mol Med. (2020) 45:1017–26. doi: 10.3892/ijmm.2020.4478
48. Vlad-Fiegen, A, Langerak, A, Eberth, S, and Muller, O. The Wnt pathway destabilizes adherens junctions and promotes cell migration via beta-catenin and its target gene cyclin D1. FEBS Open Bio. (2012) 2:26–31. doi: 10.1016/j.fob.2012.02.004
49. Bachmann, IM, Straume, O, Puntervoll, HE, Kalvenes, MB, and Akslen, LA. Importance of P-cadherin, beta-catenin, and Wnt5a/frizzled for progression of melanocytic tumors and prognosis in cutaneous melanoma. Clin Cancer Res. (2005) 11:8606–14. doi: 10.1158/1078-0432.CCR-05-0011
50. Bertrand, JU, Petit, V, Hacker, E, Berlin, I, Hayward, NK, Pouteaux, M, et al. UVB represses melanocyte cell migration and acts through beta-catenin. Exp Dermatol. (2017) 26:875–82. doi: 10.1111/exd.13318
51. Yamada, T, Hasegawa, S, Inoue, Y, Date, Y, Yamamoto, N, Mizutani, H, et al. Wnt/beta-catenin and kit signaling sequentially regulate melanocyte stem cell differentiation in UVB-induced epidermal pigmentation. J Invest Dermatol. (2013) 133:2753–62. doi: 10.1038/jid.2013.235
52. Keswell, D, Kidson, SH, and Davids, LM. Melanocyte migration is influenced by E-cadherin-dependent adhesion of keratinocytes in both two- and three-dimensional in vitro wound models. Cell Biol Int. (2015) 39:169–76. doi: 10.1002/cbin.10350
53. Yoshida, H, Kunisada, T, Grimm, T, Nishimura, EK, Nishioka, E, and Nishikawa, SI. Review: melanocyte migration and survival controlled by SCF/c-kit expression. J Investig Dermatol Symp Proc. (2001) 6:1–5. doi: 10.1046/j.0022-202x.2001.00006.x
54. Czyz, M. HGF/c-MET signaling in melanocytes and melanoma. Int J Mol Sci. (2018) 19:3844. doi: 10.3390/ijms19123844
55. Hara, M, Yaar, M, and Gilchrest, BA. Endothelin-1 of keratinocyte origin is a mediator of melanocyte dendricity. J Invest Dermatol. (1995) 105:744–8. doi: 10.1111/1523-1747.ep12325522
56. Srivastava, N, Gupta, S, and Parsad, D. Melanocyte adhesion and apoptosis in vitiligo: linking puzzle blocks. Curr Mol Med. (2022) 22. doi: 10.2174/1566524022666220621125552 [Epub ahead of print].
57. Gauthier, Y, Cario-Andre, M, Lepreux, S, Pain, C, and Taieb, A. Melanocyte detachment after skin friction in non lesional skin of patients with generalized vitiligo. Br J Dermatol. (2003) 148:95–101. doi: 10.1046/j.1365-2133.2003.05024.x
58. Gauthier, Y, Cario Andre, M, and Taieb, A. A critical appraisal of vitiligo etiologic theories. Is melanocyte loss a melanocytorrhagy? Pigment Cell Res. (2003) 16:322–32. doi: 10.1034/j.1600-0749.2003.00070.x
59. Alikhan, A, Felsten, LM, Daly, M, and Petronic-Rosic, V. Vitiligo: a comprehensive overview part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J Am Acad Dermatol. (2011) 65:473–91. doi: 10.1016/j.jaad.2010.11.061
60. Alkhateeb, A, Fain, PR, Thody, A, Bennett, DC, and Spritz, RA. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigment Cell Res. (2003) 16:208–14. doi: 10.1034/j.1600-0749.2003.00032.x
61. Zhang, XJ, Liu, JB, Gui, JP, Li, M, Xiong, QG, Wu, HB, et al. Characteristics of genetic epidemiology and genetic models for vitiligo. J Am Acad Dermatol. (2004) 51:383–90. doi: 10.1016/j.jaad.2003.12.044
62. Roberts, GHL, Santorico, SA, and Spritz, RA. The genetic architecture of vitiligo. Pigment Cell Melanoma Res. (2020) 33:8–15. doi: 10.1111/pcmr.12848
63. Hyter, S, Coleman, DJ, Ganguli-Indra, G, Merrill, GF, Ma, S, Yanagisawa, M, et al. Endothelin-1 is a transcriptional target of p53 in epidermal keratinocytes and regulates ultraviolet-induced melanocyte homeostasis. Pigment Cell Melanoma Res. (2013) 26:247–58. doi: 10.1111/pcmr.12063
64. Hirobe, T, Shinpo, T, Higuchi, K, and Sano, T. Life cycle of human melanocytes is regulated by endothelin-1 and stem cell factor in synergy with cyclic AMP and basic fibroblast growth factor. J Dermatol Sci. (2010) 57:123–31. doi: 10.1016/j.jdermsci.2009.11.006
65. Takata, T, Tarutani, M, and Sano, S. A failure in endothelin-1 production from vitiligo keratinocytes in response to ultraviolet B irradiation. J Dermatol Sci. (2013) 71:210–2. doi: 10.1016/j.jdermsci.2013.04.030
66. Kim, HJ, Choi, CP, Uhm, YK, Kim, YI, Lee, JW, Yoon, SH, et al. The association between endothelin-1 gene polymorphisms and susceptibility to vitiligo in a Korean population. Exp Dermatol. (2007) 16:561–6. doi: 10.1111/j.1600-0625.2007.00561.x
67. Bingul, I, Aydingoz, IE, Vural, P, Dogru-Abbasoglu, S, and Uysal, M. The evaluation of Endothelin-1 and endothelin receptor type a gene polymorphisms in patients with vitiligo. Indian J Dermatol. (2016) 61:118. doi: 10.4103/0019-5154.174076
68. Smith, KA, Tong, X, Abu-Yousif, AO, Mikulec, CC, Gottardi, CJ, Fischer, SM, et al. UVB radiation-induced beta-catenin signaling is enhanced by COX-2 expression in keratinocytes. Mol Carcinog. (2012) 51:734–45. doi: 10.1002/mc.20840
69. Leong, J, Hughes-Fulford, M, Rakhlin, N, Habib, A, Maclouf, J, and Goldyne, ME. Cyclooxygenases in human and mouse skin and cultured human keratinocytes: association of COX-2 expression with human keratinocyte differentiation. Exp Cell Res. (1996) 224:79–87. doi: 10.1006/excr.1996.0113
70. Li, M, Gao, Y, Li, C, Liu, L, Li, K, Gao, L, et al. Association of COX2 functional polymorphisms and the risk of vitiligo in Chinese populations. J Dermatol Sci. (2009) 53:176–81. doi: 10.1016/j.jdermsci.2008.09.010
71. Salem, SAM, Aly, DG, Amr, KS, and Abdel-Hamid, MF. Study of the relation between two common cyclooxygenase 2 gene polymorphisms with risk of developing and subtypes of vitiligo in Egyptian patients. Indian J Dermatol Venereol Leprol. (2018) 84:696–700. doi: 10.4103/ijdvl.IJDVL_813_16
72. Jin, Y, Riccardi, SL, Gowan, K, Fain, PR, and Spritz, RA. Fine-mapping of vitiligo susceptibility loci on chromosomes 7 and 9 and interactions with NLRP1 (NALP1). J Invest Dermatol. (2010) 130:774–83. doi: 10.1038/jid.2009.273
73. Taieb, A. NALP1 and the inflammasomes: challenging our perception of vitiligo and vitiligo-related autoimmune disorders. Pigment Cell Res. (2007) 20:260–2. doi: 10.1111/j.1600-0749.2007.00393.x
74. Faustin, B, Lartigue, L, Bruey, JM, Luciano, F, Sergienko, E, Bailly-Maitre, B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. (2007) 25:713–24. doi: 10.1016/j.molcel.2007.01.032
75. Jin, Y, Mailloux, CM, Gowan, K, Riccardi, SL, LaBerge, G, Bennett, DC, et al. NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med. (2007) 356:1216–25. doi: 10.1056/NEJMoa061592
76. Alkhateeb, A, and Qarqaz, F. Genetic association of NALP1 with generalized vitiligo in Jordanian Arabs. Arch Dermatol Res. (2010) 302:631–4. doi: 10.1007/s00403-010-1064-1
77. Jin, Y, Birlea, SA, Fain, PR, and Spritz, RA. Genetic variations in NALP1 are associated with generalized vitiligo in a Romanian population. J Invest Dermatol. (2007) 127:2558–62. doi: 10.1038/sj.jid.5700953
78. Boiten, W, Helder, R, van Smeden, J, and Bouwstra, J. Selectivity in cornified envelop binding of ceramides in human skin and the role of LXR inactivation on ceramide binding. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:1206–13. doi: 10.1016/j.bbalip.2019.05.003
79. Shehata, WA, Maraee, A, Mehesin, M, Tayel, N, and Azmy, R. Genetic polymorphism of liver X receptor gene in vitiligo: does it have an association? J Cosmet Dermatol. (2021) 20:1906–14. doi: 10.1111/jocd.13772
80. Agarwal, S, Kaur, G, Randhawa, R, Mahajan, V, Bansal, R, and Changotra, H. Liver X receptor-alpha polymorphisms (rs11039155 and rs2279238) are associated with susceptibility to vitiligo. Meta Gene. (2016) 8:33–6. doi: 10.1016/j.mgene.2016.02.001
81. Kumar, R, Parsad, D, Kanwar, AJ, and Kaul, D. Altered levels of LXR-alpha: crucial implications in the pathogenesis of vitiligo. Exp Dermatol. (2012) 21:853–8. doi: 10.1111/exd.12017
82. Bakry, OA, Shoeib, M, El Kady, N, and Attalla, S. Re-appraisal of keratinocytes' role in vitiligo pathogenesis. Indian J Dermatol. (2018) 63:231–40. doi: 10.4103/ijd.IJD_520_17
83. Castrillo, A, Joseph, SB, Marathe, C, Mangelsdorf, DJ, and Tontonoz, P. Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J Biol Chem. (2003) 278:10443–9. doi: 10.1074/jbc.M213071200
84. Marionnet, C, Pierrard, C, Lejeune, F, Sok, J, Thomas, M, and Bernerd, F. Different oxidative stress response in keratinocytes and fibroblasts of reconstructed skin exposed to non extreme daily-ultraviolet radiation. PLoS One. (2010) 5:e12059. doi: 10.1371/journal.pone.0012059
85. Mosenson, JA, Flood, K, Klarquist, J, Eby, JM, Koshoffer, A, Boissy, RE, et al. Preferential secretion of inducible HSP70 by vitiligo melanocytes under stress. Pigment Cell Melanoma Res. (2014) 27:209–20. doi: 10.1111/pcmr.12208
86. Mosenson, JA, Zloza, A, Klarquist, J, Barfuss, AJ, Guevara-Patino, JA, and Poole, IC. HSP70i is a critical component of the immune response leading to vitiligo. Pigment Cell Melanoma Res. (2012) 25:88–98. doi: 10.1111/j.1755-148X.2011.00916.x
87. Rashighi, M, Agarwal, P, Richmond, JM, Harris, TH, Dresser, K, Su, MW, et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med. (2014) 6:223ra23. doi: 10.1126/scitranslmed.3007811
88. Mosenson, JA, Zloza, A, Nieland, JD, Garrett-Mayer, E, Eby, JM, Huelsmann, EJ, et al. Mutant HSP70 reverses autoimmune depigmentation in vitiligo. Sci Transl Med. (2013) 5:174ra28. doi: 10.1126/scitranslmed.3005127
89. Henning, SW, Fernandez, MF, Mahon, JP, Duff, R, Azarafrooz, F, Guevara-Patino, JA, et al. HSP70i(Q435A)-encoding DNA repigments vitiligo lesions in Sinclair swine. J Invest Dermatol. (2018) 138:2531–9. doi: 10.1016/j.jid.2018.06.186
90. Marks, MS, Theos, AC, and Raposo, G. Melanosomes and MHC class II antigen-processing compartments: a tinted view of intracellular trafficking and immunity. Immunol Res. (2003) 27:409–26. doi: 10.1385/IR:27:2-3:409
91. Elela, MA, Hegazy, RA, Fawzy, MM, Rashed, LA, and Rasheed, H. Interleukin 17, interleukin 22 and FoxP3 expression in tissue and serum of non-segmental vitiligo: a case- controlled study on eighty-four patients. Eur J Dermatol. (2013) 23:350–5. doi: 10.1684/ejd.2013.2023
92. Cho, KA, Suh, JW, Lee, KH, Kang, JL, and Woo, SY. IL-17 and IL-22 enhance skin inflammation by stimulating the secretion of IL-1beta by keratinocytes via the ROS-NLRP3-caspase-1 pathway. Int Immunol. (2012) 24:147–58. doi: 10.1093/intimm/dxr110
93. Harris, JE, Harris, TH, Weninger, W, Wherry, EJ, Hunter, CA, and Turka, LA. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-gamma for autoreactive CD8(+) T-cell accumulation in the skin. J Invest Dermatol. (2012) 132:1869–76. doi: 10.1038/jid.2011.463
94. Richmond, JM, Masterjohn, E, Chu, R, Tedstone, J, Youd, ME, and Harris, JE. CXCR3 depleting antibodies prevent and reverse vitiligo in mice. J Invest Dermatol. (2017) 137:982–5. doi: 10.1016/j.jid.2016.10.048
95. Rashighi, M, and Harris, JE. Interfering with the IFN-gamma/CXCL10 pathway to develop new targeted treatments for vitiligo. Ann Transl Med. (2015) 3:343. doi: 10.3978/j.issn.2305-5839.2015.11.36
96. Shiu, J, Zhang, L, Lentsch, G, Flesher, JL, Jin, S, Polleys, C, et al. Multimodal analyses of vitiligo skin identify tissue characteristics of stable disease. JCI Insight. (2022) 7:e154585. doi: 10.1172/jci.insight.154585
97. Kovacs, D, Bastonini, E, Briganti, S, Ottaviani, M, D'Arino, A, Truglio, M, et al. Altered epidermal proliferation, differentiation, and lipid composition: novel key elements in the vitiligo puzzle. Sci Adv. (2022) 8:eabn9299. doi: 10.1126/sciadv.abn9299
98. Vaccaro, M, Cicero, F, Mannucci, C, Calapai, G, Spatari, G, Barbuzza, O, et al. IL-33 circulating serum levels are increased in patients with non-segmental generalized vitiligo. Arch Dermatol Res. (2016) 308:527–30. doi: 10.1007/s00403-016-1675-2
99. Li, P, Ma, H, Han, D, and Mou, K. Interleukin-33 affects cytokine production by keratinocytes in vitiligo. Clin Exp Dermatol. (2015) 40:163–70. doi: 10.1111/ced.12464
100. Richmond, JM, Strassner, JP, Zapata, L Jr, Garg, M, Riding, RL, Refat, MA, et al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. (2018) 10:eaam7710. doi: 10.1126/scitranslmed.aam7710
101. Richmond, JM, Strassner, JP, Rashighi, M, Agarwal, P, Garg, M, Essien, KI, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol. (2019) 139:769–78. doi: 10.1016/j.jid.2018.10.032
102. Li, S, Zhu, G, Yang, Y, Jian, Z, Guo, S, Dai, W, et al. Oxidative stress drives CD8(+) T-cell skin trafficking in patients with vitiligo through CXCL16 upregulation by activating the unfolded protein response in keratinocytes. J Allergy Clin Immunol. (2017) 140:177–189.e9. doi: 10.1016/j.jaci.2016.10.013
103. Hsueh, YC, Wang, Y, Riding, RL, Catalano, DE, Lu, YJ, Richmond, JM, et al. A keratinocyte-tethered biologic enables location-precise treatment in mouse vitiligo. J Invest Dermatol. (2022) 142:3294–303. doi: 10.1016/j.jid.2022.06.007
104. Hamzavi, I, Rosmarin, D, Harris, JE, Pandya, AG, Lebwohl, M, Gottlieb, AB, et al. Efficacy of ruxolitinib cream in vitiligo by patient characteristics and affected body areas: descriptive subgroup analyses from a phase 2, randomized, double-blind trial. J Am Acad Dermatol. (2022) 86:1398–401. doi: 10.1016/j.jaad.2021.05.047
Keywords: vitiligo, growth factors, chemokines, antigen presentation, apoptosis, stress, adhesion, melanosome transfer
Citation: Touni AA, Shivde RS, Echuri H, Abdel-Aziz RTA, Abdel-Wahab H, Kundu RV and Le Poole IC (2023) Melanocyte-keratinocyte cross-talk in vitiligo. Front. Med. 10:1176781. doi: 10.3389/fmed.2023.1176781
Edited by:
Reinhart Speeckaert, Ghent University Hospital, BelgiumReviewed by:
Shahnawaz Jadeja, University College Dublin, IrelandCopyright © 2023 Touni, Shivde, Echuri, Abdel-Aziz, Abdel-Wahab, Kundu and Le Poole. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed Ahmed Touni, YWhtZWR0b3VuaTE5OTBAZ21haWwuY29t; I. Caroline Le Poole, Y2Fyb2xpbmUubGVwb29sZUBub3J0aHdlc3Rlcm4uZWR1