 Simona Granata1
Simona Granata1 Dario Troise
Dario Troise Loreto Gesualdo
Loreto Gesualdo Giovanni Stallone
Giovanni Stallone Gianluigi Zaza
Gianluigi Zaza
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 12 May 2023
Sec. Nephrology
Volume 10 - 2023 | https://doi.org/10.3389/fmed.2023.1168967
The mammalian target of rapamycin inhibitors (mTOR-Is, Sirolimus, and Everolimus) are immunosuppressive drugs widely employed in kidney transplantation. Their main mechanism of action includes the inhibition of a serine/threonine kinase with a pivotal role in cellular metabolism and in various eukaryotic biological functions (including proteins and lipids synthesis, autophagy, cell survival, cytoskeleton organization, lipogenesis, and gluconeogenesis). Moreover, as well described, the inhibition of the mTOR pathway may also contribute to the development of the post-transplant diabetes mellitus (PTDM), a major clinical complication that may dramatically impact allograft survival (by accelerating the development of the chronic allograft damage) and increase the risk of severe systemic comorbidities. Several factors may contribute to this condition, but the reduction of the beta-cell mass, the impairment of the insulin secretion and resistance, and the induction of glucose intolerance may play a pivotal role. However, although the results of several in vitro and in animal models, the real impact of mTOR-Is on PTDM is still debated and the entire biological machinery is poorly recognized. Therefore, to better elucidate the impact of the mTOR-Is on the risk of PTDM in kidney transplant recipients and to potentially uncover future research topics (particularly for the clinical translational research), we decided to review the available literature evidence regarding this important clinical association. In our opinion, based on the published reports, we cannot draw any conclusion and PTDM remains a challenge. However, also in this case, the administration of the lowest possible dose of mTOR-I should also be recommended.
New-onset diabetes after transplantation (NODAT) refers to the occurrence of diabetes in previously non-diabetic clinically stable kidney transplant recipients who had been discharged from the hospital and tapered to their maintenance immunosuppressive therapy (1).
It occurs in almost 4% to 27% of kidney transplant recipients (2–6) and it may induce the development of the chronic allograft damage by activating several pro-fibrotic mediators [including transforming growth factor beta (TGF-β)] and promoting mesangial matrix expansion and cell hyperplasia (7), accelerating the onset of severe clinical complications/comorbidities.
To diagnose NODAT, the International Consensus Guidelines published in 2003 recommended to use the same diagnostic criteria adopted by the American Diabetes Association and the World Health Organization (ADA/WHO) for type 2 diabetes in non-transplant patients: fasting glucose ≥126 mg/dL (7 mmol/L) on more than one occasion, random glucose ≥ 200 mg/dL (11.1 mmol/L) with symptoms, two-hour glucose after 75 g oral glucose tolerance test of ≥200 mg/dL (11.1 mmol/L), and hemoglobin A1C (HbA1c) ≥ 6.5% (3).
However, since the difficulty to screen all patients before transplantation and to identify pre-existing forms of diabetes, in 2013, a second international consensus changed the nomenclature from NODAT to post-transplantation diabetes mellitus (PTDM; a more inclusive term for diabetes that is diagnosed after transplantation without specific reference to any previous history of diabetes) (3).
Risk factors for PTDM are similar to those for type 2 diabetes mellitus: increased age (>40 years), family history of type 2 diabetes, ethnicity (African-American, Asian, and Hispanic patients are at higher risk compared to Caucasian), abnormal glucose tolerance (expressed by fasting blood sugar levels between 90 and 100 mg/dL), and specific genetic factors (8–12).
Other risk factors for PTDM include the metabolic syndrome and the obesity, responsible for the insulin resistance and the glucose intolerance. Indeed, the adipose tissue from obese subjects increases the expression of Tumor Necrosis Factor-α (TNF-α) (13), which downregulates genes involved in the insulin action (such as glucose transporter (GLUT)-4, insulin receptor, and insulin receptor substrate (IRS)-1) (14, 15), reduces adiponectin release and stimulates adipocyte lipolysis (16).
Also, viral infections may increase the risk of PTDM. Hepatitis C virus (HCV) (17) infection may trigger an immune-mediated reaction against β cells with consequent cytopathic effects, glucose uptake reduction (18), and gluconeogenesis augmentation (19). Likewise, cytomegalovirus (CMV) may directly damage beta-cells by the upregulation of large pro-apoptotic machinery or, indirectly, by facilitating the release of pro-inflammatory cytokines (20).
However, in kidney transplant recipients, the administration of immunosuppressive drugs may further impact the incidence of PTDM. Corticosteroids (administered at high dosages as part of the induction therapy and/or for treating acute rejection and as main constituent of the maintenance immunosuppressive protocol) and calcineurin inhibitors (CNIs; Tacrolimus and Cyclosporine A) are the main responsible for this complication, while the diabetogenic effects of mammalian target of rapamycin inhibitors (mTOR-Is) are still debated (21).
mTOR inhibitors (mTOR-Is, Sirolimus, and Everolimus) exert their pharmacological effects via inhibition of the serine/threonine kinase mammalian target of rapamycin (mTOR). mTOR integrates signals from growth factors, hormones, nutrients, and cellular energy levels to regulate protein translational and cell growth, proliferation, and survival (22). mTOR exists in two complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (23).
mTORC1 also includes RAPTOR (24, 25), MLST8 (26), PRAS40 (27), and DEPTOR, while mTORC2 includes RICTOR, MAPKAP1, PRR5/PRR5L, Mlst8, and Deptor (28–30).
This complex is less sensitive to the acute treatment with mTOR-Is, while long-term administration of these medications inhibits mTORC2 activity by acting on complex integrity (31, 32).
The use of mTOR-Is as immunosuppressants in solid organ transplantation is mostly based on their ability to block cell cycle progression from G1 to the S phase and cellular proliferation (33). Additionally, these drugs may have further therapeutic effects by modifying: (a) protein synthesis, (b) cell cycle, (c) lipid metabolism, (d) energy metabolism, (e) autophagy, (f) angiogenesis, (g) cytoskeleton remodeling, and (h) epithelial to mesenchymal transition (23, 34–39).
Because of their specific pharmacological characteristics, mTOR-Is are highly effective in renal transplantation, and owing to their relative lack of nephrotoxicity, these inhibitors are a valid alternative to CNIs for the maintenance of immunosuppression in renal transplant recipients with chronic allograft nephropathy (40).
Although the clinical utility of this drug category is clear, as other immunosuppressive drugs, mTOR-Is may persuade the development of some adverse effects that need to be immediately recognized and treated to avoid severe illness in kidney transplant recipients.
Numerous clinical and translational studies have highlighted that mTOR-Is-treated kidney transplant recipients may develop metabolic disorders, including PTDM (41–43). As proven in in vitro and in animal models, mTOR-Is may decrease beta-cell mass through an increment of the rate of apoptosis (44–47), induce impairment of glucose-induced insulin secretion (45–49), and facilitate glucose intolerance and insulin resistance (50–52) (Table 1).
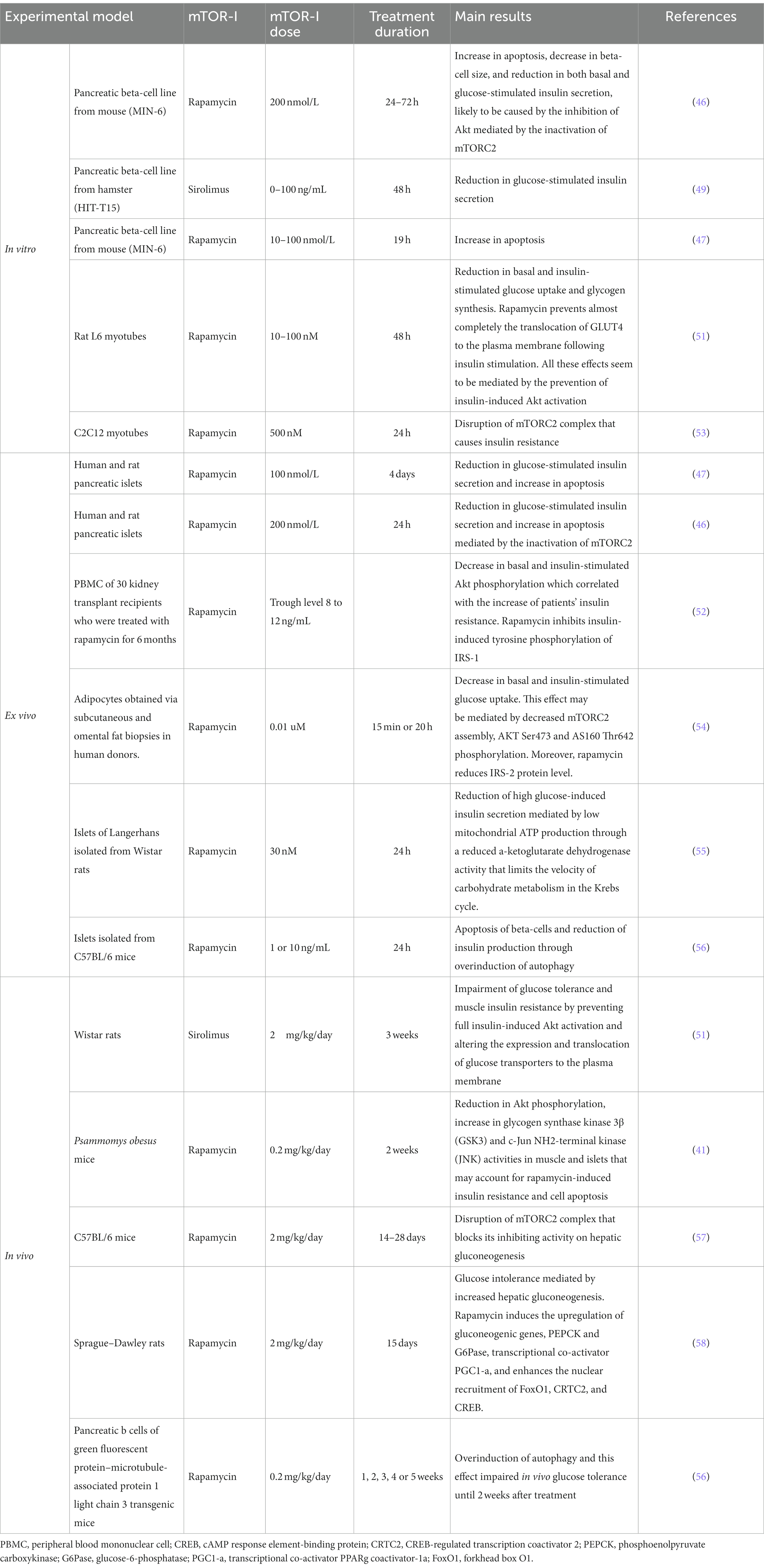
Table 1. mTOR-Is-mediated biological mechanisms involved in glucose dysmetabolism.
The impact of mTOR-Is on glucose metabolism is also mediated by its interference with insulin signal transduction. Physiologically, insulin and insulin-like growth factors (IGF) activate mTORC1 through the IRS/PI3K/Akt pathway (50). The mTOR-Is bind to mTOR and suppress the PI3K/AKT pathway (23). The reduction of Akt phosphorylation causes an increment in beta-cell apoptosis with consequent reduced beta-cell mass, impairment of glucose-stimulated insulin secretion, and proinsulin biosynthesis (48). This leads to augmented gluconeogenesis (58), reduced glucose uptake (54), glycolysis, and glycogen synthesis (41, 59) (Table 1).
However, these properties seem to depend on the metabolic context in which they are studied. mTOR-I administered to an animal model of nutrition-dependent type 2 diabetes (diabetic Psammomys obesus) worsened the metabolic state of the diabetic animals: augmented insulin resistance, β-cell dysfunction, and death, thereby preventing β-cell adaptation to hyperglycemia (41).
Hyperglycemia and glucose intolerance after mTOR-Is treatment is due to the upregulation of several gluconeogenic genes in the liver via the coordinated activation of peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1alpha, cAMP response element-binding protein (CREB), CREB-regulated transcription coactivator 2 (CRTC2), and forkhead box O1 (FoxO1) (58).
Reduced obesity and hyperlipidemia are frequently associated with the mTOR-I treatment (58, 60). Both in vitro and in vivo studies have revealed the association between adipogenesis and the mTOR pathway (61–64). Notably, obesity and overnutrition trigger chronic hyperactivation of mTOR activity in multiple tissues (50, 61). In humans, increased S6K activity (65, 66) and overphosphorylation of translation suppressor 4EBP have been observed in obesity (67). Furthermore, accelerated adipogenesis and obesity have been reported in mice lacking 4E-BP1 and 4E-BP2 genes (68). Indeed, targeting the mTOR pathway has been suggested as a treatment for obesity. Consistently, S6K knockout mice were protected against obesity due to increased lipolysis levels and energy expenditure (61, 69, 70).
mTOR-Is treatment may, then, reduce the cell number in adipose tissue with a small contribution from reduced adipocyte size. Reduced lipid uptake and fat cell number impairs the capacity of adipose tissue for plasma lipid clearance, which likely contributes to hyperlipidemia (58).
The inhibition of the mTOR pathway may also play a role in insulin secretion in pancreatic β-cells. Several studies have reported the inhibition of glucose-induced insulin secretion in clonal β-cell lines and in islets (41), but the exact mechanism is unclear (44).
The control of insulin secretion in beta-cells involves several signaling pathways. One proposed mechanism is that inhibition of mTORC1 decreases mitochondrial function, specifically, the activity of α-ketoglutarate dehydrogenase. This results in reduced carbohydrate metabolism and therefore, reduced mitochondrial ATP production (55), which regulates insulin secretion in β-cells (71). Other explanation that rapamycin promotes autophagy, and the intracellular degradation of cytoplasmic proteins involved in the insulin production and secretion (56).
Moreover, several in vitro studies have reported a leading role of mTORC2 in the function and survival of beta-cells (46, 72) and insulin resistance (53). Chronic treatment with mTOR-I also inactivates mTORC2 with subsequent inhibition of AKT signaling (46) and other mTORC2 substrates such as protein kinases PKCα, SGK1 substrate NDRG1 in the liver, muscle, and white adipose tissue (57, 73).
It has also been observed that long-term treatment with mTOR-Is (20 weeks) partially restored the detrimental effects on metabolism with enhanced insulin sensitivity, increased oxygen consumption, and improved serum lipid profile with a certain degree of glucose intolerance (74).
Furthermore, in several maintenance immunosuppressive therapeutic protocols, mTOR-I are combined with CNIs.
Numerous observations have reported that CNIs treatment may lead to PTDM by a multifactorial mechanism, which includes impaired insulin secretion, insulin resistance, altered glucokinase function, mitochondrial impairment, and pancreatic β-cell apoptosis (75–79).
The mechanism in pancreatic insulin-secreting β cells seems to be mediated by the inhibition of two targets of calcineurin: nuclear factor of activated T-Lymphocytes (NFAT) and cAMP responsive element binding protein (CREB) (80). Both transcription factors mediate the expression of IRS-2 which promotes β-pancreatic cell growth, proliferation, and survival, insulin secretion by mediating phosphorylation of Akt in response to insulin and insulin-like growth factor (IGF)-1 (81). Through the inhibition of these signaling pathways, CNIs diminish β-cell survival and replication and promote β-cell failure (81–84).
Additionally, pancreatic islets treated with CNIs showed significant morphological alterations in the form of cytoplasmic swelling and vacuolization, degranulation, and immunohistochemical and ultrastructural loss of secretory granules (85, 86).
Tacrolimus (TAC) appears also to reduce insulin secretion through a downregulation of the production of ATP and glycolysis due to a reduced activity of glucokinase, a rate-limiting enzyme in glycolysis that represents an important glucose sensor in pancreatic β-cells (78).
The diabetogenic effect of TAC can also be enhanced by mitochondrial dysfunction through a decrease in both mitochondrial respiration activity and mitochondrial mass (87).
Most studies comparing the diabetogenic effects of the CNIs report higher rates of PTDM among patients receiving TAC compared to cyclosporine A (CsA) (88–90).
During the last two decades, mTOR-Is (mainly Everolimus) have been widely used as part of the maintenance immunosuppressive therapy of kidney transplant recipients and clinical studies/trials have investigated their possible pathogenetic impact on PTDM (Table 2).
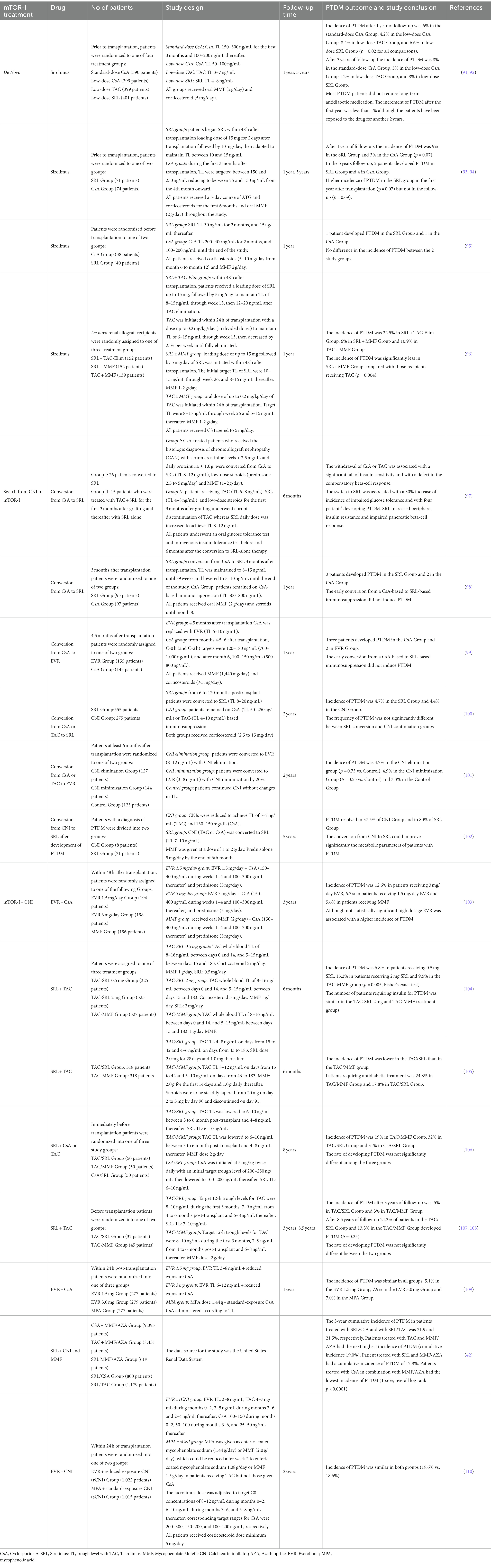
Table 2. List of major clinical trials investigating the impact of mTOR-I on PTDM.
As reported by the SYMPHONY study after 1 year and 3 years of follow-up (91, 92), patients treated with low-dose sirolimus (SRL) plus Mycophenolate Mofetil (MMF) presented a higher incidence of PTDM than those treated with low dose of CsA plus MMF (6.6% vs. 4.2% after the first year and 8% vs. 5% after 3 years of follow-up). No differences were observed in the comparison between the standard dose of CsA vs. low dose SRL (in both groups the incidence was 8%). This study suggested that a low-dose CsA-based maintenance immunosuppressive treatment or a switch from TAC to low dose of CsA or SRL could be beneficial for kidney transplant recipients at high risk of PTDM. However, in our opinion, this therapeutic strategy should be undertaken only in highly selected patients after weighing the risk of rejection or in the absence of additional adverse events. In the prospective randomized SPIESSER study, which compared the safety and efficacy of a SRL plus MMF-based immunosuppressive regimen with a CsA plus MMF-based regimen after an induction therapy with polyclonal antilymphocyte antibodies and withdrawal of steroids at 6 months’ post-transplantation, it was observed a higher incidence of PTDM in the SRL group in the first-year post-transplantation (9% vs. 3%, p = 0.07) (93). Instead, no differences were reported between the 2 study groups after 5 years of follow-up (2% vs. 4%, p = 0.69) (94). This study revealed potential diabetogenic effects of the mTOR-Is in the early post-transplant phase (probably induced by the high-dosages of these drugs in association with corticosteroids). However, the relatively low number of patients with a diagnosis of PTDM, the similar risk of PTDM between the two study groups, and the high rate of conversion from the randomized immunosuppression to other regimens in the SRL group may not allow reaching a definitive conclusion revealing the need of a larger trial on this specific topic.
Kreis et al. (ORION Study) also described no differences in the incidence of hyperglycemia and insulin-dependent PTDM in patients treated with SRL compared to CsA (95). The evaluation of two SRL-based regimens, one with CNI withdrawal (SRL + TAC-Elim) and the other with complete CNI avoidance (SRL + MMF), compared with a CNI-based regimen containing TAC + MMF in de novo renal allograft recipients demonstrated higher incidence of PTDM in TAC recipients confirming a diabetogenic effect of TAC compared to SRL (96). These results could be partially explained by the relatively high trough level of TAC used in this group.
Also in the conversion trials, the switch from CNI to mTOR-I has not shown clear diabetogenic effects. However, some authors have reported an increased risk of PTDM in mTOR-I-treated patients due to a drug-related enhancement of peripheral insulin resistance and impairment of the compensatory beta-cell response (97).
In both CONCEPT and ZEUS studies, the early conversion from CsA-based to SRL-based therapeutic regimen (3 or 4.5 months after transplantation) did not induce PTDM in a 12 months-period post-transplantation (98, 99).
Similarly, in the late conversion (CONVERT) study, where the renal allograft recipients were randomly assigned (2:1) to undergo conversion from CsA- or TAC-based immunosuppression to SRL or to continue receiving CNI-based therapy for 2 years, the frequency of PTDM was similar between the two study regimens (4.7% vs. 4.4%, p = 1.000) (100).
Holdaas et al., in the ASCERTAIN study, which included kidney transplant patients with allograft impairment (GFR 30–70 mL/min) who underwent the minimization of CNI or conversion to Everolimus (EVR), reported a percentage of patients with PTDM of 4.7% in the CNI withdrawal group, 4.9% in the minimization group and 3.3% in the control group (101).
All these studies (98–101), although performed using SRL alone in a heterogeneous patients’ population, encourage clinicians to reduce the dosages of this immunosuppressive drug.
Nevertheless, a recent systematic review and meta-analysis established that the conversion from CNIs to mTOR-Is did not significantly decrease the risk of PTDM (111).
Instead, Veroux et al. (102) showed that the conversion from CNI to mTOR-I-based therapy in patients with PTDM had a positive effect on insulin-stimulated glucose uptake. In this study, it was observed an improved glucose balance in 80% of patients converted to SRL compared with those patients (37.5%) in whom a reduction in CNI dose was carried out. No change was found in the incidence of acute rejection. According to these authors, such beneficial effects of mTOR-Is on the glycemic homeostasis could be explained by the chronic inhibition of mTORC1 (a biological/pharmacological effect such as that observed after metformin administration) (112). In this single-center study, the small sample size (particularly of patients treated with mTOR-Is) and the low incidence of PTDM (probably due to an early reduction of immunosuppression) cannot allow to draw any definitive conclusions. In all cases, a conversion from CNIs to mTOR-Is should be achieved only after a carefully benefit–risk evaluation.
Unfortunately, also the impact of the combined therapy of mTOR-Is plus CNIs on PTDM is still argued.
Vitko et al. in a 36-month, multicenter, randomized, parallel-group equivalence trial of two oral doses of EVR (1.5 or 3 mg/day) vs. MMF (2 g/day) along with CsA microemulsion (Neoral) and corticosteroids in de novo renal transplant recipients, reported a higher incidence of PTDM in patients receiving 3 mg/day EVR (12.6%) compared to those receiving a low dose of EVR (6.7%) and MMF (5.6%) (103).
In another 6-month, randomized, open-label, parallel-group, comparative trial comparing two regimens of TAC plus SRL (with either 0.5 or 2 mg) with a TAC plus MMF immunosuppressive schema, authors found that a larger number of patients treated with TAC plus SRL at 2 mg developed PTDM (p = 0.005). However, the number of patients requiring insulin for PTDM was similar in the TAC/SRL 2 mg and TAC/MMF groups (p > 0.05) (104).
In a multicenter trial, in which renal transplant recipients were randomized to TAC with fixed-dose SRL (N = 318) or TAC with MMF (N = 316), 6 months’ creatinine clearance was comparable between the 2 immunosuppressive schemas. Biopsy-confirmed acute rejection was 15.1% (TAC/SRL) and 12.3% (TAC/MMF). In both groups, graft survival was 93% and patient survival was 99%. Premature withdrawal due to an adverse event was twice as high in the TAC/SRL group (15.1% vs. 6.3%). The incidence of any antidiabetic treatment for >30 consecutive days in previously nondiabetic patients was 17.8% in TAC/SRL, and 24.8% in TAC/MMF (105).
Guerra et al., have, then, studied a long-term follow-up post-transplant (8 years) to compare TAC/SRL, TAC/MMF, and CsA/SRL. In this report, the incidence of PTDM was not significantly different among the three groups (p = 0.37), while a slightly smaller percentage of PTDM was registered in the TAC/MMF group (19%) than TAC/SRL (32%) and CsA/SRL (31%) group. However, difference did not reach any statistical difference (p = 0.16) (106). These results were confirmed by Gallon et al. and by Chhabra et al. (107, 108).
Moreover, in a 24-month, open-label study, 833 de novo renal-transplant recipients were randomized to EVR 1.5 or 3 mg/day (target troughs 3–8 and 6–12 ng/mL, respectively) with reduced-exposure CsA, or mycophenolic acid (MPA) 1.44 g/day plus standard-exposure CsA. The overall incidence of PTDM and adverse events were comparable between the groups. Corticosteroids were used in more than 99% of patients in each group during the study, with more than 70% receiving corticosteroids without discontinuation throughout the 24-month study period (109). These studies (104–109) demonstrated a similar impact of the combined therapy of CNIs and mTOR-Is on PTDM.
Johnston et al. demonstrated that the incidence of PTDM was 21.9% in patients treated with a combination of SRL plus CsA, 21.5% in those treated with SRL plus TAC, and 17.8% in the group of patients received SRL plus MMF/AZA, showing that rapamycin was an independent variable involved in the development of PTDM (42). This study, although performed on a large dataset, has major limitations including the inherent limitations of retrospective analyses of administrative data sets, the absence of information regarding the dosage of SRL and CNI used, the enrolment of patients who had Medicare as the primary payer, which may limit the applicability of its findings to other patient populations.
In the recent TRANSFORM (Transplant efficacy and safety outcomes with an EVR-based regimen) study, a 24-month, prospective, open-label trial in 2037 de novo renal transplant recipients randomized (1:1) within 24 h of transplantation to receive EVR with reduced-exposure CNI (EVR + rCNI) or mycophenolate with standard-exposure CNI, the incidence of PTDM was similar in both the arms (19.6% vs. 18.6%) (110). Even if PTDM was not included as a primary endpoint, this study confirmed the no specific diabetogenic effects of the combined therapy with CNIs plus mTOR-Is (particularly administered at low dosages), as previously suggested. Finally, a recent network meta-analysis involving 206 eligible studies that identified 75,595 patients on TAC, 51,242 on CsA, and 3,020 on SRL, demonstrated that TAC tended to exhibit higher diabetogenicity in the short-term (2–3 years post-transplant), whereas SRL exhibits higher diabetogenicity in the long-term (5–10 years post-transplant) (113). This study is quite difficult to interpreter due to the clinical heterogeneity of the immunosuppression protocols utilized in the included studies (such as co-treatments and therapy used for rejections), variability of the criteria used to define PTDM, and absence of control of several clinical/therapeutic confounding factors.
After reviewing the available literature on this topic area, we cannot draw any definite conclusions about the diabetogenic impact of the mTOR-Is. However, we can encourage clinicians to lower the dose of these immunosuppressive drugs in patients at high risk of PTDM. Moreover, our paper shows that the transplant scientific community should undertake more research programs to better study this important topic.
SG, SM, DT, and GZ searched the literature and wrote the manuscript. GZ, GS, and LG revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kasiske, BL, Snyder, JJ, Gilbertson, D, and Matas, AJ. Diabetes mellitus after kidney transplantation in the United States. Am J Transplant. (2003) 3:178–85. doi: 10.1034/j.1600-6143.2003.00010.x
2. Davidson, J, Wilkinson, A, Dantal, J, Dotta, F, Haller, H, Hernández, D, et al. Wheeler DC; international expert panel. New-onset diabetes after transplantation: 2003 international consensus guidelines. Proceedings of an international expert panel meeting. Barcelona, Spain, 19 February 2003. Transplantation. (2003) 75:SS3–SS24. doi: 10.1097/01.TP.0000069952.49242.3E
3. Sharif, A, Hecking, M, de Vries, AP, Porrini, E, Hornum, M, Rasoul-Rockenschaub, S, et al. Proceedings from an international consensus meeting on posttransplantation diabetes mellitus: recommendations and future directions. Am J Transplant. (2014) 14:1992–2000. doi: 10.1111/ajt.12850
4. Peev, V, Reiser, J, and Alachkar, N. Diabetes mellitus in the transplanted kidney. Front Endocrinol. (2014) 5:141. doi: 10.3389/fendo.2014.00141
5. Yates, CJ, Fourlanos, S, Colman, PG, and Cohney, SJ. Screening for new-onset diabetes after kidney transplantation: limitations of fasting glucose and advantages of afternoon glucose and glycated hemoglobin. Transplantation. (2013) 96:726–31. doi: 10.1097/TP.0b013e3182a012f3
6. Shabir, S, Jham, S, Harper, L, Ball, S, Borrows, R, and Sharif, A. Validity of glycated haemoglobin to diagnose new onset diabetes after transplantation. Transpl Int. (2013) 26:315–21. doi: 10.1111/tri.12042
7. Ponticelli, C, Favi, E, and Ferraresso, M. New-onset diabetes after kidney transplantation. Medicina. (2021) 57:250. doi: 10.3390/medicina57030250
8. Weir, MR, and Fink, JC. Risk for posttransplant diabetes mellitus with current immunosuppressive medications. Am J Kidney Dis. (1999) 34:1–13. doi: 10.1016/S0272-6386(99)70101-0
9. Yang, J, Hutchinson, II, Shah, T, and Min, DI. Genetic and clinical risk factors of new-onset diabetes after transplantation in Hispanic kidney transplant recipients. Transplantation. (2011) 91:1114–9. doi: 10.1097/TP.0b013e31821620f9
10. Ahmed, SH, Biddle, K, Augustine, T, and Azmi, S. Post-transplantation diabetes mellitus. Diabetes Ther. (2020) 11:779–801. doi: 10.1007/s13300-020-00790-5
11. Walczak, DA, Calvert, D, Jarzembowski, TM, Testa, G, Sankary, HN, Thielke, J, et al. Increased risk of post-transplant diabetes mellitus despite early steroid discontinuation in Hispanic kidney transplant recipients. Clin Transpl. (2005) 19:527–31. doi: 10.1111/j.1399-0012.2005.00383.x
12. Peracha, J, Nath, J, Ready, A, Tahir, S, Parekh, K, Hodson, J, et al. Risk of post-transplantation diabetes mellitus is greater in south Asian versus Caucasian kidney allograft recipients. Transpl Int. (2016) 29:727–39. doi: 10.1111/tri.12782
13. Hotamisligil, GS, Shargill, NS, and Spiegelman, BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. (1993) 259:87–91. doi: 10.1126/science.7678183
14. Stephens, JM, Lee, J, and Pilch, PF. Tumor necrosis factor-alpha-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J Biol Chem. (1997) 272:971–6. doi: 10.1074/jbc.272.2.971
15. Rydén, M, and Arner, P. Tumour necrosis factor-alpha in human adipose tissue—from signalling mechanisms to clinical implications. J Intern Med. (2007) 262:431–8. doi: 10.1111/j.1365-2796.2007.01854.x
16. Ryden, M, Dicker, A, van Harmelen, V, Hauner, H, Brunnberg, M, Perbeck, L, et al. Mapping of early signaling events in tumor necrosis factor-alpha -mediated lipolysis in human fat cells. J Biol Chem. (2002) 277:1085–91. doi: 10.1074/jbc.M109498200
17. Fabrizi, F, Messa, P, Martin, P, and Takkouche, B. Hepatitis C virus infection and post-transplant diabetes mellitus among renal transplant patients: a meta-analysis. Int J Artif Organs. (2008) 31:675–82. doi: 10.1177/039139880803100801
18. Bose, SK, Shrivastava, S, Meyer, K, Ray, RB, and Ray, R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J Virol. (2012) 86:6315–22. doi: 10.1128/JVI.00050-12
19. Kasai, D, Adachi, T, Deng, L, Nagano-Fujii, M, Sada, K, Ikeda, M, et al. HCV replication suppresses cellular glucose uptake through down-regulation of cell surface expression of glucose transporters. J Hepatol. (2009) 50:883–94. doi: 10.1016/j.jhep.2008.12.029
20. Hjelmesaeth, J, Müller, F, Jenssen, T, Rollag, H, Sagedal, S, and Hartmann, A. Is there a link between cytomegalovirus infection and new-onset posttransplantation diabetes mellitus? Potential mechanisms of virus induced beta-cell damage. Nephrol Dial Transplant. (2005) 20:2311–5. doi: 10.1093/ndt/gfi033
21. Kesiraju, S, Paritala, P, Rao Ch, UM, and Sahariah, S. New onset of diabetes after transplantation—an overview of epidemiology, mechanism of development and diagnosis. Transpl Immunol. (2014) 30:52–8. doi: 10.1016/j.trim.2013.10.006
22. Wullschleger, S, Loewith, R, and Hall, MN. TOR signaling in growth and metabolism. Cells. (2006) 124:471–84. doi: 10.1016/j.cell.2006.01.016
23. Zaza, G, Granata, S, Caletti, C, Signorini, L, Stallone, G, and Lupo, A. mTOR inhibition role in cellular mechanisms. Transplantation. (2018) 102:S3–S16. doi: 10.1097/TP.0000000000001806
24. Hara, K, Maruki, Y, Long, X, Yoshino, K, Oshiro, N, Hidayat, S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cells. (2002) 110:177–89. doi: 10.1016/S0092-8674(02)00833-4
25. Kim, DH, Sarbassov, DD, Ali, SM, King, JE, Latek, RR, Erdjument-Bromage, H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cells. (2002) 110:163–75. doi: 10.1016/S0092-8674(02)00808-5
26. Kim, DH, Sarbassov, DD, Ali, SM, Latek, RR, Guntur, KV, Erdjument-Bromage, H, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. (2003) 11:895–904. doi: 10.1016/S1097-2765(03)00114-X
27. Thedieck, K, Polak, P, Kim, ML, Molle, KD, Cohen, A, Jenö, P, et al. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One. (2007) 2:e1217. doi: 10.1371/journal.pone.0001217
28. Jacinto, E, Loewith, R, Schmidt, A, Lin, S, Rüegg, MA, Hall, A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. (2004) 6:1122–8. doi: 10.1038/ncb1183
29. Pearce, LR, Huang, X, Boudeau, J, Pawłowski, R, Wullschleger, S, Deak, M, et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. (2007) 405:513–22. doi: 10.1042/BJ20070540
30. Wälchli, M, Berneiser, K, Mangia, F, Imseng, S, Craigie, LM, Stuttfeld, E, et al. Regulation of human mTOR complexes by DEPTOR. elife. (2021) 10:e70871. doi: 10.7554/eLife.70871
31. Sarbassov, DD, Ali, SM, Sengupta, S, Sheen, JH, Hsu, PP, Bagley, AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. (2006) 22:159–68. doi: 10.1016/j.molcel.2006.03.029
32. Huang, K, and Fingar, DC. Growing knowledge of the mTOR signaling network. Semin Cell Dev Biol. (2014) 36:79–90. doi: 10.1016/j.semcdb.2014.09.011
33. Baroja-Mazo, A, Revilla-Nuin, B, Ramírez, P, and Pons, JA. Immunosuppressive potency of mechanistic target of rapamycin inhibitors in solid-organ transplantation. World J Transplant. (2016) 6:183–92. doi: 10.5500/wjt.v6.i1.183
34. Lupinacci, S, Perri, A, Toteda, G, Vizza, D, Lofaro, D, Pontrelli, P, et al. Rapamycin promotes autophagy cell death of Kaposi's sarcoma cells through P75NTR activation. Exp Dermatol. (2022) 31:143–53. doi: 10.1111/exd.14438
35. Zaza, G, Leventhal, J, Signorini, L, Gambaro, G, and Cravedi, P. Effects of antirejection drugs on innate immune cells after kidney transplantation. Front Immunol. (2019) 10:2978. doi: 10.3389/fimmu.2019.02978
36. Tomei, P, Masola, V, Granata, S, Bellin, G, Carratù, P, Ficial, M, et al. Everolimus-induced epithelial to mesenchymal transition (EMT) in bronchial/pulmonary cells: when the dosage does matter in transplantation. J Nephrol. (2016) 29:881–91. doi: 10.1007/s40620-016-0295-4
37. Granata, S, Dalla Gassa, A, Carraro, A, Brunelli, M, Stallone, G, Lupo, A, et al. Sirolimus and Everolimus pathway: reviewing candidate genes influencing their intracellular effects. Int J Mol Sci. (2016) 17:735. doi: 10.3390/ijms17050735
38. Zaza, G, Granata, S, Tomei, P, Masola, V, Gambaro, G, and Lupo, A. mTOR inhibitors and renal allograft: yin and Yang. J Nephrol. (2014) 27:495–506. doi: 10.1007/s40620-014-0103-y
39. Saxton, RA, and Sabatini, DM. mTOR signaling in growth, metabolism, and disease. Cells. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
40. Paoletti, E, Ratto, E, Bellino, D, Marsano, L, Cassottana, P, and Cannella, G. Effect of early conversion from CNI to sirolimus on outcomes in kidney transplant recipients with allograft dysfunction. J Nephrol. (2012) 25:709–18. doi: 10.5301/jn.5000044
41. Fraenkel, M, Ketzinel-Gilad, M, Ariav, Y, Pappo, O, Karaca, M, Castel, J, et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. (2008) 57:945–57. doi: 10.2337/db07-0922
42. Johnston, O, Rose, CL, Webster, AC, and Gill, JS. Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J Am Soc Nephrol. (2008) 19:1411–8. doi: 10.1681/ASN.2007111202
43. Gyurus, E, Kaposztas, Z, and Kahan, BD. Sirolimus therapy predisposes to new-onset diabetes mellitus after renal transplantation: a long-term analysis of various treatment regimens. Transplant Proc. (2011) 43:1583–92. doi: 10.1016/j.transproceed.2011.05.001
44. Asahara, SI, Inoue, H, Watanabe, H, and Kido, Y. Roles of mTOR in the regulation of pancreatic β-cell mass and insulin secretion. Biomol Ther. (2022) 12:614. doi: 10.3390/biom12050614
45. Barlow, AD, Nicholson, ML, and Herbert, TP. Evidence for rapamycin toxicity in pancreatic β-cells and a review of the underlying molecular mechanisms. Diabetes. (2013) 62:2674–82. doi: 10.2337/db13-0106
46. Barlow, AD, Xie, J, Moore, CE, Campbell, SC, Shaw, JA, Nicholson, ML, et al. Rapamycin toxicity in MIN6 cells and rat and human islets is mediated by the inhibition of mTOR complex 2 (mTORC2). Diabetologia. (2012) 55:1355–65. doi: 10.1007/s00125-012-2475-7
47. Bell, E, Cao, X, Moibi, JA, Greene, SR, Young, R, Trucco, M, et al. Rapamycin has a deleterious effect on MIN-6 cells and rat and human islets. Diabetes. (2003) 52:2731–9. doi: 10.2337/diabetes.52.11.2731
48. Ardestani, A, Lupse, B, Kido, Y, Leibowitz, G, and Maedler, K. mTORC1 signaling: a double-edged sword in diabetic β cells. Cell Metab. (2018) 27:314–31. doi: 10.1016/j.cmet.2017.11.004
49. Paty, BW, Harmon, JS, Marsh, CL, and Robertson, RP. Inhibitory effects of immunosuppressive drugs on insulin secretion from HIT-T15 cells and Wistar rat islets. Transplantation. (2002) 73:353–7. doi: 10.1097/00007890-200202150-00007
50. Mao, Z, and Zhang, W. Role of mTOR in glucose and lipid metabolism. Int J Mol Sci. (2018) 19:2043. doi: 10.3390/ijms19072043
51. Deblon, N, Bourgoin, L, Veyrat-Durebex, C, Peyrou, M, Vinciguerra, M, Caillon, A, et al. Chronic mTOR inhibition by rapamycin induces muscle insulin resistance despite weight loss in rats. Br J Pharmacol. (2012) 165:2325–40. doi: 10.1111/j.1476-5381.2011.01716.x
52. Di Paolo, S, Teutonico, A, Leogrande, D, Capobianco, C, and Schena, PF. Chronic inhibition of mammalian target of rapamycin signaling downregulates insulin receptor substrates 1 and 2 and AKT activation: a crossroad between cancer and diabetes? J Am Soc Nephrol. (2006) 17:2236–44. doi: 10.1681/ASN.2006030196
53. Ye, L, Varamini, B, Lamming, DW, Sabatini, DM, and Baur, JA. Rapamycin has a biphasic effect on insulin sensitivity in C2C12 myotubes due to sequential disruption of mTORC1 and mTORC2. Front Genet. (2012) 3:177. doi: 10.3389/fgene.2012.00177
54. Pereira, MJ, Palming, J, Rizell, M, Aureliano, M, Carvalho, E, Svensson, MK, et al. mTOR inhibition with rapamycin causes impaired insulin signalling and glucose uptake in human subcutaneous and omental adipocytes. Mol Cell Endocrinol. (2012) 355:96–105. doi: 10.1016/j.mce.2012.01.024
55. Shimodahira, M, Fujimoto, S, Mukai, E, Nakamura, Y, Nishi, Y, Sasaki, M, et al. Rapamycin impairs metabolism-secretion coupling in rat pancreatic islets by suppressing carbohydrate metabolism. J Endocrinol. (2010) 204:37–46. doi: 10.1677/JOE-09-0216
56. Tanemura, M, Nagano, H, Taniyama, K, Kamiike, W, Mori, M, and Doki, Y. Role of rapamycin-induced autophagy in pancreatic islets. Am J Transplant. (2012) 12:1067. doi: 10.1111/j.1600-6143.2011.03933.x
57. Lamming, DW, Ye, L, Katajisto, P, Goncalves, MD, Saitoh, M, Stevens, DM, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. (2012) 335:1638–43. doi: 10.1126/science.1215135
58. Houde, VP, Brûlé, S, Festuccia, WT, Blanchard, PG, Bellmann, K, Deshaies, Y, et al. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. (2010) 59:1338–48. doi: 10.2337/db09-1324
59. Vergès, B, and Cariou, B. mTOR inhibitors and diabetes. Diabetes Res Clin Pract. (2015) 110:101–8. doi: 10.1016/j.diabres.2015.09.014
60. Kasiske, BL, de Mattos, A, Flechner, SM, Gallon, L, Meier-Kriesche, HU, Weir, MR, et al. Mammalian target of rapamycin inhibitor dyslipidemia in kidney transplant recipients. Am J Transplant. (2008) 8:1384–92. doi: 10.1111/j.1600-6143.2008.02272.x
61. Tavares, MR, Pavan, IC, Amaral, CL, Meneguello, L, Luchessi, AD, and Simabuco, FM. The S6K protein family in health and disease. Life Sci. (2015) 131:1–10. doi: 10.1016/j.lfs.2015.03.001
62. Cho, HJ, Park, J, Lee, HW, Lee, YS, and Kim, JB. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem Biophys Res Commun. (2004) 321:942–8. doi: 10.1016/j.bbrc.2004.07.050
63. Gagnon, A, Lau, S, and Sorisky, A. Rapamycin-sensitive phase of 3T3-L1 preadipocyte differentiation after clonal expansion. J Cell Physiol. (2001) 189:14–22. doi: 10.1002/jcp.1132
64. Yeh, WC, Bierer, BE, and McKnight, SL. Rapamycin inhibits clonal expansion and adipogenic differentiation of 3T3-L1 cells. Proc Natl Acad Sci U S A. (1995) 92:11086–90.
65. Catalán, V, Gómez-Ambrosi, J, Rodríguez, A, Ramírez, B, Andrada, P, Rotellar, F, et al. Expression of S6K1 in human visceral adipose tissue is upregulated in obesity and related to insulin resistance and inflammation. Acta Diabetol. (2015) 52:257–66. doi: 10.1007/s00592-014-0632-9
66. Kim, JE, and Chen, J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes. (2004) 53:2748–56. doi: 10.2337/diabetes.53.11.2748
67. Lastres-Becker, I, Nonis, D, Eich, F, Klinkenberg, M, Gorospe, M, Kötter, P, et al. Mammalian ataxin-2 modulates translation control at the pre-initiation complex via PI3K/mTOR and is induced by starvation. Biochim Biophys Acta. (2016) 1862:1558–69. doi: 10.1016/j.bbadis.2016.05.017
68. Le Bacquer, O, Petroulakis, E, Paglialunga, S, Poulin, F, Richard, D, Cianflone, K, et al. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J Clin Invest. (2007) 117:387–96. doi: 10.1172/JCI29528
69. Kim, K, Pyo, S, and Um, SH. S6 kinase 2 deficiency enhances ketone body production and increases peroxisome proliferator-activated receptor alpha activity in the liver. Hepatology. (2012) 55:1727–37. doi: 10.1002/hep.25537
70. Um, SH, Frigerio, F, Watanabe, M, Picard, F, Joaquin, M, Sticker, M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. (2004) 431:200–5. doi: 10.1038/nature02866
71. Maechler, P, and Wollheim, CB. Mitochondrial function in normal and diabetic beta-cells. Nature. (2001) 414:807–12. doi: 10.1038/414807a
72. Yuan, T, Lupse, B, Maedler, K, and Ardestani, A. mTORC2 signaling: a path for pancreatic β Cell's growth and function. J Mol Biol. (2018) 430:904–18. doi: 10.1016/j.jmb.2018.02.013
73. Kumar, A, Lawrence, JC Jr, Jung, DY, Ko, HJ, Keller, SR, Kim, JK, et al. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes. (2010) 59:1397–406. doi: 10.2337/db09-1061
74. Fang, Y, Westbrook, R, Hill, C, Boparai, RK, Arum, O, Spong, A, et al. Duration of rapamycin treatment has differential effects on metabolism in mice. Cell Metab. (2013) 17:456–62. doi: 10.1016/j.cmet.2013.02.008
75. Duijnhoven, EMV, Boots, JMM, Christiaans, MHL, Wolffenbuttel, BHR, and Hooff, JPV. Influence of tacrolimus on glucose metabolism before and after renal transplantation: a prospective study. J Am Soc Nephrol. (2001) 12:583–8. doi: 10.1681/ASN.V123583
76. Larsen, JL, Bennett, RG, Burkman, T, Ramirez, AL, Yamamoto, S, Gulizia, J, et al. Tacrolimus and sirolimus cause insulin resistance in normal Sprague dawley rats. Transplantation. (2006) 82:466–70. doi: 10.1097/01.tp.0000229384.22217.15
77. Oetjen, E, Baun, D, Beimesche, S, Krause, D, Cierny, I, Blume, R, et al. Inhibition of human insulin gene transcription by the immunosuppressive drugs cyclosporin a and tacrolimus in primary, mature islets of transgenic mice. Mol Pharmacol. (2003) 63:1289–95. doi: 10.1124/mol.63.6.1289
78. Radu, RG, Fujimoto, S, Mukai, E, Takehiro, M, Shimono, D, Nabe, K, et al. Tacrolimus suppresses glucose-induced insulin release from pancreatic islets by reducing glucokinase activity. Am J Physiol Endocrinol Metab. (2005) 288:E365–71. doi: 10.1152/ajpendo.00390.2004
79. Redmon, JB, Olson, LK, Armstrong, MB, Greene, MJ, and Robertson, RP. Effects of tacrolimus (FK506) on human insulin gene expression, insulin mRNA levels, and insulin secretion in HIT-T15 cells. J Clin Invest. (1996) 98:2786–93. doi: 10.1172/JCI119105
80. Chakkera, HA, and Mandarino, LJ. Calcineurin inhibition and new-onset diabetes mellitus after transplantation. Transplantation. (2013) 95:647–52. doi: 10.1097/TP.0b013e31826e592e
81. Soleimanpour, SA, Crutchlow, MF, Ferrari, AM, Raum, JC, Groff, DN, Rankin, MM, et al. Calcineurin signaling regulates human islet {beta}-cell survival. J Biol Chem. (2010) 285:40050–9. doi: 10.1074/jbc.M110.154955
82. Dai, C, Hang, Y, Shostak, A, Poffenberger, G, Hart, N, Prasad, N, et al. Age-dependent human β cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J Clin Invest. (2017) 127:3835–44. doi: 10.1172/JCI91761
83. Heit, JJ, Apelqvist, AA, Gu, X, Winslow, MM, Neilson, JR, Crabtree, GR, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. (2006) 443:345–9. doi: 10.1038/nature05097
84. Johnson, JD, Ao, Z, Ao, P, Li, H, Dai, LJ, He, Z, et al. Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transplant. (2009) 18:833–45. doi: 10.3727/096368909X471198
85. Drachenberg, CB, Klassen, DK, Weir, MR, Wiland, A, Fink, JC, Bartlett, ST, et al. Islet cell damage associated with tacrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation. (1999) 68:396–402. doi: 10.1097/00007890-199908150-00012
86. Ajabnoor, MA, El-Naggar, MM, Elayat, AA, and Abdulrafee, A. Functional and morphological study of cultured pancreatic islets treated with cyclosporine. Life Sci. (2007) 80:345–55. doi: 10.1016/j.lfs.2006.09.034
87. Rostambeigi, N, Lanza, IR, Dzeja, PP, Deeds, MC, Irving, BA, Reddi, HV, et al. Unique cellular and mitochondrial defects mediate FK506-induced islet β-cell dysfunction. Transplantation. (2011) 91:615–23. doi: 10.1097/TP.0b013e3182094a33
88. Vincenti, F, Friman, S, Scheuermann, E, Rostaing, L, Jenssen, T, Campistol, JM, et al. DIRECT (diabetes incidence after renal transplantation: Neoral C monitoring versus tacrolimus) Investigators. Results of an international, randomized trial comparing glucose metabolism disorders and outcome with cyclosporine versus tacrolimus. Am J Transplant. (2007) 7:1506–14. doi: 10.1111/j.1600-6143.2007.01749.x
89. Ciancio, G, Burke, GW, Gaynor, JJ, Ruiz, P, Roth, D, Kupin, W, et al. A randomized long-term trial of tacrolimus/sirolimus versus tacrolimums/mycophenolate versus cyclosporine/sirolimus in renal transplantation: three-year analysis. Transplantation. (2006) 81:845–52. doi: 10.1097/01.tp.0000203894.53714.27
90. Chakkera, HA, Kudva, Y, and Kaplan, B. Calcineurin inhibitors: pharmacologic mechanisms impacting both insulin resistance and insulin secretion leading to glucose dysregulation and diabetes mellitus. Clin Pharmacol Ther. (2017) 101:114–20. doi: 10.1002/cpt.546
91. Ekberg, H, Tedesco-Silva, H, Demirbas, A, Vítko, S, Nashan, B, Gürkan, A, et al. ELITE-Symphony study. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med. (2007) 357:2562–75. doi: 10.1056/NEJMoa067411
92. Ekberg, H, Bernasconi, C, Tedesco-Silva, H, Vítko, S, Hugo, C, Demirbas, A, et al. Calcineurin inhibitor minimization in the Symphony study: observational results 3 years after transplantation. Am J Transplant. (2009) 9:1876–85. doi: 10.1111/j.1600-6143.2009.02726.x
93. Büchler, M, Caillard, S, Barbier, S, Thervet, E, Toupance, O, Mazouz, H, et al. Sirolimus versus cyclosporine in kidney recipients receiving thymoglobulin, mycophenolate mofetil and a 6-month course of steroids. Am J Transplant. (2007) 7:2522–31. doi: 10.1111/j.1600-6143.2007.01976.x
94. Lebranchu, Y, Snanoudj, R, Toupance, O, Weestel, PF, Hurault de Ligny, B, Buchler, M, et al. Five-year results of a randomized trial comparing de novo sirolimus and cyclosporine in renal transplantation: the SPIESSER study. Am J Transplant. (2012) 12:1801–10. doi: 10.1111/j.1600-6143.2012.04036.x
95. Kreis, H, Cisterne, JM, Land, W, Wramner, L, Squifflet, JP, Abramowicz, D, et al. Sirolimus in association with mycophenolate mofetil induction for the prevention of acute graft rejection in renal allograft recipients. Transplantation. (2000) 69:1252–60. doi: 10.1097/00007890-200004150-00009
96. Flechner, SM, Glyda, M, Cockfield, S, Grinyó, J, Legendre, C, Russ, G, et al. The ORION study: comparison of two sirolimus-based regimens versus tacrolimus and mycophenolate mofetil in renal allograft recipients. Am J Transplant. (2011) 11:1633–44. doi: 10.1111/j.1600-6143.2011.03573.x
97. Teutonico, A, Schena, PF, and Di Paolo, S. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol. (2005) 16:3128–35. doi: 10.1681/ASN.2005050487
98. Lebranchu, Y, Thierry, A, Toupance, O, Westeel, PF, Etienne, I, Thervet, E, et al. Efficacy on renal function of early conversion from cyclosporine to sirolimus 3 months after renal transplantation: concept study. Am J Transplant. (2009) 9:1115–23. doi: 10.1111/j.1600-6143.2009.02615.x
99. Budde, K, Becker, T, Arns, W, Sommerer, C, Reinke, P, Eisenberger, U, et al. Everolimus-based, calcineurin-inhibitor-free regimen in recipients of de-novo kidney transplants: an open-label, randomised, controlled trial. Lancet. (2011) 377:837–47. doi: 10.1016/S0140-6736(10)62318-5
100. Schena, FP, Pascoe, MD, Alberu, J, del Carmen Rial, M, Oberbauer, R, Brennan, DC, et al. Conversion from calcineurin inhibitors to sirolimus maintenance therapy in renal allograft recipients: 24-month efficacy and safety results from the CONVERT trial. Transplantation. (2009) 87:233–42. doi: 10.1097/TP.0b013e3181927a41
101. Holdaas, H, Rostaing, L, Serón, D, Cole, E, Chapman, J, Fellstrøm, B, et al. Conversion of long-term kidney transplant recipients from calcineurin inhibitor therapy to everolimus: a randomized, multicenter, 24-month study. Transplantation. (2011) 92:410–8. doi: 10.1097/TP.0b013e318224c12d
102. Veroux, M, Tallarita, T, Corona, D, Sinagra, N, Giaquinta, A, Zerbo, D, et al. Conversion to sirolimus therapy in kidney transplant recipients with new onset diabetes mellitus after transplantation. Clin Dev Immunol. (2013) 2013:496974:1–7. doi: 10.1155/2013/496974
103. Vítko, S, Margreiter, R, Weimar, W, Dantal, J, Kuypers, D, Winkler, M, et al. Three-year efficacy and safety results from a study of everolimus versus mycophenolate mofetil in de novo renal transplant patients. Am J Transplant. (2005) 5:2521–30. doi: 10.1111/j.1600-6143.2005.01063.x
104. Vitko, S, Wlodarczyk, Z, Kyllönen, L, Czajkowski, Z, Margreiter, R, Backman, L, et al. Tacrolimus combined with two different dosages of sirolimus in kidney transplantation: results of a multicenter study. Am J Transplant. (2006) 6:531–8. doi: 10.1111/j.1600-6143.2005.01193.x
105. Van Gurp, E, Bustamante, J, Franco, A, Rostaing, L, Becker, T, Rondeau, E, et al. Comparable renal function at 6 months with tacrolimus combined with fixed-dose Sirolimus or MMF: results of a randomized multicenter trial in renal transplantation. J Transp Secur. (2010) 2010:731426:1–10. doi: 10.1155/2010/731426
106. Guerra, G, Ciancio, G, Gaynor, JJ, Zarak, A, Brown, R, Hanson, L, et al. Randomized trial of immunosuppressive regimens in renal transplantation. J Am Soc Nephrol. (2011) 22:1758–68. doi: 10.1681/ASN.2011010006
107. Gallon, L, Perico, N, Dimitrov, BD, Winoto, J, Remuzzi, G, Leventhal, J, et al. Long-term renal allograft function on a tacrolimus-based, pred-free maintenance immunosuppression comparing sirolimus vs. MMF Am J Transplant. (2006) 6:1617–23. doi: 10.1111/j.1600-6143.2006.01340.x
108. Chhabra, D, Skaro, AI, Leventhal, JR, Dalal, P, Shah, G, Wang, E, et al. Long-term kidney allograft function and survival in prednisone-free regimens: tacrolimus/mycophenolate mofetil versus tacrolimus/sirolimus. Clin J Am Soc Nephrol. (2012) 7:504–12. doi: 10.2215/CJN.06940711
109. Tedesco Silva, H, Jr, CD, Johnston, T, Lackova, E, Mange, K, Panis, C, et al. Everolimus plus reduced-exposure CsA versus mycophenolic acid plus standard-exposure CsA in renal-transplant recipients. Am J Transplant. (2010) 10:1401–13. doi: 10.1111/j.1600-6143.2010.03129.x
110. Berger, SP, Sommerer, C, Witzke, O, Tedesco, H, Chadban, S, Mulgaonkar, S, et al. Two-year outcomes in de novo renal transplant recipients receiving everolimus-facilitated calcineurin inhibitor reduction regimen from the TRANSFORM study. Am J Transplant. (2019) 19:3018–34. doi: 10.1111/ajt.15480
111. Murakami, N, Riella, LV, and Funakoshi, T. Risk of metabolic complications in kidney transplantation after conversion to mTOR inhibitor: a systematic review and meta-analysis. Am J Transplant. (2014) 14:2317–27. doi: 10.1111/ajt.12852
112. Howell, JJ, Hellberg, K, Turner, M, Talbott, G, Kolar, MJ, Ross, DS, et al. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. (2017) 25:463–71. doi: 10.1016/j.cmet.2016.12.009
Keywords: post-transplant diabetes mellitus, kidney transplantation, immunosuppressive drugs, allograft co-morbidities, mTOR-inhibitor
Citation: Granata S, Mercuri S, Troise D, Gesualdo L, Stallone G and Zaza G (2023) mTOR-inhibitors and post-transplant diabetes mellitus: a link still debated in kidney transplantation. Front. Med. 10:1168967. doi: 10.3389/fmed.2023.1168967
Edited by:
David Cucchiari, Hospital Clinic of Barcelona, SpainReviewed by:
Yuan Gui, University of Connecticut, United StatesCopyright © 2023 Granata, Mercuri, Troise, Gesualdo, Stallone and Zaza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gianluigi Zaza, Z2lhbmx1aWdpLnphemFAb3V0bG9vay5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.